

Abstract
The dopamine D3 receptor has been implicated as a potential target for drug development in various complex psychiatric disorders including psychosis, drug dependence, and Parkinson’s disease. In our overall goal to develop molecules with preferential affinity at D3 receptors, we undertook a hybrid drug development approach by combining a known dopamine agonist moiety with a substituted piperazine fragment. In the present study, three compounds produced this way with preferential D3 agonist activity, were tested at D3 receptors with mutations in the agonist binding pocket of three residues known to be important for agonist binding activity. At S192A and T369V, the hybrid agonist compounds produced an interaction profile in [3H]spiperone binding assays similar to that of the parent 5-OH-DPAT and 7-OH-DPAT molecules. The loss of affinity at the S192A mutant was most prominent for 5-OH-DPAT and its corresponding hybrid compound D-237. D110N did not show any radioligand binding. Homology modeling indicated that 7-OH-DPAT-derived D-315 uniquely shares H-bonding with Tyr365 which produced favorable interaction and no loss of H-bonding in the S192A mutant, suggesting that agonist activity may not be solely controlled by residues in the binding pocket.
Keywords: Dopamine D3 receptor, Parkinson’s disease, hybrid dopamine agonists, 7-OH-DPAT, 5-OH-DPAT, homology modeling
1. Introduction
Dopamine (DA) receptors have been targeted for drug development for the treatment of various Central Nervous System (CNS)-related psychiatric illnesses, neurodegeneration, drug abuse, and other disorders[1, 2]. DA receptors belong to a class of G-protein coupled receptors (GPCRs), are found in the CNS and in the periphery [3], and can be classified as being either D1-like or D2-like. The D1 and D5 subtypes belong to the D1-like class, and the D2, D3, and D4 subtypes are D2-like receptors. These classifications have been made on the basis of receptor pharmacology and function. Both D1-like and D2-like DA receptors share the same effector molecule, adenylate cyclase. Upon receptor activation, D1-like receptors activate adenylate cyclase, whereas D2-like receptors inhibit it [4].
We employed a novel hybrid structure approach by combining known DA agonist moieties with the N-arylpiperazine moiety derived from known D3 antagonists (Figure-1) [5, 6]. This design was based on the assumption that the aminotetralin moiety would interact with the agonist binding site in the DA receptor and the arylpiperazine fragment would interact with the accessory binding site residues in the D3 receptor to impart selectivity. Based on this hybrid approach we were able to develop highly potent DA D2/D3 agonists, with some compounds being very selective for the D3 receptor [6–8]. Some of the representative molecules are shown in Figure 1. Our results prompted the proposal of a unique pharmacophore structure for hybrid derivatives consisting of three pharmacophoric centers.[9]
Fig. 1.
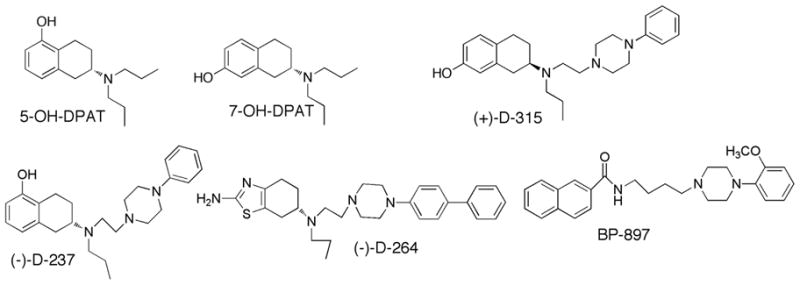
Structures of D3 dopamine receptor ligands.
In order to delineate binding of agonists at a molecular level to D2 and D3 subtype receptors, site-directed mutagenesis studies have been conducted [10–14]. These studies, concentrating on potential binding residues in trans-membrane (TM) domains 3, 5, and 7 for receptor activation, found one aspartate residue in TM3 critical for activation by agonists in both D2 and D3 receptors; depending on agonist and receptor subtype, three serine residues in TM5 played a role; and a threonine in TM7 of D3 was important. Specifically for D3, Asp110 in TM3 interacted with the basic N-atom in DA and aminotetralin molecules, whereas Ser192 in TM5 appeared to be crucial for receptor interaction and activation by a ligand through hydrogen bonding with its hydroxyl group. Another D3 mutation in TM7, T369V, significantly enhanced binding affinity for (+)-7-OH-DPAT without significantly changing DA binding [12]. The increase of binding of (+)-7-OH-DPAT to this mutant was attributed to an increase in hydrophobicity as a result of replacing threonine by valine. This observation is supported by a study modeling the D3 receptor based on the rhodopsin structure determined at 2.8 Å resolution [15]. Recently, homology models of D2, D3, and D4 receptors were developed based on the more closely related crystal structure of the β2-adrenergic receptor.[16] Docking in these models of compounds in which phenylpiperazines are linked to 7α-azaindole again pointed to the crucial role of binding of the basic N-atom in the linker to the aspartate in TM3 (Asp110 in D3). The present study focused on the role of three amino acids namely Asp110, Ser192 and Thr369 in the agonist binding sites on the D3 receptor as discussed above, and their proposed interactions with our D3 preferring hybrid agonists, D-315 (related to 7-OH-DPAT), D237 (related to 5-OH-DPAT), and D-264 (bioisosteric version). In this study, three mutants of the D3 receptor were prepared, D110N, S192A, and T369V, and studied for the ability of ligands to inhibit [3H]spiperone binding. Finally, the mode of binding of these compounds were analyzed by docking the compounds to homology models of D3 wild-type (WT) and mutant forms that were modeled based on the β2-adrenergic receptor.
