

Abstract
The human secretory leukocyte protease inhibitor (SLPI) is an 11.7 kD cysteine-rich protein that has been shown to possess anti-protease, anti-inflammatory, and antimicrobial properties. By using a Pichia pastoris strain that overproduces protein disulfide isomerase (PDI), we obtained greater than fivefold higher levels of SLPI than in strains expressing normal levels of PDI and containing multiple copies of the SLPI gene. Elevated levels of PDI also enhanced the specific activity of the secreted SLPI by helping it achieve a proper tertiary structure. Mass spectrometry analysis indicated a greater number of disulfide bonds in the SLPI produced by the PDI overexpresssion strain compared to the SLPI produced in strains with normal PDI levels. Although others have utilized a similar strategy to increase yield, we believe that this is the first example of PDI overexpression being demonstrated to enhance the folding and thus increase the biological activity of a protein produced in the yeast Pichia pastoris.
Keywords: Pichia pastoris, recombinant expression, protein disulfide isomerase, secretory leukocyte protease inhibitor, selectable markers
Introduction
The human secretory leukocyte protease inhibitor (SLPI) gene encodes an 11.7 kDa secreted inhibitor whose main role is to protect epithelial tissues from serine proteases such as trypsin and cathepsin G [1]. The SLPI peptide has been shown to also have antiviral (including HIV), antibacterial and antifungal properties. Additionally, it plays a role in wound healing and a variety of other medically important processes [2]. Because only low levels of SLPI are found in body fluids, heterologous expression is required to provide sufficient amounts of the recombinant peptide for biochemical characterization and biomedical applications. Recently, we reported the expression and purification of human SLPI in the methylotropic yeast Pichia pastoris [3]. Pichia pastoris is a popular host for heterologous protein expression in academia and industry, with over 800 recombinant proteins having been produced in this system. Although the recombinant SLPI was affinity-purified and shown to have biological activity, the yield of SLPI was low. Attempts to increase expression levels, by generating multicopy strains and optimizing culturing parameters, did not significantly raise yields.
We hypothesized that the tertiary protein structure of SLPI was partly responsible for limiting its secretion from P. pastoris. Crystal structure analysis of native SLPI indicated that each of its two domains is stabilized by four disulfide bonds with identical bridging [4]. Although P. pastoris carries out many posttranslational modifications of higher eukaryotic proteins, including the formation of disulfide bonds, it is possible that the high concentration of cysteines in such a small protein would overwhelm the secretory network, resulting in mispaired cysteines and improper folding [5]. Misfolded recombinant proteins could either aggregate inside the ER or be degraded, lowering the yield of the recombinant protein.
Critical to the folding process, protein disulfide isomerase (PDI) is a multifunctional protein that is involved in the folding, assembly, and post-translational modification of proteins in various eukaryotes [6]. A resident of the ER, PDI acts as an enzyme by increasing the rate of oxidation and disulfide bond formation and as a chaperone by facilitating the proper folding of proteins. The P. pastoris PDI gene was recently cloned, and several groups have investigated whether overexpression of the chaperone elevated expression yields of heterologous proteins in the yeast [5, 7, 8]. In one such study, PDI overexpressing strains of P. pastoris yielded a 2–5 fold increase in the levels of exported Pfs25, a Plasmodium falciparum vaccine candidate protein, when compared to a strain containing normal levels of PDI [9]. Not only did the PDI overexpressing strain produce higher amounts of Pfs25, but it also significantly reduced the degree of O-linked glycosylation. However, in other studies, PDI overexpression had little effect, and in one instance; it actually reduced secretion levels of a recombinant protein in a strain that had elevated levels of another chaperone, immunoglobulin binding protein (BiP) [10]. Thus, it is difficult to predict whether PDI can improve secretion of heterologous peptides in P. pastoris [11]. In addition, most studies do not examine how PDI overexpression affects the biological activity or the structure of the secreted protein [6].
