

Abstract
Pompe disease is a lysosomal storage disorder caused by the deficiency of acid alpha-glucosidase, the enzyme that degrades glycogen in the lysosomes. The disease manifests as a fatal cardiomyopathy and skeletal muscle myopathy in infants; in milder late-onset forms skeletal muscle is the major tissue affected. We have previously demonstrated that autophagic inclusions in muscle are prominent in adult patients and the mouse model. In this study we have evaluated the contribution of the autophagic pathology in infants before and 6 months after enzyme replacement therapy. Single muscle fibers, isolated from muscle biopsies, were stained for autophagosomal and lysosomal markers and analyzed by confocal microscopy. In addition, unstained bundles of fixed muscles were analyzed by second harmonic imaging. Unexpectedly, the autophagic component which is so prominent in juvenile and adult patients was negligible in infants; instead, the overwhelming characteristic was the presence of hugely expanded lysosomes. After 6 months on therapy, however, the autophagic buildup becomes visible as if unmasked by the clearance of glycogen. In most fibers, the two pathologies did not seem to coexist. These data point to the possibility of differences in the pathogenesis of Pompe disease in infants and adults.
Keywords: Myopathy, Glycogen storage disease, Autophagy, Lysosomal storage
1. Introduction
In a recent interview with the Editor-in-Chief of Autophagy, the Nobel Prize laureate Dr. Christian deDuve, who discovered the lysosome and also coined the term autophagy, reminisced about his hypothesis of lysosomes as cellular “suicide bags”–meaning that under certain conditions “lytic enzymes released from ruptured lysosomes might play a role in [autolysis]” [1]. He went on to say, “It is clear from the work on autophagy, that lysosomes are often involved in the self-destruction of cells. But whether this ever happens through rupture of the organelles, as my suicide-bag hypothesis implied, remains unsettled, as far as I know.” Recent data on the pathogenesis of the lysosomal storage disorder, Pompe disease, has brought this hypothesis to life.
Pompe disease (glycogen storage disease type II) is a rare autosomal recessive metabolic myopathy that affects individuals at any age [2,3]. The disorder is caused by the deficiency of the lysosomal enzyme acid alpha glucosidase (GAA) and results in the accumulation of glycogen in multiple tissues. Clinically, the disease presents as a wide spectrum of phenotypes ranging from the severe rapidly progressive infantile form to milder late-onset variants [2,4,5]. The disease in infants, who have little or no enzyme activity, is characterized by muscle weakness, feeding difficulties, and hypertrophic cardiomyopathy leading to death within the first year of life [4,6]. The late-onset forms, caused by partial enzyme deficiency, manifest with slowly progressive muscle weakness leading to wheelchair and ventilator dependence, and premature death from respiratory insufficiency [5]. The recently approved enzyme replacement therapy (ERT) shows an impressive improvement of cardiac size and function, but the reversal of pathology in skeletal muscle remains a challenge [7].
Skeletal muscle damage and resistance to therapy in Pompe disease have been attributed to lysosomal rupture and release of glycogen and lysosomal enzymes into the cytoplasm [8,9]; in this scenario, the ruptured glycogen-filled lysosomes play the role of de Duve's “suicide bags.” On the other hand, we have shown that dysfunctional autophagy is no less, if not more, critical in the pathogenesis of the disease and in the response of muscle to therapy [10].
Macroautophagy (often referred to as autophagy) is a major intracellular, lysosome-dependent, degradative pathway. This process involves the sequestration of a portion of cytoplasm by double-membrane vesicles, called autophagosomes, which deliver their contents to lysosomes for degradation and recycling. Autophagy is rapidly up-regulated when cells need to generate energy and nutrients during starvation [11,12]. In addition, constitutive autophagy is required to rid the cells of damaged proteins, pathogens, and entire organelles such as defective mitochondria [13]. Autophagy has been implicated in neurodegenerative diseases, malignancy, inflammatory diseases, as well in other lysosomal storage disorders [12,14–16].
We first showed dysfunctional autophagy in our mouse model of Pompe disease, in which skeletal muscle fibers contained large areas of autophagic debris in addition to expanded glycogen-filled lysosomes [10]. Analysis of single muscle fibers from late-onset patients with Pompe disease confirmed that the autophagic buildup is a prominent feature in humans as well [17].
Although both lysosomal expansion and autophagic accumulation are at play in Pompe disease, we now demonstrate that the relative contribution of these two processes in infants and adults is quite different: the first is the main feature in infants, whereas the latter is the predominant feature in juvenile/adults.
