

Abstract
Estrogens regulate growth and development through the action of two distinct estrogen receptors (ERs), ERα and ERβ, which mediate proliferation and differentiation of cells. For decades, ERα mediated estrogen signaling has been therapeutically targeted to treat breast cancer, most notably with the selective estrogen receptor modulator (SERM) tamoxifen. Selectively targeting ERs occurs at two levels: tissue selectivity and receptor subtype selectivity. SERMs have been developed with emphasis on tissue selectivity to target ER signaling for breast cancer treatment. Additionally, new approaches to selectively target the action of ERα going beyond ligand-dependent activity are under current investigation. As evidence of the anti-proliferative role of ERβ accumulates, selectively targeting ERβ is an attractive approach for designing new cancer therapies with the emphasis shifted to designing ligands with subtype selectivity. This review will present the mechanistic and structural features of ERs that determine tissue and subtype selectivity with an emphasis on current approaches to selectively target ERα and ERβ for cancer treatment.
Keywords: estrogen receptor alpha, estrogen receptor beta, SERMs, SERDs, selective agonist, antagonist, breast cancer, prostate cancer, colon cancer, ovarian cancer
1. Introduction
Two distinct estrogen receptors (ERs), ERα and ERβ, mediate estrogen signaling and distinctly regulate transcription driving growth, proliferation, and differentiation, among many cellular processes. ERα is well characterized as a mediator of cell proliferation, especially in breast cancer cells, driving cell proliferation in the presence of estrogen [1]. ERβ opposes ERα and inhibits ERα mediated proliferation in many cells [2–8]. Because ERs can strongly regulate cell proliferation, they can be targeted therapeutically to inhibit cancer growth. ERα specifically has been implicated as a key factor in breast cancer growth and has been effectively targeted in breast cancer with the development of selective estrogen receptor modulators (SERMs) such as tamoxifen or raloxifene [1, 9]. SERMs function to target ER signaling in a tissue specific manner and the tissue selectivity of SERMs is determined by structural features induced by SERM binding to the receptors and cell type specific factors. Approximately 30% of breast cancers develop resistance after extended exposure to SERMs [10]. SERMs target the ligand dependent activation of ER but alternative methods of targeting ER activity are emerging to overcome resistance. Selective estrogen receptor down-regulators (SERDs) have been developed to inhibit ER signaling through degradation of the receptor. Alternative approaches to inhibit ERα activity go beyond the ligand binding domain and target ER-DNA or ER-cofactor interactions. In this review we will present current methods of targeting ERα for cancer treatment and discuss the mechanistic and structural components that contribute to the tissue selectivity of SERMs.
Additionally, we will discuss the development of ER subtype selective ligands. With the identification of a second estrogen receptor subtype, ERβ, the design of compounds which selectively target ERs has shifted towards subtype selectivity. Ligands with selectivity for ERβ show promise as cancer treatments given the anti-proliferative role of ERβ in many tissues. ERβ is not yet targeted clinically for cancer treatment, but ERβ selective ligands hold therapeutic promise in breast cancer, as well as prostate, ovarian, and colon cancers. Such compounds could promote ERβ mediated growth inhibition while avoiding proliferative side affects mediated by ERα. Here, we will discuss known ERβ selective compounds with an emphasis on structural features that contribute to subtype selectivity. We will also present recent approaches to identifying novel ERβ selective ligands and discuss future directions for identifying ERβ selective compounds with therapeutic potential. Selectively targeting ERs can provide effective cancer treatments and new techniques to inhibit ERα and selectively activate ERβ are emerging to improve the effectiveness of identifying tissue and subtype selective ER ligands for cancer therapy.
2. Estrogen receptor action in normal and cancerous tissues
2.1 Estrogen receptors and normal development
ERs have important roles in normal development and function of reproductive tissues as well as non-reproductive tissues including the lungs, colon, prostate, and cardiovascular system. ERα and ERβ show overlapping and distinct tissue distributions suggesting the receptors have distinct biological roles. Both receptors are expressed in the uterus, breast, lung, heart, intestine, and brain. ERα is expressed in the absence of ERβ in hepatocytes and the hippocampus while ERβ shows unique expression patterns in the prostate, vagina, and cerebellum [11]. Much of our understanding regarding the developmental roles of ERs has been gleaned from observations of ERα and ERβ knockout mice (αERKO, βERKO, and α/βERKO mice). It is necessary to briefly discuss the functional roles of ERα and ERβ in normal development in order to understand the contributions of ER signaling in cancerous tissues. We will present a brief overview of the roles of ERs as demonstrated by αERKO and βERKO mice with a narrowed focus on tissues that may develop cancers which could benefit from selective ER therapies such as reproductive tissues, breast, prostate, and colon. The phenotypes of ERKO mice have been reviewed extensively elsewhere [12–14].
2.1.1 Reproductive development
ERα and ERβ are important mediators of normal ovarian and uterine development and function; the most obvious developmental impairment in αERKO and βERKO mice is found in reproductive structures including the ovary and uterus. ERα is required for normal reproductive development and both male and female αERKO mice are infertile [15, 16]. In normal development, estrogen stimulates proliferation of the uterine epithelium. αERKO females develop rudimentary estrogen insensitive uteri demonstrating the role for ERα in mediating estrogen induced proliferation in the uterus. In the ovaries, both ERα and ERβ are expressed though their distributions among cell types are markedly different. ERα is primarily expressed in thecal and interstitial cells whereas ERβ is primarily expressed in granulosa cells. ERα is critical for normal ovary function and αERKO mice develop abnormal ovaries in which the follicles remain immature [15, 16].
βERKO mice generated in different laboratories do not have consistent phenotypes and conflicting evidence for the role of ERβ in reproductive development is present in the literature (reviewed in [14]). In some models, βERKO females are subfertile suggesting ERβ has a less critical role in reproductive and ovarian development [16, 17]. More recently, ERβ null mice have been generated using Cre/LoxP mediated disruption of the ERβ gene past exon 3, and both males and females are infertile [18]. In these mice, follicle development proceeds normally but does not completely proceed to ovulation due to high rates of termination during atresia demonstrating a critical role for ERβ in development of functional ovaries. Unlike αERKO mice, estrogen responsiveness in the uterus and ovaries appears normal in βERKO mice. In α/βERKO mice, in which both ERα and ERβ are null, reproductive development is similar to that observed in αERKO mice demonstrating the dominant role of ERα [16].
2.1.2 Mammary gland development
Mammary gland development is also dependent on functional estrogen signaling. The mammary gland grows during puberty and completely differentiates during pregnancy and lactation. In the mammary gland, ERα is a key regulator of proliferation in response to hormone signaling and is expressed in the stroma and epithelial cells. The mammary glands of adult female αERKO mice remain immature similar to those found in newborn female mice, suggesting ERα is necessary for ductal growth. ERα has a critical role in ductal elongation which occurs via cap cell proliferation at the terminal end bud of each duct. Ductal elongation does not occur in αERKO mice and glands do not develop terminal end buds [19]. ERα is required for adequate signaling between terminal end buds and the stroma as loss of ERα prevents normal end bud development and invasion into the stroma. When wild type 3 week old female mice are implanted with the fat pad of 3 week old female αERKO mice, developing ducts of wild type mice do not elongate into the implant suggesting ERα is required for normal stromal interaction with terminal end buds [20]. ERα is also required in epithelial cells for normal alveolar development. Specific deletion of ERα in mammary epithelial cells during pre-pubertal development leads to impaired terminal end bud formation and duct elongation [21].
As mentioned previously, the reproductive phenotypes of βERKO mice are inconsistent across independently derived knockout mice. Similarly, mammary gland phenotypes of βERKO mice derived and maintained in different laboratories are inconsistent. Normal ductal structure and differentiation have been reported in ERβ null mice [13], while others have observed impaired side branching in virgin mice and impaired alveolar development in lactating mice [17, 22]. In vitro experiments with the normal mouse mammary epithelial cell line HC11, which expresses endogenous ERα and ERβ, have suggested a role for ERβ in cell adhesion and regulation of proliferation. Treatment with an ERα selective agonist, propyl pyrazole triol (PPT), stimulated proliferation while the ERβ selective agonist diarylpropionitrile (DPN) inhibited proliferation [2]. Additionally, loss of ERβ expression resulted in loss of E-cadherin suggesting a role for ERβ in cell adhesion and differentiation [23]. Though the role of ERβ in mammary development and differentiation has not been clearly defined, evidence is accumulating that ERβ regulates normal proliferation and differentiation in the mammary gland.
2.1.3 Prostate and colon development
Phenotypes of ER knockout mice also suggest roles for estrogen signaling in colon and prostate development. In the prostate, ERβ is highly expressed in epithelial cells while ERα is expressed in the stroma during early development. αERKO mice develop longer ducts in the ventral prostate with fewer side branches, in contrast to the phenotype observed in αERKO mammary glands which exhibit shorter ducts [24]. Despite high expression of ERβ in the prostate, the role of ERβ in prostate development is unclear. Discrepancies in the phenotypes of βERKO mice complicate interpretations regarding the potential role of ERβ in prostate development [25]. Epithelial hyperplasia has been observed in βERKO mice, but observations are not consistent among laboratories suggesting that external factors can influence hyperplastic development in βERKO mice. Most recently, βERKO mice generated with Cre/LoxP technology did not show significant phenotypic differences from wild-type mice and prostates appeared normal [18]. There is indirect evidence suggesting a role for ERβ in prostate cell differentiation. First, the pattern of ERβ expression during human prostate development suggests a role for ERβ in morphogenesis and differentiation [26]. Second, markers of differentiation in the ventral prostate are significantly reduced in βERKO mice that develop epithelial hyperplasia [27]. Prostate development does not have a critical dependence on ERα or ERβ but the potential role of ERβ in differentiation may prove an effective therapeutic target in prostate cancer.
Similarly, ERβ may also regulate growth and differentiation in normal colon tissue but evidence derived from βERKO mice are inconsistent. The most recent βERKO mice generated by Antal and coworkers do not exhibit abnormal colon phenotypes, but previous reports document increased rates of proliferation and migration in colon epithelial cells [18, 28]. ERβ is the predominant ER expressed in colon epithelial cells and epidemiological studies suggest that estrogen or hormone replacement therapy significantly reduces the risk of colon cancer [29]. Taken together, there is evidence suggesting ERβ may play a role in regulating proliferation in colon epithelial cells but the role of ERβ in maintaining normal colon development remains unclear.
2.2 Estrogen receptors and cancer
Given the roles of ERα and ERβ in regulating proliferation and differentiation in normal tissue, it is clear that ER signaling may be important in the dysregulation of these processes in cancer cells. Indeed, ERα has been implicated in breast cancer progression and has been an effective therapeutic target for decades. The role of ERβ in cancer cells and the therapeutic potential of ERβ are not clear, but some evidence suggests ERβ may be targeted to regulate growth of breast, colon, prostate, and ovarian cancers given its role in differentiation of normal tissue. In order to effectively utilize ERβ as a target in cancer treatment, compounds must be designed with high selectivity for ERβ to avoid the proliferative action of ERα.
