

Abstract
Objective
Dipeptidyl Peptidase 2 (DPP2/DPP7) is a regulator of quiescence as inhibition of DPP2 results in apoptosis of resting, but not activated lymphocytes. The purpose of the present study was to investigate the prognostic value of DPP2 inhibition and the role of DPP2 in cell cycle in chronic lymphocytic leukemia (CLL).
Methods
We screened 152 peripheral blood samples from patients with CLL in an apoptosis assay with AX8819, a DPP2-specific inhibitor. The apoptotic response was correlated with B-cell receptor signaling and cell cycle, and molecular prognostic factors.
Results
We categorized CLL into two prognostic subgroups. Inhibition of DPP2 induced apoptosis in 60% of CLL, while 40% were resistant to apoptosis. Resistance to apoptosis correlated with unmutated IgVH and increased ZAP-70 expression and was associated with an unfavorable clinical outcome. Sensitive CLL B-cells expressed high p27, low c-Myc protein levels and decreased Syk phosphorylation, indicative of a resting phenotype. DPP2 inhibition in those cells resulted in apoptosis accompanied by enhanced phosphorylation of Syk, degradation of p27 and p130 and upregulation of c-Myc, indicative of activation and inappropriate cell cycle entry. Resistant CLL demonstrated baseline low p27 and high c-Myc protein levels and increased pSyk, indicative of an activated phenotype. Inhibition of hsp90 in this subset of CLL partially reversed apoptosis resistance.
Conclusion
The DPP2 apoptosis assay provides a reliable prognostic factor in CLL. CLL B-cells sensitive to DPP2 inhibition are in true G0, while resistant CLL B-cells are partially activated. DPP2 inhibition alone or with concomitant inhibition of hsp90 warrants investigation as a therapeutic modality in CLL.
INTRODUCTION
Chronic lymphocytic leukemia (CLL) is the most common hematologic malignancy in the Western hemisphere. CLL is characterized by the heterogeneity of clinical outcomes. While some patients follow an indolent course, others rapidly develop disease complications, requiring treatment within a short time after diagnosis. Such heterogeneity in the clinical course points to the complexity of B-cell biology in CLL, while the prevailing pathogenic model of CLL as an accumulation of long-lived B-lymphocytes arrested in G0 does not explain this phenomenon. In fact, recent data indicate that patients with CLL experience a substantial leukemic cell turnover (1). Moreover, a proportion of peripheral blood CLL B-cells are “caught” entering cell cycle, demonstrated by Ki-67 staining (2). Furthermore, patients with a rapid turnover of leukemic stem cells, as evidenced by the incorporation of the deuterated water, manifest progressive disease (1). A number of markers have found widespread use in clinical practice to predict the course of the disease. Interphase cytogenetic abnormalities, expression of CD38 and ZAP-70, and percentage of IgVH somatic hypermutations vary between patients and have prognostic significance in CLL (3). Inappropriate expression of the T cell specific kinase ZAP-70 has been reported in some studies to be the strongest risk factor, associated with a short time interval from diagnosis to treatment in CLL (4). ZAP-70 presumably acts through modulation of B cell receptor (BCR) signaling. According to this model, overexpression of ZAP-70 would enhance BCR signaling and activate the key mediators in the BCR pathway, such as Erk and Akt (5, 6), leading to exit from the G0 state. Our laboratory has cloned and characterized Dipeptidyl Peptidase 2 (DPP2; also known as DPP7, quiescent cell proline dipeptidase), a serine protease which prevents spontaneous cell cycle entry in quiescent cells (7). Inhibition of DPP2 in resting, but not activated lymphocytes results in apoptotic cell death (8). In our pilot study we employed ValBoroPro, a non-specific inhibitor of most DPP family members, to develop DPP inhibition apoptosis assay in CLL (9, 10). Here we report on a large cohort of patients with CLL who were screened for apoptosis induction in the presence of AX8819, a DPP2-specific small molecule inhibitor. Using this assay we found that sensitivity and resistance to apoptosis correlated with known prognostic factors and disease outcome in CLL. CLL B-cells resistant to DPP2 inhibition-induced apoptosis were found to have progressed past G0 and entered early G1. Inhibition of Hsp90 led to the reversal of apoptosis resistance in CLL B-cells. Our data indicate that in prognostically poor CLL, the malignant B-cells undergo activation and advance through cell cycle, contributing to cell survival and disease propagation.
METHODS
Patients and samples
We studied 152 patients with B-CLL between 2001 and 2009. 99 (65.1%) were men and 53 (34.9%) were women. Median age was 63 years (range 38 to 89). At diagnosis, 99 (65.1%) patients were in the low clinical risk group (Rai stage 0–1). The median time from diagnosis to study entry was 5.0 years. 95 patients were recruited at the Hematology clinics at Tufts Medical Center (Boston, MA) and the Lahey Clinic (Burlington, MA), 57 samples were obtained from the CLL Program at Dana-Farber Cancer Institute. All samples were obtained under IRB approved protocol at each respective institution. The diagnosis of CLL was based on standard morphologic and immunophenotypic criteria and treatment was initiated for symptomatic disease according to standard clinical practice at each institution. When the study samples were obtained, 107 patients (70.4%) were untreated. The median follow-up from study entry was 6 years. Standard Ficoll-Hypaque (Amersham, Piscataway, NJ) techniques were used to isolate peripheral blood mononuclear cells (PBMCs) from healthy donors and consenting CLL patients. Such CLL samples had more than 90% CD19+/CD5+ cells by flow cytometry. Cells were suspended in fetal bovine serum (FBS, Atlanta Biologicals, Norcross, GA) containing 10% dimethylsulfoxide for storage in liquid nitrogen. IgVH mutational status (11), ZAP-70 expression (12), CD38 expression (13), disease stage according to Rai criteria, and history of treatment were analyzed.
Cell apoptosis studies
To investigate apoptosis induction, cells were cultured in RPMI supplemented with 10% FCS, 100 U/ml penicillin, 0.1 mg/ml streptomycin and 2 mM/L L-glutamine in 96 well plates at a concentration of 1×106/ml in the presence of ValboroPro (VbP) at a final concentration of 10 µM, AX8819 at a final concentration of 30 µM, or DMSO, a vehicle control. Cells were harvested at 16–18 h, washed twice in PBS and resuspended in 48 µl of Annexin V binding buffer (BD PharMingen); 2 µl of Annexin V-APC (BD PharMingen) and 0.2 µl of CD19-FITC mAbs (BD PharMingen) were added. Cells were kept for 15 minutes on ice, after which 150 µl of binding buffer and propidium iodide at a final concentration of 10 µg/ml were added and immediately followed by flow cytometry on FACSCalibur (Becton Dickinson, Palo Alto, CA). The fluorescence intensity of each stained cell population was compared with that of the same population treated with an isotype control, IgG2α. Apoptosis was assessed in the CD19+ population.
SDS-PAGE, Western blot and Antibodies
PBMCs from patients with CLL were washed with PBS and lysed in a modified RIPA buffer (20 mM Tris, 150 mM NaCl, 1% NP-40, 1 mM NaF, 1 mM Sodium phosphate, 1 mM NaVO3, 1 nM EDTA, 1 nM EGTA), complemented with protease inhibitor cocktail (Roche, Indianapolis, IN) and 1 mM PMSF. Cell lysates were cleared by centrifugation for 10 min at 13,000 rpm at 4° C. Protein concentration was measured using BCA Reagent (Pierce, Rockford, IL). 20 µg of protein lysate was run on 4–10% continuous gradient polyacrylamide gels (Criterion-XT Gels, BioRad, Hercules, CA) and transferred onto PVDF membranes (Millipore, Billerica, MA). Primary antibody incubations were done at 4° C overnight; secondary antibody (HRP-conjugates) incubations were carried out for 1 h at room temperature. The following antibodies and the dilutions were used: p27 (C-19, Santa Cruz Biotechnology, Santa Cruz, CA, 1:200), p130 (C-20, Santa Cruz Biotechnology, 1:100), c-myc (Upstate, Temecula, CA, 1:50), actin (AC-15, Sigma, St. Louis, MO, 1:5000), HRP-conjugated anti-mouse and anti-rabbit antibodies (BioRad, Hercules, CA). Blots were developed using HRP catalyzed chemiluminescent substrate (ECL, Amersham) and exposed by autoradiography on Biomax MR film (Kodak, Rochester, NY).
qRT-PCR
CLL B-cells were isolated to greater than 98% purity by means of a MoFlow (Dako Cytomation, Carpinteria, CA), using FITC-conjugated CD19-specific mAbs (BD Biosciences, San Jose, CA). Purified cells were immediately lysed, and RNA was extracted using RNeasy Mini Kit (Qiagen, Valencia, CA). Complementary DNA (cDNA) was synthesized from 1 µg of RNA with the use of SuperScript RT Kit (Invitrogen, Carlsbad, CA).
cDNA was used for quantitative real-time reverse-transcription PCR (qRT-PCR), using a human TaqMan MGB probe (#MGB4316034) and RT-PCR reagents (#4304971) (Applied Biosystems, Foster City, CA), run and analyzed on ABI 7200 Sequence Detection System (Applied Biosystems). All samples were run in triplicates. Amplification of the sequence of interest was compared to a reference probe (18S RNA, #4308329, Applied Biosystems). To calculate the relative amount of DPP2 transcripts, we used the comparative Ct method for the relative quantitation employing the formula 2−ΔΔCt, where ΔΔCt=ΔCtP-ΔCtK; P=Probe and K=reference sample (14).