Because of the eight intramolecular disulfide bonds in the protease inhibitor peptide, we hypothesized that SLPI would require PDI function for successful folding and export from the cell. Our main goal was to determine if PDI overexpression could enhance the secretion of SLPI as well as influence its structure and functional activity. Another objective was to use a new selectable marker system, based on primary selection with G418 resistance, to efficiently create the strains needed in this study.
Materials and Methods
Media, strains, and microbial techniques
Pichia pastoris strain yJC100 (wild-type) and yGS115 (his4) are derivatives of the original wild-type P. pastoris strain NRRL Y11430 (North Regional Research Laboratories, US Department of Agriculture, Peoria, IL) and have been described previously [12]. P. pastoris cells were cultured at 30°C in either YPD medium (1% yeast extract, 2% peptone, 2% glucose), BMGY (buffered minimal medium with 1% glycerol, 2% peptone, and 1% yeast extract) or BMMY (buffered minimal medium with 0.5% methanol, 2% peptone, and 1% yeast extract). When needed, histidine was added to a final concentration of 50 μg/ml.
Recombinant DNA manipulations were carried out in Escherichia coli strain TOP10 (Invitrogen Corp., Carlsbad, CA). Antibiotics were added to LB medium at the following final concentrations: 100 μg/ml ampicillin and 25 μg/ml Zeocin for plasmid selection. Transformation and other standard recombinant DNA techniques were performed essentially as described in [13]. Plasmid DNA was purified from E. coli cultures using a QIAprep Spin Miniprep Kit (Qiagen, Chatsworth, CA). DNA fragments were purified from agarose gels by using the Geneclean II kit (Qbiogene, Carlsbad, CA). Chromosomal DNA from P. pastoris transformants was prepared using the OmniPrep™ kit from GenoTechnology, Inc. (St. Louis, MO). All ligation junctions in newly synthesized vectors were confirmed by DNA sequencing (Geneway Research, Hayward, CA). Oligonucleotides were synthesized by Sigma Genosys (Plano, TX). Bacterially-produced, recombinant SLPI protein was purchased from R&D Systems (Minneapolis, MN).
Vectors
The 320 bp SLPI coding sequence from pET24a-SLPI [12] was amplified using the primers slpi5ecor1 CAGGAATTCACATGTCTGGTAAAAGC and slpi3not1 CTGGCGGCCGCTGCTTTTACCGGGGA. The PCR product was digested with EcoRI and NotI and ligated into an EcoRI-NotI digested pPICZαB or pBLHIS-SX vector to create pABU1 and pAM20, respectively. The SLPI coding sequence was also PCR amplified with primers slpi5pst1 GAAGCTGCAGGAATGTCTGGTAAAAG and slpi3sac2 GCCGCGGCTGCTTTTACCGGGGAAAC, digested with PstI and SacII, and inserted in the respective sites of pKanB alpha to create pAM1. pPDI was generously provided by Dr. David Narum (National Institute of Allergy and Infectious Diseases, Bethesda, MD).
Pichia pastoris transformation
P. pastoris was electrotransformed as previously described [14]. Transformed cells were allowed to recover for one hour in 1 ml of a 50% 1M sorbitol/50% YPD solution at 30°C and then plated on selective medium containing Zeocin at 100 μg/ml. The posttransformational vector amplification technique was used to isolate multicopy strains [15]. Transformed colonies were purified by streaking for isolated colonies on selective medium, and rapid colony PCR was used to confirm the presence of the SLPI coding sequence in transformed cells [16].
Real-time PCR
Real-time PCR reactions, used to estimate SLPI gene copy number, were performed as described previously [11]. Primers used to amplify the SLPI target gene were allieslpirtfow TTGATACCCCGAACCCGACTC and allieslpirtrev TTTGCCACACATACCCATACAGC. Primers used to amplify the MET2 [16] reference gene were mets100 CGTTCTCGCAACTCTTTCGAA and metas100 CAATGGCATCAGTTATGACGGAAG. Q-gene software was utilized to analyze real-time PCR data [17].
