

Abstract
Background
Pompe disease (acid α-glucosidase deficiency) is one of several lysosomal storage diseases amenable to treatment with enzyme replacement therapy (ERT). While echocardiography (echo) has been the standard method to evaluate the cardiac response to ERT, cardiac magnetic resonance imaging (CMR) has the advantage of better tissue definition and characterization of myocardial fibrosis. However, CMR for Pompe disease is not frequently performed due to the high risk of sedation. We report the first use of CMR in a feasible protocol to quantify left ventricular (LV) mass, function, and presence of myocardial fibrosis in the Pompe population.
Methods
Children with Pompe disease on ERT were assessed with transthoracic echo and CMR over a 3 year period at a single institution. Echocardiography was performed using standard techniques without sedation. CMR was performed using retrospectively gated and real-time imaging, with and without sedation. LV mass indexed to body surface area (LVMI) and ejection fraction (EF) were measured by both echo and CMR, and evaluated for change over time. Myocardial fibrosis was assessed with CMR by delayed enhancement imaging 5-10 min after gadolinium contrast using single-shot inversion recovery sequences with inversion time set to null the myocardium.
Results
Seventeen CMR scans were successfully performed in 10 subjects with Pompe disease (median age at first CMR 9 months, range 1-38 months, 80% male), with sedation only performed for 4 studies. There was a median interval of 5 months (range 0-34 months) from start of ERT to first CMR (baseline). At baseline, median indexed LVMI by CMR (140.0 g/m2, range 43.8-334.0) tended to be lower than that assessed by echo (median 204.0 g/m2, range 52.0-385.0), but did not reach statistical significance. At baseline, CMR EF was similar to that assessed by echo (55% vs. 55%). Overall, there was not a significant decrease in CMR measured LVMI over time (CMR median LVMI at baseline 94 g/m2 (range 43.8-334) vs. CMR median at most recent study 44.5 g/m2 (range 34-303), p=0.44). In 5 patients with serial CMR scans over time, LVMI decreased in 2, was similar in 2, and increased in 1 patient with high sustained antibodies to exogenous enzyme. Delayed enhancement was noted in only l separate patient who also had high sustained antibodies to exogenous enzyme.
Conclusion
CMR is a useful imaging tool that is feasible to use to serially follow LVMI and EF in children with Pompe disease on ERT. Real-time imaging is adequate for quantification purposes in these patients and minimizes the need for sedation. Quantitative CMR LVMI is generally lower than echo derived LVMI. Delayed enhancement appears to be a rare finding by CMR in Pompe Disease. Further follow-up is necessary to better understand the long term effects of ERT in infantile Pompe survivors, especially those with high sustained antibody titers or advanced cardiac disease at treatment outset.
Keywords: Pompe Disease, Enzyme Replacement Therapy, Cardiac Magnetic Resonance Imaging, Echocardiography, Delayed Enhancement
1. Introduction1
Pompe Disease (acid α-glucosidase deficiency, also known as glycogen storage disease type II) is a progressive, debilitating disease resulting from accumulation of lysosomal glycogen, especially in skeletal and myocardial cells. In the infantile form, patients are severely affected, with rapid progression of disease, severe functional limitations and rare survival beyond the first year of life (1-4). A recombinant form of acid alpha-glucosidase (rhGAA) derived from Chinese hamster ovary cells (CHO) cells was approved by the US FDA and European Union as the first treatment for Pompe disease. This was based on data from pivotal clinical trials in infants (5). In the largest comprehensive trial of 18 infants, enzyme replacement therapy (ERT) with rhGAA dramatically extended survival and reduced ventilator associated morbidity (5). Several other clinical trials of ERT with rhGAA have shown similar results (6-9).
Concomitant with the improved survival, there was a significant reduction in left ventricular (LV) hypertrophy as measured by echocardiography (echo) (5, 10) and electrocardiography (11) in infantile patients on ERT. There have been reports of cardiac arrhythmias which can be life threatening in treated infantile patients (12), similar to adult patients with Fabry Disease (13-15), who have also been reported to have myocardial fibrosis by cardiac magnetic resonance imaging (CMR) (16-18). CMR imaging, with its better quantification of left ventricular mass and ability to assess myocardial fibrosis, would be an ideal tool to serially follow Pompe patients. However, CMR has not been routinely performed in this patient group due to the high risk of anesthetic complications noted in infantile Pompe disease (19, 20) and the need for sedation for CMR studies. We therefore report the first feasibility experience and systematic analysis of the use of CMR imaging in Pompe patients receiving ERT.
2. Methods
All children with infantile Pompe Disease (defined as infants with hypotonia in the first year of life, <1% activity of GAA on skin fibroblasts and cardiac hypertrophy) receiving ERT who underwent both CMR and echo imaging from 2004 to 2007 at Duke University Medical Center were included in this study. This study was part of a broader single institutional study investigating cardiac structure and function in infantile Pompe Disease subjects receiving ERT enrolled in various clinical trials. Results of CMR imaging were reviewed for type of imaging sequence, measurement of left ventricular mass, function, presence of myocardial fibrosis as detected by delayed enhancement and use of sedation. The results from the echo most closely coinciding with the CMR scan were reviewed for left ventricular mass and function. Patient demographic data were also collected, along with data regarding the status of cross reactive immunologic material (CRIM), and titers of anti-rhGAA antibodies. The study was approved by the Duke University Medical Center Institutional Review Board and written informed consent was obtained from the parent/guardian of the subject prior to participation.