Multiplexed bead analysis
CD19+ CLL cell pellets were lysed in Nonindet P-40 lysis buffer. Sodium dodecyl sulfate (SDS) was added at a final concentration of 1%. A cytometric bead array (CBA) (BD Biosciences, San Jose, California) was used to measure total Syk and ZAP-70 and tyrosine phosphorylated pSykY352 and pZAP-70Y319, as described elsewhere (5) and according to the manufacturer instructions. The fluorescence was determined using a dual-laser FACSCalibur (BD Biosciences, San Jose, CA).
siRNAs and transfections
Sequences for siRNA against DPP2 were designed using the Dharmacon website http://www.dharmacon.com. siRNA oligos were synthesized by Dharmacon (Lafayette, CO). Sense strand against human DPP2: 5’-AAC CUG AGU GCC UCA GUC AUC -3’. Sense strand against mouse DPP2: 5’-GGU UCC UAG UGU CAG AUA A-3’. siRNA oligo electroporations were performed immediately following PBMC isolation, using Amaxa Human B-cell Nucleofection Kit (Amaxa, Cologne, Germany). Transfection efficiency, assessed by transfection with 2 µg pMaxGFP plasmid, varied between patients and fell in the range of 30–60% with cell viability of 50–80%. 1×107 PBMCs were mixed with 100 µL of Amaxa solution, and 2 µg of siRNA was nucleofected. Apoptosis assay was performed as described above.
Statistical analysis
A two-group t-statistic and a χ2 test were used to measure the ability of a parameter to discriminate between the CLL subtypes. Time to treatment initiation measured from diagnosis of CLL was estimated by the Kaplan-Meier method and compared by the log-rank test. Differences with P-values of <0.05 were considered to be significant.
RESULTS
Differential response to DPP2 inhibition and siRNA downregulation identifies two subsets of CLL predictive of disease outcome
We previously reported that inhibition of DPP2 with VbP, a non-specific inhibitor of DPPs, induces apoptosis of PBMCs (8). In this study we validated this assay using AX8819, a novel specific inhibitor of DPP2 (ActivX, La Jolla, CA) (10).
By screening peripheral blood samples from 152 CLL patients, two subsets of CLL were identified: sensitive and resistant to DPP2 inhibition-induced apoptosis. VbP induced apoptosis of >10% CD19+ CLL B-cells in 91/152 samples, identifying a sensitive CLL subgroup (S-CLL). Due to its high affinity for DPP2, AX8819 is a more potent apoptosis-inducing compound than VbP, triggering cell death of 36.05±1.05% cells in S-CLL samples (Fig. 1a and 1b). Treatment of VbP-resistant samples with AX8819 resulted in apoptosis of 15.56±1.92% CLL B-cells. We established a cutoff of at least 25% cell death in CD19+ population to categorize subjects as S-CLL, based on AX8819-mediated inhibition of DPP2. Using this cutoff, 11 (7.2%) samples were discordant: six were VbP-resistant but AX8819-sensitive, while five were VbP-sensitive but AX8819-resistant. In all subsequent experiments, sensitivity to AX8819 was used to categorize CLL samples. Thus, employing an apoptotic assay with AX8819, a DPP2-specific inhibitor, in a large cohort of patients with CLL, we identified 91 cases of S-CLL (59.9%) and 61 cases of R-CLL (40.1%).
Figure 1.

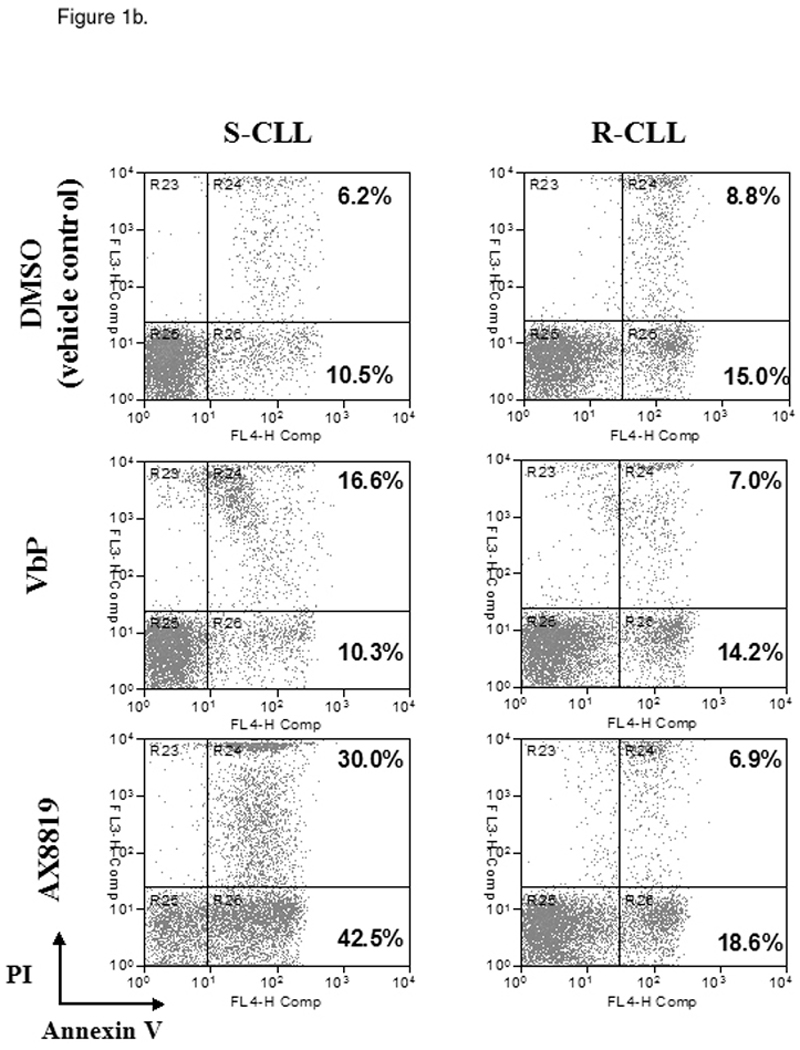
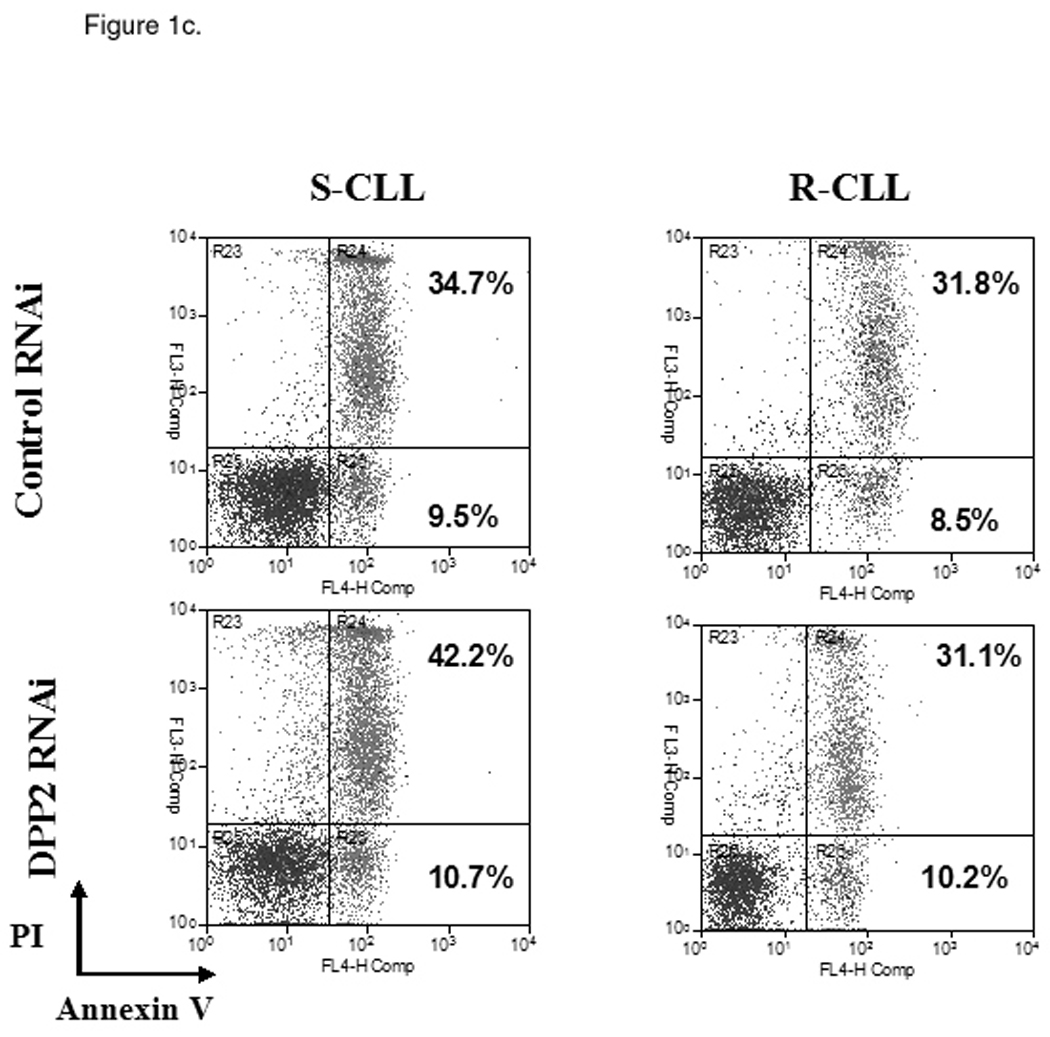

Identification of two subsets of CLL – sensitive and resistant to DPP2 inhibition-induced apoptosis. PBMCs from CLL patients were incubated with either 10 µM/L VbP or 30 µM/L AX8819 for 16 h, stained with CD19, PI and Annexin and assayed on flow cytometry. A, DPP2 inhibition-induced apoptosis in S-CLL (left two bars) and R-CLL (right two bars) CD19+ B-cells. The boxes extend from the 25th percentile to the 75th percentile, with a line at the median. The whiskers show the highest and lowest values. Vehicle control (DMSO)-induced death was subtracted. B, VbP and AX8819 induce apoptosis in S-CLL, but not in R-CLL (representative samples shown). C, Interference of DPP2 via siRNA causes apoptosis in S-CLL, but not in R-CLL (representative samples shown). PBMCs from patients with CLL were nucleofected with siRNA oligos specific for either mouse or human DPP2. D, Interference of DPP2 with human, but not with mouse, siRNA results in a decrease in DPP2 transcripts.
These results were confirmed using small interfering RNA (siRNA) to block expression of DPP2. The siRNA effectiveness against DPP2 was previously tested in 293T cells (15). Mouse DPP2-targeting siRNA was used as a negative control. When three S-CLL and three R-CLL samples were tested, no B-cell death over control siRNA was observed among R-CLL samples, whereas introduction of human DPP2-specific siRNA resulted in apoptosis of 5 to 10% S-CLL CD19+ B-cells at 48 h (Fig. 1c and 1d).
