

Abstract
Platelets are small, anucleate, discoid shaped blood cells that play a fundamental role in hemostasis. Platelets contain a large number of biologically active molecules within cytoplasmic granules that are critical to normal platelet function. Because platelets circulate in blood through out the body, release biological molecules and mediators on demand, and participate in hemostasis as well as many other pathophysiologic processes, targeting expression of proteins of interest to platelets and utilizing platelets as delivery systems for disease treatment would be a logical approach. This paper reviews the genetic therapy for inherited bleeding disorders utilizing platelets as delivery system, with a particular focus on platelet-derived FVIII for hemophilia A treatment.
Keywords: Gene therapy, Platelet-specific promoter, Platelet, Hemostasis
1. Introduction
Genetic manipulation of cells to produce proteins of interest for disease treatment is an attractive research area. Platelets, which are anucleate blood cells produced by megakaryocytes, are replete with secretory granules. These granules accumulate their stored contents from both megakaryocyte synthesis and endocytosis of plasma proteins. When platelets are activated, a large number of bioactive proteins are released from their granules by exocytosis participating in myriad physiologic and pathologic processes including hemostasis, thrombosis, inflammation, atherosclerosis, wound healing, antimicrobial defense, and malignancy [1]. Given platelets innate storage, trafficking, and release capacities, taking advantage of them as delivery systems for proteins of interest would be a logical and reasonable approach for disease treatment. The feasibility and efficacy of this novel approach, previously carried out in experimental animals, will be reviewed in this article.
2. Overview of platelet production and function
In hematopoietic stem cell linage commitment, two types of blood cell lines are derived: the lymphoid linage, which includes lymphocytes, and the myeloid linage, which includes myelocytes, red cells, and megakaryocytes. Pluripotent hematopoietic stem cells produce a progenitor committed to megakaryocyte differentiation that is still capable of mitotic division. Eventually proliferating diploid megakaryocyte progenitors lose their capacity to divide and enter endomitosis. During the endomitotic phase, cells still maintain their ability to replicate DNA and cytoplasmic maturation continues, but neither cytoplasm nor nucleus divides, resulting in cells with ploidy ranges from 8 N to 128 N in a single, highly lobated nucleus and a cytoplasmic mass containing a complex of internal membrane systems, granules, and organelles [2-6]. When the megakaryocyte matures, the polyploid nucleus becomes horseshoe-shaped and the cytoplasm expands, resulting in a low nuclear to cytoplasmic ratio, and platelet organelles and the demarcation membrane system are robustly amplified [7-9]. The cytoplasmic mass from differentiated megakaryocytes forms proplatelets and eventually gives rise to circulating platelets [10-12].
Platelets are involved in the cellular mechanisms of primary hemostasis leading to the formation of blood clots, as well as participating in many physiologic and pathologic processes including inflammation, wound healing, atherosclerosis, antimicrobial host defense, angiogenesis, and malignancy [1]. Platelets are produced by megakaryocytes in a process that consumes nearly the entire cytoplasmic complement of membranes, organelles, granules, and soluble macromolecules. Currently, there are two proposed mechanisms for platelet production. In one scenario, platelets are produced by cytoplasmic fragmentation from megakaryocytes [13-18]. The other scenario of platelet biogenesis is that platelets are budded off the tips of proplatelets, which operate like assembly lines, resulting in platelet production at the end of each proplatelet [10-12,19,20]. In either scenario, each megakaryocyte is estimated to give rise to 2000 to 5000 nascent platelets [21-23]. It is estimated that megakaryocytes regenerate at a rate of 108 cells per day [24-26]. Thus, each day the adult human produces approximately 2 × 1011 to 5 × 1011 platelets and this number can increase tenfold with demand [24,27]. Production of such a large number of cells circulating in the blood, each with a lifespan of 9 to 10 days, could potentially offer an excellent delivery system for disease treatment.
Platelets contain a large number of biologically active proteins within cytoplasmic granules including α-granules, dense granules, primary lysomes, and peroxisomes [28]. The α-granules are the most abundant granules in platelets. There are about 50 to 80 α-granules per platelet, which is 10-fold more than dense granules. They contain a wide variety of coagulation/adhesive proteins, growth factors and protease inhibitors involved in both primary and secondary hemostatic mechanisms [29,30]. Proteins present in α-granules either arise from megakaryocyte synthesis (e.g. glycoproteins (GP)Ib/V/IX, GPIV, integrin αIIbβ3 (GPIIb/IIIa), von Willebrand factor (VWF), P-selectin, thromboglobulin, platelet-derived factor), or endocytosis through cell surface membrane receptor-mediated uptake from the plasma environment (e.g. fibrinogen, fibronectin, factor V). Dense granules contain ADP, ATP, calcium ions, and serotonin [31]. Besides a vast number of molecules are stored in platelet granules, platelets have several surface receptors [32]. There are two important surface receptors that can bind adhesive glycoproteins (GP) including the GPIb/V/IX complex, which supports platelet adhesive by binding VWF, and the αIIbβ3 receptor, which mediates platelet aggregation by binding fibrinogen, collagen, and VWF. Other receptors include the serpentine receptors for ADP, thrombin, epinephrine, and thromboxane A2 (TXA2) and the Fc receptor FcγRIIA.
Platelets are the principle cells responsible for primary hemostasis. Upon vessel injury, subendothelium portions of the blood vessel wall that normally are concealed from circulating platelets by an intact endothelial lining are exposed, providing the initiation signal for platelet deposition [33]. At sites of injury, platelets adhere to the vessel wall by interactions with VWF and collagen and become activated and undergo degranulation, releasing a variety of potent functional molecules and several mediators that affect platelet function, inflammation, vascular tone, fibrinolysis, and wound healing [34-39]. Thus, platelets are packed with bioactive proteins and circulate in blood, serving as both storage “depot” and trafficking “vehicle” in the circulation. Due to these special characteristics, platelets can be a unique target for gene therapy of diseases that result from defects of proteins that are normally synthesized by megakaryocytes, such as Glanzmann Thrombasthenia, which results from deficiency of GPIIb/IIIa, and Bernard-Soulier Syndrome, which is caused by defects of GPIb/V/IX. This strategy can also be used for other diseases in which the protein of interest normally circulates in blood and is needed at the sites of vascular injury, but is not synthesized by megakaryocytes, e.g. factor VIII (FVIII) for hemophilia A and factor IX (FIX) for hemophilia B. In the later two cases, the protein of interest could be ectopically expressed in platelets where transgene protein could be stored in releasable granules, circulate through the body, and ultimately released locally at sites of platelet activation, such as at sites of vascular injury.
3. Platelet manipulation for disease treatment
Since platelets are anucleate cells with a limited life span, direct molecular manipulation can not serve as a reliable means for intervention. Although megakaryocytes are amenable to molecular manipulation [40], they also have a finite life span and can not serve as a means for long-term expression of the target protein. In contrast, hematopoietic stem cells, which give rise to all blood lineages including the megakaryocyte/platelet lineage, are preferable targets for genetic transfer to establish in vivo long-term expression of the target protein in platelets. Hematopoietic stem cells are an attractive target for gene therapy because they have a high capacity for clonal expansion including both self-renewal and differentiation into all blood lineages, thus supporting hematopoiesis throughout the lifetime, and because these cells are easily accessible and can be expanded and genetically modified ex vivo, and then reimplanted. Since the transgene is introduced by ex vivo transduction of hematopoietic stem cells, followed by transplantation, anti-vector immune response should be minimized in contrast to in vivo systemic transduction. Gene transfer into hematopoietic stem cells can potentially provide a cure for inherited and acquired diseases of hematopoietic [41,42].
To restrict transcription of the transgene to the platelet/megakaryocyte lineage rather than in all cells, the transgene has to be driven by a platelet lineage-specific promoter. Besides offering tissue-specific gene expression, there are other advantages to use tissue-specific promoters to direct transgene expression. One is that using the promoters of normal cellular genes to direct expression of the therapeutic gene under physiologic regulation, rather than strong viral promoters, may reduce the risks of activating adjacent cellular genes and thereby reduce the potential of insertional onco-mutagenesis, particularly since cell-specific promoters will remain inactivate in the vast majority of transduced cells. The other is that they are less likely to activate the host-cell defense machinery than constitutive viral promoters and therefore, the development of immune response to transgene product may be reduced or circumvented [43]. The promoters that have been employed to direct megakaryocyte/platelet lineage-specific transgene expression include those that control the expression of genes encoding GPIIb (αIIb) [44-54], GPIbα [55-57], GPVI [57], platelet factor 4 (PF4) [58-60], and c-mpl [61,62]. All of these promoters are activate in both megakaryocytes and platelets. Most reported studies have used either the GPIIb promoter or the GPIbα promoter for megakaryocyte/platelet-specific gene expression. The αIIb promoter directs high-level expression of GPIIb, which forms a complex with GPIIIa with a surface density of 80 000 copies per platelets [63]. This promoter is active throughout megakaryocytopoiesis, including very early stage of differentiation [64,65]. Recent studies done by Frampton and co-workers have showed that GPIIb is also expressed on bone marrow mast cells [66]. In contrast, the GPIbα promoter drives expression of GPIbα, which forms a complex with GPIX and V with approximately 25 000 copies per platelet, and this promoter is active at slightly later stages of megakaryoctopoiesis [67]. It has been reported that the GPIbα is also expressed on endothelial cells [68-70], dendritic cells [71] and breast carcinoma cell lines [72]. The lineage-specific expression of the endogenous genes regulated by these promoters is summarized in Table 1. Although the ability to limit transgene expression to a specific tissue is becoming more feasible due to a better understanding of the activity of gene promoters, the specificity and efficiency of transgene expression under control of a manipulated promoter might still not be exactly the same as endogenous gene expression. Many factors can affect transgene expression, including the region of the promoter used, the genomic integration site, copy number of transgene, and other regulatory elements that affect gene expression, e.g. enhancers, silencers, insulators, matrix attachment regions, and locus control regions [73]. Ohmori and co-workers[57] examined the expression of a luciferase reporter gene cassette under the transcriptional control of fragments of several platelet-specific gene promoters including αIIb, GPIbα, and GPVI. Results from that work indicate that the GPIbα gene promoter may deliver the highest level of megakaryocytic-specific transgene expression. However, the use of incomplete promoters, which is universal in transgene constructs, is likely to alter their activity and tissue specificity.