2. Materials and methods
2.1. Materials
Sources of commercially available chemicals used in this study are listed in the method descriptions in parentheses in sections 2.2 through 2.4 below. If sources are not listed, chemicals were from Fisher Scientific, Pittsburgh, PA. Three compounds were synthesized by us; the synthetic routes and compound characterization are described in detail for D-237 ((S)-6-((2-(4-phenylpiperazin-1-yl)ethyl)(propyl)amino)-5,6,7,8-tetrahydronaphthalen-1-ol) in ref. 7, for D-264 ((S)-N 6-(2-(4-([1,1′-biphenyl]-4-yl)piperazin-1-yl)ethyl)-N 6-propyl-4,5,6,7-tetrahydrobenzo[d]thiazole-2,6-diamine) in ref. 8, and for D-315 ((R )-7-((2-(4-phenylpiperazin-1-yl)ethyl)(propyl)amino)-5,6,7,8-tetrahydronaphthalen-2-ol) in ref. 9.
2.2. Site-directed mutagenesis
A clone (pCMV6:DRD3) containing a WT human DA receptor D3 cDNA (NM_000796) was purchased from OriGene Technologies (Rockville, MD, USA). Site-directed mutagenesis was performed to generate DRD3 D110N, S192A, and T369V mutants. Two complementary oligonucleotides incorporating the desired point mutations were synthesized and purchased from Integrated DNA Technologies (Coralville, IA, USA). The following primers were used: D110N, 5′-GTT TTT GTC ACC CTG AAT GTC ATG ATG TGT ACA GCC-3′; S192A, 5′-GAT TTT GTC ATC TAC GCT TCA GTG GTG TCC TTC TAC-3′; T369V, 5′-GAG CTT TAC AGT GCC ACG GTA TGG CTG GGC TAC GTG-3′. Mutagenesis was performed with the QuikChange mutagenesis kit (Stratagene, Cedar Creek, TX, USA) according to the manufacturer’s protocol. In brief, 50 ng of double-stranded DNA template (pCMV6:DRD3) was mixed with a primer and its complementary primer (0.4 μM each), 0.4 mM dNTPs, reaction buffer, and 2 units of Pfu DNA polymerase in the final 25 μl reaction mixture. The mixture was amplified by polymerase chain reaction (PCR). Initially the reaction mix was incubated at 95 °C for 1 min. Cycles were as follows: denaturation for 30 s at 95 °C, annealing for 1 min at 55 °C, elongation for 14 min at 68 °C for 16 cycles. PCR products were digested with 10 units of DpnI to remove the parental strands which were not mutagenized. The digested PCR products were transformed into Escherichia coli XL-1-blue cells by the heat shock method. Mutagenesis was verified by DNA sequence analysis (New York University, Skirball Institue of Biomolecular Medicine, DNA Sequencing Facility, NY, USA).