2.2.1 Breast cancer
Estrogen exposure and breast cancer risk have been associated in both epidemiological and experimental studies [1]. Two hypotheses have been proposed to explain this association: 1) products of estrogen metabolism are genotoxic and cause increased risk of direct DNA damage; 2) estrogen induced activity of ERs stimulates proliferation which leads to increased risk of DNA mutations due to high rates of DNA replication [30]. ERα is the dominant mediator of mammary development so it is not surprising that ERα is a prognostic marker and therapeutic target in breast cancer. ERα is expressed in approximately 70% of all human breast cancers and clinical evidence strongly supports a role of ERα in breast cancer [31]. Tamoxifen, which inhibits ERα transcriptional activity in mammary cells, effectively reduces the risk of recurrence of invasive or in situ ERα positive breast cancer, independent of age [32]. Direct evidence for the role of ERα in breast cancer progression and/or development comes from experiments conducted with Neu/ErbB2 knock-in mice which develop mammary tumors after a long latency period. When crossed with ERα null mice, mammary tumors did not develop suggesting ERα mediated signaling is a required component of carcinogenesis in this model [30].
The role of ERβ in breast cancer is less clear and the prognostic value of ERβ is still under debate [31]. It is estimated that ERβ is expressed in approximately half of human primary breast cancers, but ERβ expression is lost during breast cancer progression, most likely due to promoter hypermethylation [33]. Many breast cancer cell lines do not express ERβ, so in vitro experiments have been limited to cell lines expressing exogenous ERβ making it difficult to extrapolate results to human breast cancers. Despite limitations associated with in vitro experiments, accumulating evidence suggests ERβ is a potential tumor suppressor that promotes differentiation and inhibits ERα mediated proliferation. Inducible expression of ERβ in T47D breast cancer cells inhibited estrogen stimulated proliferation and tumor angiogenesis and growth in xenograft experiments [7, 8]. In the same experimental model, estrogen treatment and induction of ERβ led to down regulation of genes involved in cell cycle progression and DNA replication suggesting ERβ activation can negatively regulate breast cancer proliferation [34]. Inducible overexpression of ERβ in ERα positive MCF7 breast cancer cells inhibited ERα mediated proliferation and enhanced the inhibitory effects of tamoxifen [5]. In the same cell line, ERβ expression and estrogen treatment led to G2 cell cycle arrest and limited tumor formation in xenograft experiments [6]. ERβ may also regulate cell adhesion suggesting a role in inhibiting metastases. ERβ inducible expression in T47D cells led to upregulation of integrin α1 and β1, which was further enhanced by treatment with the ERβ selective ligand DPN. Cells expressing ERβ showed greater adhesion to extracellular matrix proteins like laminin and reduced cell mobility in wound healing assays [35]. The data implicate ERβ as a therapeutic target in breast cancer but it is not clear if such results will translate to clinical application due to varied levels of ERβ in patients.
2.2.2 Prostate cancer
The developing prostate is sensitive to estrogenic effects and in utero exposure to estrogens stimulates squamous metaplasia in prostate epithelium. After birth, estrogen levels decline and such effects regress, but estrogen exposure during prostate development may contribute to increased risk of prostate cancer [25]. It is difficult to interpret the roles of ERs in prostate cancer given the hormonal sensitivity of the tissue and the interactions among estrogen treatments and endogenous hormone levels. Indirect evidence implicates ERα in prostate cancer development. Estrogen accumulates in the nuclei of stromal cells in benign prostatic hyperplasia samples, suggesting ERα, which is primarily expressed in stromal cells, mediates proliferative effects of estrogen in the prostate [25]. More directly, experiments conducted with αERKO and βERKO mice treated with testosterone and estrogen demonstrate ERα may promote prostate carcinogenesis in the presence of estrogen. Wild-type and βERKO mice treated with testosterone and estrogen develop similar incidence of prostatic hyperplasia, atypical hyperplasia, and prostatic intraepithelial neoplasia. In contrast, αERKO mice have slightly reduced incidence of hyperplasia and do not develop atypical hyperplasia or prostatic intraepithelial neoplasia, suggesting ERα contributes to prostate carcinogenesis [36].
ERβ is expressed in normal prostate epithelium but expression is lost during cancer progression and re-expression is observed in metastatic prostate cancer [37]. It is not yet clear if ERβ has an anti-proliferative tumor suppressor role in prostate cancer or if ERβ expression promotes metastasis. In support of the tumor suppressor role for ERβ, proliferation and invasion were reduced in prostate cancer cell lines in which ERβ was overexpressed using adenoviral constructs [38]. Additionally, ERβ selective ligands have been shown to have anti-proliferative effects on prostate epithelium. McPherson and coworkers utilized transplants from aromatase inhibitor knockout (ArKO) mice to overcome the difficulties associated with central manipulation of hormone levels in the organism. Stromal or epithelial transplants from ArKO mice cannot produce local estrogens and ER signaling is essentially blocked when transplanted into male wild type mice. ArKO stroma induced hyperplasia in surrounding normal epithelium due to impaired estrogen production in the stroma. When treated with ERβ selective ligands, hyperplastic development was attenuated likely due to the anti-proliferative role of ERβ [39]. Thus, ERβ selective agonists may prove effective in prostate cancer prevention or early treatment.
2.2.3 Colon cancer
Epidemiological studies have shown that colon cancer incidence and risk is reduced in postmenopausal females taking estrogen replacement therapy and overall rates of colon cancer incidence are lower in females compared to males, providing indirect evidence that ER signaling may inhibit colon cancer development [29]. As mentioned previously, ERβ is the predominant ER expressed in colon epithelial cells suggesting it may mediate the effects of estrogen replacement therapy on colon cancer risk [40]. Loss of ERβ expression is associated with advanced stages of colon cancer and greater degrees of dedifferentiation, suggesting ERβ plays a role in maintaining differentiation and regulating cell proliferation [41, 42]. However, in vitro experiments have shown that tamoxifen or raloxifene treatment can inhibit proliferation of colon cancer cells providing support for targeting ER in colon cancer [43]. Raloxifene treatment reduced proliferation of colon cancer cells expressing ERβ, but had little effect on the growth of colon cancer cells that do not express ERβ. ERβ may be an effective target for colon cancer prevention. Using the model of azoxymethane (AOM) induced colon cancer in F344 rats, Janakiram and coworkers showed that raloxifene treatment effectively reduced the number of aberrant crypt foci when administered before AOM treatment [44]. In this model, rats develop colon tumors that express ERβ, suggesting ERβ may mediate protective effects of raloxifene. These data suggest that selectively targeting ERβ may have preventive or therapeutic potential in colon cancer though much more evidence is required to clarify the role of ERβ in colon cancer.
2.2.4 Ovarian cancer
Development of effective treatments for ovarian cancer is a field that is actively pursued, as it is one of the most lethal cancers in women. Up to 90% of ovarian cancers have epithelial origin and 5–10% originate from granulosa cells. Approximately two-thirds of ovarian tumors express ERs; ERα is predominantly expressed in tumors of epithelial origin and ERβ expression is more prevalent in tumors of granulosa cell origin [30]. Epidemiological studies implicate estrogens in ovarian cancer as women who used long-term estrogen replacement therapy showed higher incidence of ovarian cancer [33]. In vitro experiments provide evidence for a role of estrogens and possibly ERs in ovarian cancer, but the link between ER signaling and the growth and progression of ovarian tumors is not clearly defined. In culture, ovarian cancer cell growth and proliferation is stimulated by estrogens and inhibited by antiestrogen treatment, suggesting ER signaling can regulate ovarian cancer proliferation [45]. Additionally, overexpression of ERβ in ovarian cancer cells lacking ERα expression led to reduced rates of proliferation in response to estrogen treatment. Cell migration was significantly reduced as measured by wound healing assays and increased apoptosis occurred in cells overexpressing ERβ [46]. Given the potential tumor suppressor function of ERβ in ovarian cancer, highly selective and potent ERβ agonists may provide new therapeutic options for a disease where few targeted treatments are available.
3. General structure and signaling pathways of ERs
3.1 Classical nuclear receptor domain structure
ERα and ERβ are encoded by distinct genes on separate chromosomes [47–49] and exhibit structural differences that may provide clues as to how the receptors differentially regulate transcription. ERs are members of the nuclear receptor superfamily and thus contain five domains conserved throughout this family of transcription factors (Figure 1A). Both receptors contain two activation functions that mediate protein-protein interactions, specifically with co-regulators that can modify the transcriptional potential of the receptors. The N terminal A/B domain contains an activation function (AF-1) that mediates ligand independent activation of the receptor. The AF-1 region of ERα can activate transcription independent of ligand but the transcriptional activity of ERβ AF-1 is negligible; indeed, ERα and ERβ share only 17% similarity in this region [50]. Additionally, removal of the AF-1 region of ERβ increases the overall transcriptional activity of the receptor [51].
Figure 1.

A) Domain structures of ERα and ERβ. Percent homology shared between full length ERα and ERβ is given in parentheses. Functional regions of the receptors are shown below the domain structures. B) Domain structures of isoforms of ERβ, all of which share the same N terminal sequence and only differ in the C terminus.
Ligand dependent transcriptional activity of ERα and ERβ is determined by the structures of the receptors. A ligand dependent AF-2 region is located within the E domain, which also harbors the ligand binding domain (LBD). ERα and ERβ share 59% homology in the E domain and bind 17β-estradiol (E2) with similar affinities [52]. The DNA binding domain (DBD) in the central C domain contains two highly conserved zinc fingers. The receptors bind similar DNA sequences known as estrogen response elements (EREs) in the promoters or other regulatory regions of target genes. The consensus sequence of the ERE is a 13 base pair inverted repeat: GGTCAnnnTGACC. Despite high homology in the DBD of ERα and ERβ, the receptors regulate unique genes in many cells [53–55]. The D domain is a flexible hinge region between the DBD and LBD. Two regions in the C and E domains mediate receptor dimerization and there is a short F domain at the C terminus, the function of which may also involve dimerization or protein-protein interactions [56, 57].
3.2 Ligand dependent transcriptional regulation
ER mediated transcriptional regulation can occur through ligand dependent and ligand independent pathways. Traditional antiestrogens used to inhibit ERα action in breast cancer target the ligand dependent signaling pathway, but new approaches that target common features of all pathways, DNA-binding for example, are currently being explored as potential therapeutic strategies. For the purpose of this review, we will briefly present the ligand dependent mechanism of ER action, but the molecular pathways of ER signaling are complex and can occur independent of ligands (reviewed extensively in [58, 59]). Each level of ER signaling (ligand binding, dimerization, DNA binding, and cofactor recruitment) can be targeted for selective modulation of ER action in cancer cells and each process must be considered when designing selective ER therapies (Figure 2).
Figure 2.

Ligand dependent activity of ERs. When bound to an agonist, ERs dimerize, bind DNA, and recruit coactivators to stimulate transcription. Antagonists lead to corepressor recruitment which prevents transcription. Each step of ligand dependent activation (ligand binding, dimerization, DNA binding, and cofactor recruitment) may be selectively targeted for cancer treatment.