Small scale SLPI expression
Cultures were first grown overnight in YPD medium to stationary phase. On the second day, the optical density was measured, and 1.0 OD600 units of each culture were suspended in 10 mls of BMGY medium and grown overnight. On the third day, the optical density was measured, and 10 OD600 units of each culture were pelleted for 30 seconds at 2000×g at room temperature. The cells were suspended in 10 mls of BMMY medium. The cultures were induced for 48 and 72 hours at 30°C with shaking (225 rpm), adding methanol every 24 hours to compensate for losses from evaporation and metabolism. At each time point, the OD600 of each culture was measured, and 1.0 ml was spun down at 10,000×g for 1 minute to separate cells from extracellular supernatant. The supernatants were transferred to a new microcentrifuge tube, and Protease Arrest protease inhibitor (G Biosciences, St. Louis, MO) was added. Pellets and supernatants were immediately frozen and stored at −80°C. The protocol was scaled up to 100 ml volumes when needed.
Purification of SLPI using cobalt affinity chromatography
The purification protocol was performed according to manufacturer specifications of the Pierce HisPur Cobalt Spin Columns (Rockford, IL) with some modifications, as described previously [3].
Western analysis
Protein concentrations were determined using the Pierce (Rockford, IL) Bicinchoninic Acid (BCA) Protein Assay kit with bovine serum albumin as a standard. Either equivalent amounts of proteins or volumes of extracellular medium from equivalent numbers of cells were loaded onto stacked sodium dodecyl sulfate-polyacrylamide gels (SDS-PAGE) for electrophoresis. For westerns, proteins were transferred onto nitrocellulose using a Mini Trans-Blot Electrophoretic Transfer Cell (Bio-Rad, Hercules, CA) according to the manufacturer’s instructions. Immunoblots were visualized with the Tropix Western SuperStar Immunodetection System (Applied Biosystems, Foster City, CA). The anti-SLPI antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA), and the anti-PDI antibodies were generously provided by Dr. Carl Batt (Cornell University, Ithaca, NY). Secondary antibodies, conjugated to alkaline phosphatase, were purchased from Applied Biosystems. Chemiluminescent signals resulting from alkaline phosphatase activity were captured with a ChemiImager 5500 (Alpha Innotech, San Leandro, CA).
SLPI ELISA
SLPI quantitation was performed using the Quantikine Human SLPI ELISA kit (R&D Systems, Minneapolis, MN) according to the manufacturer’s procedure. Sample absorption was monitored at 450nm using an ELx800 Universal Microplate Reader (Bio-Tek, Inc, Winooski, VT).
SLPI anti-protease fluorescence assay
The Protease Fluorescent Detection Kit (Sigma, St. Louis, MO) was modified to measure SLPI anti-protease activity. Briefly, for the blank sample, 30 μL of incubation buffer (20 mM sodium phosphate with 150 mM sodium chloride, pH 7.6) and 20 μL of FITC-casein substrate were added to a microcentrifuge tube. For the control sample, 25 μL of incubation buffer and 5 μL of trypsin solution in incubation buffer were added to a microcentrifuge tube. The amounts of bacterial and yeast-derived SLPI used in these assays were determined by ELISA and diluted appropriately. For each SLPI test sample, 20 μL incubation buffer, 5 μL SLPI in PBS and 5 μL of trypsin in incubation buffer were added to a microcentrifuge tube. Then 20 μL of FITC-casein substrate was added to the tubes, mixed gently and incubated at 37°C in the dark for 60 minutes. After incubation, 150 μL of the 0.6 N trichloroacetic acid solution was added to each tube. The solution was mixed gently and incubated at 37°C in the dark for 30 minutes. After the tubes were spun for 10 minutes at 10,000×g, 5 μL of the supernatant was added to 500 μL of assay buffer (500 mM Tris buffer, pH 8.5) and mixed gently. 70 μL of the mixture was transferred to a minicell and the fluorescence intensity was measured with excitation at 485 nm and emission wavelength of 535 nm with a TBS-380 Mini-Fluorometer (Turner Biosystems, Sunnyvale, CA). Three trials were performed with each SLPI sample.