2.3. Assessment of D3 binding affinity
Human embryonic kidney cells (HEK-293, ATCC CRL 1573) were grown in Dulbecco’s modified Eagle’s medium (Sigma, St. Louis, MO) supplemented with 10% bovine calf serum and 2 mM l-glutamine at 37°C and 5% CO2. For transient expression, cells were seeded into100-mm petri dishes and allowed to grow to 80% confluence. Transfection was initiated by the addition of 16 μg of the wild-type or mutant pCMV6-DRD3 construct along with 40 μl Lipofectamine 2000 (Invitrogen, Carlsbad, CA) in 2 ml Opti-MEM® I reduced serum medium (Invitrogen, Carlsbad, CA). Receptor binding assays were performed approximately 24 hours after transfection. Transiently transfected cells were the source for preparation of crude membranes as described before [6]. Binding assay conditions conformed to those described as high-protein level in our recent work [17]. In a total reaction volume of 0.2 ml, [3H]spiperone (2 – 4 nM, final concentration, 15 Ci/mmol, Perkin Elmer, Waltham, MA) and crude membranes (~ 40μg of protein) were incubated together with test compounds in assay buffer (50mM Tris–HCl, pH 7.4, with 0.9% NaCl, 0.025% ascorbic acid and 0.001% bovine serum albumin) for 1 h at 30 °C in 96-well plates on a temperature-controlled shaker (Boekel scientific, Feasterville, PA). (+)-Butaclamol (2μM) was used as non-specific binding definition. Assays were terminated by filtering through an ice-cold glass fiber filtermat (PerkinElmer, Waltham, MA) in the Brandel-96 cell harvester, followed by liquid scintillation counting of radioactivity trapped onto the filters [17]. Test compounds were dissolved in pure dimethylsulfoxide (DMSO), and diluted down in 10% DMSO stocks, with a final DMSO concentration in the assay of 1%, which, by itself, did not inhibit binding. IC50 values were fitted with Origin program and converted to inhibition constants (Ki) by the Cheng–Prusoff equation [18].
2.4. Molecular modeling and simulations
The sequence of the human dopamine D3 receptor was aligned with that of the beta adrenergic receptor using the sequence alignment program ClustalW [19]. All the transmembrane regions and IC1, IC2, and EC1 loop regions aligned well with the corresponding regions of the beta adrenergic receptor (Fig. 2). However, the extracellular loop (ECL) 2 and intracellular loop (ICL) 3 in the beta adrenergic receptor are longer than the corresponding loops of the D3 receptor. These loops were modeled subsequent to imposing the B2AR structure onto D3 only for the aligned regions between D3 and B2AR (shown in Fig. 2). Structural models of the dopamine D3 receptor and the mutants S192A and T369V were derived based on the crystal structure of the beta adrenergic receptor (PDB code: 2RH1) [20] as a template with the homology modeling program Modeller (ver 9.4) [21]. For the wild type and the two mutant models, hundred starting models were built with modeller’s objective function. The structures were then clustered using MOE (ver 2008.10, http://www.chemcomp.com) and a representative member from the highly populated cluster was chosen for further studies. These best ranking conformations were subjected to 5000 energy minimization steps to relieve steric and geometric strains using NAMD (ver.2) [22] with charmm force field parameters [23]. Further refinement was performed using molecular dynamics (MD) simulations with periodic boundary conditions, under constant pressure and temperature (1.01325 bar at 310 K) using the Nosé – Hoover Langevin piston algorithm. The particle-mesh Ewald algorithm was used for calculation of electrostatics. The integration time step was 1 fs and structure recording interval was every 2000 steps (=2.0 ps in MD). The models were subjected to 2ns long MD simulations using Charmm force field adopted in NAMD program. The first half of the MD simulation was performed with harmonic constraints of K=1000kj/mol/nm on all heavy atoms which were relaxed during the next half of the simulation. The final 500ps of the production run was obtained without any constraints. The wild type and mutant models were then subject to normal mode analysis to compute the most flexible conformations obtained by perturbations along the low frequency normal modes. The elastic network model was used to compute the models by perturbing the system along the chosen low frequency vibrational mode [24]. The elNemo webserver (http://igs-server.cnrs-mrs.fr/elnemo/start.html) was used for computation of these models with the default values for displacement vectors. The first five low frequency modes were considered for each model and all the resulting conformations were used for docking studies. Ligands D237, D264, D315, D323 and 7-OH-DPAT were built using builder module and their structures were minimized using Merck force field (MMFF94) [25] and Gasteiger charges [26] as adopted in MOE (Ver 2008.10). Further conformational analysis for each of the five compounds was performed using stochastic search method adopted in MOE.
Fig. 2.

Pairwise sequence alignment of the dopamine D3 receptor (DRD3-HUMAN) and beta adrenergic receptor (BADRE-2RH1). Transmembrane (TM) forming residues are shown in red (marked as TM1 through 8). Sequence similarity is shown with: or. and identity is shown with *.