2.1 Cardiac Magnetic Resonance Imaging
Cardiac magnetic resonance imaging was performed using a 1.5 Tesla scanner (Siemens Medical Solutions; Erlangen, Germany). Sedation was performed by a pediatric cardiac anesthesiologist if necessary due to patient activity or clinical condition. Non-ECG gated single-shot steady-state free-precession (SSFP) “real-time” cine imaging, retrospectively ECG gated segmented SSFP cine imaging, or both were performed to assess wall thickness and function. Non-ECG gated single-shot SSFP “real-time” cine images were acquired using previously validated methods (21). Briefly, an imaging matrix of 90×128, 200×140 mm FOV, 5 mm slice thickness with 5 mm gap and 80 degree flip angle were used to acquire a cine image with a temporal resolution of 55-60 msec and a voxel size of 2.0 × 1.1 × 5 mm. Retrospectively ECG gated segmented SSFP cine images were acquired during free-breathing with multiple averages with an imaging matrix of 92×128, 200×140 mm FOV resulting in a voxel size of 2.7 × 1.7 × 5 mm and a temporal resolution of approximately 30 msec. The flip angle of 80 degrees and the slice thickness of 5 mm with a 5 mm gap were similar to real-time imaging.
The presence of delayed enhancement and myocardial fibrosis was evaluated using either inversion recovery single shot SSFP or segmented gradient-echo sequences in multiple short axis and the three long axis views. The inversion time was set to null normal myocardium, with sequences acquired approximately 5-10 minutes after IV administration of 0.2 mMol/kg of gadolinium.
LV mass and ejection fraction (EF) were measured from the interpolation of endocardial and epicardial contours from a stack of short axis slices of the left ventricle, with inclusion of the trabeculations and mitral papillary muscles (22). Echo LV mass was measured using the area-length method (23). For both techniques, results were indexed to body surface area to produce the left ventricular mass index (LVMI). Echo EF was calculated using the area-length method.
2.2 Statistical Analysis
Data were described using standard summary statistics, including median and range for continuous variables, and frequencies and percentages for categorical variables. To evaluate CMR vs. echo measurements of LVMI and systolic function, median LVMI and EF as assessed by CMR were compared to that assessed by echo using the Wilcoxon rank sum test for both the baseline and most recent follow-up studies. To evaluate change in these parameters over the treatment period, the Wilcoxon signed rank test was used to compare LVMI and EF at baseline and most recent follow-up for the 5 patients with a baseline study and at least one follow up study. This was performed for both echo and CMR measurements. The relationship between anti-rhGAA antibody titers, and change in LVMI and delayed enhancement was also described. All analyses were performed using SAS version 9.1 (SAS Institute Inc. Cary, NC). All p-values are two-tailed and a p-value <0.05 was considered statistically significant.
3. Results
3.1 Patient Characteristics
Patient characteristics are presented in Table 1. Ten subjects (80% male) underwent a total of 17 CMR studies during the 3 year study period. The median age at start of ERT was 4 months (range 1-10 months), with a median of 5 months elapsing until the first CMR (range 0-34 months). The median age at first CMR study was 9 months (range 1-38 months)
Table 1. Patient Characteristics.
| Subject | Sex | Age (months), start of ERT | CMR, months post-ERT | CMR Sedation | CMR Technique | LVMI (gm/m2) | LV EF | DHE | CRIM Status | Anti-rhGAA titer |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | F | 4 | 7 | No | ECG-gated segmented | 135 | 25% | No | Neg | 400 - 25600* |
| 1 | 9 | No | Single shot “real-time” | 180.6 | 58% | No | ||||
| 1 | 13 | No | Single shot “real-time” | 179.6 | 55% | No | ||||
| 2 | M | 7 | 15 | No | Single shot “real-time” | 51.2 | 55% | No | Pos | 1600 - 25600 |
| 2 | 26 | No | Single shot “real-time” | 57 | 57% | No | ||||
| 2 | 47 | Yes | Both | 44.5 | 68% | No | ||||
| 3 | M | 6 | 1 | No | Single shot “real-time” | 334 | 18% | No | Pos | 800 - 25600 |
| 3 | 2 | No | Single shot “real-time” | 303 | 25% | No | ||||
| 4 | M | 3 | 6 | No | Single shot “real-time” | 43.8 | 55% | No | Pos | 200 - 1600 |
| 4 | 36 | Yes | Single shot “real-time” | 43 | 69% | No | ||||
| 5 | M | 1 | 0 | No | Single shot “real-time” | 94 | 73% | No | Pos | 100 - 200 |
| 5 | 20 | Yes | Both | 34 | 57% | No | ||||
| 6 | M | 4 | 34 | No | ECG-gated segmented | 47.0 | 60% | No | Pos | 800 - 6400 |
| 7 | M | 6 | 2 | No | ECG-gated segmented | 252.2 | 65% | No | Pos | <100 - 400 |
| 8 | M | 4 | 3 | No | Single shot “real-time” | 180.6 | 29% | No | Pos | 800 - 3200 |
| 9 | F | 10 | 1 | No | Single shot “real-time” | 324.4 | 55% | No | Pos | <100 - 1600 |
| 10 | M | 3 | 31 | Yes | Both | 145.0 | 73% | Yes | Neg | 800 - 204,800* |
Legend: CMR=Cardiac Magnetic Resonance; CRIM=Cross-Reactive Immunologic Material; DHE=Delayed HyperEnhancement; EF=Ejection Fraction; ERT=Enzyme Replacement Therapy; LVMI=Left Ventricle Mass Indexed; Neg=CRIM Negative; Pos=CRIM Positive;
=sustained titer on ≥ 2 separate measurements
3.2 Sedation for CMR
The first 13 CMR studies were performed without sedation, while sedation supervised by pediatric cardiac anesthesia was provided for the last 4 studies. Sedation for CMR was achieved in 3 of the patients spontaneously breathing with low dose intravenous midazolam 0.1mg/kg together with fentanyl 1 mcg/kg and 1 mg/kg boluses of ketamine every 20 - 30 minutes. Nasal cannula oxygen was supplied at 2 l/min. In one patient a tracheotomy was present and in addition to the sedative medications prescribed above, sevoflurane was administered at 0.4 - 0.8% in 50% oxygen via a CMR compatible anesthetic delivery system. There were no complications during the sedated or non-sedated CMR studies.