2. Materials and methods
2.1. Subjects
Ten Taiwanese patients with a confirmed diagnosis of Pompe disease were included in the study. Seven of them had the most severe infantile form of the disease; of the seven, four were identified through the newborn screening program (NBS3, NBS4, NBS5, and NBS6), and their age of diagnosis ranged from 12 to 33 days. The remaining three infantile patients were diagnosed clinically–their age at diagnosis was 2 months (CLIN5), 2.9 months (CLIN3), and 3.5 months (CLIN4). All infants were CRIM (cross-reactive immunologic material) positive, all were either homozygous or compound heterozygous for a common (p.D645E) mutation, and all are described in detail [18]. In addition, a muscle biopsy was obtained from a 1-month-old infant (NBSL9) who was identified through the newborn screening program but whose genetic makeup (D645E/W746C) was consistent with the late-onset juvenile form of the disease [19].
Two juvenile patients have been included in the study. One of them was diagnosed through newborn screening (NBSL2); the biopsy was obtained prior to the start of ERT when the patient was 3 years of age. The other patient (NBSL9a, a sibling of NBSL9) was identified through a family study at the age of seven. Two previously described Italian late-onset patients whose muscles contained a large amount of autophagic debris [17] are revisited in the current study for fiber typing analysis. All patients are currently being treated with alglucosidase alfa (Myozyme, Genzyme Corp., Framingham, MA) by intravenous injections of 20 mg/kg every other week.
Muscle biopsies were taken before and 6 months after the initiation of therapy in infants [NBS4, NBS5, NBS6, NBSL9, CLIN4, and CLIN5]. For patients NBS3 and CLIN3, muscle biopsies were available only after therapy. Only baseline (before ERT) muscle biopsies were available from juvenile patients. Incisional biopsies were obtained from quadriceps. Biopsies were fixed as soon as possible after collection and transferred to PBS for shipment to the NIH. Local institutional review boards at all sites approved the protocol and all patients or parents provided written informed consent.
2.2. Isolation of fixed single muscle fibers and immunofluorescence microscopy
Muscle fixation, isolation of single fibers, and immunostaining are described in detail [15]. Briefly, muscle was pinned to Sylgard-coated dishes for fixation with 2% paraformaldehyde in 0.1 M phosphate buffer for 1 h, followed by fixation in methanol (−20 °C) for 6 min. Single fibers were obtained by manual teasing. Fibers were placed in a 24-well plate in Blocking Reagent (Vector Laboratories, Burlingame, CA) for 1 h. The fibers were then permeabilized, incubated with primary antibody overnight at 4 °C, washed, incubated with secondary antibody for 2 h, washed again, and mounted in Vectashield (Vector Laboratories, Burlingame, CA) on a glass slide. Immunostained fibers were analysed by confocal microscopy (Zeiss 510 META). Characterization of autofluorescence with 405 nm laser excitation was done with lambda scans using the Zeiss 510 META spectral detector, covering 410–800 nm at 10 nm increments. The dip around 520 nm (shaded area in Fig. 3) is an artefact of the microscope dichroic filter. For confocal analysis, at least 30 fibers were isolated from each biopsy. The following primary antibodies were used: rabbit anti-LC3B (microtubule-associated protein 1 light chain 3) (Sigma, St. Louis, MO); mouse anti-human LAMP-2 (Lysosomal-Associated Membrane Protein 2) (BD Pharmingen, San Diego, CA); mouse anti-myosin slow and anti-myosin fast (both from Sigma). Alexa Fluor-conjugated 488 and 568 secondary antibodies were purchased from Molecular Probes, Eugene, OR.
Fig. 3.

Fiber from juvenile patient NBSL9a was stained for LAMP-2 (red)/LC3 (green), but not with DAPI. Autophagic areas contain autofluorescent structures (blue; arrows); arrowheads—autofluorescence-negative nuclei. (A) DIC (Differential Interference Contrast), (B) DIC and autofluorescence, (C) LAMP-2/LC3 staining and autofluorescence, (D) Characterization of autofluorescence signal in autophagic area. Bar: 10 microns. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
2.3. Second harmonic generation (SHG) imaging microscopy
Two-photon excited fluorescence (2PEF) and SHG images [20] were collected on a Leica SP5 NLO confocal microscope equipped with a 3W Mai Tai HP Ti:sapphire laser. A 40 × 1.25NA oil immersion objective was used; the excitation wavelength was 870 nm. SHG in the forward direction was imaged in the transmitted light detector after filtration with a 435/20 bandpass filter. Backscattered auto-fluorescence was collected in a non-descanned detector. Confocal images were assembled into montages and linearly enhanced, when needed, with Adobe CS4 on a Macintosh computer.
3. Results
We have previously demonstrated in our mouse model of Pompe disease the presence of large areas of autophagic accumulation in myofibers [10]. The areas are located in the core of the fibers, often spanning their entire length and growing as the disease progresses. Inside these areas we can identify enlarged clustered LAMP-positive lysosomes and LC3-positive vesicles called autophagosomes (LC3 is a highly specific marker for autophagosomes [21,22]). In adult patients (Fig. 1A), even more so than in mice (Fig. 1B), the enormous autophagic buildup often dwarfs the enlarged glycogen-filled lysosomes that lie outside the autophagic region [17].