Conformations are generated by random rotation of the bonds (in this study, the ring bonds were excluded) using a bias of ~30° for dihedral angle rotation. In addition a Cartesian perturbation of 0.4Ǻ was also applied and the search was terminated when the number of failures reached 1000 in a row. Only those conformations that succeeded these stringent criteria were stored and clustered based on their energy. Representative member from the most populated cluster was chosen for further optimization using the AM1 semi-empirical quantum chemical method adopted in MOE. The optimized conformations were used for further docking experiments. Compounds D237, D264, D315, D323 and 7-OH-DPAT were docked to all five conformations of each model (wt, S192A, T369V) using docking software Gold (ver 4.1) [27]. Twenty independent runs were performed for each ligand and all the docked complexes (3*5*20 = 300) were scored using Goldscore [27], chemscore [28] and customized scoring scheme [29]. The customized scoring scheme was designed to identify correct binding modes of the ligand by scoring the known interactions such as the salt bridge with Asp110 and aromatic interactions with the aromatic residues from TM5, TM6 and TM7 positively and penalizing reversed binding modes. Finally the docked complexes were ranked based on a consensus scoring scheme described in detail in Kortagere et al. [29]. The consensus docking score of an active compound j with i conformations was computed as
| (equation 1) |
The values of Si,j was computed for each of the five receptor conformations for the wt, S192A and T369V models and sij is the original goldscore for the compound i in its jth conformation and wij is the custom score for compound i in jth conformation. The best ranking conformation was chosen based on the final consensus score that produced the maximum value for Si,j..
3. Results and Discussion
3.1. Ligand interactions with WT and mutant hD3 receptor
Mutation of Asp110 to Asn abolished the capability of the hD3 receptor to bind [3H]spiperone (data collected in 3 independent experiments, Table 1). This correlates with the lack of agonist or antagonist binding to the hD2 receptors in which the corresponding Asp114 was mutated to either Asn or Gly [14]. Mutation of Ser192 to Ala (S192A) in hD3, while not changing the Kd of [3H]spiperone binding, appreciably decreased the affinity (increased Ki) of all agonists 3- to 18-fold (Table 1). This suggests a direct role for Ser192 in agonist binding, borne out in homology modeling described below for each compound separately (sections 3.3–3.7). In contrast, Thr369 appeared much less important for agonist interaction as judged from little or no effect of the T369V mutation on inhibition by agonists of [3H]spiperone binding (Table 1). This mutation also did not affect the Kd of antagonist binding. The Ki observed here for D-237 in interacting with hD3 is somewhat higher than our previously published value for its interaction with rD3 [7], possibly due to the species difference or the different batch of radioligand.
Table 1.
Affinity for human D3 receptors transiently expressed in HEK-293 cells, as measured by inhibition of [3H]spiperone binding.
| Kd (spiperone) or Ki (other compounds) (nM) in inhibiting [3H]spiperone binding | ||||
|---|---|---|---|---|
| Compound | WT | S192A | T369V | D110N |
| Spiperone | 0.329±0.131 | 0.332±0.136 | 0.203±0.093 | NA |
| 7-OH-DPAT | 5.64 ± 0.50 | 28.1± 2.7* | 2.40 ± 0.08* | NA |
| D315 | 0.503 ± 0.082 | 3.16 ± 0.55* | 0.310 ± 0.064 | NA |
| 5-OH-DPAT (D323) | 5.94 ± 1.80 | 110 ± 32* | 5.29 ± 1.54 | NA |
| D237 | 3.65 ± 0.32 | 40.0 ± 2.2* | 3.28 ± 0.72 | NA |
| D264 | 5.81 ± 1.22 | 15.5 ± 2.0* | 9.14 ± 1.21 | NA |
NA; Not Active: [3H]spiperone binding was not detectable in 3 independent experiments.
Data are expressed as Mean ± S.E.M. for 3 to 4 independent experiments, each performed in triplicate.
P<0.05 compared with WT (one-way ANOVA followed by Dunnett’s multiple comparisons test)
3.2. Overall findings in homology docking studies
The WT and the mutant D3 receptor were modeled based on the crystal structure of the beta adrenergic receptor (B2AR). A structural superpositioning of the backbone atoms from WT and mutant models with the B2AR resulted in a root mean square deviation of 2.6 and 2.8Ǻ respectively (Fig. 3A). The largest variation in the structures was observed in loop regions and in the binding site residues. The ECL2 folds over the transmembrane region in the B2AR and is significantly different in structure from that of rhodopsin [30]. Certain residues from ECL2 are also known to interact with the ligand in other biogenic amine receptors [31, 32]. In our models for WT and mutant forms, the ECL2 folds over the ligand binding domain and interacts with the ligands Fig. 3A). D315 binds in a slightly modified conformation in the WT and mutant forms (Fig. 3B). This could be due to the flexibility of the extended ligand structure versus the native 7-OH-DPAT and also could be due to insufficient sampling of the receptor-ligand complex. Long time scale MD simulations are needed to derive effective conclusions about the nature of binding of the ligand in WT and mutant models.
Fig. 3.