Table 1. The lineage-specific expression of genes.
| GPIIb | GPIbα | GPVI | PF4 | c-mpl | |
|---|---|---|---|---|---|
| hematopoietic progenitors | Yes [65] | Yes [62] | |||
| megakaryocyte precursors | Yes [142] | Yes [143] | Yes [144] | Yes [145] | Yes [62] |
| mature megakaryocytes | Yes [146] | Yes [143] | Yes [144] | Yes [145] | Yes [62] |
| platelets | Yes [146] | Yes [143] | Yes [144] | Yes [145] | Yes [62] |
| other cells | mast cells [66] | endothelial cells [68-70]; dendritic cells [71]; breast carcinoma cell lines [72] | endothelial cells [147] | endothelial cells [148] |
Efficiently transduction and long-term stable integration of the target gene in hematopoietic stem cells is important for long-term expression of transgene in the continuously replenished platelets that are derived from these cells. Due to the relatively quiescent nature of hematopoietic stem cells, they are considered poor targets for conventional retroviral vectors, which require cell division for integration. Lentiviral vectors derived from the human immunodeficiency virus have an advantage over oncoretroviruses in that they also target non-dividing cells, including quiescent hematopoietic stem cells [74-78]. It has been shown that gene-transfer vectors used in most clinical studies to date, derived from murine γ-retroviruses, have a high predisposition toward integrating near the 5′ ends of cellular genes placing them in close proximity to transcriptional control elements of cellular genes [79]. Newer vectors developed from lentiviruses tend to integrate across broader regions of the genome, which may decrease their potential to transactivate cellular genes [80,81]. Lentivirus vectors represent a promising tool for gene therapy of many diseases through the use of hematopoietic stem cells as a target. Introduction of the target cassette under control of a platelet-specific promoter using lentivirus-mediated gene transfer into hematopoietic stem cells could potentially provide patients with a self-replicating pool of stem cells for long-term transgene expression in megakaryocytes and the platelets derived from them. This will establish an in vivo “manufacturing plant” for therapeutic protein production, together with a pre-established delivery system to shuttle the protein of interest to sites where and when it is needed.
4. Gene therapy of platelet-mediated bleeding disorders using platelets and megakaryocytes as a target
4.1. Glanzmann Thrombasthenia
Glanzmann Thrombasthenia is a rare inherited hemorrhagic disorder characterized by severely reduced or absent agonist-induced platelet aggregation, caused by qualitative or quantitative abnormalities of platelet glycoprotein αIIb (GPIIb) and/or β3 (GPIIIa). Transfusion of platelets is the mainstay of therapy for serious bleeding in Glanzmann Thrombasthenia and as prophylaxis prior to surgery or other major hemostatic stresses. However, platelet alloimmunization and antibodies directed at platelet proteins have the potential to make further platelet transfusion ineffectual. Gene therapy could be an attractive alternative to correct the genetic defect in Glanzmann Thrombasthenia. Studies by Wilcox and co-workers have demonstrated that when the human GPIIIa gene driven by the αIIb promoter was introduced into CD34+ hematopoietic stem cells from a Glanzmann Thrombasthenia patient with defects in GPIIIa by retroviral vector, normal human GPIIb/IIIa was detected on the surface of megakaryocytes derived from transduced hematopoietic stem cells, resulting in ex vivo correction of fibrin clot retraction [52,53]. Further studies from the same group have demonstrated that targeting human GPIIIa expression to platelets under control of the GPIIb gene promoter using lentivirus-mediated gene transfer system can correct the bleeding diathesis in animal models including murine and canine Glanzmann Thrombasthenia models [48,82]. Their studies have shown that the small proportion (10 – 20%) of total circulating platelets that were genetically modified was able to effectively mediate normal hemostasis, even among the large majority of defective platelets. This is a promising approach with the potential to develop a strategy for long-term Glanzmann Thrombasthenia gene therapy in humans.
4.2. Bernand-Soulier Syndrome
Bernand-Soulier Syndrome is a severe congenital platelet disorder that results in a markedly prolonged bleeding time, very large platelets, and thrombocytopenia [83]. The molecular basis of Bernand-Soulier Syndrome is a deficiency of the platelet membrane glycoprotein (GP) Ib/IX complex, which is composed of four subunits (GPIbα, GPIbβ, GPIX, and GPV), forming the receptor for VWF on the platelet surface [84]. Mutations in GPIbα, GPIbβ, or GPIX can each result in Bernand-Solier Syndrome, with many of the known mutations occurring in GPIbα. Treatment of hemorrhage in patients with Bernand-Soulier Syndrome usually requires transfusion with normal platelets, but this only transiently improves bleeding time. Ware and co-workers have demonstrated that transgenic expression of human GPIbα can correct Bernand-Soulier Syndrome in a GPIbα deficient mouse model [85]. Shi and co-workers have used lentiviral vector to introduce human GPIbα with an HA tag into human primary megakaryocytes, under control of the platelet-specific αIIb promoter [50]. Human CD34+ hematopoietic stem cells were transduced with αIIb-HA-GPIbα lentivirus and cultured for 9 days with media containing a cocktail of cytokines known to induce differentiation into a population comprised of several lineages including megakaryocytes. Flow cytometric analysis indicated that 51% of the megakaryocytes derived from these CD34+ stem cells were expressing HA-tagged GPIbα transgene protein. Immuno-precipitation and western blot showed that transgene protein was associated with endogenous GPIbβ and GPIX, forming a complete GPIb/IX complex. Whether this lentivirus-mediated gene transfer can introduce efficient GPIbα transgene expression and ameliorate the bleeding diathesis associated with Bernand-Soulier Syndrome still needs to be further investigated.
5. Ectopic expression of coagulation factors in megakaryocytes and platelets for gene therapy of hemophilia
5.1. Hemophilia A
Hemophilia A is an X-chromosome linked recessive genetic bleeding disorder resulting from a deficiency of, or defective factor VIII (FVIII) molecules [86-88]. Currently, hemophilia A is treated with protein replacement therapy using either plasma-derived or recombinant FVIII. Although bleeding episodes are prevented or controlled quickly after FVIII infusion, its expensive often limits its universal availability [89-91]. Furthermore, up to 30% of patients with severe hemophilia A develop antibodies that inactivate the FVIII procoagulant activity and are referred to as FVIII inhibitors [92-94]. The clinical hallmark of inhibitor development in hemophilia A patients is failure to respond to routine replacement therapy for bleeding episodes [95-97]. Gene therapy could be an attractive alternative treatment for hemophilia A patients, but it may be more challenging for patients with inhibitors because inhibitory antibodies can inactivate FVIII if it is constitutively secreted into the circulation [98].
5.1.1. The impact of VWF on hemophilia A gene therapy
FVIII is a large and relatively unstable protein that is naturally confined to the circulation. Unless it is bound to its carrier protein, VWF, it is highly susceptible to proteolytic degradation [99-102]. Therefore, it has been thought in the past that, for FVIII gene therapy to be successful, FVIII must be secreted directly into the circulation to allow rapid binding to circulating VWF. The problems in previous clinical gene therapy trials [103-105] in which transgene FVIII was directly secreted into the circulation has induced a redoubling of efforts to explore the site (or sites) of FVIII synthesis and the importance of its relationship to VWF biosynthesis.
VWF is a large adhesive glycoprotein that performs an essential role in hemostasis, initiating platelet adhesion and aggregation at sites of vascular injury, and it also serves as the carrier protein for FVIII. The non-covalent VWF/FVIII complex protects FVIII from rapid proteolytic degradation and delivers FVIII to sites of platelet plug formation [106]. It is clear that VWF is synthesized exclusively in two cell types within the body (megakaryocytes and endothelial cells) and it is stored in α-granules in megakaryocytes and platelets and in Weibel-Palade bodies in endothelial cells [107-109]. In contrast, the site(s) FVIII synthesized is still controversial (Figure 1) and how the regulated releasable pool of FVIII is established is still unclear [110-112], although it has been suspected that synthesis of FVIII in a subpopulation of endothelial cells seems most likely [113-116]. To date there is no evidence showing either FVIII synthesis or release from platelets in normal individuals.
Figure 1.