Classical estrogen signaling is ligand dependent and ERs can directly or indirectly regulate transcription of target genes in response to ligand binding. In direct ligand dependent ER action, ERs form dimers upon ligand binding and directly bind EREs, thereby initiating recruitment of coregulator proteins that promote or prevent transcription. Ligands may act as agonists, stimulating the formation of transcriptionally active dimers, or antagonists, which bind the receptor and render it transcriptionally inactive. Crystal structures of the liganded ERα and ERβ LBDs reveal that the 3D conformation induced upon ligand binding determines the agonistic or antagonistic properties of a molecule [60–62]. The magnitude of transcriptional activation is mediated by the recruitment of coactivators and corepressors that initiate or inhibit transcription, respectively. Over 300 coregulators have been described in the literature and the coregulator complexes involved in ER mediated transcriptional regulation are complex [63]. In general, members of the p160 or SRC family of coactivators bridge the receptors to a transcriptional complex that includes p300/CBP, which induces chromatin remodeling, and RNA polymerase II recruitment.
ERs can indirectly regulate transcription of target genes that do not contain consensus EREs in their promoters in a ligand dependent manner. For example, SERMs like tamoxifen, can induce ER mediated transcription at AP-1 sites through AF-2 dependent and independent mechanisms [64–66]. ERα mediates E2-stimulated transcription at AP-1 sites by forming a transcriptional complex with Fos/Jun transcription factors; functional AF-1 and AF-2 regions are required for E2 action at AP-1 sites [64, 65]. ERβ can also mediate transcriptional activation at AP-1 sites, but the mechanism seems to be independent of functional AF-1 and AF-2 regions [65]. Additionally, ERs mediate indirect transcription through interaction with Sp1 at Sp1 binding sites at the promoters. This interaction requires the AF-1 domain of ERα but ERβ AF-1 does not have a comparable transcriptional effect at Sp1 sites demonstrating the differential transcriptional mechanisms of ERα and ERβ [67].
3.3 ERα and ERβ signaling interactions
ERα and ERβ form homodimers or heterodimers that have unique transcriptional properties [68, 69]. Many splice variants of ERα and ERβ have been identified (Figure 1B). There are five known isoforms of ERβ [70]. ERβcx, or ERβ2, contains 26 amino acids in place of 60 amino acids found in the C terminus of ERβ1 which renders the LBD non functional [71]. ERβ 4 and ERβ 5 also lack functional LBDs [72]. In fact, only the longest isoform, ERβ1, exhibits ligand dependent transcriptional activity. However, the other isoforms can form heterodimers with ERα or ERβ to negatively impact transcriptional activation [71–74]. Thus, interactions among the isoforms and the splice variants of each isoform must be considered when designing therapies that target ERα or ERβ as coexpression of the receptors and their splice variants can modulate the transcriptional activity of the receptors.
4. ERα or ERβ selective ligands
Designing receptor selective ligands has been challenging due to the high similarity between the ERα and ERβ LBD. In order to compare selectivity, two approaches are used to quantify ligand affinity: relative binding affinity (RBA) and Ki values. RBA is defined as the binding relative to E2 as measured by radiometric or fluorometric ligand-binding assays, typically expressed as a percent:
Ki values are also determined using IC50 measurements obtained in competitive ligand-binding assays in which the tracer is E2:
Conversion between RBA and Kd values is possible given standard Kd values for E2 (typically 0.2 nM for ERα and 0.5 nM for ERβ) [75].
Ligand binding does not necessarily correlate with transcriptional activity, and ER ligands may display selectivity for one receptor subtype in a concentration dependent manner (i.e. selective only at low concentrations) or ligands may display selectivity at the transcriptional level (i.e. ligands bind both receptors but only induce transcriptional activation of one subtype). Transcriptional selectivity is determined using cell-based assays that measure a reporter, such as luciferase, after treatment in cells transfected with the reporter typically linked to one or more EREs. It is difficult to compare EC50 values obtained from transcriptional assays because transcriptional activity is highly dependent on cell type specific variables like availability of cofactors and cell culture conditions. Ligands that display selectivity in a concentration dependent manner bind both receptors at high concentrations but induce transcriptional activity at lower concentrations for one subtype. Subtype selective ligands that are highly potent and highly selective will show greatest promise in therapeutic development aimed at selectively targeting ERβ activity in cancer.
4.1 Structural similarities of ERα and ERβ ligand binding domains
Ligand selectivity is ultimately determined by the three dimensional structure of the LBD. The three dimensional structures of liganded ERα and ERβ LBDs have similar features shared among steroid nuclear receptors [76, 77]. A hydrophobic core is created by 11 major helices arranged in three layers sandwiched in an antiparallel conformation [61, 62]. Coregulators bind the LBD at AF-2, which consists of 4 alpha helices (H3, H4, H5, and H12) that form a hydrophobic groove. Coactivators including SRC-1/NCoA1, SRC-2/TIF2/GRIP1/NCoA-2, and SRC-3/AIB1 contain NR box motifs consisting of three or four LXXLL repeats which bind the hydrophobic groove [61, 78–80]. The orientation of H12 is a critical determinant of cofactor binding and is oriented by the ligand bound to the receptor. In crystal structures of ERα or ERβ LBD bound to an agonist like E2, H12 is oriented across the hydrophobic pocket to allow binding of coactivators like SRC-2. In the antagonist orientation, H12 is oriented in the coactivator binding site and prevents recruitment of the transcriptional complex by blocking a critical residue, Lys362, which is required for coactivator recruitment [60–62].
Unlike many nuclear receptors, the hydrophobic core in the LBD of estrogen receptor is relatively larger than the endogenous ligand 17β-estradiol (E2), which allows the receptor to bind a variety of small molecules, some of which are presented in Table 1 [61, 62]. E2 is just 245 Å3 in size, while the LBD of ERα is nearly double at 450 Å3. The LBD of ERβ is slightly smaller at 390 Å3. The high affinity for E2 is determined by the hydrophobic nature of E2 and a series of hydrogen bonds with a water molecule and the hydroxyl groups of E2 that stabilize the ligand (Figure 3). Glu353 and Arg394 of ERα, corresponding to Glu305 and Arg346 of ERβ, interact with a water molecule and the hydroxyl group of E2’s A ring. At the opposite end of E2, the 17β hydroxyl group attached to the D ring forms hydrogen bonds with His524 in ERα, corresponding to His475 in ERβ. The hydrogen bonds generated with the hydroxyl groups of the A and D rings of E2 are also observed in the crystal structure of ERα LBD bound to diethylstilbestrol (DES), a high affinity ligand of ERs that has a similar distance between the opposing hydroxyls as E2 [52, 60]. DES has an even higher affinity for ERα and ERβ than E2 due to additional hydrophobic interactions which contribute to stability in the LBD.
Table 1.
ER ligands and relative binding affinities to ERα and ERβ.
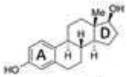
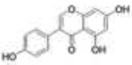
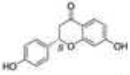
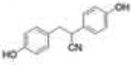
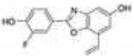
Figure 3.

Structural similarities and differences of ERα and ERβ LBD. Taken from [75] with the author’s permission. ERα and ERβ differ in two amino acids within the LBD that contribute to ligand selectivity. Leu384 of ERα is slightly larger and more inflexible than Met336 of ERβ so ligands with bulky constituents in the region tend to show selectivity for ERβ. Ile373 of ERβ is slightly for flexible than Met421 of ERα so bulky substituents in this region also contributes to ERβ selectivity. Both receptors share Glu, Arg, and His residues that participate in a series of hydrogen bonds with the hydroxyl groups of 17β-estradiol.
4.2 Structural differences of ERα and ERβ ligand binding domains
The tissue distributions of ERα and ERβ are strong determinants for tissue selectivity of ligand action while ER subtype selectivity is ultimately determined by structural differences in the LBDs of ERα and ERβ. The sizes of ERα and ERβ LBDs contribute to ligand selectivity but ERα and ERβ share a high degree of similarity in residues that line the binding cavity making the design of highly potent and selective ligands difficult. Within the residues that line the binding cavity, ERα and ERβ differ in only two amino acids: in helix 5, Leu384 of ERα corresponds to Met336 of ERβ and Met421 of ERα corresponds to Ile373 of ERβ in loop 6–7. Met421 of ERα and Ile373 of ERβ lie below the D ring of E2 and Leu384 of ERα and Met336 of ERβ are above the D ring of E2 (Figure 3). Subtype selective ligands and structural modeling have shown the differences in the flexibility and size of Met421/Ile373 and Leu384/Met336 are the major determinants for subtype selectivity. A detailed discussion of the structural determinants of subtype selective ligands is presented in section 6.1.
5. Current approaches for targeting ERα for cancer therapy
The therapeutic potential of ERα has been utilized for breast cancer treatment for decades and approximately 70% of breast cancers express ERα [31]. Though ERα and ERβ are expressed in mammary cells, the therapeutic value of ERβ is still under debate and current therapies for targeting ER signaling in breast cancer aim to impair ERα activity. Current treatments, most notably tamoxifen, exhibit tissue selectivity in terms of agonistic and antagonistic properties. Such compounds, called selective estrogen receptor modulators (SERMs), act as antagonists in mammary tissue and agonists in tissues such as bone and the uterus. Selective estrogen receptor down-regulators (SERDs), like fulvestrant, are also used clinically to treat ERα positive breast cancer. Despite the success of treatments that target the LBD of ERα, many breast cancers develop resistance to current therapies [10]. New strategies for inhibiting ERα action in breast cancer include targeting ER-cofactor interactions and ER-DNA interactions. Such strategies may prove effective in treating ERα positive breast cancers that develop resistance to LBD targeted therapies. Treatments targeting ERα in breast cancer do not necessarily display subtype selectivity. Though tamoxifen is currently used to inhibit ERα activity in breast cancers it has recently been shown that tamoxifen can also impair ERβ mediated gene expression regulation suggesting adverse impacts on normal ERβ activity [81]. The impacts of current treatments initially designed to target ERα on ERβ action in breast cancer patients is not clear and the discussion of SERMs and SERDs will focus on their tissue selectivity and mechanisms of action in the context of ERα.
5.1 SERMs – Determinants of tissue selectivity
Tissue selectivity of SERMs is determined by conformation changes in ER, cofactor recruitment, and promoter context, but the exact mechanisms and interactions between the components that determine tissue selectivity are unclear and it is difficult to predict the tissue specific effects SERMs may elicit. In general, SERMs act as agonists in bone, liver, and the cardiovascular system and antagonists in the breast. In the uterus, SERMs can show mixed antagonist and agonist activities [82]. The ideal SERM will act as an antagonist in hormone responsive tissues like breast, uterus, and ovaries thereby reducing the risks of hormonal cancers in these tissues and act as an agonist in bone and the cardiovascular system thereby reducing risk of osteoporosis, cardiovascular disease and strokes. The therapeutic potential and activity of SERMs have been reviewed extensively [9, 83, 84] so we will briefly discuss some SERMs currently used or in development for cancer treatment with an emphasis on the mechanisms that determine tissue selectivity.