Resistance to DPP2 inhibition-induced apoptosis was associated with a more aggressive clinical course of CLL. 41.8% of patients with R-CLL presented with Rai stage 2–4 at diagnosis, as compared with 23.3% of patients with S-CLL (χ2=5.45, p=0.02). After a median follow-up of 6 years, treatment was initiated in 43 (70.5%) R-CLL patients vs 24 (26.4%) S-CLL patients (χ2=28.8, p<0.0001). Patients with R-CLL initiated treatment earlier (median time to treatment 4.0 vs 20.0 years; HR=4.41, 95% CI, 4.00 to 12.46, p<0.0001; Fig. 2).
Figure 2.

The prognostic effect of resistance vs. susceptibility to DPP2 inhibition-induced apoptosis on the clinical course of CLL. Rate of disease progression was assessed by the time interval measured in years from diagnosis to treatment initiation.
DPP2 apoptotic assay correlates with established prognostic markers in CLL
DPP2 and BCR IgVH mutational status
In our cohort, the results of IgVH mutational status analysis were available on 48 patients. In accordance with the previous reports, a cutoff of >98% homology to germline was used to define the unmutated pattern of IgVH gene (11). 13/14 R-CLL samples carried an unmutated IgVH gene (ranging between 98.6% and 100% homology), with 100% homology in eight. The remaining sample had 96% homology in the IgVH gene. This patient manifested progressive disease at diagnosis and received four different chemotherapy regimens over the following 9 years. Some experts suggest that a less stringent cutoff of <5% IgVH mutations may be used as an adverse prognostic indicator in CLL (16).
Of 34 S-CLL cases, 31 (91.2%) had mutated IgVH, with homology ranging between 85% and 97.9%. Three remaining cases had unmutated IgVH gene with 98.6%, 99.3% and 100% homology to germline. Of those three cases, two did not require treatment of CLL over the follow-up of 6 and 14 years. The third patient began treatment after 10 years of follow-up. Thus, we demonstrate a 92% correlation between IgVH mutational status and resistance/susceptibility to DPP2 inhibition-induced apoptosis in CLL (χ2=31.5, p<0.001; Table 1).
Table 1.
Resistance to DPP2 inhibition-induced apoptosis correlates with known prognostic factors
| R-CLL | S-CLL | P value | |
|---|---|---|---|
| Unmutated IgVH | 13/14 (92.9%) | 3/34 (8.8%) | χ2=31.5, p<0.001 |
| ZAP-70 >20% | 16/16 (100%) | 8/38 (21.1%) | χ2=28.4, p<0.001 |
| CD38 >30% | 11/33 (33.3%) | 6/32 (18.8%) | χ2=1.11, p<0.29 |
DPP2 and ZAP-70
ZAP-70 protein expression was determined in 54 CLL cases by FACS analysis. All R-CLL samples had high levels of ZAP-70 expression (>20%), ranging between 21% and 99% (mean 52.4%). An average of 12.2% of S-CLL B-cells expressed ZAP-70. Eight S-CLL samples had high ZAP-70, with a mean expression of 39.1%. We found an 85.2% correlation between ZAP-70 expression levels and sensitivity to DPP2 inhibition-induced apoptosis (χ2=28.4, p<0.001). In our cohort, the discordance between IgVH mutational status and ZAP-70 level was 14.9%, which is comparable to what has been reported in the literature (17).
DPP2 and del17p
Deletion of chromosome 17p in CLL leads to inactivation of the p53 apoptotic pathway and is, thus, a poor prognostic indicator in CLL, independent of the other criteria. del17p was observed in 10 (6.6%) patients with clone size of 3 to 64% by FISH. 8/10 samples exhibiting del17p were resistant to DPP2 inhibition-induced apoptosis. The remaining 2/10 samples were S-CLL, however in those samples del17p was observed in a small fraction of nuclei only (3% and 4%). These findings confirm our observation that DPP2 inhibition-induced apoptosis is dependent on p53 (15).
DPP2 apoptosis assay reveals a disparity in cell cycle distribution between the CLL subsets
Accumulation of p16Ink4a and p27Kip1, hypophosphorylated forms of retinoblastoma (Rb) protein, along with absence of active forms of cyclins and cyclin-dependent kinases, define the G0 state (18). p27Kip1 prevents activation of Cyclin E-cdk2 and Cyclin D-cdk4 complexes and thus acts as a cell cycle inhibitor. Cell activation and exit from the resting state are accompanied by degradation of p27Kip1, which is followed by recruitment of c-Myc (19). Upregulation of c-Myc heralds cell cycle entry (20). DPP2 is essential for maintenance of G0 in human fibroblasts and lymphocytes, and inhibition of DPP2 is accompanied by loss of p27Kip1 (15). It was previously noted that the p27Kip1 protein level is elevated in CLL compared with other hematologic malignancies (21). We analyzed p27Kip1 and c-Myc protein levels in the two subsets of CLL. Expression of p27Kip1 was increased in S-CLL compared with R-CLL (Fig. 3a). In contrast to that, c-Myc protein level was elevated in R-CLL samples (Fig. 3b). This suggests that S-CLL B-cells remain in a true quiescent state, while R-CLL B-cells progressed past the G0 state. Expression of p130 varied between samples, however its expression was somewhat higher in the R-CLL samples (Fig. 3b).