Cyanogen bromide digestion and MALDI MS analysis
At room temperature 20 μL of a 0.25 M cyanogen bromide (CNBr) solution in 70% formic acid was added to the protein solution (1 μL, c ~ 1 μg/μL). The mixture was allowed to sit in the dark at room temperature for 12 h. The solvents were evaporated, and the remainder was dissolved in 50 μL of water and evaporated to remove traces of formic acid. The dry sample was re-dissolved in MeCN/water (1:1, 20 μL) and was subjected to MALDI-TOF mass spectrometry.
The dried-droplet method was used for MALDI-TOF sample preparation. The sample solution (1 μL) was mixed with matrix solution (1 μL, saturated CHCA solution in 50% MeCN in 0.1% TFA) on the MALDI-plate. External calibration of MALDI mass spectra was carried out with the ProteoMass™ MALDI calibration kit (Sigma-Aldrich, St. Louis, MO). Ionization was achieved with an N2-laser (λ = 337 nm). MALDI spectra were recorded in positive reflectron mode on an AXIMA curved-field reflectron (CFR) MALDI-TOF mass spectrometer (Kratos/Shimadzu, Columbia, MD)
Results and Discussion
Creation of single and multicopy SLPI strains
We initially created several strains that expressed SLPI protein in a wild type P. pastoris strain (yJC100), which contains one copy of the endogenous P. pastoris PDI gene. The SLPI gene was transformed into the wild type yJC100 strain on plasmids containing either the Zeocin resistance gene (pABU1) or the G418 resistance gene (pAM1) (Table 1). In these plasmids, the SLPI gene was transcribed from the methanol-inducible alcohol oxidase I (AOX1) promoter, which is used for high level expression of proteins in P. pastoris [18]. The AOX1 promoter is repressed by glycerol or glucose but is induced over 1,000 fold in medium containing methanol. Multicopy strains, designated as PB1 and PB2, were also constructed that carried at least 1 copy each of the plasmids pABU1 and pAM1 utilizing both Zeocin and G418 resistance markers in a stepwise fashion (Table 1). Additionally, we created a strain (yJC100:pPDI:pAM1) containing both the SLPI and PDI genes. In the pPDI vector, the P. pastoris PDI gene was under the control of the AOX1 promoter as well. Lastly the his4 strain yGS115 was transformed with a vector harboring SLPI and the HIS4 marker (pAM20).
Table 1.
Comparison of copy number and expression level of SLPI in P .pastoris strains.
| Strain | 1st Selectable Marker | 2nd Selectable Marker | Protein Expressed | SLPI Copy number | SLPI Concentration (mg/L) ELISA |
|---|---|---|---|---|---|
| yGS115:pAM20 | HIS4 | none | SLPI | 1 | 0.16 |
| yJC100:pABU1 | ZeoR | none | SLPI | 1 | 0.20 |
| yJC100:pAM1 | G418R | none | SLPI | 2 | 0.34 |
| yJC100:pABU1:pAM1 (PB1) | ZeoR | G418R | SLPI | 3 | 0.21 |
| yJC100:pAM1:pABU1 (PB2) | G418R | ZeoR | SLPI | 3 | 0.26 |
| yJC100:pPDI:pAM1 | G418R | ZeoR | SLPI, PDI | 1 | 1.70 |
In all strains the expression cassette, containing the AOX1 promoter fused to the SLPI coding sequence, was integrated into the AOX1 locus. Colony PCR analysis confirmed the insertion of PDI and SLPI genes into the P. pastoris genome (data not shown). Real-time PCR was used to estimate copy number of the SLPI gene in each strain, which indicated that the strains ranged from 1 to 3 copies of the SLPI gene (Table 1).