Fig. 1.
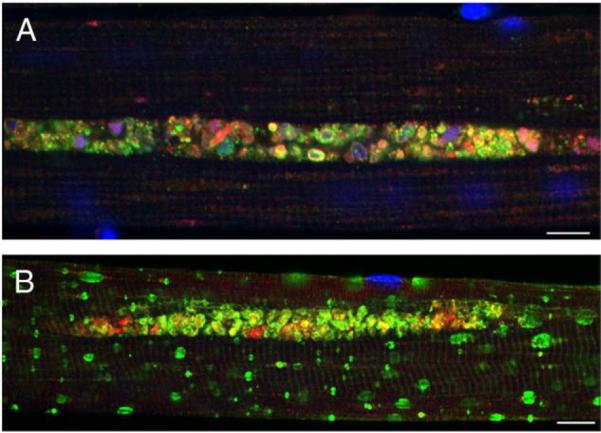
Single fibers from an adult Italian patient (A) and a Pompe mouse (B) were stained for LAMP-2 (green) and LC3 (red). Nuclei were stained with DAPI. Autophagic accumulation appears to be the only pathology in human fibers. Bar: 10 microns. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
We have extended these data by adding two additional late-onset juvenile patients (ages 3 and 7 years old) from Taiwan. They too, contain autophagic accumulation in many fibers as shown by the presence of LC3-positive vesicles in the vicinity of expanded lysosomes (Fig. 2A). Similar to what is observed in adult patients, this area of autophagic accumulation is located in the core of the muscle fiber (occasionally the area is found closer to the sarcolemma) (Fig. 2A–C, E). The lysosomal pathology is strikingly negligible in the surrounding region although a small proportion of the fibers do contain expanded lysosomes (most are less than 1μm in length; a few reach 5 μm) that are distributed throughout the fiber, a pathology presumed to be typical for this disease (Fig. 2D). In addition, many fibers have bizarre lakes of amorphous material with barely identifiable lysosomes and autophagosomes; these regions contain structures that resemble nuclei but, in fact, represent autofluorescent material as indicated by analysis of unstained fibers (Fig. 3).
Fig. 2.

Single fibers from Taiwanese juvenile patients (before ERT) were stained with LAMP-2 (red) and LC3 (green). Autophagic accumulation is a predominant pathology in juvenile patients (shown for NBSL9a in A, B, C, E); some fibers exhibit only lysosomal pathology (shown for NBSL2 in D). Bar: 10 microns. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
We have also checked if the accumulation of debris is limited to type II fast glycolytic fibers—a phenomenon observed in the Pompe mouse model [23,24]. Unlike our findings in mice, the autophagic accumulation is found in fast fibers from one adult patient (not shown) and slow fibers from the other (Fig. 4). Of the two juvenile patients from the current study, one contains the debris in fast fibers (NBSL2) and the other in both fast and slow fibers (NBSL9a) (not shown).
Fig. 4.
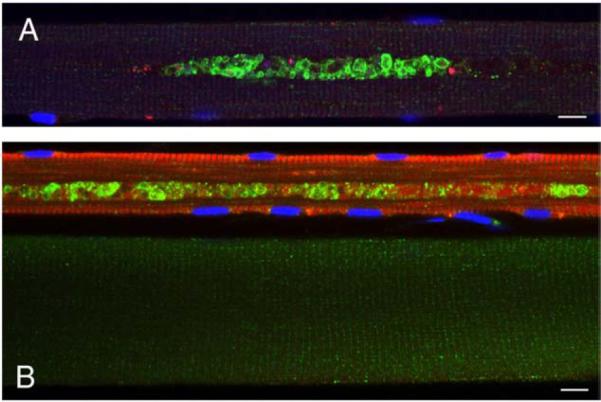
Autophagic accumulation in slow fibers; fibers from an adult Italian patient were stained for LC3 (green) and fast myosin (red) in A or slow myosin in B. Bar: 10 microns. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Since the autophagic pathology and the extent of the area occupied by the debris are so prominent in fibers from late-onset juvenile and adult patients, we expected an even greater contribution in the more severe infantile form of the disease. Human muscle biopsies, in particular infantile samples, are extremely hard to come by, but we had a rare opportunity to obtain them through a newborn screening program for lysosomal storage diseases in Taiwan. Of note, the isolation of single fibers from infantile biopsies is a daunting task considering the extent of fiber damage and their small diameter (often less than 10 μm).