Figure 3.
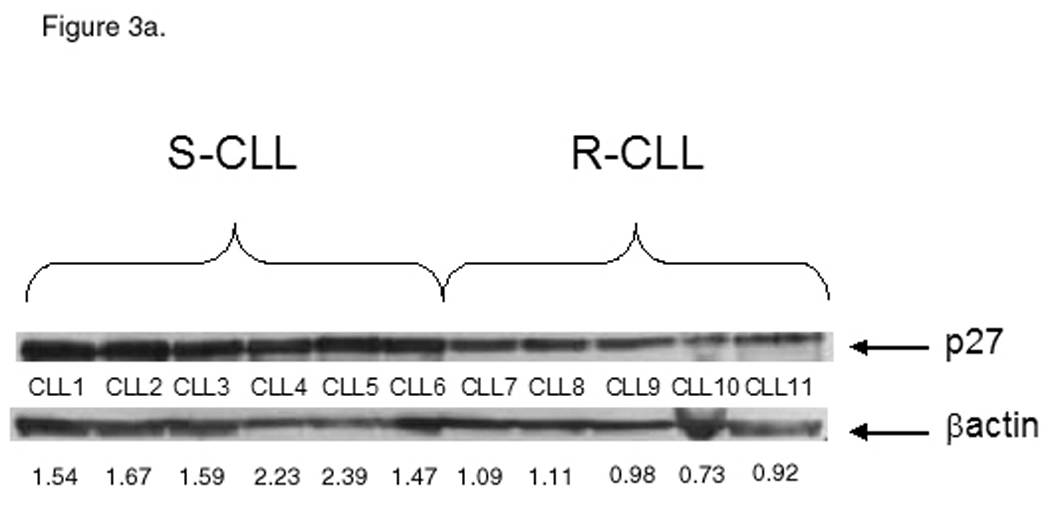
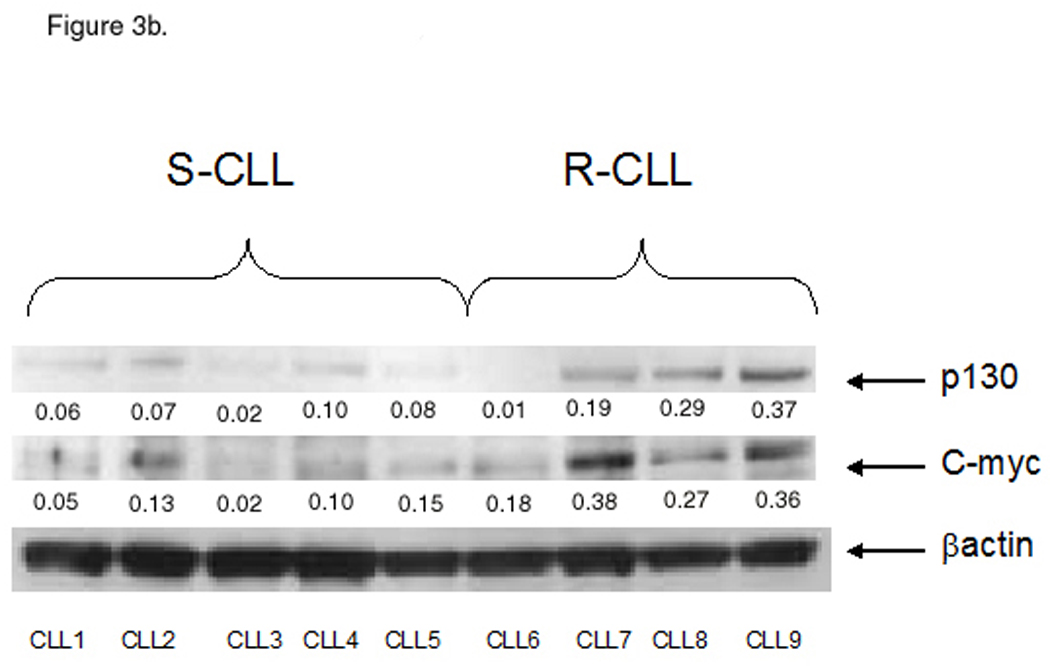
Differential cell cycle distribution between the CLL subsets. A, S-CLL samples (1 through 6) express high baseline protein level of p27 compared with R-CLL (samples 7 through 11). B, S-CLL samples (1 through 5) express low baseline protein level of c-Myc and p130 compared with R-CLL (samples 6 through 9). Representative data of three independent experiments are shown.
Normal B-cells depend on BCR-signaling for their survival and proliferation (22). CLL B-cells vary in their ability to respond to outside stimuli through the BCR (23). However, BCR-signaling may promote CLL B-cell survival, particularly in the subgroup of CLL which features unmutated IgVH genes (24). Moreover, an ability to respond to BCR ligation was associated with cell activation and survival and correlated with disease aggressiveness in CLL (25). The spleen tyrosine kinase, Syk, is a key mediator of the equilibrium between the activation and inhibitory signals through the BCR complex, known as tonic BCR-signaling (26). Syk protein expression and phosphorylation is enhanced in CLL, and inhibition of Syk leads to apoptosis of CLL B-cells (27). To assess whether R-CLL B-cells exit G0 due to increased BCR-signaling, we measured Syk phosphorylation levels in the two CLL subsets. Indeed, R-CLL CD19+ B-cells had a higher level of SykY352 phosphorylation compared with S-CLL (p=0.03; Fig. 4a), implying that BCR-signaling is enhanced in this subset of CLL. On the other hand, we detected no difference in ZAP-70Y319 phosphorylation between the CLL subsets (Fig. 4b).
Figure 4.


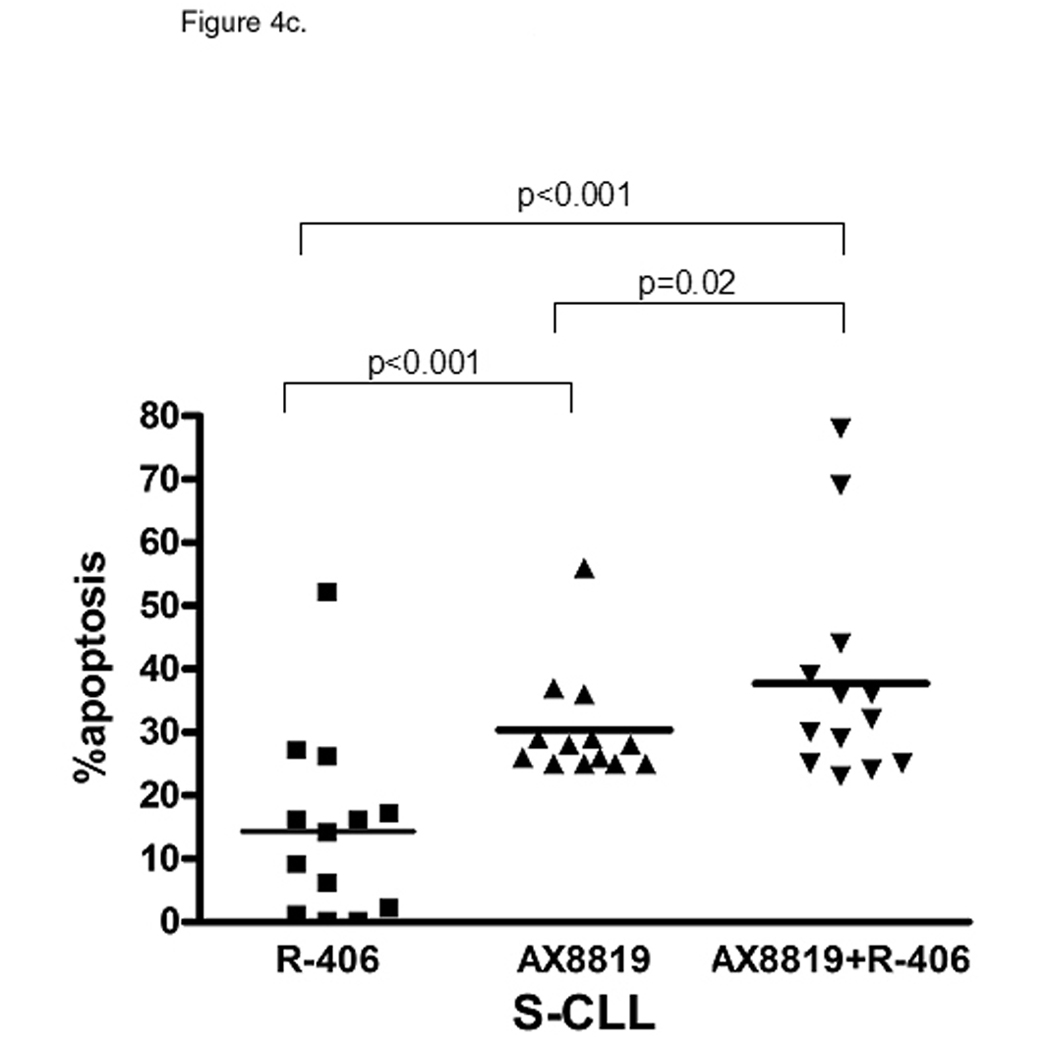
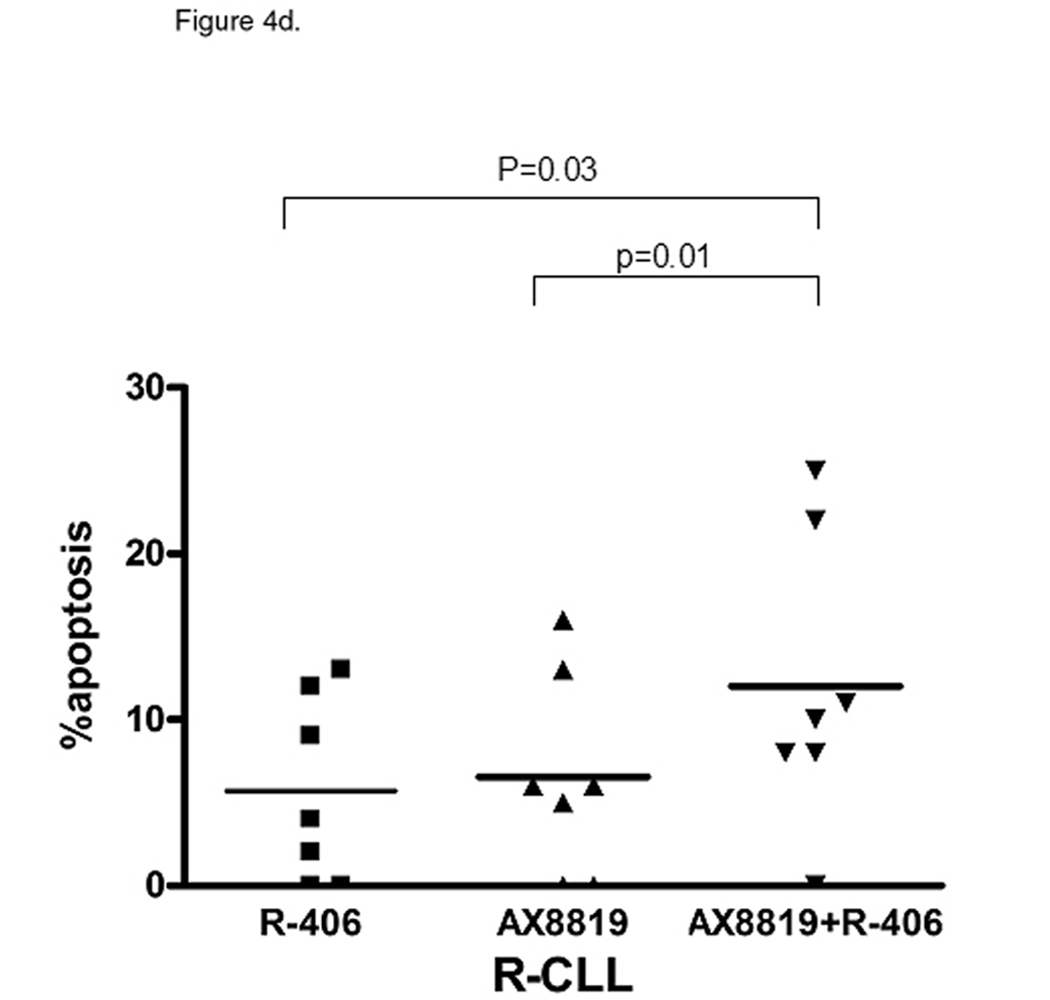