3.3 CMR vs. Echo Measurements
CMR and echo measurements of LVMI and EF are presented in Table 2. CMR measurements of LVMI tended to be lower compared to echo at both baseline and most recent study, but this did not reach statistical significance. EF was similar by CMR and echo.
Table 2. CMR vs. Echocardiographic Measurements.
| CMR LVMI (g/m2) | Echo LVMI (g/m2) | p-value | |
|---|---|---|---|
| Baseline | 140.0 (43.8-334.0) | 204.0 (52.0-385.0) | 0.38 |
| Most Recent | 44.5 (34.0-303.0) | 71.0 (46.0-286.0) | 0.43 |
| CMR EF (%) | Echo EF (%) | p-value | |
| Baseline | 55 (18-73) | 55 (25-84) | 0.38 |
| Most Recent | 57 (25-69) | 55 (16-60) | 0.48 |
Baseline = all patients at time of first study (n=10)
Most recent = data from last study for those with serial studies (n=5)
Data presented as Median (range)
Legend: CMR=Cardiac Magnetic Resonance; EF=Ejection Fraction; LVMI=Left Ventricle Mass Indexed
3.4 Change in CMR and Echo Measurements over time
Five subjects underwent follow-up CMR and echo scans during the study period (Table 3 and Figure 1). Overall, there was no decrease in CMR measured LVMI over time. Figure 1 shows the change in LVMI over time for each patient as assessed by CMR. Two subjects demonstrated a decrease in LVMI, LVMI remained stable in 2 other subjects, and increased in one CRIM negative subject. LVMI as assessed by echo was unchanged from baseline to follow-up study. There was also no significant change in EF over time as assessed by either echo or CMR.
Table 3. Change in CMR and Echo Measurements over Time (n=5).
| Baseline | Most Recent | p-value | |
|---|---|---|---|
| CMR LVMI (g/m2) | 94.0 (43.8-334.0) | 44.5 (34.0-303.0) | 0.44 |
| Echo LVMI (g/m2) | 69.9 (52.0-334.0) | 71.0 (46.0-286.0) | 1.0 |
| CMR EF (%) | 55 (18-73) | 57 (25-69) | 0.44 |
| Echo EF (%) | 51 (25-60) | 55 (16-60) | 0.69 |
Legend: CMR=Cardiac Magnetic Resonance; EF=Ejection Fraction; LVMI=Left Ventricle Mass Indexed
Figure 1.
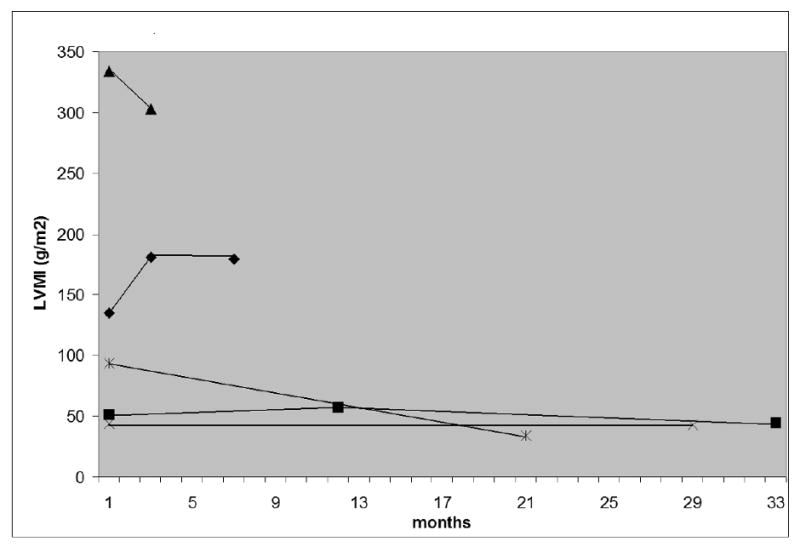
Change in CMR LVMI over time.
Legend: CMR=Cardiac Magnetic Resonance; LVMI=Left Ventricular Mass Indexed
Data shown as months from baseline study for those with serial CMR scans (n=5). Legend: CMR=Cardiac Magnetic Resonance; LVMI=Left Ventricular Mass Indexed
3.5 Anti-rhGAA titer, CRIM status and delayed enhancement
Anti-rhGAA titer range, CRIM status and presence of delayed enhancement are shown in Table 1. Four subjects had a peak anti-rhGAA titer of 25,600, but in only 2 subjects was this elevation sustained on 2 or more measurements separated by 4 week intervals. Both subjects with sustained high anti-rhGAA titers were CRIM negative. One of these subjects (subject 1) demonstrated the increase in LVMI despite ERT, while the second (subject 10) was the single subject to demonstrate delayed enhancement in the region of the basal to mid anterior and anterolateral walls on CMR. None of the other subjects demonstrated myocardial fibrosis on delayed enhancement imaging. A typical delayed enhancement image from this study, contrasted with that from a patient with Fabry disease, is shown in Figure 2.
Figure 2.