Immunostaining of muscle fibers from infants before ERT shows occasional autophagosomes (arrows in Fig. 5A) or clusters of autophagosomes (arrows in Fig. 5B) in some areas of the fibers, suggesting that in infants, as in the adults, autophagy is up-regulated. The number of fibers containing autophagosomes varies among the patients, but never reaches more than 20–25%, and none of the fibers have the massive autophagic buildup which is so prominent in the adult patients. Instead, the overwhelming characteristic is the presence of hugely expanded (10–15 μm) often clustered, glycogen-filled lysosomes, which almost overtake the entire fiber. These lysosomes have an irregular shape, broken borders, and are often seen joined together (Fig. 5C–F and Video 1); the coalesced structures can reach up to 30 μm (arrows in Fig. 5F). The degree of the lysosomal pathology in fibers from every infant varies dramatically ranging from minimally affected fibers (Fig. 5D and F top) to those totally devoid of the contractile elements (Fig. 5C dashed line).
Fig. 5.

Lysosomal pathology is predominant in infants. Fibers were stained for LAMP-2 (red)/LC3 (green). Clusters of autophagosomes are seen in some fibers [A (projection) and B]. Fibers from CLIN4 are shown in A, B, C, and F; fibers from NBS6 are shown in D and E. The dashed line marks a fiber that appears to be devoid of contractile elements. Bar: 10 microns. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Although there has been clinical improvement in most patients after 6 months on ERT [18], the lysosomal pathology in some fibers appeared unchanged (Fig. 6A–D and Video 2). Remarkably, autophagic accumulation becomes prominent in infants treated with ERT as shown by the presence of LC3 positive autophagosomes adjacent to lysosomes in the core of the fiber (Fig. 6E, F, and I); many of these vesicles contain dense autofluorescent material within balloon-like structures that reach up to 5–7 μm (Fig. 6G, H, and J). These abnormalities closely resemble what was described above for the two juvenile patients as well as for the previously described adult patients. Interestingly, the autophagic pathology in fibers from ERT-treated infants seems to develop in the fibers in which the lysosomal defect is minimal or absent.
Fig. 6.

Autophagic accumulation is noticeable on ERT. Fibers from infants were stained for LAMP-2 (red)/LC3 (green). Left: a variable response to ERT in NBS6 (A), CLIN3 (B, D), and CLIN5 (C). Right: autophagic buildup emerges upon ERT [CLIN3 (E, G, H, J) and NBS4 (F, I)]. Bar: 10 microns. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
The results from single fiber immunofluorescence were reinforced by SHG and 2PEF imaging of fiber bundles (Fig. 7). SHG visualizes myosin while 2PEF visualizes autofluorescence. We have previously demonstrated that the combination of these techniques highlights the defects found in muscles of Pompe patients and of the knockout mouse model [20]. In an untreated infant (Fig. 7A) the fibers are small with numerous interruptions of the myosin bands, mostly of lysosomal appearance (holes, arrowheads); the areas of autophagy (arrows) are not frequent and are typically limited in length. In a patient after 6 months on therapy (Fig. 7B), the myosin pattern is more regular and there are few holes (arrowheads); however highly damaged fibers can still be seen (arrow). Both an infant on ERT and a juvenile patient (Fig. 7C and D) show large highly autofluorescent particles (tick signs) and autophagic accumulations (arrow) that span a significant portion of the fibers.
Fig. 7.

Combination of SHG (green) and 2PEF (red) microscopies highlights lysosomal holes (arrowheads), autophagic accumulations (arrows, except in B, damaged fiber), nucleus (asterisk) and autofluorescent particles (tick marks) in 3 mo-old pre-ERT infant (A; CLIN4), post-ERT infants (B; CLIN3, C; NBS3) and juvenile (D; NBLS2). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
We also had a chance to analyze by confocal microscopy an unusual case (NBSL9) before (1 month old) and 6 months after therapy. This patient was identified through the newborn screening program, was asymptomatic, had an elevated CK level, and carried a mutation which would predict a milder late-onset variant of the disease. Before therapy the fibers were well preserved, had no autophagic buildup, and contained minimally enlarged (1–2 μm) lysosomes (Fig. 8A). After therapy all fibers analyzed looked normal (Fig. 8B).
Fig. 8.

Effect of ERT in a patient (NBSL9) who started therapy at 1 month of age although his genetic defect would predict a late-onset form of the disease. Fibers were stained with LAMP-2 (red) and LC3 (green). (A) Before ERT. (B) 6m after ERT. Bar: 10 microns. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
In conclusion, autophagic buildup is initially negligible in myofibers from infantile patients and therefore cannot be used as an early prognostic factor; however, since the buildup emerges with time, its rate and extent may prove helpful in predicting the later course of the disease.