Structural superposition of hD3 wildtype (green) and hD3 S192A mutant (purple) onto the beta adrenergic receptor crystal structure (red). Receptors are rendered in ribbons. The ligand D315 is docked to the wildtype and S192A mutant and is rendered in ball and stick model and colored atom type (C- cyan, O-red and N-blue). A) Mode parallel to the membrane. B) Top view from the membrane (to show the position of the ligand, the loops regions were sectioned out).
In many cases, analysis of the docking modes of the present agonists to WT type receptor maintained conserved interactions such as the salt bridge with Asp110 and aromatic stacking interactions with His349, and hydrogen bonded interactions with S192 and Y365 (Fig. 4). Mutation of Asp110 also led to loss of binding of the antagonist [3H]spiperone consistent with reported lack of antagonist/agonist binding following mutation of the corresponding Asp residue in hD2.[14] Interactions with T369 were found to be mainly between the main chain carbon atoms of T369 with the hydrophobic groups of the ligands. Hence mutation of T369 to Val had no significant effect on the binding affinity of the ligands except in the case of 7-OH-DPAT which showed a modest gain (2-fold) in potency. The latter gain was less than the 9-fold gain reported by Lundstrom et al. [12], and the present modest effect is in line with our homology modeling results (see section 3.8 below). It was proposed by Varaday et. al. [15] that a 16-fold gain in affinity (in fact it was 9-fold [12]) might be due to the close proximity of the methyl group of valine to the propyl group of 7-OH-DPAT resulting in production of hydrophobic interaction, but the difference between their homology modeling and ours lies in it being based on the then available rhodopsin crystal versus the more recently published β2-adrenergic receptor which more closely resembles the mammalian hD3 receptor. In addition, our binding studies did not uncover the pronounced effect of T369V mutation as reported by Lundstrom et al. [12].
Fig. 4.

Schematic description of the binding mode and interactions of A) hD3 wild type with 7-OH-DPAT, B) hD3 S192A mutant with 7-OH-DPAT, C) hD3 wildtype with 5-OH-DPAT (D323), D) hD3 S192A mutant with 5-OH-DPAT (D323), E) hD3 wildtype with D-237, F) hD3 S192A mutant with D237, G) hD3 wildtype with D264 and H) hD3 S192A mutant with D264. The legend describes the color codes for the schematics. The figures were generated using the LIGX module of MOE program.
3.3. Docking of 7-OHDPAT and effect of S192A mutation
The agonist 7-OH-DPAT binds to the WT hD3 receptor with a Ki of 5.6 nM. It maintains a conserved salt bridge with Asp110 and aromatic stacking interactions with His349 and a hydrogen bond with Ser192 (Fig. 4A). In the S192A mutated form the compound reorients itself to form hydrogen bond with Ser196, while maintaining salt bridge interaction with Asp110 (Fig. 4B). However, due to the reorientation, the ligand loses the favorable pi-stacking interaction with His349 (Fig. 4B). In the binding experiments, a loss of approximately 5-fold in activity was observed (Table 1).
3.4. Docking of 5-OH-DPAT and effect of S192A mutation
5-OH-DPAT has favorable interactions with His349 and Ser192, and maintains the critical Asp110 salt bridge (Fig. 4C). In the S192A mutant, there is a loss of hydrogen bond with the hydroxyl group that is not compensated by other residues and the reorientation also leads to uncompensated loss of stacking interactions with His349 (Fig. 4D). The binding experiments indicate an 18-fold decrease in activity (Table 1).
3.5. Docking of D-237 and effect of S192A mutation
D-237 which is a 5-hydroxy derived derivative, exhibited a similar profile as 5-OH-DPAT with the rest of the molecule adding marginally to its affinity for the D3 receptor compared to 5-OH-DPAT. However, we see a bifurcated salt bridge with the piperazine nitrogen and the protonated tertiary amine (Fig. 4E). Similar to 5-OH-DPAT, there is an uncompensated loss of a favorable hydrogen bonded interaction in the S192A mutant receptor (Fig. 4F). However, both Ser196 and His349 are positioned at 3.9 and 3.7Ǻ respectively and may contribute to some favorable electrostatic interactions, but not hydrogen bond (Fig. 4F). A loss of 10-fold in binding activity was observed (Table 1).
3.6. Docking of D-264 and effect of S192A mutation
D264 has a similar activity profile in the WT receptor to that of 7-OH-DPAT with favorable interactions with aromatic cluster from TM5 and TM6 namely His349 and Phe162 (Fig. 4G). There is a bifurcated salt bridge with piperazine nitrogen and the tertiary protonated nitrogen with Asp110 of the receptor. Similarly, a bifurcated hydrogen bond interaction is seen with the nitrogen atoms of the thiazolidium ring with Ser192. In the S192A mutant forms, the ligand loses the hydrogen bond with S192 but maintains the same with Thr369 (Fig. 4H). Overall, its binding activity shows a minor loss of 2-fold (Table 1).