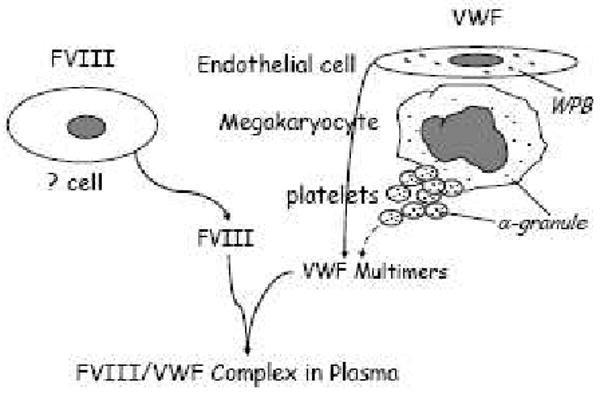
The sources of VWF and FVIII synthesis. It is clear that VWF is synthesized by endothelial cells and megakaryocytes, but the site of FVIII synthesis is still controversial. Recent reports have demonstrated that some endothelial cells make FVIII [115,116].
Early studies of the in vitro production of recombinant FVIII demonstrated the enhancement of FVIII synthesis when performed in a cell that is also synthesizing VWF [117,118]. Thus, directing FVIII expression to cells that synthesize VWF (megakaryocyte and endothelial cell) in vivo could potentially enhance the stability of FVIII compared to cells not synthesizing VWF (hepatocyte, fibroblast). Current experience suggests the normal plasma levels of FVIII will probably not be achieved with early gene therapy, so the protective effect of producing FVIII in a milieu of VWF production might be crucial to enhance the stability of the potentially limited amount of nascent FVIII produced [103,104]. Furthermore, in platelets or endothelial cells both proteins would be stored together in the protective confines of secretory granules (α-granules in platelets and Weibel Palade bodies in endothelial cells). This is especially beneficial in platelet-derived FVIII gene therapy because FVIII will be delivered together with VWF to the right place at the right time, where and when it is needed.
5.1.2. Platelet-derived FVIII for hemophilia A gene therapy
The hypothesis of this novel approach is that targeting synthesis of FVIII to megakaryocytes under control of a platelet-specific promoter will direct the storage of FVIII with VWF in the α-granules of platelets derived from these cells establishing a pool of FVIII together with VWF. At sites of injury, platelets will activate and the stored FVIII will be released to locally improve hemostatic response. This is of particular importance for hemophilia A with inhibitors because the inhibitory antibodies will inactivate the FVIII if transgene protein is constitutively secreted into the circulation [98]. Since platelet-derived FVIII would be released at hemostatic sites, the time-dependent inactivation by antibodies might be circumvented, with improved hemostasis being a potential outcome. Additionally, more tightly regulated release by platelets may limit immune exposure to the exogenous FVIII, reducing the incidence of inhibitor formation.
5.1.2.1. Studies from transgenic mouse models
Several laboratories have been instrumental in developing unique strategies for inducing FVIII synthesis in platelets. Shi and co-workers have developed a vector (2bF8) that targets human B-domain deleted FVIII (hFVIII) expression to platelets under control of the platelet-specific human integrin αIIb (GPIIb) gene promoter. Using this construct, a transgenic mouse model was generated in which functional FVIII expression was confined to platelets, at a level corresponding to about 1.2% of FVIII activity in normal mouse blood. Immunostaining demonstrated that FVIII was stored together with VWF in the α-granules of 2bF8 transgenic platelets. This platelet-derived FVIII corrected the bleeding diathesis in hemophilia A mice even in the presence of high-titer inhibitory antibodies. When 2bF8 transgene was bred into a VWF knockout background, the level of platelet-FVIII significantly decreased, indicating co-expression with VWF in platelets is important for optimal FVIII expression [49]. Further studies by Shi and co-workers using 2bF8 transgenic mice have demonstrated that even with only 1 to 5% of platelets containing FVIII, hemostasis is still improved in hemophilia A mice with pre-existing immunity in a tail-clip injury model. Sustained platelet-FVIII expression was achieved in immunized recipients that received bone marrow from 2bF8 transgenic mice, indicating that the presence of FVIII-specific immunity in recipients does not negate engraftment of 2bF8 genetically-modified hematopoietic stem cells [119].
Yarovoi and co-workers have generated a transgenic mouse model in which the platelet-specific murine GPIb promoter was used to drive FVIII expression in the megakaryocyte/platelet lineage. FVIII was detected in platelets from transgenic mice with a level equivalent to a plasma FVIII correction of 9% antigen. This platelet-FVIII corrected the clotting defect in hemophilia A mice in a FeCl3 carotid artery injury model, achieving occlusive time equivalent to that provided by infusion of FVIII to 20% of normal level [55]. Further studies done by the same group demonstrated that FVIII is stored in α-granules even in the absence of VWF and that platelet-derived FVIII without VWF still improved hemostasis in hemophilia A animals [120]. In comparisons of the efficacy of platelet-FVIII to infused rhFVIII in the presence of circulating inhibitors, studies done by by Gewirtz and co-workers showed about 6-fold greater protection of platelet-FVIII compared to infused rhFVIII followed by inhibitory antibodies in FVIIInull mice, using a FeCl3 carotid artery injury model and commercial anti-FVIII inhibitory antibodies [121]. Using the same transgenic model, Neyman et al used a laser-induced cremaster vessel injury model to study the accumulation of platelets and fibrin in clots in pFVIII/FVIIInull mice vs FVIIInull mice treated with full-length FVIII infusion, and showed that clots based on platelet-derived human B-domain deleted FVIII had a tendency toward more embolization than those generated after full-length FVIII infusion [122].
Damon and co-workers generated a transgenic mouse model in which a rat PF4 core promoter was used to direct human B-domain deleted FVIII expression. FVIII activity was detected in transgenic platelets with a level of approximately 122 mU/1 × 109 platelets. The studies demonstrated that ectopic expression of FVIII in platelets under control of the PF4 promoter had no effect on α-granule-derived platelet factor V/Va function. The amount of FVIII per platelet was decreased, with a parallel decrease in efficacy when transgenic mice were infused with thrombopoietin [60].
5.1.2.2. Lentivirus-mediated platelet-specific gene therapy for hemophilia A
To apply this platelet-derived FVIII approach to “real life” gene therapy of hemophilia A, efficient gene transfer is essential. Shi and co-workers used lentivirus-mediated gene transfer to transduce bone marrow cells from FVIIInull mice with 2bF8 lentivirus, which were then transplanted into littermates. The results demonstrate that platelet FVIII expression was maintained for at least 5 months in primary transplant recipients, indicating successful treatment of the desired hematopoietic stem cells. Stable integration of 2bF8 transgene was confirmed by secondary transplants, which showed long-term expression of the 2bF8 cassette resulting in storage of functional FVIII protein in the platelets of recipients with no inhibitor development [51]. Further studies (performed by Shi and co-workers) have demonstrated that lentivirus-mediated platelet-derived FVIII gene transfer into hematopoietic stem cells can provide sustained improvement of hemostasis in hemophilic mice with pre-existing immunity, indicating that this approach may be a promising strategy for gene therapy of hemophilia A with inhibitory antibodies in humans [123]. The clinical efficacy of platelet-derived FVIII has been examined using large animal model by Wilcox and co-workers. Their studies demonstrated that autologous transplantation of 2bF8 lentivirus-transduced hematopoietic stem cells resulted in FVIII expression exclusively in platelets with levels approximately 5 mU/108 platelets from 20 weeks through more than one year after transplant and that platelet-FVIII ameliorated the bleeding phenotype in hemophilia A dogs [124].
Ohmori and co-workers used a simian immunodeficiency virus-based vector harboring GPIb promoter to direct FVIII expression for efficient platelet-targeted gene transduction, and efficient expression of FVIII in platelets and phenotypic correction in hemophilia A mice were achieved [57]. Instead of FVIII, Ohmori and coworkers also inserted a FVII constructed under control of the GPIb promoter into a lentiviral vector. Following transduction and transplantation of bone marrow cells using that lentiviral vector, FVII was expressed in platelets and localized in the cytoplasm. The platelet-derived FVII improved hemostasis in hemophilia A mice even in the presence of anti-FVIII inhibitory antibodies probably by a mechanism that by pass the need for FVIII to activate FX via the tissue factor pathway [56].
These studies have demonstrated that targeting platelets as delivery systems for FVIII or other coagulation factors can improve hemostasis in hemophilia A and hemophilia A with inhibitors in animal models. This could be a promising approach for gene therapy of hemophilia A patients and patients with inhibitors.
5.2. Hemophilia B
Similar to hemophilia A, hemophilia B is also an X-linked, recessive bleeding disorder resulting from the deficiency of another coagulation factor, factor IX (FIX). Replacement therapy using either plasma-derived or recombinant FIX is the current standard treatment, but therapy is fraught with the same complications of recurrent hemorrhages as hemophilia A. Although only about 3% of hemophilia B patients develop inhibitory antibodies response to factor IX concentrates in replacement therapy, a frequency about 10-fold lower than in hemophilia A, an allergic reaction or anaphylaxis is very common in hemophilia B patients with inhibitors [125-127].