Tonic Syk signaling is essential for CLL cell survival. A, R-CLL B-cells demonstrate constitutive phosphorylation of Syk. pSyk Y352 and total Syk were measured with BD CBA. B, ZAP-70 phosphorylation is similar between the CLL subsets. pZAP-70Y319 and total ZAP-70 were measured with BD CBA. C and D, Inhibition of Syk enhances DPP2 inhibition-induced apoptosis in CLL. PBMCs were incubated with 2 µM R406, AX8819 or both and assayed by flow cytometry. Representative data of three independent experiments are shown. The whiskers show standard deviation.
We further investigated whether blocking Syk and hence abrogating the BCR-signal in CLL B-cells will increase their susceptibility to inhibition of DPP2. After 16 h incubation with Syk inhibitor, R406 (Rigel Pharmaceuticals, San Francisco, CA), apoptosis was more prominent in S-CLL than in R-CLL B-cells (14.30±3.65% vs. 5.71±2.65%, p=0.04; Fig. 4c and 4d). Increased BCR-signaling and activation of downstream survival programs in R-CLL may ultimately decrease dependence and sensitivity to the inhibition of Syk. In S-CLL AX8819 caused more apoptosis than R406 at 16 h of incubation (p<0.001, Fig. 4c). Concomitant inhibition of DPP2 and Syk resulted in additive apoptotic effect in both subsets of CLL (Fig. 4c and 4d).
Inhibition of DPP2 in S-CLL results in inappropriate cell cycle entry with ensuing apoptosis
Inhibition of DPP2 resulted in deregulation of cell cycle components in S-CLL. Treatment with AX8819 led to degradation of p27Kip1 and p130 (Fig 5a), accompanied by upregulation of c-Myc (Fig. 5a, 5b) in S-CLL samples. S-CLL B-cells demonstrated a dramatic phosphorylation of SykY352 after 16 h incubation with AX8819 (Fig. 5c). Thus, inhibition of DPP2 leads to activation of S-CLL B-cells, which begin to enter cell cycle. Since this occurs under inappropriate cell cycle conditions, upregulation of c-Myc leads to apoptosis induction through several mechanisms (28).
Figure 5.