Quantitation of SLPI expression
These strains were grown initially on glycerol-based medium in shake flasks (10 ml volume) to generate biomass and then shifted to methanol-based medium to induce the AOX1 promoter and drive the expression of the SLPI protein. The OD600 of the strains were monitored, and volumes of extracellular supernatant corresponding to equivalent numbers of cells were collected after 72 hours of induction. The extracellular fractions were analyzed by ELISA and western analysis.
The ELISA data indicated that, in strains harboring normal levels of PDI, the SLPI protein was secreted at approximately equivalent levels in all single copy strains but accumulated to only slightly higher levels in strains containing multiple copies of the SLPI gene (Table 1). The choice of selectable markers had no significant effect because there was little difference in the yield of SLPI in these strains (Figure 1, lanes 6, 7 and 9 and Table 1). However, the overexpression of PDI in a strain containing only one copy of the SLPI gene (yJC100:pPDI:pAM1) increased secretion levels over eight-fold compared with strains containing a single copy of SLPI and expressing normal levels of PDI (yGS115:pAM20, yJC100:pABU1) (Table 1). Furthermore, yJC100:pPDI:pAM1 secreted approximately five to eight times as much SLPI as some multicopy SLPI strains possessing wild type levels of PDI in shake flask cultures. While the 1.70 mg/L expression level is low, it would be higher in fermenter conditions.
Figure 1.
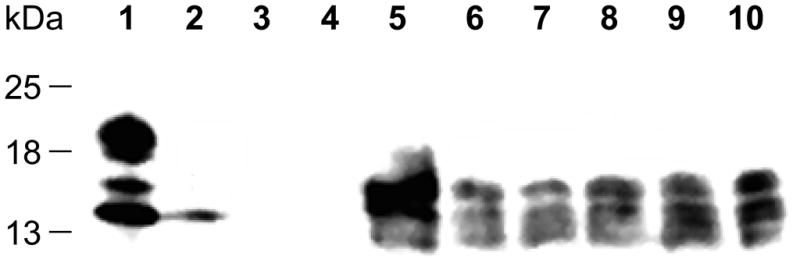
Western blot analysis of SLPI in extracellular fractions of SLPI expression cultures. Volumes of extracellular supernatant corresponding to equivalent numbers of cells were collected after 72 hours of induction and run on a 15% SDS-PAGE gel. Samples were probed with a mouse anti-SLPI polyclonal antibody. Lane 1: purified recombinant SLPI from P. pastoris. Lane 2: purified recombinant SLPI from E. coli (purchased from R&D Systems, Minneapolis, MN). Lane 3: ProSieve protein marker. Lane 4: Untransformed yJC100 strain. Lane 5: yJC100:pPDI:pAM1. Lane 6: yGS115:pAM20. Lane 7: yJC100:pABU1. Lane 8: yJC100:pABU1:pAM1 (PB1). Lane 9: yJC100:pAM1. Lane 10: yJC100:pAM1:pABU1 (PB2).
Western analysis confirmed this ELISA data: the SLPI level of the PDI overexpression strain (Figure 1, lane 5) surpassed that produced by single and multicopy SLPI strains containing only the wild type levels of PDI protein (Figure 1, lanes 6–10). Furthermore, westerns using an anti-PDI antibody confirmed that the 58 kDa PDI was overproduced from an AOX1-PDI expression cassette as expected (Figure 2). The level of PDI overexpression was comparable to that observed in other studies [10].
Figure 2.

Western blot analysis of PDI in intracellular fractions of wild type and PDI overexpression strains. Equivalent numbers of induced cells were loaded into each lane and analyzed with a mouse anti-PDI polyclonal antibody. Lane 1: yJC100:pAM1. Lane 2: yJC100:pPDI:pAM1.