3.7. Docking of D-315 and effect of S192A mutation
D-315 which is a 7-hydroxy derived compound, exhibited a 10-fold higher activity compared to 7-OH-DPAT at WT (Table 1). Although D-315 was designed based on 7-OH-DPAT with a similar structural component of a tetralin agonist binding moiety in the molecule, the extended structure seems to have an effect on the overall conformation of the ligand. This seems to be facilitated by the favorable positioning of the OH group that has favorable hydrogen bonded interactions with Tyr365 and aromatic – pi interactions with His349 and conserved salt bridge with Asp110 (Fig. 5a). In a small fraction of the conformations, the OH group had hydrogen bonded interactions with Ser193 (not shown). Ser192 is positioned at 3.6Ǻ from the ligand and has polar interactions with tertiary nitrogen. In the S192A mutant receptor, the ligand maintains its hydrogen bonded interactions with Tyr365and the salt bridge with Asp110 (Fig. 5b). Although the ligand retains the hydrogen bond interaction it loses the stacking interactions with His349 albeit some hydrophobic interactions with His349 which could explain a six fold loss in activity (Table 1).
Fig. 5.

Schematic comparison between D315 docked to A) wild type dopamine D3 receptor and B) S192A mutant dopamine D3 receptor. The color code for the schematic is the same as in Fig. 4. The figure was generated using LIGX module of MOE program.
3.8. Effect of T369V and S192A mutation
In contrast to S192A mutation, T369V mutation had little or no impact on agonist binding to hD3. For Ser192, comparison of docking studies between WT and S192A mutant receptor shows that in addition to loss of hydrogen bonding interactions with Ser192 when mutated to Ala, the receptor also seem to undergo conformational changes that affect other residues in the binding pocket such as His349 that contribute significantly to the agonist activity of the ligands [12]. In the case of D-315, our model suggests that the hydroxyl group tends to form hydrogen bonded interactions with Tyr365 in the wild type and S192A mutant receptor (Fig. 5). This could suggest that the agonist activity of the ligand may not be solely controlled by serine residues in the binding pocket and that other hydrogen bonding partners could provide such interactions. This hypothesis will be further confirmed by a follow-up future mutant study.
Conclusion
In this report, we have carried out site-directed mutagenesis studies with the DA D3 receptor to study molecular interactions of D3 preferring hybrid agonist molecules at the receptor level. Docking of molecules in both WT and mutant D3 receptors was carried out by homology modeling, and binding affinity of the compounds was assessed by radioligand receptor binding studies. Our studies demonstrated that the mutation of Ser192 to Ala led to a loss of activity for all agonist compounds with the most pronounced loss occurring in 5-hydroxy derived molecules D-237 and 5-OH-DPAT. In this regard, S192 in the D3 receptor has been implicated in critical H-bonding with the catechol moiety in the DA molecule [11]. On the other hand, T369V mutation resulted in little or no change in binding affinity compared with WT except in the case of 7-OH-DPAT where it produced a two-fold increase of binding affinity. This is contrary to an earlier reported finding where a much higher affinity was found in T369V than in WT for 7-OH-DPAT [12]. The mutant D110N did not bind [3H]spiperone at all. Homology modeling study indicated that 7-OH-DPAT-derived D-315 might uniquely share H-bonding with Tyr365 which produced favorable interaction and no loss of H-bonding in the S192A mutant. Overall, the results indicate that our hybrid agonist molecules, similar to the original 7-OH-DPAT and 5-OH-DPAT molecules, display interaction profiles with the residues in the D3 receptor previously shown to be critical for agonist binding. Further site-directed mutagenesis studies will be carried out in the future to map out more interaction sites beyond the agonist binding pocket for these hybrid D3 preferring agonist compounds.