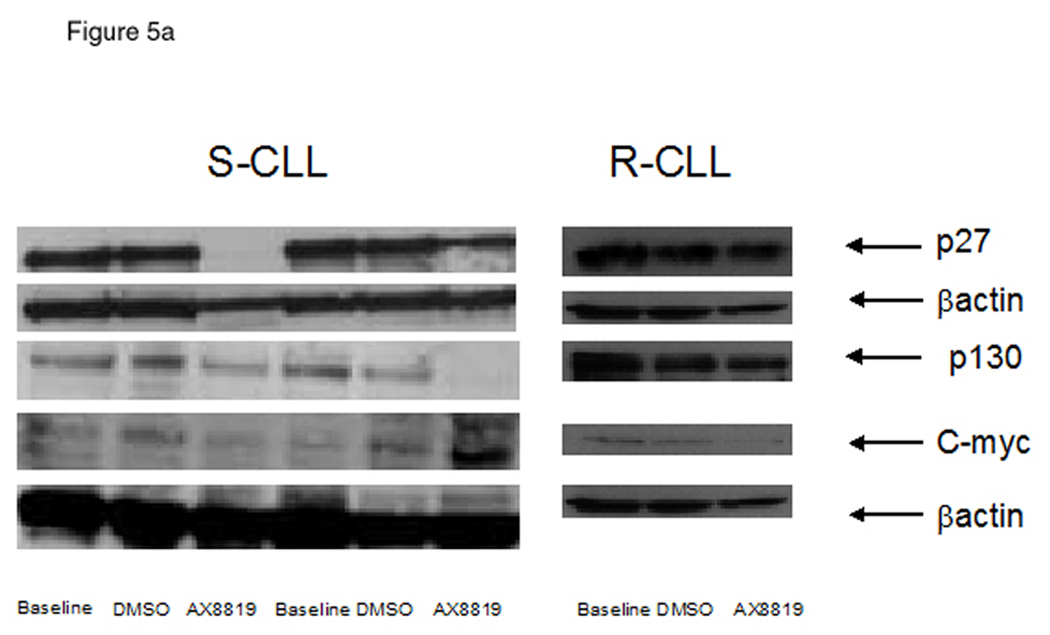

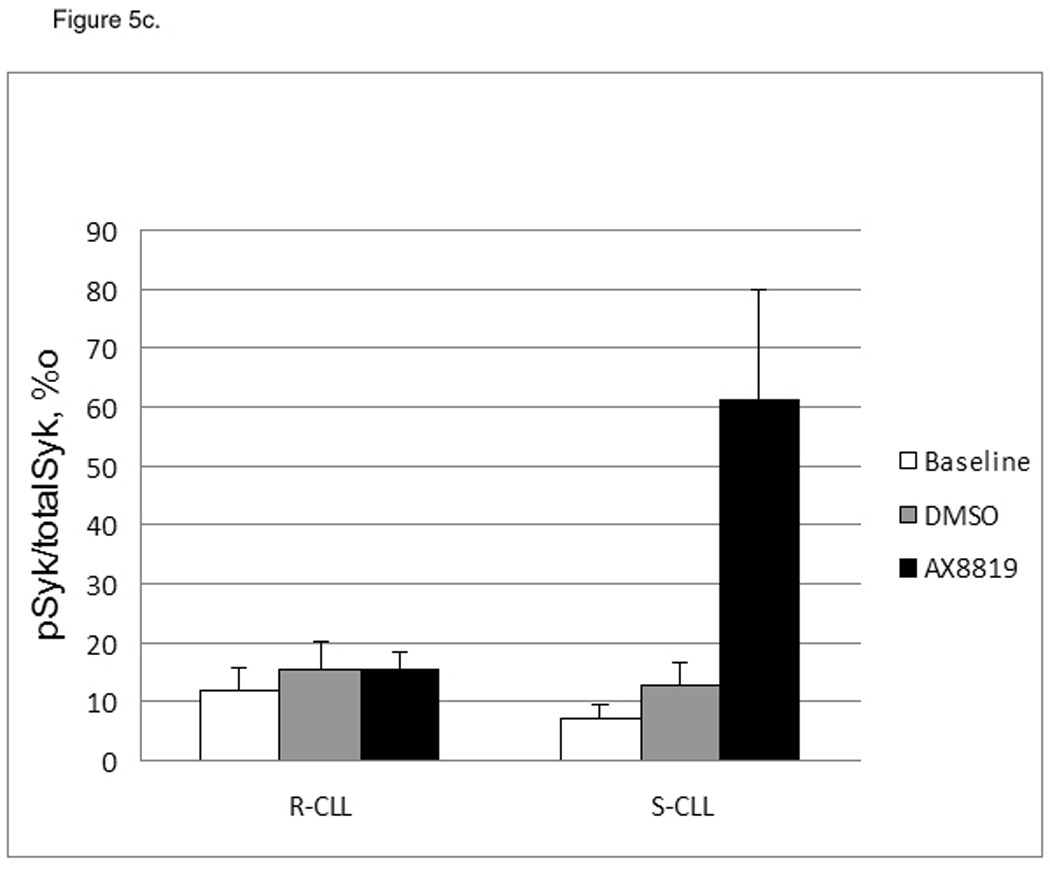
Differential cell cycle regulation in CLL sensitive and resistant to DPP2 inhibition-induced apoptosis. A, Inhibition of DPP2 results in degradation of p27 and p130 and accumulation of c-Myc in S-CLL, but not R-CLL, indicating inappropriate cell cycle entry. PBMCs were incubated with either DMSO or AX8819 for 16 h. Ten samples were tested, three representative samples are shown. B, Interference of DPP2 via siRNA oligos causes an elevation of c-myc mRNA. S-CLL PBMCs were nucleofected with either mouse or human siDPP2. qRT-PCR was run in triplicates, C, S-CLL, but not R-CLL B-cells phosphorylate Syk at position Y352 upon inhibition of DPP2. pSyk and total Syk were measured with BD CBA.
We previously demonstrated that sensitivity to DPP2 inhibition is lost upon cell activation (8). Enhanced BCR-signaling in R-CLL results in partial CLL B-cell activation, exit from G0 and therefore loss of sensitivity to inhibition of DPP2. Inhibition of DPP2 in R-CLL did not affect Syk phosphorylation or protein levels of p27Kip1, p130 or c-Myc (Fig. 5a).
Inhibition of hsp90 reverses resistance to DPP2 inhibition-induced apoptosis
Targeting Hsp90 interferes with CLL cell survival. Inhibition of Hsp90 in CLL B-cells leads to Akt depletion (29), alters p53 function (30) and modulates BCR signaling through destabilization of ZAP-70 (31). The Hsp90 inhibitor, 17-allylaminogendanamycin (17-AAG), has been shown to induce apoptosis in CLL at 100 nM, an effect which was more pronounced in cells that had an unmutated IgVH gene (31). We observed a very modest apoptotic effect when CLL B-cells were incubated with 100 nM 17-AAG for 24 h. Treatment of S-CLL samples with both AX8819 and AAG-17 did not augment the pro-apoptotic effect of DPP2 inhibition (Fig. 6a). However, concomitant exposure of R-CLL to AX8819 and AAG-17 enhanced apoptosis, resulting in death of 17.8±3.0% CD19+ CLL B-cells over background (range, 0–23%; Fig. 6b).
Figure 6.


Inhibition of hsp90 enhances apoptotic response to AX8819 in R-CLL, but not in S-CLL. Cells were treated with 30 µM AX8819, a DPP2 inhibitor, in presence or absence of 100 nM AAG-17, an hsp90 inhibitor, stained with CD19, PI and Annexin and assayed on flow cytometry. A, Co-incubation of S-CLL B-cells with AX8819 and AAG-17 did not result in enhanced apoptosis. B, Co-incubation of R-CLL B-cells with AX8819 and AAG-17 reversed resistance to apoptosis.
DISCUSSION
The prevailing view is that CLL is an accumulative disease with B-cells arrested in G0 (32, 33). However, heterogeneity of clinical outcome in CLL points to the complexity of the disease biology. Rapid clonal renewal in patients with aggressive disease suggests that the neoplastic B-cells may exit the resting state (1). Lymphocyte quiescence is not a default state, but is maintained through active signaling mechanisms (34). We have previously identified DPP2 as an important regulator of quiescence (9, 15). In a large CLL patient cohort we demonstrate that interference with DPP2 protease activity via AX8819, a novel DPP2-specific inhibitor, identifies two subsets of CLL, sensitive and resistant to apoptosis. Of 152 patients with CLL, 91 (59.9%) had S-CLL and 61 (40.1%) had R-CLL. This apoptotic assay demonstrates substantial prognostic significance in our cohort of patients with CLL. Patients with R-CLL manifested a clinically aggressive disease phenotype, such as advanced Rai stage at presentation and a shorter time to treatment initiation (HR=4.41, p<0.0001). Resistance to DPP2 inhibition-induced apoptosis correlated strongly with other known adverse prognostic factors. Notably, R-CLL samples had unmutated IgVH genes and expressed high ZAP-70 level, both of which predict inferior outcome in CLL (11, 35, 36). Cytogenetic abnormalities, particularly del17p, representing an independent CLL subset, are strong predictors of response to therapy in CLL (37). Among the 10 patients with known del17p enrolled in our study, eight patients with large del17p clones had R-CLL, whereas patients with small del17p clones (3% of CLL cells) had S-CLL, implicating that in CLL B-cells, like in normal lymphocytes, DPP2 inhibition-mediated apoptosis depends on functional p53 (15). Our data are also consistent with recent findings that the size of the p53-deleting clone is predictive of disease outcome in CLL (38).