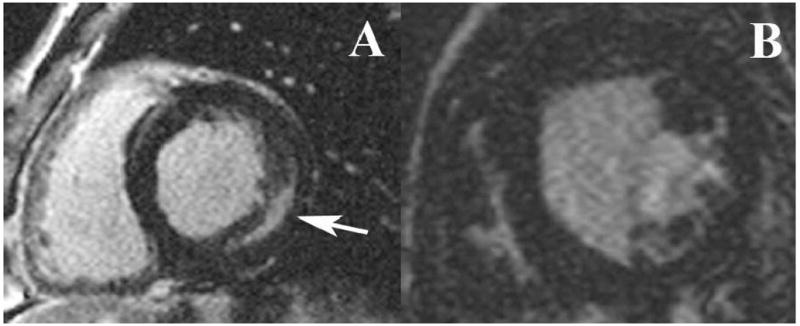
Representative short axis-images of the left ventricle in a patient with Fabry Disease (A) and a patient with Pompe disease (B). Note the presence of delayed hyperenhancement (DHE, arrow) in the mid-inferolateral wall of the Fabry patient and the absence of DHE in the Pompe patient.
4. Discussion
Prior to the development of enzyme replacement therapy, the prognosis for children with infantile Pompe disease was uniformly poor. However, the use of enzyme replacement therapy has improved both the length and quality of life for some of these children (5) (24), and permitted the use of more sophisticated imaging tools to assess the cardiac response to therapy.
For cardiac imaging, echocardiography has the advantage of portability, high temporal resolution and real-time imaging. However, transthoracic echocardiography is limited to available acoustic windows, interference from bone/lungs that prevent good definition of the endocardium/blood interface, and reliance on assumptions of left ventricular geometry in the calculation of left ventricular mass. In contrast, CMR achieves excellent definition of the myocardium distinct from the blood pool, with imaging windows optimized to provide an image of the heart independent of thoracic/pulmonary geometry. CMR images can be degraded by excessive respiratory motion and a thin myocardium providing a lower signal to noise ratio, and will have worse temporal resolution than pediatric echocardiography. For these reasons, sedation or anesthesia is frequently used in young children to control respiratory gating and patient position during the CMR study.
Sedation and anesthesia for children with infantile Pompe disease have been shown to be associated with an increased risk for perioperative cardiac arrhythmias, myocardial ischemia and sudden intraoperative cardiac arrest (19, 20). The incidence of arrhythmias or cardiopulmonary arrest in one series of 139 infantile onset Pompe patients undergoing induction of anesthesia was 8% (20). Death resulting from arrhythmias in this recent series was associated with an echo derived LVMI greater than 350 g/m2 (20), suggesting extreme hypertrophic cardiomyopathy as the pathophysiologic basis for these risks. These cardiac associated anesthetic risks are not only significantly higher in the untreated patient, but also present during the early phase of recombinant enzyme therapy before the enzyme has affected remodeling of the hypertrophic myocardium (19). Anesthetic and sedative agents administered to infantile Pompe patients may depress myocardial contractility, lower diastolic blood pressure and sometimes increase heart rate (19, 20). It has been found that a low diastolic blood pressure associated with tachycardia in this group of patients may be associated with myocardial ischemia and the risk for arrhythmias (19, 20). In patients with severe cardiac involvement associated with Pompe disease, inadequate preload as a result of preoperative fasting or decreased ventricular cavity volume in the child with significant left ventricular outflow obstruction, is also poorly tolerated and may lead to myocardial ischemia (19, 20). Mechanical ventilation, even temporarily, also poses a risk for these patients who may have baseline respiratory compromise. For these reasons, avoidance of deep sedation or anesthesia is preferable. Based on our experience, a spontaneously breathing sedation and anesthesia technique was used, as previously described (19), with no complications.
This study demonstrates that non-ECG gated single shot SSFP “real-time” cine imaging provides adequate images for calculation of LV mass and function in these patients. Anecdotally, we observed that retrospectively ECG gated segmented SSFP cine imaging (without breath-holding) also provided clinically adequate images due to the shallower respiratory pattern in the free-breathing children limiting respiratory artifact. In fact, this latter technique combined with careful conscious sedation under the supervision of a Pediatric Cardiac Anesthesia team can be especially helpful in children with Pompe disease who have survived with ERT to the age where they may be more mobile and less cooperative, as were the sedated children in this study.
Analysis of all subject data demonstrates that some patients had a decrease in LV mass with ERT, similar to prior echocardiographic reports (10) (5) (3, 24). However, this was not a uniform finding and did not reach statistical significance, likely due to the wide range of LV mass between subjects and the overall small number of subjects with serial CMR studies (5 subjects total). One subject had a modest increase in LV mass despite CMR over the study period, although this subject was also CRIM negative. In general, LV mass as measured by CMR was less than LV mass measured by echo, although again the wide range of LV mass between subjects and small sample size limited our analysis, such that these differences did not reach statistical significance.
Studies comparing left ventricular mass measurements by cardiac CMR to cardiac weights measured at autopsy have shown that CMR is an accurate method of measuring left ventricular mass with good intra-observer variability (r=0.96, standard error of estimate 11.1g) and inter-observer variability (r=0.91, standard error of estimates=17.8g)(25). Several studies comparing left ventricular mass measurements by CMR to echocardiography have revealed that echocardiography overestimates left ventricular mass in comparison to CMR and that echocardiography has a greater inter and intra-observer variability than CMR (26) (27). These differences are greater in patients with abnormal left ventricles such as patients with hypertrophic cardiomyopathy, similar to the subjects in this study, which reinforces the potential benefits of CMR imaging for this patient group.