Effect of PDI overexpression on SLPI specific activity
Although previous studies had investigated the effect of elevated PDI levels on the amount of a secreted, heterologous protein, they did not examine the specific activity of the recombinant protein compared to when it was produced in a wild type P. pastoris strain. We hypothesized that increased intracellular concentrations of the chaperone may cause each molecule of exported SLPI to be more correctly folded and thus have higher functional activity than a SLPI protein from a wild type cell. We focused on SLPI’s anti-protease activity. To explore this possibility, trypsin protease and a FITC-casein substrate were mixed with equivalent amounts of SLPI isolated from our wild type expression strain (yJC100:pAM1), our PDI overexpression strain (yJC100:pPDI:pAM1), E. coli (EcSLPI, purchased from R&D Systems, Minneapolis, MN) or no SLPI. Our results indicated that the SLPI from the PDI overexpression strain had slightly higher specific inhibitory activity (42% remaining trypsin activity) compared to the protein produced in a wild type strain (62% remaining trypsin activity) (Figure 3), approximately a 50% increase in inhibitory activity. Although others have reported that PDI overexpression improves secretion yields of recombinant proteins, we believe that this is the first example of PDI overpression being demonstrated to increase the biological activity of a protein produced in the yeast Pichia pastoris.
Figure 3.

Comparison of specific inhibitory activity of SLPI obtained from different sources. 50 ng of trypsin protease and FITC-casein substrate were mixed no inhibitor or with equivalent amounts (45 ng) of SLPI purified from our wild type expression strain (pAM1), our PDI overexpression strain (pAM1::pPDI) and E. coli (EcSLPI). The activity of the trypsin was then quantified using the Protease Fluorescent Detection Kit (Sigma, St. Louis, MO). The data represent results from two separate experiments.
Analysis of SLPI glycosylation
To determine the structural basis for this difference in specific inhibitory activity, we analyzed the glycosylation and disulfide bonding in the SLPI species obtained from the wild type and PDI overexpression strains. In our previous work, SLPI, with an expected molecular mass of 15 kDa, appeared as several species in SDS PAGE gels and westerns. Despite a consensus sequence for N-glycosylation (60NPT62), PNGAse F digestion had no effect on its mobility, suggesting that N-linked glycosylation differences had not caused this phenomenon [3]. The possibility of O-glycosylation could not be excluded. As shown in Figure 1, the SLPI proteins, produced by all the strains regardless of PDI levels, appeared as several species of approximately 15–20 kDa, as reported previously. These results suggested that increased amounts of intracellular PDI did not influence the glycosylation of SLPI.
Mass spectrometry analysis of SLPI
Mass spectrometry was then used to explore if PDI overexpression affected disulfide bonding in SLPI, which contains eight of these bonds. The SLPI produced from a wild type strain was first examined. Following cyanogen bromide digestion of SLPI (Figure 4A), Matrix-Assisted Laser Desorption/Ionization (MALDI)-Time Of Flight (TOF) mass spectrometry of the mixture revealed, besides unidentified impurities, a peptide at m/z 1555.2 (Figure 4B). This mass-to-charge ratio corresponds to the protonated peptide [108DGQCKRDLKCCMGM121+H+] including one oxidative modification, presumably at the non-terminal methionine, and consequential miss of cleavage at that point. The peptide was also obtained from in silico digestion of the completely reduced SLPI amino acid sequence (Protein Prospector, UC San Francsico). The presence of the peptide at m/z 1555.2 in the digestion mixture suggests that incomplete post-translational processing of the disulfide bonds resulted during the expression of SLPI from P. pastoris with wild type levels of PDI activity. Based on the amino acid sequence and the known disulfide bond pattern, incomplete oxidation must have affected Cys111, Cys117, and Cys118. If all disulfide bonds were formed correctly, cyanogen bromide digestion would not result in any fragments of lower mass because all peptide strands will still be held together as can be seen from the amino acid sequence (Figure 4A). Whether the peptide is indeed present in only low abundance as the mass spectral intensity implies or whether the low intensity is caused by partial ion suppression in the mixture of compounds during desorption could not be determined. The same peptide was absent in the digest from SLPI produced in P. pastoris which overexpressed PDI (data not shown). Considering the results from the protease inhibition assay, SLPI from PDI overexpression strains did show more inhibitory power than SLPI from wild type strains, consistent with complete folding and post-translational processing in the former and possibly partial misfold or partially incomplete disulfide processing in the latter.