DPP2 is a key regulator of lymphocyte quiescence (8, 15). DPP2 promoter activity is regulated by quiescence-specific elements, such as KLF2 and TOB1 (39). DPP2 recognizes a dipeptide sequence (penultimate Ala/Pro) which is common in transcription regulators (eg, KLF2 and FOXO3A). Although DPP2-specific targets have not been identified definitively, we propose that DPP2 modifies a quiescence-promoting factor via cleavage of an N-dipeptide, rendering this putative factor active. Our data indicate that DPP2 is an essential component of the quiescence program in CLL B-cells. In normal PBMCs sensitivity to DPP2 inhibition-induced apoptosis is lost upon cell activation (8). Thus, it is likely that the DPP2 apoptotic assay uncovers differences in cell cycle distribution between the indolent and aggressive forms of CLL. We postulate that enhanced BCR-signaling in R-CLL may result in a partial CLL B-cell activation, transition from G0 to an early G1 phase of cell cycle and, therefore, loss of sensitivity to inhibition of DPP2. In support of this notion, we found higher protein levels of p27Kip1 and lower levels of c-Myc in S-CLL compared with R-CLL, suggesting that S-CLL B-cells remain in true G0. Conversely, cell cycle progression beyond G0 with loss of p27Kip1 renders R-CLL cells insensitive to inhibition of DPP2. Notably, a small population of CLL B-cells remains susceptible to inhibition of DPP2 in R-CLL samples, suggesting that there is heterogeneity in cell cycle distribution in the peripheral blood CLL B-cells, with some cells remaining in true G0.
p130 acts to restrict G1-S transition in cooperation with pRb and p107 (40). We found, however, that p130 levels are elevated in R-CLL. This may be a compensatory response to cell activation to prevent full cell cycle entry. On the other hand, p130 may be undergoing cdk-cyclin-dependent hyperphosphorylation (40), resulting in availability of transcription factor E2F and progression towards an S-phase of cell cycle. The role of p130 in CLL needs to be studied in more detail.
We observed increased phosphorylation of Syk in R-CLL compared to S-CLL cells. Syk is a key mediator in the transduction of both tonic and activation signals in healthy B-lymphocytes (26), as signaling through Syk promotes B-cell survival. Syk protein expression and phosphorylation have been reported to be enhanced in CLL, leading to upregulation of anti-apoptotic pathways, while inhibition of Syk induces apoptosis (27). The qualitative differences in cell cycle distribution among CLL cells probably result from the quantitative differences in BCR-signal strength, as measured by Syk phosphorylation. In agreement with this notion we found that increased level of Syk phosphorylation corresponds with downregulated p27Kip1 and upregulated c-Myc in R-CLL. Interestingly, concomitant inhibition of Syk and DPP2 in R-CLL results in summation of the apoptotic effect. ZAP-70 kinase activity is not required for BCR-signaling (41), as phosphorylation of its positive regulatory tyrosine residues is not observed following BCR-stimulation in CLL cells (6). Consistent with this, we detected no difference in ZAP-70 phosphorylation levels between the two subsets of CLL.
Inhibition of DPP2 results in deregulation of cell cycle components involved in maintenance of a quiescent state, such as pRb, p107, p27Kip1, cyclin D etc. (15). In S-CLL apoptotic death in response to inhibition of DPP2 is due to induction of activation and inappropriate cell cycle entry, as evidenced by increase in Syk phosphorylation and upregulation of c-Myc with concomitant loss of cell cycle inhibitors, p27Kip1 and p130. In contrast, inhibition of DPP2 in R-CLL did not result in deregulation of p27Kip1, p130 or c-Myc. We propose that R-CLL cells receive a stronger baseline pSyk-mediated activation signal than S-CLL cells. This would lead to cell cycle progression in the former, rendering them independent of DPP2 for survival.
We further demonstrate that resistance to DPP2 inhibition-induced apoptosis can be reversed through inhibition of Hsp90, as AAG-17 sensitized R-CLL B-cells to apoptosis by inhibition of DPP2. Hsp90 is a molecular chaperone that stabilizes client proteins critical to BCR-signaling. Particularly, Hsp90 inhibition results in destabilization of ZAP-70 (31) and depletion of Akt (29). Furthermore, it impairs BCR-signaling in CLL cells and renders them non-responsive to BCR-stimulation with anti-µ (31). Thus, inhibition of Hsp90 in R-CLL B-cells may reinstate G0 and render such cells sensitive to DPP2 inhibition.
In conclusion, CLL can be categorized into two prognostic groups, based on the sensitivity of B-cells to DPP2 inhibition-induced apoptosis. Resistance to apoptosis correlates with unmutated IgVH and high ZAP-70 and is associated with an unfavorable disease course. We propose that this distinction stems from differences in the respective quiescence programs. While S-CLL B-cells rest in true G0, R-CLL B-cells may be partially activated due to increased Syk-mediated signaling, leading to transition from G0 to early G1 and thus escaping apoptosis. This model is strengthened by our finding that inhibition of Hsp90 converts R-CLL into S-CLL. Thus, DPP2 inhibition alone or with concomitant inhibition of Hsp90 warrants investigation as a therapeutic modality in CLL.
Acknowledgements
We would like to thank the CLL Research Consortium Tissue Bank for determination of IgVH and ZAP-70 on the DFCI CLL samples. Support: BTH - DPP2 RO1AI43469. OVD - BD Biosciences Research Grant. JRB was supported in part by NIH grants K23 CA115682 and PO1-CA081534 for the CLL Research Consortium (CRC).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Disclosure: No financial interest/relationships with financial interest relating to the topic of this article have been declared.
References
- 1.Messmer BT, Messmer D, Allen SL, et al. In vivo measurements document the dynamic cellular kinetics of chronic lymphocytic leukemia B cells. J Clin Invest. 2005;115:755–764. doi: 10.1172/JCI23409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Damle RN, Temburni S, Calissano C, et al. CD38 expression labels an activated subset within chronic lymphocytic leukemia clones enriched in proliferating B cells. Blood. 2007;110:3352–3359. doi: 10.1182/blood-2007-04-083832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moreno C, Montserrat E. New prognostic markers in chronic lymphocytic leukemia. Blood Rev. 2008;22:211–219. doi: 10.1016/j.blre.2008.03.003. [DOI] [PubMed] [Google Scholar]
- 4.Rassenti LZ, Jain S, Keating MJ, et al. Relative value of ZAP-70, CD38, and immunoglobulin mutation status in predicting aggressive disease in chronic lymphocytic leukemia. Blood. 2008;112:1923–1930. doi: 10.1182/blood-2007-05-092882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen L, Apgar J, Huynh L, et al. ZAP-70 directly enhances IgM signaling in chronic lymphocytic leukemia. Blood. 2005;105:2036–2041. doi: 10.1182/blood-2004-05-1715. [DOI] [PubMed] [Google Scholar]
- 6.Gobessi S, Laurenti L, Longo PG, Sica S, Leone G, Efremov DG. ZAP-70 enhances B-cell-receptor signaling despite absent or inefficient tyrosine kinase activation in chronic lymphocytic leukemia and lymphoma B cells. Blood. 2007;109:2032–2039. doi: 10.1182/blood-2006-03-011759. [DOI] [PubMed] [Google Scholar]
- 7.Underwood R, Chiravuri M, Lee H, et al. Sequence, purification, and cloning of an intracellular serine protease, quiescent cell proline dipeptidase. J Biol Chem. 1999;274:34053–34058. doi: 10.1074/jbc.274.48.34053. [DOI] [PubMed] [Google Scholar]
- 8.Chiravuri M, Schmitz T, Yardley K, Underwood R, Dayal Y, Huber BT. A novel apoptotic pathway in quiescent lymphocytes identified by inhibition of a post-proline cleaving aminodipeptidase: a candidate target protease, quiescent cell proline dipeptidase. Immunol. 1999;163:3092–3099. [PubMed] [Google Scholar]