CMR also permits the assessment of ventricular fibrosis by imaging the heart approximately 10 minutes after the infusion of gadolinium contrast (28). Gadolinium, as an extracellular molecule, has limited accumulation in a normal heart with closely spaced myocytes and a small extracellular volume of distribution. However, gadolinium will accumulate in regions of myocardial injury, where there is cell destruction, or myocardial fibrosis where there is an increase in interstitial tissue, both processes which increase the extracellular volume of distribution. By imaging the heart later in time, taking advantage of the difference in inversion recovery times between gadolinium and normal myocardium, and minimizing the signal from healthy myocardium, any region of fibrosis will appear “enhanced” due to greater signal intensity. This technique is particularly useful in assessing myocardial viability, as it adds more information beyond standard tissue characterization and wall motion abnormalities (29, 30)
Different disease processes have been demonstrated to create myocardial fibrosis in different myocardial locations. As an example, myocardial infarction due to coronary artery disease produces fibrosis in a subendocardial to transmural distribution (31). In contrast, viral myocarditis often produces a mid-myocardial defect (32), while Fabry disease (α-galactosidase deficiency) produces mid-myocardial or subepicardial defects in characteristic basal inferolateral locations (33, 34) (35). Patchy myocardial fibrosis may also be seen in patients with more typical hypertrophic cardiomyopathy and likely represents pathologic ventricular remodeling in this group as well as Fabry patients. The presence of myocardial fibrosis has been proposed as a risk factor for sudden death due to ventricular arrhythmias in the hypertrophic cardiomyopathy (36) and Fabry populations, with myocardial fibrosis present at autopsy (18).
As an example of tissue characterization by CMR, Figure 2 demonstrates the presence of delayed enhancement in a patient with Fabry disease compared to one of our subjects with Pompe disease, in whom there is no delayed enhancement. Since Pompe disease, as a lysosomal storage disorder, is similar to Fabry disease but with more severe ventricular hypertrophy, we would also expect these subjects to be at higher risk for myocardial fibrosis. However, only one subject had fibrosis detected by CMR, in contrast to the relatively common finding of delayed enhancement in approximately 50% of patients with Fabry disease (34). The presence of delayed enhancement in the Fabry population predicts a poor response to ERT, with no significant regression in LV mass in these patients, while patients without delayed enhancement demonstrated a significant regression in LV mass and no progression to delayed enhancement (34). This dichotomy may help explain 2 recent reports suggesting that ERT for Fabry disease does not produce a significant reduction in LV mass after 24 months of treatment, since the presence of delayed enhancement was not assessed at the start or during either study (37) (38). For our study, the relative lack of delayed enhancement may be due to either the younger age and shorter follow-up time in the Pompe population, as myocardial fibrosis may be a cumulative process and may be ameliorated by earlier ERT, or the higher dose of ERT used for Pompe patients (20 mg/kg as compared to 1 mg/kg for Fabry disease). Less likely, the difference in the presence of delayed enhancement could be due to an unexpected difference between the two disease processes.
Interestingly, myocardial T2 relaxation time has been reported to be significantly prolonged in patients with Fabry disease, with or without LV hypertrophy, when compared to normal controls and patients with hypertrophic cardiomyopathy unrelated to Fabry disease (16). This difference in T2 relaxation time has been attributed to changes in the tissue water and lipid characteristics, with glycolipid deposition and accumulation known to occur in cardiac myocytes (16) (5) (24). Unfortunately this analysis was not included in the imaging protocol of our study, but may be a fruitful avenue for further investigation.
One additional factor that may affect the individual response to ERT is the cross reactive immunologic material status of patients (CRIM positive or negative). In CRIM negative subjects there is no detectable acid α-glucosidase activity, and therefore an immune response to exogenous enzyme. These individuals mount a greater antibody mediated immune response to ERT, with significantly worse clinical response to therapy (39). Clinically, a sustained anti-rhGAA titer of >12,800 has been suggested to lead to a poorer response to treatment (Myozyme Package Insert, Genzyme Corporation)(40). In the present study, there was no correlation between peak anti-rhGAA antibody titer and LV mass regression for either CRIM positive and negative patients. However, subjects with high sustained anti-rhGAA titers (the 2 CRIM negative subjects) had notable CMR differences on CMR compared to the CRIM positive subjects. One CRIM negative subject had a paradoxical increase in LV mass despite ERT, with a sustained titer of 25,600. The other CRIM negative subject had an almost 10-fold higher sustained antibody response (204,800) and was the single subject who demonstrated the presence of myocardial fibrosis. While the number of CRIM negative patients in this study is too small to draw more definitive conclusions, these two findings concur with other studies suggesting a worse clinical outcome in these patients (41). Further investigation is necessary into factors such as age and stage of disease at start of ERT, role of antibody titers and CRIM status. It is also possible that myocardial fibrosis formation represents a cumulative insult, which will require longer follow-up to detect in these surviving patients.
4.1 Limitations
The major limitation of this study is the small sample size, which reflects the relatively low incidence of Pompe disease for any center, as well as our evolving clinical skill in the safe use of CMR in this patient population. There is also a wide range of initial LV mass, a wide range in age at diagnosis, a wide range of age at initiation of ERT, variation in time to first CMR after start of ERT, and variable timing of CMR and echo studies. While children with Pompe disease on ERT have significantly improved survival, longer follow-up is necessary to determine long-term cardiac function and risks for fibrosis.
5. Conclusions
CMR is a feasible and useful technique to longitudinally follow children with Pompe disease and their myocardial response to enzyme replacement therapy As shown by others, CMR LV mass calculations have the advantage of less reliance on echocardiographic windows, better definition of the blood-myocardial interface and greater reproducibility with less inter and intraobserver variability. Most importantly, CMR can be performed safely with or even without sedation in this high risk patient population. CMR also has the unique ability to characterize tissue and detect myocardial fibrosis, which further increases its utility as a tool to longitudinally assess cardiac status in patients with Pompe disease.