Acknowledgments
Role of the funding source
This research was supported by the National Institutes of Health (NIH), National Institute of Neurological Disorders and Stroke (NINDS), grant R01 NS047198. In this study, NIH or NINDS had no role in in the collection, analysis, and interpretation of data; in the writing of the report; and in the decision to submit the paper for publication. NIH reviewed the grant application in a study section that commented on study design.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Emilien G, Maloteaux J-M, Geurts M, Hoogenberg K, Cragg S. Dopamine receptors--physiological understanding to therapeutic intervention potential. Pharmacology & Therapeutics. 1999;84:133–56. doi: 10.1016/s0163-7258(99)00029-7. [DOI] [PubMed] [Google Scholar]
- 2.Missale C, Nash SR, Robinson SW, Jaber M, Caron MG. Dopamine receptors: from structure to function. Physiol Rev. 1998;78:189–225. doi: 10.1152/physrev.1998.78.1.189. [DOI] [PubMed] [Google Scholar]
- 3.D’Ischia M, Prota G. Biosynthesis, Structure, and Function of Neuromelanin and its Relation to Parkinson’s Disease: A Critical Update. Pigment Cell Research. 1997;10:370–6. doi: 10.1111/j.1600-0749.1997.tb00694.x. [DOI] [PubMed] [Google Scholar]
- 4.Gurevich EV, Joyce JN. Distribution of Dopamine D3 Receptor Expressing Neurons in the Human Forebrain: Comparison with D2 Receptor Expressing Neurons. Neuropsychopharmacology. 1999;20:60–80. doi: 10.1016/S0893-133X(98)00066-9. [DOI] [PubMed] [Google Scholar]
- 5.Dutta AK, Fei X-S, Reith MEA. A novel series of hybrid compounds derived by combining 2-aminotetralin and piperazine fragments: binding activity at D2 and D3 receptors. Bioorganic & Medicinal Chemistry Letters. 2002;12:619–22. doi: 10.1016/s0960-894x(01)00820-4. [DOI] [PubMed] [Google Scholar]
- 6.Dutta AK, Venkataraman SK, Fei X-S, Kolhatkar R, Zhang S, Reith MEA. Synthesis and biological characterization of novel hybrid 7-{[2-(4-phenyl-piperazin-1-yl)-ethyl]-propyl-amino}-5,6,7,8-tetrahydro-naphthalen-2-ol and their heterocyclic bioisosteric analogues for dopamine D2 and D3 receptors. Bioorganic & Medicinal Chemistry. 2004;12:4361–73. doi: 10.1016/j.bmc.2004.06.019. [DOI] [PubMed] [Google Scholar]
- 7.Biswas S, Zhang S, Fernandez F, Ghosh B, Zhen J, Kuzhikandathil E, et al. Further Structure– Activity Relationships Study of Hybrid 7-{[2-(4-Phenylpiperazin-1-yl)ethyl]propylamino}-5,6,7,8-tetrahydronaphthalen-2-ol Analogues: Identification of a High-Affinity D3-Preferring Agonist with Potent in Vivo Activity with Long Duration of Action. J Med Chem. 2008;51:101–17. doi: 10.1021/jm070860r. [DOI] [PubMed] [Google Scholar]
- 8.Biswas S, Hazeldine S, Ghosh B, Parrington I, Kuzhikandathil E, Reith ME, et al. Bioisosteric Heterocyclic Versions of 7-{[2-(4-Phenyl-piperazin-1-yl)ethyl]propylamino}-5,6,7,8-tetrahydronaphth alen-2-ol: Identification of Highly Potent and Selective Agonists for Dopamine D3 Receptor with Potent in Vivo Activity. J Med Chem. 2008;51:3005–19. doi: 10.1021/jm701524h. [DOI] [PubMed] [Google Scholar]
- 9.Brown DA, Kharkar PS, Parrington I, Reith ME, Dutta AK. Structurally constrained hybrid derivatives containing octahydrobenzo[g or f]quinoline moieties for dopamine D2 and D3 receptors: binding characterization at D2/D3 receptors and elucidation of a pharmacophore model. J Med Chem. 2008;51:7806–19. doi: 10.1021/jm8008629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cox BA, Henningsen RA, Spanoyannis A, Neve RL, Neve KA. Contributions of Conserved Serine Residues to the Interactions of Ligands with Dopamine D2 Receptors. Journal of Neurochemistry. 1992;59:627–35. doi: 10.1111/j.1471-4159.1992.tb09416.x. [DOI] [PubMed] [Google Scholar]
- 11.Sartania N, Strange PG. Role of conserved serine residues in the interaction of agonists with D3 dopamine receptors. J Neurochem. 1999;72:2621–4. doi: 10.1046/j.1471-4159.1999.0722621.x. [DOI] [PubMed] [Google Scholar]
- 12.Lundstrom K, Turpin MP, Large C, Robertson G, Thomas P, Lewell XQ. Mapping of dopamine D3 receptor binding site by pharmacological characterization of mutants expressed in CHO cells with the Semliki Forest virus system. J Recept Signal Transduct Res. 1998;18:133–50. doi: 10.3109/10799899809047741. [DOI] [PubMed] [Google Scholar]