Sustained phenotypic correction of hemophilia B by gene transfer technology has been achieved in animal models using several different gene delivery systems, including retroviral vectors, adenoviral vectors and adeno-associated viral vectors containing FIX cDNA targeted at either muscle cells or hepatocytes, leading to increased significant levels of plasma FIX [128-136]. Clinical trials of gene therapy for hemophilia B using AAV vectors were started in humans but have been stopped [133,137-139].
Studies have demonstrated that transgene FVIII can store in platelet α-granules even in the absence of VWF [49,120]. Therefore, targeting FIX expression to platelets might also enable storage of FIX in platelet α-granules where it might be released locally at the site of vascular injury to improve hemostasis in hemophilia B. Zhang and coworkers (personnel communication) have constructed a FIX expression cassette (2bF9) in which FIX cDNA was placed under the control of the platelet αIIb promoter. Using a 2bF9 construct, a platelet-derived FIX transgenic mouse model was developed in which about 90% of FIX is stored in platelet α-granules and is releasable by agonist stimulation. The study demonstrated that platelet-derived FIX can ameliorate murine hemophilia B phenotype in a tail clip injury model. Clinical efficacy can be transferred by either platelet transfusion or bone marrow transplantation. These studies demonstrated that targeting FIX expression in platelets can correct the hemophilia B phenotype.
5.3. The safety issues
There are some concerns arising from the ectopic expression of coagulation factors in genetic therapy as well. These include 1) whether platelet-specific expression of transgene interferes with expression of the endogenous protein that is driven by the same promoter; 2) whether platelet-derived FVIII or FIX will cause thrombogenesis; 3) whether platelet-derived FVIII will induce the immune response; and 4) the possibility of insertional mutagenesis. To address whether platelet-derived FVIII affects endogenous protein directed by the same promoter, we analyzed mouse endogenous GPIIb expression using flow cytometry in 2bF8 transgenic animals and wild type or FVIIInull control mice. The results demonstrated that GPIIb expression in 2bF8 transgenic mice is not significantly different from control mice (unpublished data). In 2bF8 or 2bF9 transgenic model, all mice with platelet-FVIII, including mice with a normal level of murine plasma FVIII plus platelet-FVIII, have normal life spans with no overt evidence of health problems related to thrombogenesis. The development of inhibitory antibodies to exogenous FVIII is not only a severe complication of FVIII infusion in hemophilia patients, but also a concern in gene therapy [140,141]. Since FVIII or FIX stores in platelet α-granules when transgene expression is targeted to platelets, this may limit immune exposure to the exogenous FVIII or FIX, reducing the incidence of inhibitor formation. Our studies have demonstrated that neither inhibitory antibodies nor non-inhibitory antibodies were detected in 2bF8-lentivirus mediated gene therapy animals [51]. Insertion site-related mutagenesis is of the major concern in gene therapy. The advanced self-inactivating design of lentiviral vectors and the differences in integration site selection between lentiviral and oncoretroviral vectors may reduce the risk of insertional mutagenesis. Oncoretroviral vectors tend to integrate preferentially into promoter-proximal regions, whereas lentiviral vectors appear to use a more random model of integration, mainly inserting into open chromatin [80]. Utilizing the protection of a suicide gene may further improve the safety of platelet-derived gene therapy. Introducing a drug-inducible suicide gene into hematopoietic stem cells along with the transgene, e.g. FVIII or FIX, may sensitize the manipulated cells to ablation on demand if cells misbehave. Further characterizing the function of platelet-expressed FVIII and continued vigilance regarding the insertional mutagenesis are important for lentivirus-mediated platelet-derived gene therapy of hemophilia.
6. Conclusion
In summary, these studies have demonstrated the feasibility of platelet delivery of proteins of interest for disease treatment through genetic manipulation of hematopoietic stem cells. Targeting gene expression to platelets is a promising approach, which might lead to a cure for genetic bleeding disorders. To apply this approach in the clinic, hematopoietic stem cells would be harvested for ex vivo introduction of the gene of interest by lentivirus-mediated gene transfer, followed by reintroduction into patients as an autologous transplant. Furthermore, if in vitro production of platelets were established as transfusion alternative in the clinic, platelets with FVIII or FIX could be used therapeutically in patients with hemophilia A or B and the half-life of that FVIII or FIX would be prolonged.
Acknowledgments
This work was supported by American Heart Association National Center Scientist Development Award (0730183N) (QS), National Hemophilia Foundation Career Development Award (QS), Hemophilia Association of New York grant (QS), National Institutes of Health grants HL-44612 (RRM), the Goerlich Foundation (RRM), and Bayer Hemophilia Award (RRM).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Blair P, Flaumenhaft R. Platelet alpha-granules: basic biology and clinical correlates. Blood Rev. 2009;23:177–189. doi: 10.1016/j.blre.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Odell TT, Jr, Jackson CW. Polyploidy and maturation of rat megakaryocytes. Blood. 1968;32:102–110. [PubMed] [Google Scholar]
- 3.Odell TT, Jr, Jackson CW, Friday TJ. Megakaryocytopoiesis in rats with special reference to polyploidy. Blood. 1970;35:775–782. [PubMed] [Google Scholar]
- 4.Ebbe S, Stohlman F., Jr Megakaryocytopoiesis in the Rat. Blood. 1965;26:20–35. [PubMed] [Google Scholar]
- 5.Tomer A, Harker LA, Burstein SA. Purification of human megakaryocytes by fluorescence-activated cell sorting. Blood. 1987;70:1735–1742. [PubMed] [Google Scholar]
- 6.Tomer A, Harker LA, Burstein SA. Flow cytometric analysis of normal human megakaryocytes. Blood. 1988;71:1244–1252. [PubMed] [Google Scholar]
- 7.Breton-Gorius J, Reyes F. Ultrastructure of human bone marrow cell maturation. Int Rev Cytol. 1976;46:251–321. doi: 10.1016/s0074-7696(08)60993-6. [DOI] [PubMed] [Google Scholar]
- 8.Ebbe S. Biology of megakaryocytes. Prog Hemost Thromb. 1976;3:211–229. [PubMed] [Google Scholar]
- 9.Nakao K, Angrist AA. Membrane surface specialization of blood platelet and megakaryocyte. Nature. 1968;217:960–961. doi: 10.1038/217960a0. [DOI] [PubMed] [Google Scholar]
- 10.Italiano JE, Jr, Lecine P, Shivdasani RA, Hartwig JH. Blood platelets are assembled principally at the ends of proplatelet processes produced by differentiated megakaryocytes. J Cell Biol. 1999;147:1299–1312. doi: 10.1083/jcb.147.6.1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Italiano JE, Jr, Shivdasani RA. Megakaryocytes and beyond: the birth of platelets. J Thromb Haemost. 2003;1:1174–1182. doi: 10.1046/j.1538-7836.2003.00290.x. [DOI] [PubMed] [Google Scholar]
- 12.Patel SR, Hartwig JH, Italiano JE., Jr The biogenesis of platelets from megakaryocyte proplatelets. J Clin Invest. 2005;115:3348–3354. doi: 10.1172/JCI26891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zucker-Franklin D, Petursson S. Thrombocytopoiesis--analysis by membrane tracer and freeze-fracture studies on fresh human and cultured mouse megakaryocytes. J Cell Biol. 1984;99:390–402. doi: 10.1083/jcb.99.2.390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mori M, Tsuchiyama J, Okada S. Proliferation, migration and platelet release by megakaryocytes in long-term bone marrow culture in collagen gel. Cell Struct Funct. 1993;18:409–417. doi: 10.1247/csf.18.409. [DOI] [PubMed] [Google Scholar]