Supplementary Material
Acknowledgments
Disclosure Statement: Priya S. Kishnani (PSK) has received research/grant support and honoraria from Genzyme Corporation. PSK is a member of the Pompe Disease and the Gaucher Disease Registry Advisory Board for Genzyme Corporation. rhGAA, in the form of Genzyme's product alglucosidase alfa, (Myozyme™/Lumizyme™) has been approved by the US FDA and the European Union as therapy for Pompe disease. Duke University and inventors for the method of treatment and predecessors of the cell lines used to generate the enzyme (rhGAA) receive royalty payments pursuant to the University's Policy on Inventions, Patents and Technology Transfer.
Sara K. Pasquali (SKP) receives support (KL2 RR024127-02) from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH), and from the American Heart Association (AHA) Mid-Atlantic Affiliate Clinical Research Program.
Raymond J. Kim (RJK) is an inventor of a US patent on Delayed Enhancement MRI, which is owned by Northwestern University.
Jennifer S. Li (JSL) has received modest research support for the evaluation of Pompe disease from Genzyme Inc.
Richard J. Ing (RJI) receives support as a Principle Investigator from CASMED.
Footnotes
Abbreviations: CMR, Cardiac Magnetic Resonance; CRIM, Cross Reactive Immunologic Material; EF, Ejection Fraction; ERT, Enzyme Replacement Therapy; LVMI, Left Ventricular Mass Index; rhGAA, recombinant Acid Alpha-Glucosidase;SSFP, Steady State Free Precession;
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hirschhorn R. Glycogen storage disease type II: acid alpha-glucosidase (acid maltase deficiency) In: CR S, editor. The Metabolic and Molecular Basis of Inherited Disease. New York: McGraw Hill; 2001. pp. 3389–420. [Google Scholar]
- 2.Kishnani PS, Howell RR. Pompe disease in infants and children. J Pediatr. 2004 May;144(5 Suppl):S35–43. doi: 10.1016/j.jpeds.2004.01.053. [DOI] [PubMed] [Google Scholar]
- 3.Kishnani PS, Hwu WL, Mandel H, Nicolino M, Yong F, Corzo D. A retrospective, multinational, multicenter study on the natural history of infantile-onset Pompe disease. J Pediatr. 2006 May;148(5):671–6. doi: 10.1016/j.jpeds.2005.11.033. [DOI] [PubMed] [Google Scholar]
- 4.van den Hout HM, Hop W, van Diggelen OP, Smeitink JA, Smit GP, Poll-The BT, Bakker HD, Loonen MC, de Klerk JB, Reuser AJ, van der Ploeg AT. The natural course of infantile Pompe's disease: 20 original cases compared with 133 cases from the literature. Pediatrics. 2003 Aug;112(2):332–40. doi: 10.1542/peds.112.2.332. [DOI] [PubMed] [Google Scholar]
- 5.Kishnani PS, Corzo D, Nicolino M, Byrne B, Mandel H, Hwu WL, Leslie N, Levine J, Spencer C, McDonald M, Li J, Dumontier J, Halberthal M, Chien YH, Hopkin R, Vijayaraghavan S, Gruskin D, Bartholomew D, van der Ploeg A, Clancy JP, Parini R, Morin G, Beck M, De la Gastine GS, Jokic M, Thurberg B, Richards S, Bali D, Davison M, Worden MA, Chen YT, Wraith JE. Recombinant human acid [alpha]-glucosidase: major clinical benefits in infantile-onset Pompe disease. Neurology. 2007 Jan 9;68(2):99–109. doi: 10.1212/01.wnl.0000251268.41188.04. [DOI] [PubMed] [Google Scholar]
- 6.Amalfitano A, Bengur AR, Morse RP, Majure JM, Case LE, Veerling DL, Mackey J, Kishnani P, Smith W, McVie-Wylie A, Sullivan JA, Hoganson GE, Phillips JA, 3rd, Schaefer GB, Charrow J, Ware RE, Bossen EH, Chen YT. Recombinant human acid alpha-glucosidase enzyme therapy for infantile glycogen storage disease type II: results of a phase I/II clinical trial. Genet Med. 2001 Mar-Apr;3(2):132–8. [PubMed] [Google Scholar]
- 7.Kishnani PS, Nicolino M, Voit T, Rogers RC, Tsai AC, Waterson J, Herman GE, Amalfitano A, Thurberg BL, Richards S, Davison M, Corzo D, Chen YT. Chinese hamster ovary cell-derived recombinant human acid alpha-glucosidase in infantile-onset Pompe disease. J Pediatr. 2006 Jul;149(1):89–97. doi: 10.1016/j.jpeds.2006.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klinge L, Straub V, Neudorf U, Schaper J, Bosbach T, Gorlinger K, Wallot M, Richards S, Voit T. Safety and efficacy of recombinant acid alpha-glucosidase (rhGAA) in patients with classical infantile Pompe disease: results of a phase II clinical trial. Neuromuscul Disord. 2005 Jan;15(1):24–31. doi: 10.1016/j.nmd.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 9.van der Ploeg AT, Clemens PR, Corzo D, Escolar DM, Florence J, Groeneveld GJ, Herson S, Kishnani PS, Laforet P, Lake SL, Lange DJ, Leshner RT, Mayhew JE, Morgan C, Nozaki K, Park DJ, Pestronk A, Rosenbloom B, Skrinar A, van Capelle CI, van der Beek NA, Wasserstein M, Zivkovic SA. A randomized study of alglucosidase alfa in late-onset Pompe's disease. N Engl J Med. 2010 Apr 15;362(15):1396–406. doi: 10.1056/NEJMoa0909859. [DOI] [PubMed] [Google Scholar]