Currently, raloxifene and tamoxifen are SERMs used to treat and prevent breast cancer and new SERMs that show promise for cancer treatment have been synthesized based on the core structures of tamoxifen and raloxifene. Tamoxifen is the first generation SERM that has been used to treat ERα positive breast cancers for the past 30 years and has contributed to a decline in breast cancer mortality rates. Tamoxifen also reduces the risk of breast cancer by 50% in high risk patients [9]. The primary unwanted side effects of tamoxifen are its agonistic effect in the endometrium which increases risk for endometrial cancer 2–5 fold and an increased risk of thromboembolic disease in postmenopausal women [9]. Modifications of the triphenylethylene core of tamoxifen have led to the development of new SERMs that show potential for clinical treatment of breast cancer. For example, toremifene is a chlorinated analog of tamoxifen that has been approved for treatment of metastatic breast cancer and is as effective as tamoxifen with the advantage of fewer genotoxic metabolites and a slightly reduced risk of endometrial cancers [83, 85]. Despite a slightly safer profile in epidemiology studies, toremifene can stimulate endometrial cancer tumors in xenograft experiments suggesting the tissue selective action of toremifene is very similar to that of tamoxifen [86]. Considering the structural similarities of tamoxifen and toremifene, it is likely that tamoxifen-resistant cancers will not be susceptible to toremifene treatment. Raloxifene is a second generation SERM that is as effective as tamoxifen at reducing the risk of invasive breast cancer but unlike tamoxifen, the risk of endometrial cancer is not increased [83]. Like tamoxifen, unwanted side effects of raloxifene include hot flashes and blood clots. The raloxifene core structure has been modified to synthesize SERMs with greater pharmacokinetic properties or tissue and subtype selectivity. Many of the raloxifene analogs are currently under investigation as preventives for breast cancer.
The mechanisms that determine tissue selectivity of SERMs have primarily been studied with tamoxifen and raloxifene, though the exact determinants of tissue selectivity remain undefined. Coactivator recruitment is an ultimate requirement for agonist activity of SERMs in certain tissues and this is determined by the availability of cofactors and the conformational changes induced by SERM binding. As discussed previously, ligand binding induces conformational changes that alter the orientation of H12 which is a critical determinant of cofactor binding. Crystal structures of ERα and ERβ LBD bound to tamoxifen and raloxifene show that the orientation of H12 is determined by interaction between Asp351 of ERα and the long side chains of tamoxifen or raloxifene [61, 62]. Mutation of Asp351 to a glycine leads to pure antagonistic effects with tamoxifen or raloxifene treatment, effectively abolishing the agonist properties of these SERMs [87]. Coactivator availability is tissue and cell type specific and contributes to tissue selective activity of SERMs. Tamoxifen stimulates cell cycle progression of Ishikawa endometrial cancer cells and the proliferative effects of tamoxifen require SRC-1 [88]. SRC-1 expression is higher in Ishikawa cells compared to MCF-7 breast cancer cells, a trend observed across a number of endometrial and breast cell lines, and knockdown of SRC-1 abolished the proliferative effects of tamoxifen in the endometrial cell line. This effect was specific to SRC-1, suggesting the availability of SRC-1 can mediate the tissue selective agonist effects of tamoxifen.
Additionally, promoter context determines the agonist and antagonist activity of SERMs. In tissues in which SERMs act as agonists, SERMs can stimulate transcription at non-conventional ERE regulatory elements such as AP-1 or Sp1 sites, emphasizing the importance of promoter context in determining tissue selectivity. Raloxifene and tamoxifen can induce transcription at AP-1 sites through ERβ independent of functional AF-1 and AF-2 regions, possibly by sequestering corepressors and histone deacetylases (HDACs) that can repress transcription at distant sites [64]. Ligands can act as agonists or antagonists on AP-1 sites in a receptor-specific manner highlighting the transcriptional differences between ERα and ERβ. E2 acts as an agonist with ERα at AP-1 sites but inhibits transcription with ERβ. Tamoxifen and raloxifene act as agonists with ERβ at AP-1 sites while maintaining antagonist effects with ERα [89]. ER transcriptional regulation at AP-1 sites is also dependent on the cell context. In fact, tamoxifen stimulates transcription through AP-1 sites in uterine cells but not in breast cancer cells suggesting this mechanism may contribute to unwanted uterotrophic side effects of tamoxifen [66]. Thus, the mechanism of ER mediated transcription can contribute to tissue selectivity of ER ligands.
5.2 SERDs
Selective estrogen receptor down-regulators (SERDs) provide a second line of treatment in breast cancers that develop resistance to commonly used therapies such as tamoxifen [90]. SERDs bind the LBD of ER and induce rapid proteosomal degradation to inhibit ER signaling. Fulvestrant (ICI 182,780) is a SERD currently used in the clinic to treat metastatic breast cancer in patients with recurring or progressive disease despite tamoxifen or aromatase inhibitor treatment [91]. Fulvestrant acts through multiple mechanisms; it is a complete antagonist to ER and also promotes ER ubiquitination, likely on Lys302 and 303 found in the hinge region, targeting the receptor for degradation via the ubiquitin-26S proteosomal pathway [92]. ERα and ERβ homo- and heterodimers form upon fulvestrant treatment [69], but nuclear localization is impaired [93]. Unlike tamoxifen, the inhibitory effects of fulvestrant on ER activity are not tissue specific and ER degradation occurs in both mammary and uterine tissues [94]. Another SERD currently under investigation is GW5638. Though it promotes ER degradation like fulvestrant, GW5638 induced degradation may not follow the same mechanism as fulvestrant [95]. Upon fulvestrant treatment, ERα is only found in the insoluble fraction of cell lysate after 30 minutes. Subcellular localization of ER upon GW5638 treatment follows a time course similar to that observed after E2 treatment; initially ERα is found in cytoplasmic, nuclear, and insoluble fractions but shifts solely to the insoluble fraction after 2 hours. Crystal structure analysis shows that GW5638 induces a conformational change in the orientation of H12 thereby exposing hydrophobic side chains of Leu536, Leu539, Leu540, and Met543, which are buried in the hydrophobic core when the receptor is bound to a partial agonist like tamoxifen. Exposure of hydrophobic residues in the LBD is greater when bound to GW5638 when compared to fulvestrant [96]. Greater exposure of hydrophobic residues may stimulate ER degradation by reducing stability, contributing to the differing mechanisms of degradation observed for GW5638 and fulvestrant. Further development of SERDs will require consideration of the ER conformation induced upon SERD binding and exposure of hydrophobic residues may be a critical factor for the efficient degradation of ER.
5.3 New approaches to selectively target ER signaling
Novel approaches for targeting ER action go beyond the development of compounds that target the LBD. Recently, the electrophile disulfide benzamide (DIBA) was identified as a molecule that inhibits ER-DNA interactions independent of ligand binding [97]. Interestingly, DIBA showed selectivity for ERα zinc fingers that mediate DNA binding. In MCF-7/LCC2 breast cancer cells which express ERα but are tamoxifen resistant, DIBA treatment restored sensitivity to tamoxifen suggesting that combined treatment may be an option for patients that develop tamoxifen resistant breast cancer [98]. Shapiro and coworkers also recently identified theophylline, 8-[(benzylthio)methyl]-(7CI,8CI) (TPBM) as a small molecule inhibitor of ER-DNA interaction [99]. Fluorescein-labeled ERE DNA was used in a high throughput approach to screen small molecule libraries for inhibition of binding between ERE and recombinant ERα. TPBM exhibited moderate selectivity for inhibiting ERα-ERE interactions; IC50 concentrations were 3 mM for ERα, compared to 7.6 µM and 9 µM for androgen receptor and progesterone receptor, respectively. Additionally, TPBM inhibited ERα mediated transcription in T47D breast cancer cells. TPBM inhibited estrogen dependent growth of ERα positive BG-1 ovarian cancer cells (IC50∼5µM) but was not toxic in 60 other cancer cell lines, suggesting TPBM may also hold therapeutic potential as a selective ER inhibitor.
Inhibitors of ER-cofactor interactions have been designed using the known structure of the conserved coactivator NR box LXXLL motif and the hydrophobic groove to which it binds. Such inhibitors also show potential as treatments that can selectively target ER signaling. Small molecule coactivator binding inhibitors (CBIs) were described for ERα by Rodriguez and coworkers who used fluorescence polarization assays to identify pyrimidine compounds that inhibited binding of labeled SRC-1 NR box peptide [100]. Such compounds were further refined by Parent and coworkers and relatively potent CBIs were identified using fluorescence resonance energy transfer (FRET) assays in which energy transfer between labeled ER and SRC-3 NR domain was measured [101]. Surprisingly, many of the compounds identified in the experiments showed greater subtype selectivity for inhibiting ERα over ERβ, demonstrating cofactor recruitment can be a determinant of subtype selectivity. Pyrimidine CBIs inhibited ERβ mediated transcription in cell based assays but the biological effects on cancer cells have not yet been fully characterized. Amphipathic benzenes have also been designed as CBIs to mimic the NR box conserved in coactivators and effectively inhibited ERα coactivator interactions at low micromolar concentrations in FRET assays and cell based transcriptional assays [102]. Again, the biological effects of amphipathic benzene CBIs have not been characterized in cancer cells but they show promise as a new approach to overcome ligand independent ER signaling.
6. Selectively targeting ERβ for cancer treatment
As previously discussed, selective ERβ agonists may be used to stimulate the tumor suppressor function of ERβ in breast, ovarian, and prostate cancers. Both natural and synthetic ERβ selective ligands have been described [75, 103]. A selection of ERβ selective ligands is presented in Table 1 with corresponding RBA values for ERα and ERβ. A comprehensive review of ERβ ligands was recently published [75] so we will narrow our focus to natural and synthetic ligands that display high ERβ selectivity and discuss the structural characteristics of the molecules that contribute to ERβ selectivity. Additionally, we will present current approaches for identifying novel ERβ selective ligands.
6.1 Structural determinants of ERβ selective ligands
Developing selective ERβ ligands has been a field of active research since the identification of ERβ in 1996 [47]. Structural features shared among ERβ selective ligands have allowed the characterization of five features fundamental to ERβ selectivity and affinity [75]. Two hydroxyl groups on opposing ends of the ligand ensure binding affinity but do not enhance selectivity as ERα and ERβ undergo similar hydrogen bonding with E2. A phenolic hydroxyl is required for establishing the network of hydrogen bonds with Arg346 and Glu305. An opposing phenol, alcohol, or pseudophenol is a common feature of ERβ ligands because the molecule is stabilized by hydrogen bonds with His475. This second hydroxyl is not an absolute requirement as many ERβ selective ligands do not have this feature. Three structural components contribute to the subtype selectivity of a ligand due to interactions with Met336 and Ile373 of ERβ, which are not shared with ERα. Compounds with structurally bulky groups near Met 336 and Ile373 tend to have higher selectivity for ERβ. Met336 of ERβ is not as large and inflexible as Leu384 of ERα. An additional bulky protrusion towards Ile373 can contribute to ERβ selectivity since Met421 of ERα is longer and sterically clashes with ligand substituents. Finally, most ERβ ligands have space to accommodate Ile373 near the bulky substituent, described as a structural “inlet” by Minutolo and coworkers [75]. Though not all ERβ selective ligands share these structural features, all have at least one or more structural features that contribute to increased affinity and selectivity for ERβ.