Figure 4.

Structural analysis of SLPI with mass spectrometry. A) Cyanogen bromide digestion and formation of homoserine lactone of the amino acid sequence of SLPI expressed in P. pastoris. The disulfide bond pattern is symmetrical in the tandem repeat sequence (see UNIPROT data bank entry # P03973-1). B) MALDI-TOF mass spectrum of SLPI expressed in P. pastoris with wild type levels of protein disulfide isomerase (PDI) activity.
Conclusions
Although PDI overexpression did not influence glycosylation, it did increase the secretion levels as well as the specific activity of SLPI peptides produced in P. pastoris. The PDI overexpressing strain with a single copy of the SLPI gene gave higher yields than multicopy SLPI strains containing normal levels of the chaperone, regardless of the choice of selectable marker, in shake flask cultures. Mass spectrometry data detected complete disulfide formation in SLPI peptides from our PDI overexpression strain, demonstrating that the increase in SPLI specific activity resulted presumably from better folding. Some of these disulfide bonds were missing in the SLPI made by the strain with normal PDI levels. While it is widely known that misfolded proteins are degraded prior to secretion, our studies demonstrate that P. pastoris will secrete proteins, such as SLPI, that do not contain optimal folding. Our results suggest that there may be a heterogeneous population of secreted SLPI peptides, with varied levels of disulfide bond formation, produced by the wild type strain. However, an elevated level of PDI in the ER may lead to a higher fraction of secreted proteins that more closely resemble the native state. This finding is intriguing in relation to SLPI, which belongs to a family of low molecular weight, cysteine-rich proteins with multiple functions that have potential for therapeutic applications [19]. Our results suggest that the yield, folding and functional activity of all these proteins may be improved if they are produced in strains with elevated levels of PDI. The availability of several dominant markers in P. pastoris (Zeocin, Blasticidin, and the G418 resistance genes) should make the creation of such strains a fairly easy strategy to pursue [14].
Acknowledgments
This work was supported by NIH-AREA grant GM65882-02 and a University of the Pacific Scholarly and Artistic Activity Grant to J. L.-C. and G. P. L.-C.
Abbreviations
- PDI
protein disulfide isomerase
- SLPI
secretory leukocyte protease inhibitor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Weldon S, McGarry N, Taggart CC, McElvaney NG. The role of secretory leucoprotease inhibitor in the resolution of inflammatory responses. Biochem Soc Trans. 2007;35:273–276. doi: 10.1042/BST0350273. [DOI] [PubMed] [Google Scholar]
- 2.Doumas S, Kolokotronis A, Stefanopoulos P. Anti-inflammatory and antimicrobial roles of secretory leukocyte protease inhibitor. Infect Immun. 2005;73:1271–1274. doi: 10.1128/IAI.73.3.1271-1274.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li Z, Moy A, Sohal K, Dam C, Kuo P, Whittaker J, Whittaker M, Duzgunes N, Konopka K, Franz AH, Lin-Cereghino J, Lin-Cereghino GP. Expression and characterization of recombinant human secretory leukocyte protease inhibitor (SLPI) protein from Pichia pastoris. Protein Expr Purif. 2009;67:175–181. doi: 10.1016/j.pep.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grutter MG, Fendrich G, Huber R, Bode W. The 2.5 A X-ray crystal structure of the acid-stable proteinase inhibitor from human mucous secretions analysed in its complex with bovine alpha-chymotrypsin. Embo J. 1988;7:345–351. doi: 10.1002/j.1460-2075.1988.tb02819.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Inan M, Aryasomayajula D, Sinha J, Meagher MM. Enhancement of protein secretion in Pichia pastoris by overexpression of protein disulfide isomerase. Biotechnol Bioeng. 2006;93:771–778. doi: 10.1002/bit.20762. [DOI] [PubMed] [Google Scholar]