- 15.Caine YG, Vlodavsky I, Hersh M, Polliack A, Gurfel D, Or R, Levine RF, Eldor A. Adhesion, spreading and fragmentation of human megakaryocytes exposed to subendothelial extracellular matrix: a scanning electron microscopy study. Scan Electron Microsc. 1986:1087–1094. [PubMed] [Google Scholar]
- 16.Deutsch VR, Olson TA, Nagler A, Slavin S, Levine RF, Eldor A. The response of cord blood megakaryocyte progenitors to IL-3, IL-6 and aplastic canine serum varies with gestational age. Br J Haematol. 1995;89:8–16. doi: 10.1111/j.1365-2141.1995.tb08917.x. [DOI] [PubMed] [Google Scholar]
- 17.Kosaki G. Platelet production by megakaryocytes: protoplatelet theory justifies cytoplasmic fragmentation model. Int J Hematol. 2008;88:255–267. doi: 10.1007/s12185-008-0147-7. [DOI] [PubMed] [Google Scholar]
- 18.Kosaki G. In vivo platelet production from mature megakaryocytes: does platelet release occur via proplatelets? Int J Hematol. 2005;81:208–219. doi: 10.1532/IJH97.04177. [DOI] [PubMed] [Google Scholar]
- 19.Zucker-Franklin D. The submembranous fibrils of human blood platelets. J Cell Biol. 1970;47:293–299. doi: 10.1083/jcb.47.1.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Choi ES, Nichol JL, Hokom MM, Hornkohl AC, Hunt P. Platelets generated in vitro from proplatelet-displaying human megakaryocytes are functional. Blood. 1995;85:402–413. [PubMed] [Google Scholar]
- 21.Long MW. Megakaryocyte differentiation events. Semin Hematol. 1998;35:192–199. [PubMed] [Google Scholar]
- 22.Harker LA, Finch CA. Thrombokinetics in man. J Clin Invest. 1969;48:963–974. doi: 10.1172/JCI106077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stenberg PE, Levin J. Mechanisms of platelet production. Blood Cells. 1989;15:23–47. [PubMed] [Google Scholar]
- 24.Kaushansky K. The molecular mechanisms that control thrombopoiesis. J Clin Invest. 2005;115:3339–3347. doi: 10.1172/JCI26674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Deutsch VR, Tomer A. Megakaryocyte development and platelet production. Br J Haematol. 2006;134:453–466. doi: 10.1111/j.1365-2141.2006.06215.x. [DOI] [PubMed] [Google Scholar]
- 26.Becker RC, Smyth S. The evolution of platelet-directed pharmacotherapy. J Thromb Haemost. 2009;7 1:266–271. doi: 10.1111/j.1538-7836.2009.03428.x. [DOI] [PubMed] [Google Scholar]
- 27.Branehog I, Ridell B, Swolin B, Weinfeld A. Megakaryocyte quantifications in relation to thrombokinetics in primary thrombocythaemia and allied diseases. Scand J Haematol. 1975;15:321–332. doi: 10.1111/j.1600-0609.1975.tb01087.x. [DOI] [PubMed] [Google Scholar]
- 28.Bentfeld ME, Bainton DF. Cytochemical localization of lysosomal enzymes in rat megakaryocytes and platelets. J Clin Invest. 1975;56:1635–1649. doi: 10.1172/JCI108246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Corash L, Costa JL, Shafer B, Donlon JA, Murphy D. Heterogeneity of human whole blood platelet subpopulations. III. Density-dependent differences in subcellular constituents. Blood. 1984;64:185–193. [PubMed] [Google Scholar]
- 30.Vicic WJ, Weiss HJ. Evidence that platelet alpha-granules are a major determinant of platelet density: studies in storage pool deficiency. Thromb Haemost. 1983;50:878–880. [PubMed] [Google Scholar]
- 31.Harrison P, Cramer EM. Platelet alpha-granules. Blood Rev. 1993;7:52–62. doi: 10.1016/0268-960x(93)90024-x. [DOI] [PubMed] [Google Scholar]
- 32.Tomer A. Human marrow megakaryocyte differentiation: multiparameter correlative analysis identifies von Willebrand factor as a sensitive and distinctive marker for early (2N and 4N) megakaryocytes. Blood. 2004;104:2722–2727. doi: 10.1182/blood-2004-02-0769. [DOI] [PubMed] [Google Scholar]
- 33.Ruggeri ZM. Platelets in atherothrombosis. Nat Med. 2002;8:1227–1234. doi: 10.1038/nm1102-1227. [DOI] [PubMed] [Google Scholar]
- 34.Parker RI, Gralnick HR. Identification of platelet glycoprotein IIb/IIIa as the major binding site for released platelet-von Willebrand factor. Blood. 1986;68:732–736. [PubMed] [Google Scholar]
- 35.Atri SC, Misra J, Bisht D, Misra K. Use of homologous platelet factors in achieving total healing of recalcitrant skin ulcers. Surgery. 1990;108:508–512. [PubMed] [Google Scholar]
- 36.Gawaz M, Langer H, May AE. Platelets in inflammation and atherogenesis. J Clin Invest. 2005;115:3378–3384. doi: 10.1172/JCI27196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weber C. Platelets and chemokines in atherosclerosis: partners in crime. Circ Res. 2005;96:612–616. doi: 10.1161/01.RES.0000160077.17427.57. [DOI] [PubMed] [Google Scholar]
- 38.Anitua E, Andia I, Ardanza B, Nurden P, Nurden AT. Autologous platelets as a source of proteins for healing and tissue regeneration. Thromb Haemost. 2004;91:4–15. doi: 10.1160/TH03-07-0440. [DOI] [PubMed] [Google Scholar]
- 39.Klinger MH, Jelkmann W. Role of blood platelets in infection and inflammation. J Interferon Cytokine Res. 2002;22:913–922. doi: 10.1089/10799900260286623. [DOI] [PubMed] [Google Scholar]
- 40.Ohmori T, Kashiwakura Y, Ishiwata A, Madoiwa S, Mimuro J, Sakata Y. Silencing of a targeted protein in in vivo platelets using a lentiviral vector delivering short hairpin RNA sequence. Arterioscler Thromb Vasc Biol. 2007;27:2266–2272. doi: 10.1161/ATVBAHA.107.149872. [DOI] [PubMed] [Google Scholar]
- 41.Medin JA, Karlsson S. Viral vectors for gene therapy of hematopoietic cells. Immunotechnology. 1997;3:3–19. doi: 10.1016/s1380-2933(96)00059-0. [DOI] [PubMed] [Google Scholar]
- 42.Medin JA, Karlsson S. Selection of retrovirally transduced cells to enhance the efficiency of gene therapy. Proc Assoc Am Physicians. 1997;109:111–119. [PubMed] [Google Scholar]
- 43.Weeratna RD, Wu T, Efler SM, Zhang L, Davis HL. Designing gene therapy vectors: avoiding immune responses by using tissue-specific promoters. Gene Ther. 2001;8:1872–1878. doi: 10.1038/sj.gt.3301602. [DOI] [PubMed] [Google Scholar]
- 44.Tronik-Le RD, Roullot V, Schweitzer A, Berthier R, Marguerie G. Suppression of erythro-megakaryocytopoiesis and the induction of reversible thrombocytopenia in mice transgenic for the thymidine kinase gene targeted by the platelet glycoprotein alpha IIb promoter. J Exp Med. 1995;181:2141–2151. doi: 10.1084/jem.181.6.2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Poujol C, Tronik-Le RD, Tropel P, Roullot V, Nurden A, Marguerie G, Nurden P. Ultrastructural analysis of bone marrow hematopoiesis in mice transgenic for the thymidine kinase gene driven by the alpha IIb promoter. Blood. 1998;92:2012–2023. [PubMed] [Google Scholar]
- 46.Tropel P, Roullot V, Vernet M, Poujol C, Pointu H, Nurden P, Marguerie G, Tronik-Le RD. A 2.7-kb portion of the 5′ flanking region of the murine glycoprotein alphaIIb gene is transcriptionally active in primitive hematopoietic progenitor cells. Blood. 1997;90:2995–3004. [PubMed] [Google Scholar]
- 47.Berridge MV, Ralph SJ, Tan AS. Cell-lineage antigens of the stem cell-megakaryocyte-platelet lineage are associated with the platelet IIb-IIIa glycoprotein complex. Blood. 1985;66:76–85. [PubMed] [Google Scholar]
- 48.Fang J, Hodivala-Dilke K, Johnson BD, DU LM, Hynes RO, White GC, Wilcox DA. Therapeutic expression of the platelet-specific integrin, alphaIIbbeta3, in a murine model for Glanzmann thrombasthenia. Blood. 2005;106:2671–2679. doi: 10.1182/blood-2004-12-4619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shi Q, Wilcox DA, Fahs SA, Weiler H, Wells CW, Cooley BC, Desai D, Morateck PA, Gorski J, Montgomery RR. Factor VIII ectopically targeted to platelets is therapeutic in hemophilia A with high-titer inhibitory antibodies. J Clin Invest. 2006;116:1974–1982. doi: 10.1172/JCI28416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shi Q, Wilcox DA, Morateck PA, Fahs SA, Kenny D, Montgomery RR. Targeting platelet GPIbalpha transgene expression to human megakaryocytes and forming a complete complex with endogenous GPIbbeta and GPIX. J Thromb Haemost. 2004;2:1989–1997. doi: 10.1111/j.1538-7836.2004.00961.x. [DOI] [PubMed] [Google Scholar]