Met336 is a major determinant of ERβ selectivity for some ligands, as shown by structural modeling and site directed mutagenesis [104]. Diarylpropionitrile (DPN) is a synthetic ERβ selective ligand that has a 70-fold selectivity for ERβ in binding assays and 78-fold selectivity in transcriptional assays. When Met336 of ERβ is replaced with a leucine, the transcriptional dose-response curve for DPN shifts toward that of ERα and selectivity is essentially lost. Structural models show Met336 interacts positively with the cyano group of DPN to stabilize the bound ligand [75]. The bulky cyano group can be accommodated by Met336 of ERβ but clashes with Leu384 of ERα. Conversely, the Leu384/Met336 transition in ERα and ERβ can contribute to ERα selectivity; propyl pyrazole triol (PPT) has approximately 400-fold greater binding affinity for ERα than ERβ. Structural modeling suggests that Met 336 of ERβ sterically hinders ligand binding which does not occur at Leu384 of ERα [105]. Met421 of ERα and Ile373 of ERβ also determine selectivity. The benzoxazole ERB-041 has a nearly 250 fold selectivity for ERβ in competitive binding assays and structural analysis shows selectivity is due to the interaction of the vinyl substituent of ERB-041 with Met421/Ile373 of ERα/ERβ [106, 107]. In ERα, Met421 sterically clashes with the vinyl substituent but Ile373 of ERβ is small enough to accommodate the vinyl group and possibly participate in hydrophobic attraction.
Many phytoestrogens display ERβ selectivity, particularly compounds with flavone or isoflavone core structures. Genistein is an isoflavone prevalent in soy that shows 22 fold selectivity for ERβ with ERβ RBA of 87% [52]. Initial crystal structure analysis showed genistein induces a shift in H12 toward the antagonist orientation when bound to ERβ LBD [62]. More recently, crystal structure analysis and computational modeling suggest genistein induces similar conformation changes when bound to ERα and ERβ in the presence of coactivator peptide fragments containing LXXLL motifs, suggesting coactivator binding can stabilize the ligand bound ERβ in an active conformation [107]. Liquiritigenin is another natural ERβ selective phytoestrogen recently identified as a component of an herbal extract that has been used in clinical trials to treat menopausal hot flashes. It has a flavone core structure and binds ERβ with a 20 fold higher binding s for ERα and ERβ, respectively) [108]. Crystal structures of ERβ bound to liquiritigenin have not been reported, but the structure of liquiritigenin contains many of the components required for ERβ affinity and selectivity. It contains two opposing phenolic hydroxyl groups that can participate in hydrogen bond networks with Glu305/Arg346 and His475. It is possible that the carbonyl group provides the structural bulk which can be accommodated by Met336 or Ile373 conferring selectivity for ERβ. Structural modeling may reveal the components of liquiritigenin that confer selectivity and it may show promise as a pharmacophore from which other ERβ selective ligands may be synthesized.
6.2 Approaches for identifying ERβ selective ligands
Due to the structural features of ERα and ERβ, it has been challenging to identify ERβ selective ligands with high selectivity, potency, and binding affinity. Several approaches have been used to identify new ERβ selective ligands. First, the structural core of known ERβ selective ligands can be modified in an attempt to identify ligands with greater selectivity and/or potency. Such an approach has been used with the isoflavone core structure of genistein, but the synthesized analogs did not display the selectivity or affinity observed with genistein [109]. Similarly, the structure of DPN was used to synthesize a series of ligands with modifications of the phenolic hydroxyl groups or nitrile groups [110]. Ligands produced from modifications of the DPN structure revealed the nitrile group is an important determinant of binding affinity and selectivity but the synthesized analogs of DPN showed similar or reduced selectivity for ERβ. Additionally, high throughput screening using cells with stably integrated ERE-reporter genes may also been used to identify receptor selective ligands, but only ligands that induce or inhibit transcription are identified. Transcription based assays are dependent on the cell type used so the assay is limited by the cell specific activity of the ligand. Finally, our lab has developed a bioluminescence resonance energy transfer (BRET) assay to measure ER dimerization induced by ligand binding [69]. This assay measures energy transfer between ER proteins fused to Renilla luciferase (the donor) or yellow fluorescent protein (YFP, the acceptor) and YFP emission indicates dimerization. Using the BRET assay, ERα/α and ERβ/β homodimerization was measured in response to ligand, as well as ERα/β heterodimerization, revealing ligands that selectively induce hetero- or homodimerization. Liquiritigenin was found to selectively induce ERβ/β and ERα/β dimers. In contrast, DPN induced ERβ/β homodimers and genistein induced all three dimer pairs, suggesting ERβ selective ligands may show selectivity at the level of dimerization. Future development of SERMs or ER selective ligands may be designed with dimer selectivity as well as subtype and tissue selectivity and compounds with optimal selectivity at all levels will have great potential for effectively regulating ER action in cancer and normal tissues. Additional consideration must also be given to receptor selectivity as ER ligands may also activate other cellular targets. Recently, DPN was shown to activate the aryl hydrocarbon receptor (AhR) with high activity suggesting known ER ligands may also activate AhR and have off target effects [111].
7. Conclusions
ERs regulate growth and development in response to estrogen exposure and the importance of ER mediated signaling in normal tissues reflects the therapeutic potential of selectively targeting ER for cancer treatment. In the breast, ERα and ERβ regulate proliferation and differentiation during normal mammary gland development and dysregulation of ER signaling in breast cancer has been effectively targeted with SERMs like tamoxifen for the past three decades. SERMs target ER signaling with tissue selective activity, which is determined by conformation changes induced by ligand binding, cofactor recruitment, and promoter context. With the identification of ERβ in 1996 and the emerging role of ERβ as a tumor suppressor in many cancers, selectively targeting ERs for cancer treatment has evolved into a new field of identifying subtype selective ligands that are highly selective and potent for ERβ with minimal induction of ERα activity. Though the development of such ligands has proved challenging due to the similarities of ERα and ERβ LBDs, subtle differences in the size and amino acids lining the LBD allow subtype selective ligands. Both subtype selectivity and tissue selectivity must be optimized in order to effectively target ER signaling for cancer therapy.
Acknowledgements
This publication was made possible by grant number T32 ES007015 from the National Institute of Environmental Health Sciences (NIEHS), NIH. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIEHS, NIH. W.X. is supported by NIH grants R01CA125387 and R03MH089442 and Shaw Scientist Award from Greater Milwaukee Foundation. We would like to acknowledge Emily Powell for providing comments during the writing process.
Abbreviations
- ER
estrogen receptor
- SERM
selective estrogen receptor modulator
- SERD
selective estrogen receptor down-regulator
- ERKO
estrogen receptor knockout
- PPT
propyl pyrazole triol
- DPN
diarylpropionitrile
- ARKO
aromatase inhibitor knockout
- AOM
azoxymethane
- ERE
estrogen response element
- DBD – DNA
binding domain
- LBD
ligand binding domain
- DES
diethylstilbestrol
- CBI
coactivator binding inhibitor
- BRET
bioluminescence resonance energy transfer
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Yager JD, Davidson NE. Estrogen carcinogenesis in breast cancer. N. Engl. J. Med. 2006;354:270–282. doi: 10.1056/NEJMra050776. [DOI] [PubMed] [Google Scholar]
- 2.Helguero LA, Faulds MH, Gustafsson JA, Haldosen LA. Estrogen receptors alpha (ERα) and beta (ERβ) differentially regulate proliferation and apoptosis of the normal murine mammary epithelial cell line HC11. Oncogene. 2005;24:6605–6616. doi: 10.1038/sj.onc.1208807. [DOI] [PubMed] [Google Scholar]
- 3.Murphy LC, Peng B, Lewis A, Davie JR, Leygue E, Kemp A, Ung K, Vendetti M, Shiu R. Inducible upregulation of oestrogen receptor-β1 affects oestrogen and tamoxifen responsiveness in MCF7 human breast cancer cells. J. Mol. Endocrinol. 2005;34:553–566. doi: 10.1677/jme.1.01688. [DOI] [PubMed] [Google Scholar]
- 4.Treeck O, Lattrich C, Springwald A, Ortmann O. Estrogen receptor beta exerts growth-inhibitory effects on human mammary epithelial cells. Breast Cancer Res. Treat. 2010;120:557–565. doi: 10.1007/s10549-009-0413-2. [DOI] [PubMed] [Google Scholar]
- 5.Hodges-Gallagher L, Valentine C, Bader S, Kushner P. Estrogen receptor beta increases the efficacy of antiestrogens by effects on apoptosis and cell cycling in breast cancer cells. Breast Cancer Res. Treat. 2008;109:241–250. doi: 10.1007/s10549-007-9640-6. [DOI] [PubMed] [Google Scholar]
- 6.Paruthiyil S, Parmar H, Kerekatte V, Cunha GR, Firestone GL, Leitman DC. Estrogen receptor β inhibits human breast cancer cell proliferation and tumor formation by causing a G2 cell cycle arrest. Cancer Res. 2004;64:423–428. doi: 10.1158/0008-5472.can-03-2446. [DOI] [PubMed] [Google Scholar]