4. Discussion
The recently approved enzyme replacement therapy with recombinant human acid alpha-glucosidase has profoundly changed the natural course of Pompe disease in infants. Without therapy the affected infants usually develop symptoms by about 3 months of age; they rarely survive beyond 1 year and most die from cardiorespiratory failure at a median age of 6–7 months [4,6]. The results of the published trials in infants showed very impressive improvement in cardiac size and function, but the clearance of skeletal muscle glycogen appears to be less effective than anticipated [9,25,26]. Early diagnostics through newborn screening and early treatment of pre-symptomatic patients hold promise to significantly improve the therapy of skeletal muscle as has been recently shown in a group of Taiwanese patients [18].
The outcome of the completed and ongoing open-label clinical trials in late-onset forms, in which cardiac muscle is spared, confirmed the difficulty of treating skeletal muscle. The therapy seems to stabilize the progression of the disease, but no major effects were observed after 1 year on ERT [27]. Other studies showed the stabilization of pulmonary and muscle function and improvement in the quality of life [28,29]. The recently published first randomized, double-blind, placebo-controlled study of 90 late-onset patients has shown a modest positive effect of therapy [30].
The limitations of ERT in Pompe disease and the challenges of treating skeletal muscle include inefficient targeting of the drug to skeletal muscle, the clearance of the enzyme by the liver, and antibody responses to the exogenous protein in the CRIM-negative infantile patients [31–33]. In addition to these limitations of ERT, there is also variability in how each individual responds to therapy. The age when the first symptoms occur, the timing of intervention, and the extent of lysosomal pathology have been considered critical factors that determine the outcome of therapy [25,26,34,35].
We have demonstrated in adult patients with the disease and in our Pompe mouse model that the pathology involves not only lysosomal expansion but also massive autophagic accumulation. Furthermore, we have shown that the autophagic buildup interferes with the delivery of the recombinant enzyme in mice [24]. The current study has been undertaken to determine whether the autophagic abnormalities contribute to the pathology in infants. This study became possible through the newborn screening program for Pompe disease implemented in Taiwan where the infantile form of the disease is the most common glycogen storage disorder with an estimated frequency of one in ~33,000 [36,37]. The rationale behind this newborn screening program was to compare the efficacy of ERT in the asymptomatic infants diagnosed through the program (NBS group, who started therapy at less than 1m of age) and in infants diagnosed clinically (CLIN group, who started therapy at the age of 2–6 m); the study showed the benefits of early treatment [18].
We have analyzed baseline muscle biopsies from three patients from the NBS group and two from the CLIN group. All infants carried a mutation which is common among Taiwanese with Pompe disease and is due to a founder effect [36]. The pathology in the fibers from these infants was quite different from that in the previously described adult patients or in muscle fibers from the knockout model. The autophagic accumulation was minimal in both the NBS and CLIN groups, and therefore, could have no effect on the initial outcome of ERT. The lysosomal expansion seemed to be the main pathology, and the uneven shape of the hugely enlarged lysosomes without clear borders suggests the possibility of lysosomal rupture, a hypothesis put forward by Christian de Duve and applied to Pompe disease by JL Griffin in 1984 [38]. The rupture hypothesis was later updated by B. Thurberg and colleagues based on more recent light and electron microscopy data [9].
In characterizing the disease progression in infants, Thurberg and colleagues proposed five stages: at the early stage, muscle cells contain small, glycogen-filled lysosomes; this is followed by enlargement of the lysosomes and leakage of glycogen into the cytoplasm in some areas; as the disease progresses, lysosomal rupturing continues until the majority of glycogen is cytoplasmic, leading to a complete breakdown of muscle architecture [9].
This description, however, created an impression that lysosomal expansion and rupture is the universal pathology in Pompe disease. On the other hand, our findings in adult patients and in the mouse model, suggested a prominent role for autophagy in the pathogenesis of Pompe disease and again created an impression that this pathology is relevant to all forms.
We suggest here that both views are valid, but that these two pathologies do not usually present together in the same fiber. In untreated infants lysosomal rupture likely causes muscle damage, while in untreated late-onset patients well-confined areas of debris, without any widespread lysosomal expansion, appear to destroy the fiber. The therapy, however, changes this equation: fibers from ERT-treated infants show areas in which the lysosomal pathology is reversed, areas which still contain expanded lysosomes, and areas of autophagic buildup; curiously, it seems that the autophagic pathology emerges as the lysosomal pathology subsides. A much larger sample size, however, is required to establish if the two pathologies are indeed separate in most fibers and to what extent the fibers with predominantly lysosomal or autophagic abnormalities co-exist in muscle from the same individual. In addition, long term follow up is needed to determine if autophagic buildup remains limited in size or whether it continues to spread out in patients on ERT.
Supplementary Material
Acknowledgments
This research was supported by the Intramural Research Program of the NIAMS of the NIH.
Financial disclosure Drs. Chien and Hwu received honoraria and research grant funding from Genzyme. Dr. Hwu is a member of the Pompe disease advisory board for Genzyme. Rebecca Baum's fellowship is supported by a Cooperative Research and Development Agreement (CRADA) between NIAMS and the Genzyme Corporation for studies related to Pompe disease. The other authors have no financial interests to disclose.