- 51.Shi Q, Wilcox DA, Fahs SA, Fang J, Johnson BD, DU LM, Desai D, Montgomery RR. Lentivirus-mediated platelet-derived factor VIII gene therapy in murine haemophilia A. J Thromb Haemost. 2007;5:352–361. doi: 10.1111/j.1538-7836.2007.02346.x. [DOI] [PubMed] [Google Scholar]
- 52.Wilcox DA, Olsen JC, Ishizawa L, Griffith M, White GC. Integrin alphaIIb promoter-targeted expression of gene products in megakaryocytes derived from retrovirus-transduced human hematopoietic cells. Proc Natl Acad Sci U S A. 1999;96:9654–9659. doi: 10.1073/pnas.96.17.9654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wilcox DA, Olsen JC, Ishizawa L, Bray PF, French DL, Steeber DA, Bell WR, Griffith M, White GC. Megakaryocyte-targeted synthesis of the integrin beta(3)-subunit results in the phenotypic correction of Glanzmann thrombasthenia. Blood. 2000;95:3645–3651. [PubMed] [Google Scholar]
- 54.Rodriguez MH, Enjolras N, Plantier JL, Rea M, Leboeuf M, Uzan G, Bordet JC, Negrier C. Expression of coagulation factor IX in a haematopoietic cell line. Thromb Haemost. 2002;87:366–373. [PubMed] [Google Scholar]
- 55.Yarovoi HV, Kufrin D, Eslin DE, Thornton MA, Haberichter SL, Shi Q, Zhu H, Camire R, Fakharzadeh SS, Kowalska MA, Wilcox DA, Sachais BS, Montgomery RR, Poncz M. Factor VIII ectopically expressed in platelets: efficacy in hemophilia A treatment. Blood. 2003;102:4006–4013. doi: 10.1182/blood-2003-05-1519. [DOI] [PubMed] [Google Scholar]
- 56.Ohmori T, Ishiwata A, Kashiwakura Y, Madoiwa S, Mitomo K, Suzuki H, Hasegawa M, Mimuro J, Sakata Y. Phenotypic correction of hemophilia A by ectopic expression of activated factor VII in platelets. Mol Ther. 2008;16:1359–1365. doi: 10.1038/mt.2008.117. [DOI] [PubMed] [Google Scholar]
- 57.Ohmori T, Mimuro J, Takano K, Madoiwa S, Kashiwakura Y, Ishiwata A, Niimura M, Mitomo K, Tabata T, Hasegawa M, Ozawa K, Sakata Y. Efficient expression of a transgene in platelets using simian immunodeficiency virus-based vector harboring glycoprotein Ibalpha promoter: in vivo model for platelet-targeting gene therapy. FASEB J. 2006;20:1522–1524. doi: 10.1096/fj.05-5161fje. [DOI] [PubMed] [Google Scholar]
- 58.Ravid K, Beeler DL, Rabin MS, Ruley HE, Rosenberg RD. Selective targeting of gene products with the megakaryocyte platelet factor 4 promoter. Proc Natl Acad Sci U S A. 1991;88:1521–1525. doi: 10.1073/pnas.88.4.1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kufrin D, Eslin DE, Bdeir K, Murciano JC, Kuo A, Kowalska MA, Degen JL, Sachais BS, Cines DB, Poncz M. Antithrombotic thrombocytes: ectopic expression of urokinase-type plasminogen activator in platelets. Blood. 2003;102:926–933. doi: 10.1182/blood-2003-01-0054. [DOI] [PubMed] [Google Scholar]
- 60.Damon AL, Scudder LE, Gnatenko DV, Sitaraman V, Hearing P, Jesty J, Bahou WF. Altered bioavailability of platelet-derived factor VIII during thrombocytosis reverses phenotypic efficacy in haemophilic mice. Thromb Haemost. 2008;100:1111–1122. doi: 10.1160/th08-04-0242. [DOI] [PubMed] [Google Scholar]
- 61.Deveaux S, Filipe A, Lemarchandel V, Ghysdael J, Romeo PH, Mignotte V. Analysis of the thrombopoietin receptor (MPL) promoter implicates GATA and Ets proteins in the coregulation of megakaryocyte-specific genes. Blood. 1996;87:4678–4685. [PubMed] [Google Scholar]
- 62.Kaushansky K, Drachman JG. The molecular and cellular biology of thrombopoietin: the primary regulator of platelet production. Oncogene. 2002;21:3359–3367. doi: 10.1038/sj.onc.1205323. [DOI] [PubMed] [Google Scholar]
- 63.Wagner CL, Mascelli MA, Neblock DS, Weisman HF, Coller BS, Jordan RE. Analysis of GPIIb/IIIa receptor number by quantification of 7E3 binding to human platelets. Blood. 1996;88:907–914. [PubMed] [Google Scholar]
- 64.Prandini MH, Martin F, Thevenon D, Uzan G. The tissue-specific transcriptional regulation of the megakaryocytic glycoprotein IIb gene is controlled by interactions between a repressor and positive cis-acting elements. Blood. 1996;88:2062–2070. [PubMed] [Google Scholar]
- 65.Murray LJ, Mandich D, Bruno E, DiGiusto RK, Fu WC, Sutherland DR, Hoffman R, Tsukamoto A. Fetal bone marrow CD34+CD41+ cells are enriched for multipotent hematopoietic progenitors, but not for pluripotent stem cells. Exp Hematol. 1996;24:236–245. [PubMed] [Google Scholar]
- 66.Berlanga O, Emambokus N, Frampton J. GPIIb (CD41) integrin is expressed on mast cells and influences their adhesion properties. Exp Hematol. 2005;33:403–412. doi: 10.1016/j.exphem.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 67.Debili N, Issaad C, Masse JM, Guichard J, Katz A, Breton-Gorius J, Vainchenker W. Expression of CD34 and platelet glycoproteins during human megakaryocytic differentiation. Blood. 1992;80:3022–3035. [PubMed] [Google Scholar]
- 68.Sprandio JD, Shapiro SS, Thiagarajan P, McCord S. Cultured human umbilical vein endothelial cells contain a membrane glycoprotein immunologically related to platelet glycoprotein Ib. Blood. 1988;71:234–237. [PubMed] [Google Scholar]
- 69.Konkle BA, Shapiro SS, Asch AS, Nachman RL. Cytokine-enhanced expression of glycoprotein Ib alpha in human endothelium. J Biol Chem. 1990;265:19833–19838. [PubMed] [Google Scholar]
- 70.Wu G, Essex DW, Meloni FJ, Takafuta T, Fujimura K, Konkle BA, Shapiro SS. Human endothelial cells in culture and in vivo express on their surface all four components of the glycoprotein Ib/IX/V complex. Blood. 1997;90:2660–2669. [PubMed] [Google Scholar]
- 71.Monteiro MR, Shapiro SS, Takafuta T, Menezes DW, Murphy GF. Von Willebrand factor receptor GPIb alpha is expressed by human factor XIIIa-positive dermal dendrocytes and is upregulated by mast cell degranulation. J Invest Dermatol. 1999;113:272–276. doi: 10.1046/j.1523-1747.1999.00665.x. [DOI] [PubMed] [Google Scholar]
- 72.Oleksowicz L, Dutcher JP, DeLeon-Fernandez M, Etkind P. A GPIb alpha-related protein is expressed by fresh human breast carcinoma tissue and is regulated by a PKC-sensitive mechanism. Exp Cell Res. 1997;237:110–117. doi: 10.1006/excr.1997.3784. [DOI] [PubMed] [Google Scholar]
- 73.Pennacchio LA, Rubin EM. Genomic strategies to identify mammalian regulatory sequences. Nat Rev Genet. 2001;2:100–109. doi: 10.1038/35052548. [DOI] [PubMed] [Google Scholar]
- 74.Lewis PF, Emerman M. Passage through mitosis is required for oncoretroviruses but not for the human immunodeficiency virus. J Virol. 1994;68:510–516. doi: 10.1128/jvi.68.1.510-516.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Miller DG, Adam MA, Miller AD. Gene transfer by retrovirus vectors occurs only in cells that are actively replicating at the time of infection. Mol Cell Biol. 1990;10:4239–4242. doi: 10.1128/mcb.10.8.4239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bukrinsky MI, Haggerty S, Dempsey MP, Sharova N, Adzhubel A, Spitz L, Lewis P, Goldfarb D, Emerman M, Stevenson M. A nuclear localization signal within HIV-1 matrix protein that governs infection of non-dividing cells. Nature. 1993;365:666–669. doi: 10.1038/365666a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bukrinsky MI, Sharova N, McDonald TL, Pushkarskaya T, Tarpley WG, Stevenson M. Association of integrase, matrix, and reverse transcriptase antigens of human immunodeficiency virus type 1 with viral nucleic acids following acute infection. Proc Natl Acad Sci U S A. 1993;90:6125–6129. doi: 10.1073/pnas.90.13.6125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zennou V, Petit C, Guetard D, Nerhbass U, Montagnier L, Charneau P. HIV-1 genome nuclear import is mediated by a central DNA flap. Cell. 2000;101:173–185. doi: 10.1016/S0092-8674(00)80828-4. [DOI] [PubMed] [Google Scholar]
- 79.Bester AC, Schwartz M, Schmidt M, Garrigue A, Hacein-Bey-Abina S, Cavazzana-Calvo M, Ben-Porat N, von KC, Fischer A, Kerem B. Fragile sites are preferential targets for integrations of MLV vectors in gene therapy. Gene Ther. 2006;13:1057–1059. doi: 10.1038/sj.gt.3302752. [DOI] [PubMed] [Google Scholar]