- 10.Levine JC, Kishnani PS, Chen YT, Herlong JR, Li JS. Cardiac Remodeling After Enzyme Replacement Therapy with Acid alpha-Glucosidase for Infants with Pompe Disease. Pediatr Cardiol. 2008 Jul 26; doi: 10.1007/s00246-008-9267-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ansong AK, Li JS, Nozik-Grayck E, Ing R, Kravitz RM, Idriss SF, Kanter RJ, Rice H, Chen YT, Kishnani PS. Electrocardiographic response to enzyme replacement therapy for Pompe disease. Genet Med. 2006 May;8(5):297–301. doi: 10.1097/01.gim.0000195896.04069.5f. [DOI] [PubMed] [Google Scholar]
- 12.McDowell R, Li JS, Benjamin DK, Jr, Morgan C, Becker A, Kishnani PS, Kanter RJ. Arrhythmias in patients receiving enzyme replacement therapy for infantile Pompe disease. Genet Med. 2008 Oct;10(10):758–62. doi: 10.1097/GIM.0b013e318183722f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Frustaci A, Chimenti C. Images in cardiovascular medicine. Cryptogenic ventricular arrhythmias and sudden death by Fabry disease: prominent infiltration of cardiac conduction tissue. Circulation. 2007 Sep 18;116(12):e350–1. doi: 10.1161/CIRCULATIONAHA.107.723387. [DOI] [PubMed] [Google Scholar]
- 14.Joshi SB, Ahmar W, Lee G, Aggarwal A. Fabry's disease presenting as ventricular tachycardia and left ventricular ‘hypertrophy’. Eur J Echocardiogr. 2008 Sep;9(5):697–9. doi: 10.1093/ejechocard/jen132. [DOI] [PubMed] [Google Scholar]
- 15.Shah JS, Hughes DA, Sachdev B, Tome M, Ward D, Lee P, Mehta AB, Elliott PM. Prevalence and clinical significance of cardiac arrhythmia in Anderson-Fabry disease. Am J Cardiol. 2005 Sep 15;96(6):842–6. doi: 10.1016/j.amjcard.2005.05.033. [DOI] [PubMed] [Google Scholar]
- 16.Imbriaco M, Spinelli L, Cuocolo A, Maurea S, Sica G, Quarantelli M, Pisani A, Liuzzi R, Cianciaruso B, Sabbatini M, Salvatore M. MRI characterization of myocardial tissue in patients with Fabry's disease. AJR Am J Roentgenol. 2007 Mar;188(3):850–3. doi: 10.2214/AJR.05.0442. [DOI] [PubMed] [Google Scholar]
- 17.Moon JC, Sachdev B, Elkington AG, McKenna WJ, Mehta A, Pennell DJ, Leed PJ, Elliott PM. Gadolinium enhanced cardiovascular magnetic resonance in Anderson-Fabry disease. Evidence for a disease specific abnormality of the myocardial interstitium. Eur Heart J. 2003 Dec;24(23):2151–5. doi: 10.1016/j.ehj.2003.09.017. [DOI] [PubMed] [Google Scholar]
- 18.Moon JC, Sheppard M, Reed E, Lee P, Elliott PM, Pennell DJ. The histological basis of late gadolinium enhancement cardiovascular magnetic resonance in a patient with Anderson-Fabry disease. J Cardiovasc Magn Reson. 2006;8(3):479–82. doi: 10.1080/10976640600605002. [DOI] [PubMed] [Google Scholar]
- 19.Ing RJ, Cook DR, Bengur RA, Williams EA, Eck J, Dear Gde L, Ross AK, Kern FH, Kishnani PS. Anaesthetic management of infants with glycogen storage disease type II: a physiological approach. Paediatr Anaesth. 2004 Jun;14(6):514–9. doi: 10.1111/j.1460-9592.2004.01242.x. [DOI] [PubMed] [Google Scholar]
- 20.Wang LY, Ross AK, Li JS, Dearmey SM, Mackey JF, Worden M, Corzo D, Morgan C, Kishnani PS. Cardiac arrhythmias following anesthesia induction in infantile-onset Pompe disease: a case series. Paediatr Anaesth. 2007 Aug;17(8):738–48. doi: 10.1111/j.1460-9592.2007.02215.x. [DOI] [PubMed] [Google Scholar]
- 21.Hori Y, Yamada N, Higashi M, Hirai N, Nakatani S. Rapid evaluation of right and left ventricular function and mass using real-time true-FISP cine MR imaging without breath-hold: comparison with segmented true-FISP cine MR imaging with breath-hold. J Cardiovasc Magn Reson. 2003 Jul;5(3):439–50. doi: 10.1081/jcmr-120022260. [DOI] [PubMed] [Google Scholar]
- 22.Bellenger NG, P D. Cardiovascular Magnetic Resonance. New York: Ny Churchill Livingstone; 2002. [Google Scholar]
- 23.Snider AR, G S, Ritter SB. Echocardiography in Pediatric Heart Disease. 2nd. Mosby-Year Book; 1997. [Google Scholar]
- 24.Li JS, C Y, Morgan C, Kishnani PS. Developments in the Treatment of Pompe Disease. US Cardiovascular Disease. 2006:1–4. [Google Scholar]
- 25.Katz J, Milliken MC, Stray-Gundersen J, Buja LM, Parkey RW, Mitchell JH, Peshock RM. Estimation of human myocardial mass with MR imaging. Radiology. 1988 Nov;169(2):495–8. doi: 10.1148/radiology.169.2.2971985. [DOI] [PubMed] [Google Scholar]