Abbreviations
- GAA
acid alpha glucosidase
- ERT
enzyme replacement therapy
- SHG
second harmonic generation
- 2PEF
two-photon excited fluorescence
- LAMP-2
lysosomal-associated membrane protein 2
- LC3
microtubule-associated protein 1 light chain 3
Footnotes
Supplementary data to this article can be found online at doi:10.1016/j.ymgme.2010.08.001.
Reference
- [1].Klionsky DJ. Autophagy revisited: a conversation with Christian de Duve. Autophagy. 2008;4:740–743. doi: 10.4161/auto.6398. [DOI] [PubMed] [Google Scholar]
- [2].Hirschhorn R, Reuser AJ. Glycogen storage disease type II: acid alpha-glucosidase (acid maltase) deficiency. In: Scriver C, Beaudet A, Sly W, Valle D, editors. The Metabolic and Molecular Basis of Inherited Disease. McGraw-Hill; New York, NY: 2001. pp. 3389–3420. [Google Scholar]
- [3].Engel AG, Hirschhorn R, Huie ML. Acid Maltase Deficiency. In: Engel AG, Franzini-Armstrong C, editors. Myology. McGraw-Hill; New York, NY: 2003. pp. 1559–1586. [Google Scholar]
- [4].Kishnani PS, Hwu WL, Mandel H, Nicolino M, Yong F, Corzo D. A retrospective, multinational, multicenter study on the natural history of infantile-onset Pompe disease. J. Pediatr. 2006;148:671–676. doi: 10.1016/j.jpeds.2005.11.033. [DOI] [PubMed] [Google Scholar]
- [5].Winkel LP, Hagemans ML, Van Doorn PA, Loonen MC, Hop WJ, Reuser AJ, Van der Ploeg AT. The natural course of non-classic Pompe's disease; a review of 225 published cases. J. Neurol. 2005;252:875–884. doi: 10.1007/s00415-005-0922-9. [DOI] [PubMed] [Google Scholar]
- [6].van den Hout HM, Hop W, van Diggelen OP, Smeitink JA, Smit GP, Poll-The BT, Bakker HD, Loonen MC, de Klerk JB, Reuser AJ, Van der Ploeg AT. The natural course of infantile Pompe's disease: 20 original cases compared with 133 cases from the literature. Pediatrics. 2003;112:332–340. doi: 10.1542/peds.112.2.332. [DOI] [PubMed] [Google Scholar]
- [7].Schoser B, Hill V, Raben N. Therapeutic approaches in glycogen storage disease type II/Pompe disease. Neurotherapeutics. 2008;5:569–578. doi: 10.1016/j.nurt.2008.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Griffin JL. Infantile acid maltase deficiency. I. Muscle fiber destruction after lysosomal rupture. Virchows Arch. B Cell Pathol. Incl. Mol. Pathol. 1984;45:23–36. doi: 10.1007/BF02889849. [DOI] [PubMed] [Google Scholar]
- [9].Thurberg BL, Lynch MC, Vaccaro C, Afonso K, Tsai AC, Bossen E, Kishnani PS, O'Callaghan M. Characterization of pre- and post-treatment pathology after enzyme replacement therapy for pompe disease. Lab. Invest. 2006;86:1208–1220. doi: 10.1038/labinvest.3700484. [DOI] [PubMed] [Google Scholar]
- [10].Fukuda T, Ewan L, Bauer M, Mattaliano RJ, Zaal K, Ralston E, Plotz PH, Raben N. Dysfunction of endocytic and autophagic pathways in a lysosomal storage disease. Ann. Neurol. 2006;59:700–708. doi: 10.1002/ana.20807. [DOI] [PubMed] [Google Scholar]
- [11].He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 2009;43:67–93. doi: 10.1146/annurev-genet-102808-114910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Komatsu M, Ueno T, Waguri S, Uchiyama Y, Kominami E, Tanaka K. Constitutive autophagy: vital role in clearance of unfavorable proteins in neurons. Cell Death Differ. 2007;14:887–894. doi: 10.1038/sj.cdd.4402120. [DOI] [PubMed] [Google Scholar]
- [14].Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Raben N, Shea L, Hill V, Plotz P. Monitoring autophagy in lysosomal storage disorders. Methods Enzymol. 2009;453:417–449. doi: 10.1016/S0076-6879(08)04021-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ballabio A, Gieselmann V. Lysosomal disorders: from storage to cellular damage. Biochim. Biophys. Acta. 2009;1793:684–696. doi: 10.1016/j.bbamcr.2008.12.001. [DOI] [PubMed] [Google Scholar]