- 80.De PM, Montini E, Santoni de Sio FR, Benedicenti F, Gentile A, Medico E, Naldini L. Promoter trapping reveals significant differences in integration site selection between MLV and HIV vectors in primary hematopoietic cells. Blood. 2005;105:2307–2315. doi: 10.1182/blood-2004-03-0798. [DOI] [PubMed] [Google Scholar]
- 81.Neschadim A, McCart JA, Keating A, Medin JA. A roadmap to safe, efficient, and stable lentivirus-mediated gene therapy with hematopoietic cell transplantation. Biol Blood Marrow Transplant. 2007;13:1407–1416. doi: 10.1016/j.bbmt.2007.09.014. [DOI] [PubMed] [Google Scholar]
- 82.Fang J, Jensen ES, Du LM, Boudreaux MK, Wilcox DA. Intravenous Immunoglobulin (IVIG) Diminishes Immune-Mediated Clearance of Platelets Expressing an Integrin {alpha}IIb{beta}3 Transgene Product That Restores Hemostasis in a Canine Model for Glanzmann Thrombasthenia. ASH Annual Meeting Abstracts. 2006;108:3263. [Google Scholar]
- 83.Bernard J, Soulier JP. Sur une nouvelle variet e de dystrophie thrombocytarie-hemorragipare congenitale. Sem Hop. 1948;24:3217–3223. [PubMed] [Google Scholar]
- 84.Balduini CL, Iolascon A, Savoia A. Inherited thrombocytopenias: from genes to therapy. Haematologica. 2002;87:860–880. [PubMed] [Google Scholar]
- 85.Ware J, Russell S, Ruggeri ZM. Generation and rescue of a murine model of platelet dysfunction: the Bernard-Soulier syndrome. Proc Natl Acad Sci U S A. 2000;97:2803–2808. doi: 10.1073/pnas.050582097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Abshire T, Kenet G. Recombinant factor VIIa: review of efficacy, dosing regimens and safety in patients with congenital and acquired factor VIII or IX inhibitors. J Thromb Haemost. 2004;2:899–909. doi: 10.1111/j.1538-7836.2004.00759.x. [DOI] [PubMed] [Google Scholar]
- 87.Yoshioka A, Shima M, Fukutake K, Takamatsu J, Shirahata A. Safety and efficacy of a new recombinant FVIII formulated with sucrose (rFVIII-FS) in patients with haemophilia A: a long-term, multicentre clinical study in Japan. Haemophilia. 2001;7:242–249. doi: 10.1046/j.1365-2516.2001.00511.x. [DOI] [PubMed] [Google Scholar]
- 88.Kreuz W, Gill JC, Rothschild C, Manco-Johnson MJ, Lusher JM, Kellermann E, Gorina E, Larson PJ. Full-length sucrose-formulated recombinant factor VIII for treatment of previously untreated or minimally treated young children with severe haemophilia A: results of an international clinical investigation. Thromb Haemost. 2005;93:457–467. doi: 10.1160/TH03-10-0643. [DOI] [PubMed] [Google Scholar]
- 89.Bohn RL, Aledort LM, Putnam KG, Ewenstein BM, Mogun H, Avorn J. The economic impact of factor VIII inhibitors in patients with haemophilia. Haemophilia. 2004;10:63–68. doi: 10.1046/j.1365-2516.2003.00849.x. [DOI] [PubMed] [Google Scholar]
- 90.Berntorp E, Archey W, Auerswald G, Federici AB, Franchini M, Knaub S, Kreuz W, Lethagen S, Mannucci PM, Pollmann H, Scharrer I, Hoots K. A systematic overview of the first pasteurised VWF/FVIII medicinal product, Haemate P/Humate -P: history and clinical performance. Eur J Haematol Suppl. 2008:3–35. doi: 10.1111/j.1600-0609.2008.01049.x. [DOI] [PubMed] [Google Scholar]
- 91.Ballal RD, Botteman MF, Foley I, Stephens JM, Wilke CT, Joshi AV. Economic evaluation of major knee surgery with recombinant activated factor VII in hemophilia patients with high titer inhibitors and advanced knee arthropathy: exploratory results via literature-based modeling. Curr Med Res Opin. 2008;24:753–768. doi: 10.1185/030079908X273048. [DOI] [PubMed] [Google Scholar]
- 92.Ananyeva NM, Lacroix-Desmazes S, Hauser CA, Shima M, Ovanesov MV, Khrenov AV, Saenko EL. Inhibitors in hemophilia A: mechanisms of inhibition, management and perspectives. Blood Coagul Fibrinolysis. 2004;15:109–124. doi: 10.1097/00001721-200403000-00001. [DOI] [PubMed] [Google Scholar]
- 93.Sharathkumar A, Lillicrap D, Blanchette VS, Kern M, Leggo J, Stain AM, Brooker L, Carcao MD. Intensive exposure to factor VIII is a risk factor for inhibitor development in mild hemophilia A. J Thromb Haemost. 2003;1:1228–1236. doi: 10.1046/j.1538-7836.2003.00230.x. [DOI] [PubMed] [Google Scholar]
- 94.Key NS. Inhibitors in congenital coagulation disorders. Br J Haematol. 2004;127:379–391. doi: 10.1111/j.1365-2141.2004.05168.x. [DOI] [PubMed] [Google Scholar]
- 95.White GC, McMillan CW, Blatt PM, Roberts HR. Factor VIII inhibitors: a clinical overview. Am J Hematol. 1982;13:335–342. doi: 10.1002/ajh.2830130410. [DOI] [PubMed] [Google Scholar]
- 96.Scandella DH. Properties of anti-factor VIII inhibitor antibodies in hemophilia A patients. Semin Thromb Hemost. 2000;26:137–142. doi: 10.1055/s-2000-9815. [DOI] [PubMed] [Google Scholar]
- 97.Furie B, Limentani SA, Rosenfield CG. A practical guide to the evaluation and treatment of hemophilia. Blood. 1994;84:3–9. [PubMed] [Google Scholar]
- 98.Shi Q, Fahs SA, Wilcox DA, Weiler H, Haberichter SL, Montgomery RR. Endothelial and Platelet FVIII/VWF Expression Divergence in Clinical Effect in Murine Models of Hemophilia A with and without FVIII Inhibitory Antibodies. Blood. 2006;108:938a. [Google Scholar]
- 99.Chuah MK, Brems H, Vanslembrouck V, Collen D, Vandendriessche T. Bone marrow stromal cells as targets for gene therapy of hemophilia A. Hum Gene Ther. 1998;9:353–365. doi: 10.1089/hum.1998.9.3-353. [DOI] [PubMed] [Google Scholar]
- 100.Wise RJ, Dorner AJ, Krane M, Pittman DD, Kaufman RJ. The role of von Willebrand factor multimers and propeptide cleavage in binding and stabilization of factor VIII. J Biol Chem. 1991;266:21948–21955. [PubMed] [Google Scholar]
- 101.Kaufman RJ, Pipe SW. Regulation of factor VIII expression and activity by von Willebrand factor. Thromb Haemost. 1999;82:201–208. [PubMed] [Google Scholar]
- 102.Weiss HJ, Hoyer IW. Von Willebrand factor: dissociation from antihemophilic factor procoagulant activity. Science. 1973;182:1149–1151. doi: 10.1126/science.182.4117.1149. [DOI] [PubMed] [Google Scholar]
- 103.Roth DA, Tawa NE, Jr, O'Brien JM, Treco DA, Selden RF. Nonviral transfer of the gene encoding coagulation factor VIII in patients with severe hemophilia A. N Engl J Med. 2001;344:1735–1742. doi: 10.1056/NEJM200106073442301. [DOI] [PubMed] [Google Scholar]
- 104.Powell JS, Ragni MV, White GC, Lusher JM, Hillman-Wiseman C, Moon TE, Cole V, Ramanathan-Girish S, Roehl H, Sajjadi N, Jolly DJ, Hurst D. Phase 1 trial of FVIII gene transfer for severe hemophilia A using a retroviral construct administered by peripheral intravenous infusion. Blood. 2003;102:2038–2045. doi: 10.1182/blood-2003-01-0167. [DOI] [PubMed] [Google Scholar]
- 105.Chuah MK, Collen D, Vandendriessche T. Clinical gene transfer studies for hemophilia A. Semin Thromb Hemost. 2004;30:249–256. doi: 10.1055/s-2004-825638. [DOI] [PubMed] [Google Scholar]
- 106.Wagner DD. Cell biology of von Willebrand factor. Annu Rev Cell Biol. 1990;6:217–246. doi: 10.1146/annurev.cb.06.110190.001245. [DOI] [PubMed] [Google Scholar]
- 107.Nachman R, Levine R, Jaffe EA. Synthesis of factor VIII antigen by cultured guinea pig megakaryocytes. J Clin Invest. 1977;60:914–921. doi: 10.1172/JCI108846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Jaffe EA, Hoyer LW, Nachman RL. Synthesis of antihemophilic factor antigen by cultured human endothelial cells. J Clin Invest. 1973;52:2757–2764. doi: 10.1172/JCI107471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wagner DD. The Weibel-Palade body: the storage granule for von Willebrand factor and P-selectin. Thromb Haemost. 1993;70:105–110. [PubMed] [Google Scholar]
- 110.Xu L, Nichols TC, Sarkar R, McCorquodale S, Bellinger DA, Ponder KP. Absence of a desmopressin response after therapeutic expression of factor VIII in hemophilia A dogs with liver-directed neonatal gene therapy. Proc Natl Acad Sci U S A. 2005;102:6080–6085. doi: 10.1073/pnas.0409249102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Horita K, Matsunami H, Shimizu Y, Shimizu A, Kurimoto M, Suzuki K, Tsukadaira T, Arai M. Treatment of a patient with hemophilia A and hepatitis C virus-related cirrhosis by living-related liver transplantation from an obligate carrier donor. Transplantation. 2002;73:1909–1912. doi: 10.1097/00007890-200206270-00010. [DOI] [PubMed] [Google Scholar]