- 26.Devlin AM, Moore NR, Ostman-Smith I. A comparison of MRI and echocardiography in hypertrophic cardiomyopathy. Br J Radiol. 1999 Mar;72(855):258–64. doi: 10.1259/bjr.72.855.10396215. [DOI] [PubMed] [Google Scholar]
- 27.Stewart GA, Foster J, Cowan M, Rooney E, McDonagh T, Dargie HJ, Rodger RS, Jardine AG. Echocardiography overestimates left ventricular mass in hemodialysis patients relative to magnetic resonance imaging. Kidney Int. 1999 Dec;56(6):2248–53. doi: 10.1046/j.1523-1755.1999.00786.x. [DOI] [PubMed] [Google Scholar]
- 28.Kim RJ, Shah DJ, Judd RM. How we perform delayed enhancement imaging. J Cardiovasc Magn Reson. 2003 Jul;5(3):505–14. doi: 10.1081/jcmr-120022267. [DOI] [PubMed] [Google Scholar]
- 29.Kim HW, Farzaneh-Far A, Kim RJ. Cardiovascular magnetic resonance in patients with myocardial infarction: current and emerging applications. J Am Coll Cardiol. 2009 Dec 29;55(1):1–16. doi: 10.1016/j.jacc.2009.06.059. [DOI] [PubMed] [Google Scholar]
- 30.Klem I, Heitner JF, Shah DJ, Sketch MH, Jr, Behar V, Weinsaft J, Cawley P, Parker M, Elliott M, Judd RM, Kim RJ. Improved detection of coronary artery disease by stress perfusion cardiovascular magnetic resonance with the use of delayed enhancement infarction imaging. J Am Coll Cardiol. 2006 Apr 18;47(8):1630–8. doi: 10.1016/j.jacc.2005.10.074. [DOI] [PubMed] [Google Scholar]
- 31.Senthilkumar A, Majmudar MD, Shenoy C, Kim HW, Kim RJ. Identifying the etiology: a systematic approach using delayed-enhancement cardiovascular magnetic resonance. Heart Fail Clin. 2009 Jul;5(3):349–67. vi. doi: 10.1016/j.hfc.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sechtem U, Mahrholdt H, Vogelsberg H. Cardiac magnetic resonance in myocardial disease. Heart. 2007 Dec;93(12):1520–7. doi: 10.1136/hrt.2005.067355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.De Cobelli F, Esposito A, Belloni E, Pieroni M, Perseghin G, Chimenti C, Frustaci A, Del Maschio A. Delayed-Enhanced Cardiac MRI for Differentiation of Fabry's Disease from Symmetric Hypertrophic Cardiomyopathy. Am J Roentgenol. 2009 March 1;192(3):W97–102. doi: 10.2214/AJR.08.1201. [DOI] [PubMed] [Google Scholar]
- 34.Beer M, Weidemann F, Breunig F, Knoll A, Koeppe S, Machann W, Hahn D, Wanner C, Strotmann J, Sandstede J. Impact of enzyme replacement therapy on cardiac morphology and function and late enhancement in Fabry's cardiomyopathy. Am J Cardiol. 2006 May 15;97(10):1515–8. doi: 10.1016/j.amjcard.2005.11.087. [DOI] [PubMed] [Google Scholar]
- 35.Silva C, Moon JC, Elkington AG, John AS, Mohiaddin RH, Pennell DJ. Myocardial late gadolinium enhancement in specific cardiomyopathies by cardiovascular magnetic resonance: a preliminary experience. J Cardiovasc Med (Hagerstown) 2007 Dec;8(12):1076–9. doi: 10.2459/01.JCM.0000296538.82763.f0. [DOI] [PubMed] [Google Scholar]
- 36.Leonardi S, Raineri C, De Ferrari GM, Ghio S, Scelsi L, Pasotti M, Tagliani M, Valentini A, Dore R, Raisaro A, Arbustini E. Usefulness of cardiac magnetic resonance in assessing the risk of ventricular arrhythmias and sudden death in patients with hypertrophic cardiomyopathy. Eur Heart J. 2009 August 2;30(16):2003–10. doi: 10.1093/eurheartj/ehp152. [DOI] [PubMed] [Google Scholar]
- 37.Hughes DA, Elliott PM, Shah J, Zuckerman J, Coghlan G, Brookes J, Mehta AB. Effects of enzyme replacement therapy on the cardiomyopathy of Anderson-Fabry disease: a randomised, double-blind, placebo-controlled clinical trial of agalsidase alfa. Heart. 2008 Feb;94(2):153–8. doi: 10.1136/hrt.2006.104026. [DOI] [PubMed] [Google Scholar]
- 38.Koskenvuo JW, Hartiala JJ, Nuutila P, Kalliokoski R, Viikari JS, Engblom E, Penttinen M, Knuuti J, Mononen I, Kantola IM. Twenty-four-month alpha-galactosidase A replacement therapy in Fabry disease has only minimal effects on symptoms and cardiovascular parameters. J Inherit Metab Dis. 2008 Jun;31(3):432–41. doi: 10.1007/s10545-008-0848-3. [DOI] [PubMed] [Google Scholar]
- 39.Sun B, Bird A, Young SP, Kishnani PS, Chen YT, Koeberl DD. Enhanced response to enzyme replacement therapy in Pompe disease after the induction of immune tolerance. Am J Hum Genet. 2007 Nov;8(5):1042–9. doi: 10.1086/522236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Myozyme, package insert. Available from: http://www.myozyme.com/PDF/mz_pi.pdf
- 41.Kishnani PS, Goldenberg PC, DeArmey SL, Heller J, Benjamin D, Young S, Bali D, Smith SA, Li JS, Mandel H, Koeberl D, Rosenberg A, Chen YT. Cross-reactive immunologic material status affects treatment outcomes in Pompe disease infants. Mol Genet Metab. Jan;99(1):26–33. doi: 10.1016/j.ymgme.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.