- [17].Raben N, Takikita S, Pittis MG, Bembi B, Marie SKN, Roberts A, Page L, Kishnani PS, Schoser BGH, Chien YH, Ralston E, Nagaraju K, Plotz PH. Deconstructing Pompe disease by analyzing single muscle fibers. Autophagy. 2007;3:546–552. doi: 10.4161/auto.4591. [DOI] [PubMed] [Google Scholar]
- [18].Chien YH, Lee NC, Thurberg BL, Chiang SC, Zhang XK, Keutzer J, Huang AC, Wu MH, Huang PH, Tsai FJ, Chen YT, Hwu WL. Pompe disease in infants: improving the prognosis by newborn screening and early treatment. Pediatrics. 2009;124:e1116–e1125. doi: 10.1542/peds.2008-3667. [DOI] [PubMed] [Google Scholar]
- [19].Wan L, Lee CC, Hsu CM, Hwu WL, Yang CC, Tsai CH, Tsai FJ. Identification of eight novel mutations of the acid alpha-glucosidase gene causing the infantile or juvenile form of glycogen storage disease type II. J. Neurol. 2008;255:831–838. doi: 10.1007/s00415-008-0714-0. [DOI] [PubMed] [Google Scholar]
- [20].Ralston E, Swaim B, Czapiga M, Hwu WL, Chien YH, Pittis MG, Bembi B, Schwartz O, Plotz P, Raben N. Detection and imaging of non-contractile inclusions and sarcomeric anomalies in skeletal muscle by second harmonic generation combined with two-photon excited fluorescence. J. Struct. Biol. 2008;162:500–508. doi: 10.1016/j.jsb.2008.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kabeya Y, Mizushima N, Yamamoto A, Oshitani-Okamoto S, Ohsumi Y, Yoshimori T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J. Cell Sci. 2004;117:2805–2812. doi: 10.1242/jcs.01131. [DOI] [PubMed] [Google Scholar]
- [23].Raben N, Fukuda T, Gilbert AL, de Jong D, Thurberg BL, Mattaliano RJ, Meikle P, Hopwood JJ, Nagashima K, Nagaraju K, Plotz PH. Replacing acid alpha-glucosidase in Pompe disease: recombinant and transgenic enzymes are equipotent, but neither completely clears glycogen from type II muscle fibers. Mol. Ther. 2005;11:48–56. doi: 10.1016/j.ymthe.2004.09.017. [DOI] [PubMed] [Google Scholar]
- [24].Fukuda T, Ahearn M, Roberts A, Mattaliano RJ, Zaal K, Ralston E, Plotz PH, Raben N. Autophagy and mistargeting of therapeutic enzyme in skeletal muscle in pompe disease. Mol. Ther. 2006;14:831–839. doi: 10.1016/j.ymthe.2006.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kishnani PS, Corzo D, Nicolino M, Byrne B, Mandel H, Hwu WL, Leslie N, Levine J, Spencer C, McDonald M, Li J, Dumontier J, Halberthal M, Chien YH, Hopkin R, Vijayaraghavan S, Gruskin D, Bartholomew D, Van der PA, Clancy JP, Parini R, Morin G, Beck M, De la Gastine GS, Jokic M, Thurberg B, Richards S, Bali D, Davison M, Worden MA, Chen YT, Wraith JE. Recombinant human acid [alpha]-glucosidase: major clinical benefits in infantile-onset Pompe disease. Neurology. 2007;68:99–109. doi: 10.1212/01.wnl.0000251268.41188.04. [DOI] [PubMed] [Google Scholar]
- [26].Nicolino M, Byrne B, Wraith JE, Leslie N, Mandel H, Freyer DR, Arnold GL, Pivnick EK, Ottinger CJ, Robinson PH, Loo JC, Smitka M, Jardine P, Tato L, Chabrol B, McCandless S, Kimura S, Mehta L, Bali D, Skrinar A, Morgan C, Rangachari L, Corzo D, Kishnani PS. Clinical outcomes after long-term treatment with alglucosidase alfa in infants and children with advanced Pompe disease. Genet. Med. 2009;11:210–219. doi: 10.1097/GIM.0b013e31819d0996. [DOI] [PubMed] [Google Scholar]
- [27].Strothotte S, Strigl-Pill N, Grunert B, Kornblum C, Eger K, Wessig C, Deschauer M, Breunig F, Glocker FX, Vielhaber S, Brejova A, Hilz M, Reiners K, Muller-Felber W, Mengel E, Spranger M, Schoser B. Enzyme replacement therapy with alglucosidase alfa in 44 patients with late-onset glycogen storage disease type 2: 12-month results of an observational clinical trial. J. Neurol. 2009 doi: 10.1007/s00415-009-5275-3. [DOI] [PubMed] [Google Scholar]