- 7.Ström A, Hartman J, Foster JS, Kietz S, Wimalasena J, Gustafsson JA. Estrogen receptor β inhibits 17β-estradiol-stimulated proliferation of the breast cancer cell line T47D. Proc. Natl. Acad. Sci. U. S. A. 2004;101:1566–1571. doi: 10.1073/pnas.0308319100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hartman J, Lindberg K, Morani A, Inzunza J, Strom A, Gustafsson JA. Estrogen receptor beta inhibits angiogenesis and growth of T47D breast cancer xenografts. Cancer Res. 2006;66:11207–11213. doi: 10.1158/0008-5472.CAN-06-0017. [DOI] [PubMed] [Google Scholar]
- 9.Swaby R, Sharma C, Jordan V. SERMs for the treatment and prevention of breast cancer. Rev. Endocr. and Metab. Disord. 2007;8:229–239. doi: 10.1007/s11154-007-9034-4. [DOI] [PubMed] [Google Scholar]
- 10.Musgrove EA, Sutherland RL. Biological determinants of endocrine resistance in breast cancer. Nat. Rev. Cancer. 2009;9:631–643. doi: 10.1038/nrc2713. [DOI] [PubMed] [Google Scholar]
- 11.Taylor AH, Al-Azzawi F. Immunolocalisation of oestrogen receptor beta in human tissues. J. Mol. Endocrinol. 2000;24:145–155. doi: 10.1677/jme.0.0240145. [DOI] [PubMed] [Google Scholar]
- 12.Hewitt SC, Harrell JC, Korach KS. Lessons in estrogen biology from knockout and transgenic animals. Annu. Rev. Physiol. 2005;67:285–308. doi: 10.1146/annurev.physiol.67.040403.115914. [DOI] [PubMed] [Google Scholar]
- 13.Couse JF, Korach KS. Estrogen receptor null mice: What have we learned and where will they lead us? Endocr. Rev. 1999;20:358–417. doi: 10.1210/edrv.20.3.0370. [DOI] [PubMed] [Google Scholar]
- 14.Harris HA. Estrogen receptor-beta: Recent lessons from in vivo studies. Mol. Endocrinol. 2007;21:1–13. doi: 10.1210/me.2005-0459. [DOI] [PubMed] [Google Scholar]
- 15.Lubahn DB MJ, Golding TS, Couse JF, Korach KS, Smithies O. Alteration of reproductive function but not prenatal sexual development after insertional disruption of the mouse estrogen receptor gene. Proc. Natl. Acad. Sci. U. S. A. 1993;90:11162–11166. doi: 10.1073/pnas.90.23.11162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dupont S, Krust A, Gansmuller A, Dierich A, Chambon P, Mark M. Effect of single and compound knockouts of estrogen receptors alpha (ERalpha) and beta (ERbeta) on mouse reproductive phenotypes. Development. 2000;127:4277–4291. doi: 10.1242/dev.127.19.4277. [DOI] [PubMed] [Google Scholar]
- 17.Krege JH, Hodgin JB, Couse JF, Enmark E, Warner M, Mahler JF, Sar M, Korach KS, Gustafsson JA, Smithies O. Generation and reproductive phenotypes of mice lacking estrogen receptor β. Proc. Natl. Acad. Sci. U. S. A. 1998;95:15677–15682. doi: 10.1073/pnas.95.26.15677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Antal MC, Krust A, Chambon P, Mark M. Sterility and absence of histopathological defects in nonreproductive organs of a mouse ERbeta-null mutant. Proc. Natl. Acad. Sci. U. S. A. 2008;105:2433–2438. doi: 10.1073/pnas.0712029105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bocchinfuso WP, Korach KS. Mammary gland development and tumorigenesis in estrogen receptor knockout mice. J. Mammary Gland Biol. Neoplasia. 1997;2:323–334. doi: 10.1023/a:1026339111278. [DOI] [PubMed] [Google Scholar]
- 20.Kenney NJ, Bowman A, Korach KS, Carl Barrett J, Salomon DS. Effect of exogenous epidermal-like growth factors on mammary gland development and differentiation in the estrogen receptor-alpha knockout (ERKO) mouse. Breast Cancer Res. Treat. 2003;79:161–173. doi: 10.1023/a:1023938510508. [DOI] [PubMed] [Google Scholar]
- 21.Feng Y, Manka D, Wagner KU, Khan SA. Estrogen receptor-alpha expression in the mammary epithelium is required for ductal and alveolar morphogenesis in mice. Proc. Natl. Acad. Sci. U S A. 2007;104:14718–14723. doi: 10.1073/pnas.0706933104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Förster C, Mäkela S, Wärri A, Kietz S, Becker D, Hultenby K, Warner M, Gustafsson JA. Involvement of estrogen receptor β in terminal differentiation of mammary gland epithelium. Proc. Natl. Acad. Sci. U. S. A. 2002;99:15578–15583. doi: 10.1073/pnas.192561299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Helguero LA, Lindberg K, Gardmo C, Schwend T, Gustafsson JA, Haldosen LA. Different roles of estrogen receptors alpha and beta in the regulation of E-cadherin protein levels in a mouse mammary epithelial cell line. Cancer Res. 2008;68:8695–8704. doi: 10.1158/0008-5472.CAN-08-0788. [DOI] [PubMed] [Google Scholar]
- 24.Omoto Y, Imamov O, Warner M, Gustafsson JA. Estrogen receptor α and imprinting of the neonatal mouse ventral prostate by estrogen. Proc. Natl. Acad. Sci. U. S. A. 2005;102:1484–1489. doi: 10.1073/pnas.0409168102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Prins GS, Korach KS. The role of estrogens and estrogen receptors in normal prostate growth and disease. Steroids. 2008;73:233–244. doi: 10.1016/j.steroids.2007.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shapiro E, Huang H, Masch RJ, McFadden DE, Wilson EL, Wu X-R. Immunolocalization of estrogen receptor alpha and beta in human fetal prostate. J. Urology. 2005;174:2051–2053. doi: 10.1097/01.ju.0000176472.90432.5b. [DOI] [PubMed] [Google Scholar]
- 27.Imamov O, Morani A, Shim G-J, Omoto Y, Thulin-Andersson C, Warner M, Gustafsson JA. Estrogen receptor β regulates epithelial cellular differentiation in the mouse ventral prostate. Proc. Natl. Acad. Sci. U. S. A. 2004;101:9375–9380. doi: 10.1073/pnas.0403041101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wada-Hiraike O, Imamov O, Hiraike H, Hultenby K, Schwend T, Omoto Y, Warner M, Gustafsson JA. Role of estrogen receptor β in colonic epithelium. Proc. Natl. Acad. Sci. U. S. A. 2006;103:2959–2964. doi: 10.1073/pnas.0511271103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Newcomb PA, Storer BE. Postmenopausal hormone use and risk of large-bowel cancer. J. Natl. Cancer Inst. 1995;87:1067–1071. doi: 10.1093/jnci/87.14.1067. [DOI] [PubMed] [Google Scholar]
- 30.Deroo BJ, Korach KS. Estrogen receptors and human disease. J. Clin. Investig. 2006;116:561. doi: 10.1172/JCI27987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Skliris GP, Leygue E, Watson PH, Murphy LC. Estrogen receptor alpha negative breast cancer patients: Estrogen receptor beta as a therapeutic target. J. Steroid Biochem. Mol. Biol. 2008;109:1–10. doi: 10.1016/j.jsbmb.2007.12.010. [DOI] [PubMed] [Google Scholar]
- 32.Chia S, Bryce C, Gelmon K. Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. The Lancet. 2005;365:1687–1717. doi: 10.1016/S0140-6736(05)66544-0. [DOI] [PubMed] [Google Scholar]
- 33.Zhao C, Dahlman-Wright K, Gustafsson JA. Estrogen receptor beta: an overview and update. Nucl. Recept. Signal. 2008;6:e003. doi: 10.1621/nrs.06003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lin CY, Strom A, Li Kong S, Kietz S, Thomsen J, Tee J, Vega V, Miller L, Smeds J, Bergh J, Gustafsson JA, Liu E. Inhibitory effects of estrogen receptor beta on specific hormone-responsive gene expression and association with disease outcome in primary breast cancer. Breast Cancer Res. 2007;9:R25. doi: 10.1186/bcr1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lindberg K, Ström A, Lock JG, Gustafsson JA, Haldosén LA, Helguero LA. Expression of estrogen receptor beta increases integrin alpha1 and integrin beta1 levels and enhances adhesion of breast cancer cells. J. Cell. Physiol. 2010;222:156–167. doi: 10.1002/jcp.21932. [DOI] [PubMed] [Google Scholar]
- 36.Ricke WA, McPherson SJ, Bianco JJ, Cunha GR, Wang Y, Risbridger GP. Prostatic hormonal carcinogenesis is mediated by in situ estrogen production and estrogen receptor alpha signaling. FASEB J. 2008;22:1512–1520. doi: 10.1096/fj.07-9526com. [DOI] [PubMed] [Google Scholar]
- 37.Fixemer T, Remberger K, Bonkhoff H. Differential expression of the estrogen receptor beta (ERbeta) in human prostate tissue, premalignant changes, and in primary, metastatic, and recurrent prostatic adenocarcinoma. The Prostate. 2003;54:79–87. doi: 10.1002/pros.10171. [DOI] [PubMed] [Google Scholar]
- 38.Cheng J, Lee EJ, Madison LD, Lazennec G. Expression of estrogen receptor beta in prostate carcinoma cells inhibits invasion and proliferation and triggers apoptosis. FEBS Letters. 2004;566:169–172. doi: 10.1016/j.febslet.2004.04.025. [DOI] [PubMed] [Google Scholar]
- 39.McPherson SJ, Ellem SJ, Simpson ER, Patchev V, Fritzemeier KH, Risbridger GP. Essential role for estrogen receptor β in stromal-epithelial regulation of prostatic hyperplasia. Endocrinology. 2007;148:566–574. doi: 10.1210/en.2006-0906. [DOI] [PubMed] [Google Scholar]
- 40.Campbell-Thompson M, Lynch IJ, Bhardwaj B. Expression of estrogen receptor (ER) subtypes and ERβ isoforms in colon cancer. Cancer Res. 2001;61:632–640. [PubMed] [Google Scholar]
- 41.Konstantinopoulos PA, Kominea A, Vandoros G, Sykiotis GP, Andricopoulos P, Varakis I, Sotiropoulou-Bonikou G, Papavassiliou AG. Oestrogen receptor beta (ERβ) is abundantly expressed in normal colonic mucosa, but declines in colon adenocarcinoma paralleling the tumour's dedifferentiation. Eur. J. Cancer. 2003;39:1251–1258. doi: 10.1016/s0959-8049(03)00239-9. [DOI] [PubMed] [Google Scholar]
- 42.Jassam N, Bell SM, Speirs V, Quirke P. Loss of expression of oestrogen receptor beta in colon cancer and its association with Dukes' staging. Oncol. Rep. 2005;14:17–21. [PubMed] [Google Scholar]