- 13.Wiens BL, Nelson CS, Neve KA. Contribution of serine residues to constitutive and agonist-induced signaling via the D2S dopamine receptor: evidence for multiple, agonist-specific active conformations. Mol Pharmacol. 1998;54:435–44. doi: 10.1124/mol.54.2.435. [DOI] [PubMed] [Google Scholar]
- 14.Mansour A, Meng F, Meador-Woodruff JH, Taylor LP, Civelli O, Akil H. Site-directed mutagenesis of the human dopamine D2 receptor. Eur J Pharmacol. 1992;227:205–14. doi: 10.1016/0922-4106(92)90129-j. [DOI] [PubMed] [Google Scholar]
- 15.Varady J, Wu X, Fang X, Min J, Hu Z, Levant B, et al. Molecular modeling of the three-dimensional structure of dopamine 3 (D3) subtype receptor: discovery of novel and potent D3 ligands through a hybrid pharmacophore- and structure-based database searching approach. J Med Chem. 2003;46:4377–92. doi: 10.1021/jm030085p. [DOI] [PubMed] [Google Scholar]
- 16.Ehrlich K, Gotz A, Bollinger S, Tschammer N, Bettinetti L, Harterich S, et al. Dopamine D2, D3, and D4 selective phenylpiperazines as molecular probes to explore the origins of subtype specific receptor binding. J Med Chem. 2009;52:4923–35. doi: 10.1021/jm900690y. [DOI] [PubMed] [Google Scholar]
- 17.Zhen J, Antonio T, Dutta AK, Reith ME. Concentration of receptor and ligand revisited in a modified receptor binding protocol for high-affinity radioligands: [3H]Spiperone binding to D2 and D3 dopamine receptors. J Neurosci Methods. 188:32–8. doi: 10.1016/j.jneumeth.2010.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheng Y, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochemical pharmacology. 1973;22:3099–108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 19.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–80. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, et al. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science. 2007;318:1258–65. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sanchez R, Sali A. Evaluation of comparative protein structure modeling by MODELLER-3. Proteins. 1997;(Suppl 1):50–8. doi: 10.1002/(sici)1097-0134(1997)1+<50::aid-prot8>3.3.co;2-w. [DOI] [PubMed] [Google Scholar]
- 22.Kalé LS, Bhandarkar R, Brunner M, Gursoy R, Krawetz A, Phillips N, Shinozaki J, Varadarajan A, Schulten K. NAMD2: Greater Scalability for Parallel Molecular Dynamics. Journal of Computational Physics. 1999;151:283–312. [Google Scholar]
- 23.Brooks BRB, Olafson RE, States BD, Swaminathan DJS, Karplus M. CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J Comput Chem. 1983;4:187–217. [Google Scholar]
- 24.Tama F, Sanejouand YH. Conformational change of proteins arising from normal mode calculations. Protein Eng. 2001;14:1–6. doi: 10.1093/protein/14.1.1. [DOI] [PubMed] [Google Scholar]
- 25.Halgren TA. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. journal of computational chemistry. 1996;17:490–519. [Google Scholar]
- 26.Marsili JGaM. Iterative partial equalization of orbital electronegativity a rapid access to atomic charges. Tetrahedron. 1980;36:3219–28. [Google Scholar]
- 27.Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and validation of a genetic algorithm for flexible docking. J Mol Biol. 1997;267:727–48. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 28.Eldridge MD, Murray CW, Auton TR, Paolini GV, Mee RP. Empirical scoring functions: I. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. J Comput Aided Mol Des. 1997;11:425–45. doi: 10.1023/a:1007996124545. [DOI] [PubMed] [Google Scholar]
- 29.Kortagere S, Welsh WJ. Development and application of hybrid structure based method for efficient screening of ligands binding to G-protein coupled receptors. J Comput Aided Mol Des. 2006;20:789–802. doi: 10.1007/s10822-006-9077-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Costanzi S. On the applicability of GPCR homology models to computer-aided drug discovery: a comparison between in silico and crystal structures of the beta2-adrenergic receptor. J Med Chem. 2008;51:2907–14. doi: 10.1021/jm800044k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kortagere S, Roy A, Mehler EL. Ab initio computational modeling of long loops in G-protein coupled receptors. J Comput Aided Mol Des. 2006;20:427–36. doi: 10.1007/s10822-006-9056-0. [DOI] [PubMed] [Google Scholar]
- 32.Shi L, Javitch JA. The second extracellular loop of the dopamine D2 receptor lines the binding-site crevice. Proc Natl Acad Sci U S A. 2004;101:440–5. doi: 10.1073/pnas.2237265100. [DOI] [PMC free article] [PubMed] [Google Scholar]