- 9.Danilov AV, Klein AK, Lee HJ, Baez DV, Huber BT. Differential control of G0 programme in chronic lymphocytic leukaemia: a novel prognostic factor. Br J Haematol. 2005;128:472–481. doi: 10.1111/j.1365-2141.2004.05346.x. [DOI] [PubMed] [Google Scholar]
- 10.Danilova O, Li B, Szardenings AK, Huber BT, Rosenblum JS. Synthesis and activity of a potent, specific azabicyclo[3.3.0]-octane-based DPP II inhibitor. Bioorg Med Chem Lett. 2007;17:507–510. doi: 10.1016/j.bmcl.2006.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Damle RN, Wasil T, Fais F, et al. Ig V gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukemia. Blood. 1999;94:1840–1847. [PubMed] [Google Scholar]
- 12.Rassenti LZ, Huynh L, Toy TL, et al. ZAP-70 compared with immunoglobulin heavy-chain gene mutation status as a predictor of disease progression in chronic lymphocytic leukemia. N Engl J Med. 2004;351:893–901. doi: 10.1056/NEJMoa040857. [DOI] [PubMed] [Google Scholar]
- 13.Krober A, Seiler T, Benner A, et al. V(H) mutation status, CD38 expression level, genomic aberrations, and survival in chronic lymphocytic leukemia. Blood. 2002;100:1410–1416. [PubMed] [Google Scholar]
- 14.Wilda M, Bruch J, Harder L, et al. Inactivation of the ARF-MDM-2-p53 pathway in sporadic Burkitt's lymphoma in children. Leukemia. 2004;18:584–588. doi: 10.1038/sj.leu.2403254. [DOI] [PubMed] [Google Scholar]
- 15.Mele DA, Bista P, Baez DV, Huber BT. Dipeptidyl peptidase 2 is an essential survival factor in the regulation of cell quiescence. Cell Cycle. 2009;8:2425–2434. doi: 10.4161/cc.8.15.9144. [DOI] [PubMed] [Google Scholar]
- 16.Lin K, Sherrington PD, Dennis M, Matrai Z, Cawley JC, Pettitt AR. Relationship between p53 dysfunction, CD38 expression, and IgV(H) mutation in chronic lymphocytic leukemia. Blood. 2002;100:1404–1409. doi: 10.1182/blood-2001-11-0066. [DOI] [PubMed] [Google Scholar]
- 17.Kern W, Dicker F, Schnittger S, Haferlach C, Haferlach T. Correlation of flow cytometrically determined expression of ZAP-70 using the SBZAP antibody with IgVH mutation status and cytogenetics in 1,229 patients with chronic lymphocytic leukemia. Cytometry B Clin Cytom. 2009;76:385–393. doi: 10.1002/cyto.b.20483. [DOI] [PubMed] [Google Scholar]
- 18.Ho A, Dowdy SF. Regulation of G(1) cell-cycle progression by oncogenes and tumor suppressor genes. Curr Opin Genet Dev. 2002;12:47–52. doi: 10.1016/s0959-437x(01)00263-5. [DOI] [PubMed] [Google Scholar]
- 19.Obaya AJ, Kotenko I, Cole MD, Sedivy JM. The proto-oncogene c-myc acts through the cyclin-dependent kinase (Cdk) inhibitor p27(Kip1) to facilitate the activation of Cdk4/6 and early G(1) phase progression. J Biol Chem. 2002;277:31263–31269. doi: 10.1074/jbc.M202528200. [DOI] [PubMed] [Google Scholar]
- 20.Kelly K, Cochran BH, Stiles CD, Leder P. Cell-specific regulation of the c-myc gene by lymphocyte mitogens and platelet-derived growth factor. Cell. 1983;35(3 Pt 2):603–610. doi: 10.1016/0092-8674(83)90092-2. [DOI] [PubMed] [Google Scholar]
- 21.Vrhovac R, Delmer A, Tang R, Marie JP, Zittoun R, Ajchenbaum-Cymbalista F. Prognostic significance of the cell cycle inhibitor p27Kip1 in chronic B-cell lymphocytic leukemia. Blood. 1998;91:4694–4700. [PubMed] [Google Scholar]
- 22.Kurosaki T. Regulation of B-cell signal transduction by adaptor proteins. Nat Rev Immunol. 2002;2:354–363. doi: 10.1038/nri801. [DOI] [PubMed] [Google Scholar]
- 23.Lankester AC, Schijndel GM, Pakker NG, Van Oers RH, van Lier RA. Antigen receptor function in chronic lymphocytic leukemia B cells. Leuk Lymphoma. 1996;24:27–33. doi: 10.3109/10428199609045711. [DOI] [PubMed] [Google Scholar]
- 24.Allsup DJ, Kamiguti AS, Lin K, et al. B-cell receptor translocation to lipid rafts and associated signaling differ between prognostically important subgroups of chronic lymphocytic leukemia. Cancer Res. 2005;65:7328–7337. doi: 10.1158/0008-5472.CAN-03-1563. [DOI] [PubMed] [Google Scholar]
- 25.Deglesne PA, Chevallier N, Letestu R, et al. Survival response to B-cell receptor ligation is restricted to progressive chronic lymphocytic leukemia cells irrespective of Zap70 expression. Cancer Res. 2006;66:7158–7166. doi: 10.1158/0008-5472.CAN-06-0085. [DOI] [PubMed] [Google Scholar]
- 26.Monroe JG. ITAM-mediated tonic signalling through pre-BCR and BCR complexes. Nat Rev Immunol. 2006;6:283–294. doi: 10.1038/nri1808. [DOI] [PubMed] [Google Scholar]
- 27.Buchner M, Fuchs S, Prinz G, et al. Spleen tyrosine kinase is overexpressed and represents a potential therapeutic target in chronic lymphocytic leukemia. Cancer Res. 2009;69:5424–5432. doi: 10.1158/0008-5472.CAN-08-4252. [DOI] [PubMed] [Google Scholar]
- 28.Evan GI, Wyllie AH, Gilbert CS, et al. Induction of apoptosis in fibroblasts by c-myc protein. Cell. 1992;69:119–128. doi: 10.1016/0092-8674(92)90123-t. [DOI] [PubMed] [Google Scholar]
- 29.Johnson AJ, Wagner AJ, Cheney CM, et al. Rituximab and 17-allylamino-17-demethoxygeldanamycin induce synergistic apoptosis in B-cell chronic lymphocytic leukaemia. Br J Haematol. 2007;139:837–844. doi: 10.1111/j.1365-2141.2007.06878.x. [DOI] [PubMed] [Google Scholar]
- 30.Lin K, Rockliffe N, Johnson GG, Sherrington PD, Pettitt AR. Hsp90 inhibition has opposing effects on wild-type and mutant p53 and induces p21 expression and cytotoxicity irrespective of p53/ATM status in chronic lymphocytic leukaemia cells. Oncogene. 2008;27:2445–2455. doi: 10.1038/sj.onc.1210893. [DOI] [PubMed] [Google Scholar]
- 31.Castro JE, Prada CE, Loria O, et al. ZAP-70 is a novel conditional heat shock protein 90 (Hsp90) client: inhibition of Hsp90 leads to ZAP-70 degradation, apoptosis, and impaired signaling in chronic lymphocytic leukemia. Blood. 2005;106:2506–2512. doi: 10.1182/blood-2005-03-1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Damle RN, Ghiotto F, Valetto A, et al. B-cell chronic lymphocytic leukemia cells express a surface membrane phenotype of activated, antigen-experienced B lymphocytes. Blood. 2002;99:4087–4093. doi: 10.1182/blood.v99.11.4087. [DOI] [PubMed] [Google Scholar]
- 33.Hamblin TJ, Oscier DG. Chronic lymphocytic leukaemia: the nature of the leukaemic cell. Blood Rev. 1997;11:119–128. doi: 10.1016/s0268-960x(97)90007-2. [DOI] [PubMed] [Google Scholar]
- 34.Yusuf I, Fruman DA. Regulation of quiescence in lymphocytes. Trends Immunol. 2003;24:380–386. doi: 10.1016/s1471-4906(03)00141-8. [DOI] [PubMed] [Google Scholar]
- 35.Hamblin TJ, Davis Z, Gardiner A, Oscier DG, Stevenson FK. Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood. 1999;94:1848–1854. [PubMed] [Google Scholar]
- 36.Rosenwald A, Alizadeh AA, Widhopf G, et al. Relation of gene expression phenotype to immunoglobulin mutation genotype in B cell chronic lymphocytic leukemia. J Exp Med. 2001;194:1639–1647. doi: 10.1084/jem.194.11.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grever MR, Lucas DM, Dewald GW, et al. Comprehensive assessment of genetic and molecular features predicting outcome in patients with chronic lymphocytic leukemia: results from the US Intergroup Phase III Trial E2997. J Clin Oncol. 2007;25:799–804. doi: 10.1200/JCO.2006.08.3089. [DOI] [PubMed] [Google Scholar]
- 38.Tam CS, Shanafelt TD, Wierda WG, et al. De novo deletion 17p13.1 chronic lymphocytic leukemia shows significant clinical heterogeneity: the M. D. Anderson and Mayo Clinic experience. Blood. 2009;114:957–964. doi: 10.1182/blood-2009-03-210591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bista P, Mele DA, Baez DV, Huber BT. Lymphocyte quiescence factor Dpp2 is transcriptionally activated by KLF2 and TOB1. Mol Immunol. 2008;45:3618–3623. doi: 10.1016/j.molimm.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sun A, Bagella L, Tutton S, Romano G, Giordano A. From G0 to S phase: a view of the roles played by the retinoblastoma (Rb) family members in the Rb-E2F pathway. J Cell Biochem. 2007;102:1400–1404. doi: 10.1002/jcb.21609. [DOI] [PubMed] [Google Scholar]
- 41.Chen L, Huynh L, Apgar J, et al. ZAP-70 enhances IgM signaling independent of its kinase activity in chronic lymphocytic leukemia. Blood. 2008;111:2685–2692. doi: 10.1182/blood-2006-12-062265. [DOI] [PMC free article] [PubMed] [Google Scholar]