- 43.Picariello L, Fiorelli G, Martineti V, Tognarini I, Pampaloni B, Tonelli F, Brandi ML. Growth response of colon cancer cell lines to selective estrogen receptor modulators. Anticancer Res. 2003;23:2419–2424. [PubMed] [Google Scholar]
- 44.Janakiram NB, Steele VE, Rao CV. Estrogen receptor-β as a potential target for colon cancer prevention: Chemoprevention of azoxymethane-induced colon carcinogenesis by raloxifene in F344 Rats. Cancer Prev. Res. 2009;2:52–59. doi: 10.1158/1940-6207.CAPR-08-0140. [DOI] [PubMed] [Google Scholar]
- 45.Syed V, Ulinski G, Mok SC, Yiu GK, Ho S-M. Expression of gonadotropin receptor and growth responses to key reproductive hormones in normal and malignant human ovarian surface epithelial cells. Cancer Res. 2001;61:6768–6776. [PubMed] [Google Scholar]
- 46.Treeck O, Pfeiler G, Mitter D, Lattrich C, Piendl G, Ortmann O. Estrogen receptor {beta}1 exerts antitumoral effects on SK-OV-3 ovarian cancer cells. J. Endocrinol. 2007;193:421–433. doi: 10.1677/JOE-07-0087. [DOI] [PubMed] [Google Scholar]
- 47.Kuiper GG, Enmark E, Pelto-Huikko M, Nilsson S, Gustafsson JA. Cloning of a novel receptor expressed in rat prostate and ovary. Proc. Natl. Acad. Sci. U. S. A. 1996;93:5925–5930. doi: 10.1073/pnas.93.12.5925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Greene GL, Gilna P, Waterfield M, Baker A, Hort Y, Shine J. Sequence and expression of human estrogen receptor complementary DNA. Science. 1986;231:1150–1154. doi: 10.1126/science.3753802. [DOI] [PubMed] [Google Scholar]
- 49.Enmark E, Pelto-Huikko M, Grandien K, Lagercrantz S, Lagercrantz J, Fried G, Nordenskjold M, Gustafsson JA. Human estrogen receptor β-gene structure, chromosomal localization, and expression pattern. J. Clin. Endocrinol. Metab. 1997;82:4258–4265. doi: 10.1210/jcem.82.12.4470. [DOI] [PubMed] [Google Scholar]
- 50.Cowley SM, Parker MG. A comparison of transcriptional activation by ERα and ERβ. J. Steroid Biochem. Mol. Biol. 1999;69:165–175. doi: 10.1016/s0960-0760(99)00055-2. [DOI] [PubMed] [Google Scholar]
- 51.Hall JM, McDonnell DP. The estrogen receptor beta-isoform (ERbeta) of the human estrogen receptor modulates ER alpha transcriptional activity and is a key regulator of the cellular response to estrogens and antiestrogens. Endocrinology. 1999;140:5566–5578. doi: 10.1210/endo.140.12.7179. [DOI] [PubMed] [Google Scholar]
- 52.Kuiper GG, Carlsson B, Grandien K, Enmark E, Haggblad J, Nilsson S, Gustafsson JA. Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors alpha and beta. Endocrinology. 1997;138:863–870. doi: 10.1210/endo.138.3.4979. [DOI] [PubMed] [Google Scholar]
- 53.Klinge CM. Estrogen receptor interaction with estrogen response elements. Nucl. Acids Res. 2001;29:2905–2919. doi: 10.1093/nar/29.14.2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Secreto FJ, Monroe DG, Dutta S, Ingle JN, Spelsberg TC. Estrogen receptor alpha/beta isoforms, but not betacx, modulate unique patterns of gene expression and cell proliferation in Hs578T cells. J. Cell. Biochem. 2007;101:1125–1147. doi: 10.1002/jcb.21205. [DOI] [PubMed] [Google Scholar]
- 55.Stossi F, Barnett DH, Frasor J, Komm B, Lyttle CR, Katzenellenbogen BS. Transcriptional profiling of estrogen-regulated gene expression via estrogen receptor (ER) {alpha} or ER{beta} in human osteosarcoma cells: Distinct and common target genes for these receptors. Endocrinology. 2004;145:3473–3486. doi: 10.1210/en.2003-1682. [DOI] [PubMed] [Google Scholar]
- 56.Koide A, Zhao C, Naganuma M, Abrams J, Deighton-Collins S, Skafar DF, Koide S. Identification of regions within the F domain of the human estrogen receptor α that are important for modulating transactivation and protein-protein interactions. Mol. Endocrinol. 2007;21:829–842. doi: 10.1210/me.2006-0203. [DOI] [PubMed] [Google Scholar]
- 57.Yang J, Singleton DW, Shaughnessy EA, Khan SA. The F-domain of estrogen receptor-alpha inhibits ligand induced receptor dimerization. Mol. Cell. Endocrinol. 2008;295:294. doi: 10.1016/j.mce.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 58.Nilsson S, Makela S, Treuter E, Tujague M, Thomsen J, Andersson G, Enmark E, Pettersson K, Warner M, Gustafsson JA. Mechanisms of estrogen action. Physiol. Rev. 2001;81:1535–1565. doi: 10.1152/physrev.2001.81.4.1535. [DOI] [PubMed] [Google Scholar]
- 59.Heldring N, Pike A, Andersson S, Matthews J, Cheng G, Hartman J, Tujague M, Strom A, Treuter E, Warner M, Gustafsson JA. Estrogen receptors: How do they signal and what are their targets. Physiol. Rev. 2007;87:905–931. doi: 10.1152/physrev.00026.2006. [DOI] [PubMed] [Google Scholar]
- 60.Shiau AK, Barstad D, Loria PM, Cheng L, Kushner PJ, Agard DA, Greene GL. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell. 1998;95:927–937. doi: 10.1016/s0092-8674(00)81717-1. [DOI] [PubMed] [Google Scholar]
- 61.Brzozowski AM, Pike AC, Dauter Z, Hubbard RE, Bonn T, Engstrom O, Ohman L, Greene GL, Gustafsson JA, Carlquist M. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature. 1997;389:753–758. doi: 10.1038/39645. [DOI] [PubMed] [Google Scholar]
- 62.Pike AC, Brzozowski AM, Hubbard RE, Bonn T, Thorsell AG, Engstrom O, Ljunggren J, Gustafsson J, Carlquist M. Structure of the ligand-binding domain of oestrogen receptor beta in the presence of a partial agonist and a full antagonist. EMBO J. 1999;18:4608–4618. doi: 10.1093/emboj/18.17.4608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lonard DM, O'Malley BW. Nuclear receptor coregulators: Judges, juries, and executioners of cellular regulation. Mol. Cell. 2007;27:691–700. doi: 10.1016/j.molcel.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 64.Kushner PJ, Agard DA, Greene GL, Scanlan TS, Shiau AK, Uht RM, Webb P. Estrogen receptor pathways to AP-1. J. Steroid Biochem. Mol. Biol. 2000;74:311–317. doi: 10.1016/s0960-0760(00)00108-4. [DOI] [PubMed] [Google Scholar]
- 65.Webb P, Nguyen P, Valentine C, Lopez GN, Kwok GR, McInerney E, Katzenellenbogen BS, Enmark E, Gustafsson JA, Nilsson S, Kushner PJ. The estrogen receptor enhances AP-1 activity by two distinct mechanisms with different requirements for receptor transactivation functions. Mol. Endocrinol. 1999;13:1672–1685. doi: 10.1210/mend.13.10.0357. [DOI] [PubMed] [Google Scholar]
- 66.Webb P, Lopez GN, Uht RM, Kushner PJ. Tamoxifen activation of the estrogen receptor/AP-1 pathway: Potential origin for the cell-specific estrogen-like effects of antiestrogens. Mol. Endocrinol. 1995;9:443–456. doi: 10.1210/mend.9.4.7659088. [DOI] [PubMed] [Google Scholar]
- 67.Saville B, Wormke M, Wang F, Nguyen T, Enmark E, Kuiper G, Gustafsson JA, Safe S. Ligand,- cell,- and estrogen receptor subtype (α/β)-dependent activation at GC-rich (Sp1) promoter elements. J. Biol. Chem. 2000;275:5379–5387. doi: 10.1074/jbc.275.8.5379. [DOI] [PubMed] [Google Scholar]
- 68.Monroe DG, Secreto FJ, Subramaniam M, Getz BJ, Khosla S, Spelsberg TC. Estrogen receptor alpha and beta heterodimers exert unique effects on estrogen- and tamoxifen-dependent gene expression in human U2OS osteosarcoma cells. Mol. Endocrinol. 2005;19:1555–1568. doi: 10.1210/me.2004-0381. [DOI] [PubMed] [Google Scholar]
- 69.Powell E, Xu W. Intermolecular interactions identify ligand-selective activity of estrogen receptor α/β dimers. Proc. Natl. Acad. Sci. U. S. A. 2008;105:19012–19017. doi: 10.1073/pnas.0807274105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Moore JT, McKee DD, Slentz-Kesler K, Moore LB, Jones SA, Horne EL, Su J-L, Kliewer SA, Lehmann JM, Willson TM. Cloning and characterization of human estrogen receptor beta isoforms. Biochem. Biophys. Res. Comm. 1998;247:75–78. doi: 10.1006/bbrc.1998.8738. [DOI] [PubMed] [Google Scholar]
- 71.Ogawa S, Inoue S, Watanabe T, Orimo A, Hosoi T, Ouchi Y, Muramatsu M. Molecular cloning and characterization of human estrogen receptor betacx: a potential inhibitor of estrogen action in human. Nucl. Acids Res. 1998;26:3505–3512. doi: 10.1093/nar/26.15.3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Poola I, Abraham J, Baldwin K, Saunders A, Bhatnagar R. Estrogen receptors beta4 and beta5 are full length functionally distinct ERβ isoforms. Endocrine. 2005;27:227–238. doi: 10.1385/ENDO:27:3:227. [DOI] [PubMed] [Google Scholar]
- 73.Leung Y-K, Mak P, Hassan S, Ho S-M. Estrogen receptor (ER)-β isoforms: A key to understanding ER-β signaling. Proc. Natl. Acad. Sci. U. S. A. 2006;103:13162–13167. doi: 10.1073/pnas.0605676103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhao C, Matthews J, Tujague M, Wan J, Strom A, Toresson G, Lam EWF, Cheng G, Gustafsson JA, Dahlman-Wright K. Estrogen receptor beta2 negatively regulates the transactivation of estrogen receptor alpha in human breast cancer cells. Cancer Res. 2007;67:3955–3962. doi: 10.1158/0008-5472.CAN-06-3505. [DOI] [PubMed] [Google Scholar]
- 75.Minutolo F, Macchia M, Katzenellenbogen BS, Katzenellenbogen JA. Estrogen receptor beta ligands: Recent advances and biomedical applications. Med. Res. Rev. 2009;9999 doi: 10.1002/med.20186. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 76.Wurtz J-M, Bourguet W, Renaud J-P, Vivat V, Chambon P, Moras D, Gronemeyer H. A canonical structure for the ligand-binding domain of nuclear receptors. Nat. Struct. Mol. Biol. 1996;3:87–94. doi: 10.1038/nsb0196-87. [DOI] [PubMed] [Google Scholar]
- 77.Moras D, Gronemeyer H. The nuclear receptor ligand-binding domain: structure and function. Curr. Opin. Cell Biol. 1998;10:384–391. doi: 10.1016/s0955-0674(98)80015-x. [DOI] [PubMed] [Google Scholar]
- 78.Heery DM, Kalkhoven E, Hoare S, Parker MG. A signature motif in transcriptional co-activators mediates binding to nuclear receptors. Nature. 1997;387:733–736. doi: 10.1038/42750. [DOI] [PubMed] [Google Scholar]