- 6.Wilkinson B, Gilbert HF. Protein disulfide isomerase. Biochim Biophys Acta. 2004;1699:35–44. doi: 10.1016/j.bbapap.2004.02.017. [DOI] [PubMed] [Google Scholar]
- 7.Huo X, Liu Y, Wang X, Ouyang P, Niu Z, Shi Y, Qiu B. Co-expression of human protein disulfide isomerase (hPDI) enhances secretion of bovine follicle-stimulating hormone (bFSH) in Pichia pastoris. Protein Expr Purif. 2007;54:234–239. doi: 10.1016/j.pep.2007.03.016. [DOI] [PubMed] [Google Scholar]
- 8.Vad R, Nafstad E, Dahl LA, Gabrielsen OS. Engineering of a Pichia pastoris expression system for secretion of high amounts of intact human parathyroid hormone. J Biotechnol. 2005;116:251–260. doi: 10.1016/j.jbiotec.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 9.Tsai CW, Duggan PF, Shimp RL, Jr, Miller LH, Narum DL. Overproduction of Pichia pastoris or Plasmodium falciparum protein disulfide isomerase affects expression, folding and O-linked glycosylation of a malaria vaccine candidate expressed in P. pastoris. J Biotechnol. 2006;121:458–470. doi: 10.1016/j.jbiotec.2005.08.025. [DOI] [PubMed] [Google Scholar]
- 10.Damasceno LM, Anderson KA, Ritter G, Cregg JM, Old LJ, Batt CA. Cooverexpression of chaperones for enhanced secretion of a single-chain antibody fragment in Pichia pastoris. Appl Microbiol Biotechnol. 2007;74:381–389. doi: 10.1007/s00253-006-0652-7. [DOI] [PubMed] [Google Scholar]
- 11.Mattanovich D, Gasser B, Hohenblum H, Sauer M. Stress in recombinant protein producing yeasts. J Biotechnol. 2004;113:121–135. doi: 10.1016/j.jbiotec.2004.04.035. [DOI] [PubMed] [Google Scholar]
- 12.Cregg JM, Barringer KJ, Hessler AY, Madden KR. Pichia pastoris as a host system for transformations. Mol Cell Biol. 1985;5:3376–3385. doi: 10.1128/mcb.5.12.3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: A Laboratory Handbook. 2. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, New York: 1989. [Google Scholar]
- 14.Lin-Cereghino J, Hashimoto MD, Moy A, Castelo J, Orazem CC, Kuo P, Xiong S, Gandhi V, Hatae CT, Chan A, Lin-Cereghino GP. Direct selection of Pichia pastoris expression strains using new G418 resistance vectors. Yeast. 2008;25:293–299. doi: 10.1002/yea.1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sunga AJ, Tolstorukov I, Cregg JM. Posttransformational vector amplification in the yeast Pichia pastoris. FEMS Yeast Res. 2008;8:870–876. doi: 10.1111/j.1567-1364.2008.00410.x. [DOI] [PubMed] [Google Scholar]
- 16.Thor D, Xiong S, Orazem C, Kwan AC, Cregg J, Lin-Cereghino J, Lin-Cereghino G. Cloning and characterization of the Pichia pastoris MET2 gene as a selectable marker. FEMS Yeast Research. 2005;5:935–942. doi: 10.1016/j.femsyr.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 17.Muller PY, Janovjak H, Miserez AR, Dobbie Z. Processing of gene expression data generated by quantitative real-time RT-PCR. Biotechniques. 2002;32:1372–1374. 1376, 1378–1379. [PubMed] [Google Scholar]
- 18.Cereghino JL, Cregg JM. Heterologous protein expression in the methylotrophic yeast Pichia pastoris. FEMS Microbiol Rev. 2000;24:45–66. doi: 10.1111/j.1574-6976.2000.tb00532.x. [DOI] [PubMed] [Google Scholar]
- 19.Williams SE, Brown TI, Roghanian A, Sallenave JM. SLPI and elafin: one glove, many fingers. Clin Sci (Lond) 2006;110:21–35. doi: 10.1042/CS20050115. [DOI] [PubMed] [Google Scholar]