- 112.Lamont PA, Ragni MV. Lack of desmopressin (DDAVP) response in men with hemophilia A following liver transplantation. J Thromb Haemost. 2005;3:2259–2263. doi: 10.1111/j.1538-7836.2005.01553.x. [DOI] [PubMed] [Google Scholar]
- 113.Do H, Healey JF, Waller EK, Lollar P. Expression of factor VIII by murine liver sinusoidal endothelial cells. J Biol Chem. 1999;274:19587–19592. doi: 10.1074/jbc.274.28.19587. [DOI] [PubMed] [Google Scholar]
- 114.Montgomery RR, Gill JC. Interactions between von Willebrand factor and Factor VIII: where did they first meet. J Pediatr Hematol Oncol. 2000;22:269–275. doi: 10.1097/00043426-200005000-00017. [DOI] [PubMed] [Google Scholar]
- 115.Shahani T, Lavend'homme R, Luttun A, Saint-Remy JM, Peerlinck K, Jacquemin M. Activation of human endothelial cells from specific vascular beds induces the release of a FVIII storage pool. Blood. 2010 doi: 10.1182/blood-2009-07-232546. [DOI] [PubMed] [Google Scholar]
- 116.Jacquemin M, Neyrinck A, Hermanns MI, Lavend'homme R, Rega F, Saint-Remy JM, Peerlinck K, Van RD, Kirkpatrick CJ. FVIII production by human lung microvascular endothelial cells. Blood. 2006;108:515–517. doi: 10.1182/blood-2005-11-4571. [DOI] [PubMed] [Google Scholar]
- 117.Kaufman RJ, Wasley LC, Dorner AJ. Synthesis, processing, and secretion of recombinant human factor VIII expressed in mammalian cells. J Biol Chem. 1988;263:6352–6362. [PubMed] [Google Scholar]
- 118.Kaufman RJ, Wasley LC, Davies MV, Wise RJ, Israel DI, Dorner AJ. Effect of von Willebrand factor coexpression on the synthesis and secretion of factor VIII in Chinese hamster ovary cells. Mol Cell Biol. 1989;9:1233–1242. doi: 10.1128/mcb.9.3.1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Shi Q, Fahs SA, Wilcox DA, Kuether EL, Morateck PA, Mareno N, Weiler H, Montgomery RR. Syngeneic transplantation of hematopoietic stem cells that are genetically modified to express factor VIII in platelets restores hemostasis to hemophilia A mice with preexisting FVIII immunity. Blood. 2008;112:2713–2721. doi: 10.1182/blood-2008-02-138214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Yarovoi H, Nurden AT, Montgomery RR, Nurden P, Poncz M. Intracellular interaction of von Willebrand factor and factor VIII depends on cellular context: lessons from platelet-expressed factor VIII. Blood. 2005;105:4674–4676. doi: 10.1182/blood-2004-12-4701. [DOI] [PubMed] [Google Scholar]
- 121.Gewirtz JE, Rauova L, Poncz M. Characterizing Factor VIII Inhibitor Resistance of Platelet-Delivered Factor VIII in the Treatment of Hemophilia A. Blood. 2007;110(11):240a. [Google Scholar]
- 122.Neyman M, Gewirtz J, Poncz M. Analysis of the spatial and temporal characteristics of platelet-delivered factor VIII-based clots. Blood. 2008;112:1101–1108. doi: 10.1182/blood-2008-04-152959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Shi Q, Kuether EL, Cooley BC, Fahs SA, Schroeder JA, Wilcox DA, Montgomery RR. Sustained Phenotypic Correction of Murine Hemophilia A with Pre-Existing Anti-FVIII Immunity Using Lentivirus-Mediated Platelet-Specific FVIII Gene Transfer. ASH Annual Meeting Abstracts. 2009;114:29. [Google Scholar]
- 124.Du LM, Nichols TC, Haberichter SL, Jacobi PM, Jensen ES, Fang J, Shi Q, Montgomery RR, Wilcox DA. Platelet-Targeted Expression of Human BDD-FVIII Reduces Bleeding in Canine Hemophilia A. ASH Annual Meeting Abstracts. 2009;114:691. [Google Scholar]
- 125.Warrier I, Lusher JM. Development of anaphylactic shock in haemophilia B patients with inhibitors. Blood Coagul Fibrinolysis. 1998;9 1:S125–S128. [PubMed] [Google Scholar]
- 126.Astermark J. Treatment of the bleeding inhibitor patient. Semin Thromb Hemost. 2003;29:77–86. doi: 10.1055/s-2003-37972. [DOI] [PubMed] [Google Scholar]
- 127.Lusher JM. Inhibitor antibodies to factor VIII and factor IX: management. Semin Thromb Hemost. 2000;26:179–188. doi: 10.1055/s-2000-9821. [DOI] [PubMed] [Google Scholar]
- 128.Kay MA, Rothenberg S, Landen CN, Bellinger DA, Leland F, Toman C, Finegold M, Thompson AR, Read MS, Brinkhous KM. In vivo gene therapy of hemophilia B: sustained partial correction in factor IX-deficient dogs. Science. 1993;262:117–119. doi: 10.1126/science.8211118. [DOI] [PubMed] [Google Scholar]
- 129.Herzog RW, High KA. Adeno-associated virus-mediated gene transfer of factor IX for treatment of hemophilia B by gene therapy. Thromb Haemost. 1999;82:540–546. [PubMed] [Google Scholar]
- 130.Nathwani AC, Davidoff AM, Hanawa H, Hu Y, Hoffer FA, Nikanorov A, Slaughter C, Ng CY, Zhou J, Lozier JN, Mandrell TD, Vanin EF, Nienhuis AW. Sustained high-level expression of human factor IX (hFIX) after liver-targeted delivery of recombinant adeno-associated virus encoding the hFIX gene in rhesus macaques. Blood. 2002;100:1662–1669. doi: 10.1182/blood-2002-02-0589. [DOI] [PubMed] [Google Scholar]
- 131.Chao H, Walsh CE. AAV vectors for hemophilia B gene therapy. Mt Sinai J Med. 2004;71:305–313. [PubMed] [Google Scholar]
- 132.Niemeyer GP, Herzog RW, Mount J, Arruda VR, Tillson DM, Hathcock J, van Ginkel FW, High KA, Lothrop CD., Jr Long-term correction of inhibitor-prone hemophilia B dogs treated with liver-directed AAV2-mediated factor IX gene therapy. Blood. 2009;113:797–806. doi: 10.1182/blood-2008-10-181479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Hasbrouck NC, High KA. AAV-mediated gene transfer for the treatment of hemophilia B: problems and prospects. Gene Ther. 2008;15:870–875. doi: 10.1038/gt.2008.71. [DOI] [PubMed] [Google Scholar]
- 134.Chang AH, Stephan MT, Lisowski L, Sadelain M. Erythroid-specific human factor IX delivery from in vivo selected hematopoietic stem cells following nonmyeloablative conditioning in hemophilia B mice. Mol Ther. 2008;16:1745–1752. doi: 10.1038/mt.2008.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Bigger BW, Siapati EK, Mistry A, Waddington SN, Nivsarkar MS, Jacobs L, Perrett R, Holder MV, Ridler C, Kemball-Cook G, Ali RR, Forbes SJ, Coutelle C, Wright N, Alison M, Thrasher AJ, Bonnet D, Themis M. Permanent partial phenotypic correction and tolerance in a mouse model of hemophilia B by stem cell gene delivery of human factor IX. Gene Ther. 2006;13:117–126. doi: 10.1038/sj.gt.3302638. [DOI] [PubMed] [Google Scholar]
- 136.Xu L, O'Malley T, Sands MS, Wang B, Meyerrose T, Haskins ME, Ponder KP. In vivo transduction of hematopoietic stem cells after neonatal intravenous injection of an amphotropic retroviral vector in mice. Mol Ther. 2004;10:37–44. doi: 10.1016/j.ymthe.2004.04.010. [DOI] [PubMed] [Google Scholar]
- 137.Manno CS, Chew AJ, Hutchison S, Larson PJ, Herzog RW, Arruda VR, Tai SJ, Ragni MV, Thompson A, Ozelo M, Couto LB, Leonard DG, Johnson FA, McClelland A, Scallan C, Skarsgard E, Flake AW, Kay MA, High KA, Glader B. AAV-mediated factor IX gene transfer to skeletal muscle in patients with severe hemophilia B. Blood. 2003;101:2963–2972. doi: 10.1182/blood-2002-10-3296. [DOI] [PubMed] [Google Scholar]
- 138.Manno CS, Pierce GF, Arruda VR, Glader B, Ragni M, Rasko JJ, Ozelo MC, Hoots K, Blatt P, Konkle B, Dake M, Kaye R, Razavi M, Zajko A, Zehnder J, Rustagi PK, Nakai H, Chew A, Leonard D, Wright JF, Lessard RR, Sommer JM, Tigges M, Sabatino D, Luk A, Jiang H, Mingozzi F, Couto L, Ertl HC, High KA, Kay MA. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat Med. 2006;12:342–347. doi: 10.1038/nm1358. [DOI] [PubMed] [Google Scholar]
- 139.Murphy SL, High KA. Gene therapy for haemophilia. Br J Haematol. 2008;140:479–487. doi: 10.1111/j.1365-2141.2007.06942.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.High KA. Update on progress and hurdles in novel genetic therapies for hemophilia. Hematology Am Soc Hematol Educ Program. 2007;2007:466–472. doi: 10.1182/asheducation-2007.1.466. [DOI] [PubMed] [Google Scholar]
- 141.Ponder KP. Gene therapy for hemophilia. Curr Opin Hematol. 2006;13:301–307. doi: 10.1097/01.moh.0000239700.94555.b1. [DOI] [PubMed] [Google Scholar]