- 79.Ding XF, Anderson CM, Ma H, Hong H, Uht RM, Kushner PJ, Stallcup MR. Nuclear receptor-binding sites of coactivators glucocorticoid receptor interacting protein 1 (GRIP1) and steroid receptor coactivator 1 (SRC-1): Multiple motifs with different binding specificities. Mol. Endocrinol. 1998;12:302–313. doi: 10.1210/mend.12.2.0065. [DOI] [PubMed] [Google Scholar]
- 80.Feng W, Ribeiro RC, nbsp J, Wagner RL, Nguyen H, Apriletti JW, Fletterick RJ, Baxter JD, Kushner PJ, West BL. Hormone-dependent coactivator binding to a hydrophobic cleft on nuclear receptors. Science. 1998;280:1747–1749. doi: 10.1126/science.280.5370.1747. [DOI] [PubMed] [Google Scholar]
- 81.Levy N, Paruthiyil S, Zhao X, Vivar OI, Saunier EF, Griffin C, Tagliaferri M, Cohen I, Speed TP, Leitman DC. Unliganded estrogen receptor-[beta] regulation of genes is inhibited by tamoxifen. Mol. Cell. Endocrin. 2010;315:201–207. doi: 10.1016/j.mce.2009.08.030. [DOI] [PubMed] [Google Scholar]
- 82.Katzenellenbogen BS, Katzenellenbogen JA. Defining the 'S' in SERMs. Science. 2002;295:2380. doi: 10.1126/science.1070442. [DOI] [PubMed] [Google Scholar]
- 83.Peng J, Sengupta S, Jordan VC. Potential of selective estrogen receptor modulators as treatments and preventives of breast cancer. Anticancer Agents Med. Chem. 2009;9:481–499. doi: 10.2174/187152009788451833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jordan VC, O'Malley BW. Selective estrogen-receptor modulators and antihormonal resistance in breast cancer. J. Clin. Oncol. 2007;25:5815–5824. doi: 10.1200/JCO.2007.11.3886. [DOI] [PubMed] [Google Scholar]
- 85.Harvey HA, Kimura M, Hajba A. Toremifene: An evaluation of its safety profile. The Breast. 2006;15:142–157. doi: 10.1016/j.breast.2005.09.007. [DOI] [PubMed] [Google Scholar]
- 86.O'Regan RM, Cisnero A, England GM, MacGregor JI, Muenzner HD, Assiki VJ, Bilimoria MM, Piette M, Dragan YP, Pitot HC, Chatterton R, Jordan VC. Effects of the antiestrogens tamoxifen, toremifene, and ICI 182,780 on endometrial cancer growth. J. Natl. Cancer Inst. 1998;90:1552–1558. doi: 10.1093/jnci/90.20.1552. [DOI] [PubMed] [Google Scholar]
- 87.Levenson AS, MacGregor Schafer JI, Bentrem DJ, Pease KM, Jordan VC. Control of the estrogen-like actions of the tamoxifen-estrogen receptor complex by the surface amino acid at position 351. J. Steroid Biochem. Mol. Biol. 2001;76:61–70. doi: 10.1016/s0960-0760(00)00143-6. [DOI] [PubMed] [Google Scholar]
- 88.Shang Y, Brown M. Molecular determinants for the tissue specificity of SERMs. Science. 2002;295:2465–2468. doi: 10.1126/science.1068537. [DOI] [PubMed] [Google Scholar]
- 89.Paech K, Webb P, Kuiper GGJM, Nilsson S, Gustafsson JA, Kushner PJ, Scanlan TS. Differential ligand activation of estrogen receptors ERα and ERβ at AP1 Sites. Science. 1997;277:1508–1510. doi: 10.1126/science.277.5331.1508. [DOI] [PubMed] [Google Scholar]
- 90.Robertson JFR, Come SE, Jones SE, Beex L, Kaufmann M, Makris A, Nortier JWR, Possinger K, Rutqvist LE. Endocrine treatment options for advanced breast cancer - the role of fulvestrant. European Journal of Cancer. 2005;41:346. doi: 10.1016/j.ejca.2004.07.035. [DOI] [PubMed] [Google Scholar]
- 91.Chia S, Gradishar W, Mauriac L, Bines J, Amant F, Federico M, Fein L, Romieu G, Buzdar A, Robertson JFR, Brufsky A, Possinger K, Rennie P, Sapunar F, Lowe E, Piccart M. Double-blind, randomized placebo controlled trial of fulvestrant compared with exemestane after prior nonsteroidal aromatase inhibitor therapy in postmenopausal women with hormone receptor-positive, advanced breast cancer: Results from EFECT. J. Clin. Oncol. 2008;26:1664–1670. doi: 10.1200/JCO.2007.13.5822. [DOI] [PubMed] [Google Scholar]
- 92.Berry NB, Fan M, Nephew KP. Estrogen receptor-α hinge-region lysines 302 and 303 regulate receptor degradation by the proteasome. Mol. Endocrinol. 2008;22:1535–1551. doi: 10.1210/me.2007-0449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Dauvois S, White R, Parker MG. The antiestrogen ICI 182780 disrupts estrogen receptor nucleocytoplasmic shuttling. J. Cell Sci. 1993;106:1377–1388. doi: 10.1242/jcs.106.4.1377. [DOI] [PubMed] [Google Scholar]
- 94.Robertson JF. Estrogen receptor downregulators: New antihormonal therapy for advanced breast cancer. Clin. Ther. 2002;24:A17. doi: 10.1016/s0149-2918(02)85032-9. [DOI] [PubMed] [Google Scholar]
- 95.Wittmann BM, Sherk A, McDonnell DP. Definition of functionally important mechanistic differences among selective estrogen receptor down-regulators. Cancer Res. 2007;67:9549–9560. doi: 10.1158/0008-5472.CAN-07-1590. [DOI] [PubMed] [Google Scholar]
- 96.Wu Y-L, Yang X, Ren Z, McDonnell DP, Norris JD, Willson TM, Greene GL. Structural Basis for an unexpected mode of SERM-mediated ER antagonism. Molecular Cell. 2005;18:413–424. doi: 10.1016/j.molcel.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 97.Wang LH, Yang XY, Zhang X, Mihalic K, Fan YX, Xiao W, Howard OM, Appella E, Maynard AT, Farrar WL. Suppression of breast cancer by chemical modulation of vulnerable zinc fingers in estrogen receptor. Nat. Med. 2004;10:40–47. doi: 10.1038/nm969. [DOI] [PubMed] [Google Scholar]
- 98.Wang LH, Yang XY, Zhang X, An P, Kim H-J, Huang J, Clarke R, Osborne CK, Inman JK, Appella E, Farrar WL. Disruption of estrogen receptor DNA-binding domain and related intramolecular communication restores tamoxifen sensitivity in resistant breast cancer. Cancer Cell. 2006;10:487–499. doi: 10.1016/j.ccr.2006.09.015. [DOI] [PubMed] [Google Scholar]
- 99.Mao C, Patterson NM, Cherian MT, Aninye IO, Zhang C, Montoya JB, Cheng J, Putt KS, Hergenrother PJ, Wilson EM, Nardulli AM, Nordeen SK, Shapiro DJ. A new small molecule inhibitor of estrogen receptor α binding to estrogen response elements blocks estrogen-dependent growth of cancer cells. J. Biol. Chem. 2008;283:12819–12830. doi: 10.1074/jbc.M709936200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Rodriguez AL, Tamrazi A, Collins ML, Katzenellenbogen JA. Design, synthesis, and in vitro biological evaluation of small molecule inhibitors of estrogen receptor α coactivator binding. J. Med. Chem. 2003;47:600–611. doi: 10.1021/jm030404c. [DOI] [PubMed] [Google Scholar]
- 101.Parent AA, Gunther JR, Katzenellenbogen JA. Blocking estrogen signaling after the hormone: Pyrimidine-core inhibitors of estrogen receptor-coactivator binding. J. Med. Chem. 2008;51:6512. doi: 10.1021/jm800698b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gunther JR, Moore TW, Collins ML, Katzenellenbogen JA. Amphipathic benzenes are designed inhibitors of the estrogen receptor α/steroid receptor coactivator interaction. ACS Chem. Biol. 2008;3:282–286. doi: 10.1021/cb800056r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kuiper GG, Lemmen JG, Carlsson B, Corton JC, Safe SH, van der Saag PT, van der Burg B, Gustafsson JA. Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor β. Endocrinology. 1998;139:4252–4263. doi: 10.1210/endo.139.10.6216. [DOI] [PubMed] [Google Scholar]
- 104.Sun J, Baudry J, Katzenellenbogen JA, Katzenellenbogen BS. Molecular basis for the subtype discrimination of the estrogen receptor-{beta}-selective ligand, diarylpropionitrile. Mol. Endocrinol. 2003;17:247–258. doi: 10.1210/me.2002-0341. [DOI] [PubMed] [Google Scholar]
- 105.Stauffer SR, Coletta CJ, Tedesco R, Nishiguchi G, Carlson K, Sun J, Katzenellenbogen BS, Katzenellenbogen JA. Pyrazole ligands: Structure-affinity/activity relationships and estrogen receptor-β-selective agonists. J. Med. Chem. 2000;43:4934–4947. doi: 10.1021/jm000170m. [DOI] [PubMed] [Google Scholar]
- 106.Harris HA, Albert LM, Leathurby Y, Malamas MS, Mewshaw RE, Miller CP, Kharode YP, Marzolf J, Komm BS, Winneker RC, Frail DE, Henderson RA, Zhu Y, Keith JC., Jr Evaluation of an estrogen receptor-β agonist in animal models of human disease. Endocrinology. 2003;144:4241–4249. doi: 10.1210/en.2003-0550. [DOI] [PubMed] [Google Scholar]
- 107.Manas ES, Unwalla RJ, Xu ZB, Malamas MS, Miller CP, Harris HA, Hsiao C, Akopian T, Hum W-T, Malakian K, Wolfrom S, Bapat A, Bhat RA, Stahl ML, Somers WS, Alvarez JC. Structure-based design of estrogen receptor-β selective ligands. J. Am. Chem. Soc. 2004;126:15106–15119. doi: 10.1021/ja047633o. [DOI] [PubMed] [Google Scholar]
- 108.Mersereau JE, Levy N, Staub RE, Baggett S, Zogric T, Chow S, Ricke WA, Tagliaferri M, Cohen I, Bjeldanes LF, Leitman DC. Liquiritigenin is a plant-derived highly selective estrogen receptor beta agonist. Mol. Cell. Endocrinol. 2008;283:49–57. doi: 10.1016/j.mce.2007.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Davis DD, Díaz-Cruz ES, Landini S, Kim Y-W, Brueggemeier RW. Evaluation of synthetic isoflavones on cell proliferation, estrogen receptor binding affinity, and apoptosis in human breast cancer cells. J. Steroid Biochem. Mol. Biol. 2008;108:23–31. doi: 10.1016/j.jsbmb.2007.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Meyers MJ, Sun J, Carlson KE, Marriner GA, Katzenellenbogen BS, Katzenellenbogen JA. Estrogen receptor-b potency-selective ligands: Structure-activity relationship studies of diarylpropionitriles and their acetylene and polar analogues. J. Med. Chem. 2001;44:4230–4251. doi: 10.1021/jm010254a. [DOI] [PubMed] [Google Scholar]
- 111.DuSell CD, Nelson ER, Wittmann BM, Fretz JA, Kazmin D, Thomas RS, Pike JW, McDonnell DP. Regulation of aryl hydrocarbon receptor function by selective estrogen receptor modulators. Mol. Endocrinol. 2010;24:33–46. doi: 10.1210/me.2009-0339. [DOI] [PMC free article] [PubMed] [Google Scholar]