- [28].Winkel LP, Van den Hout JM, Kamphoven JH, Disseldorp JA, Remmerswaal M, Arts WF, Loonen MC, Vulto AG, Van Doorn PA, de Jong G, Hop W, Smit GP, Shapira SK, Boer MA, van Diggelen OP, Reuser AJ, Van der Ploeg AT. Enzyme replacement therapy in late-onset Pompe's disease: a three-year follow-up. Ann. Neurol. 2004;55:495–502. doi: 10.1002/ana.20019. [DOI] [PubMed] [Google Scholar]
- [29].Rossi M, Parenti G, Della CR, Romano A, Mansi G, Agovino T, Rosapepe F, Vosa C, Del Giudice E, Andria G. Long-term enzyme replacement therapy for pompe disease with recombinant human alpha-glucosidase derived from Chinese hamster ovary cells. J. Child Neurol. 2007;22:565–573. doi: 10.1177/0883073807302598. [DOI] [PubMed] [Google Scholar]
- [30].Van der Ploeg AT, Clemens PR, Corzo D, Escolar DM, Florence J, Groeneveld GJ, Herson S, Kishnani PS, Laforet P, Lake SL, Lange DJ, Leshner RT, Mayhew JE, Morgan C, Nozaki K, Park DJ, Pestronk A, Rosenbloom B, Skrinar A, van Capelle CI, van der Beek NA, Wasserstein M, Zivkovic SA. A randomized study of alglucosidase alfa in late-onset Pompe's disease. N Engl J. Med. 2010;362:1396–1406. doi: 10.1056/NEJMoa0909859. [DOI] [PubMed] [Google Scholar]
- [31].Raben N, Danon M, Gilbert AL, Dwivedi S, Collins B, Thurberg BL, Mattaliano RJ, Nagaraju K, Plotz PH. Enzyme replacement therapy in the mouse model of Pompe disease. Mol. Genet. Metab. 2003;80:159–169. doi: 10.1016/j.ymgme.2003.08.022. [DOI] [PubMed] [Google Scholar]
- [32].Amalfitano A, Bengur AR, Morse RP, Majure JM, Case LE, Veerling DL, Mackey J, Kishnani P, Smith W, McVie-Wylie A, Sullivan JA, Hoganson GE, Phillips III JA, Schaefer GB, Charrow J, Ware RE, Bossen EH, Chen YT. Recombinant human acid alpha-glucosidase enzyme therapy for infantile glycogen storage disease type II: results of a phase I/II clinical trial. Genet. Med. 2001;3:132–138. doi: 10.109700125817-200103000-00007. [DOI] [PubMed] [Google Scholar]
- [33].Kishnani PS, Goldenberg PC, DeArmey SL, Heller J, Benjamin D, Young S, Bali D, Smith SA, Li JS, Mandel H, Koeberl D, Rosenberg A, Chen YT. Cross-reactive immunologic material status affects treatment outcomes in Pompe disease infants. Mol. Genet. Metab. 2010;99:26–33. doi: 10.1016/j.ymgme.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kishnani PS, Corzo D, Leslie ND, Gruskin D, Van der PA, Clancy JP, Parini R, Morin G, Beck M, Bauer MS, Jokic M, Tsai CE, Tsai BW, Morgan C, O'Meara T, Richards S, Tsao EC, Mandel H. Early treatment with alglucosidase alpha prolongs long-term survival of infants with Pompe disease. Pediatr. Res. 2009;66:329–335. doi: 10.1203/PDR.0b013e3181b24e94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Kishnani PS, Nicolino M, Voit T, Rogers RC, Tsai AC, Waterson J, Herman GE, Amalfitano A, Thurberg BL, Richards S, Davison M, Corzo D, Chen YT. Chinese hamster ovary cell-derived recombinant human acid alpha-glucosidase in infantile-onset Pompe disease. J. Pediatr. 2006;149:89–97. doi: 10.1016/j.jpeds.2006.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Shieh JJ, Lin CY. Frequent mutation in Chinese patients with infantile type of GSD II in Taiwan: evidence for a founder effect. Hum. Mutat. 1998;11:306–312. doi: 10.1002/(SICI)1098-1004(1998)11:4<306::AID-HUMU8>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- [37].Chien YH, Chiang SC, Zhang XK, Keutzer J, Lee NC, Huang AC, Chen CA, Wu MH, Huang PH, Tsai FJ, Chen YT, Hwu WL. Early detection of Pompe disease by newborn screening is feasible: results from the Taiwan screening program. Pediatrics. 2008;122:e39–e45. doi: 10.1542/peds.2007-2222. [DOI] [PubMed] [Google Scholar]
- [38].Griffin JL. Infantile acid maltase deficiency. I. Muscle fiber destruction after lysosomal rupture. Virchows Arch. B Cell Pathol. Incl. Mol. Pathol. 1984;45:23–36. doi: 10.1007/BF02889849. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.