

Abstract
Retinoschisin (RS1) is a cell-surface adhesion molecule expressed by photoreceptor and bipolar cells of the retina. The 24-kDa protein encodes two conserved sequence motifs: the initial signal sequence targets the protein for secretion while the larger discoidin domain is implicated in cell adhesion. RS1 helps to maintain the structural organization of the retinal cell layers and promotes visual signal transduction. RS1 gene mutations cause X-linked retinoschisis disease (XLRS) in males, characterized by early-onset central vision loss. We analyzed the biochemical consequences of several RS1 signal-sequence mutants (c.1A>T, c.35T>A, c.38T>C, and c.52G>A) found in our subjects. Expression analysis in COS-7 cells demonstrates that they affect RS1 biosynthesis and result in an RS1 null phenotype by several different mechanisms. By comparison, discoidin-domain mutations generally lead to nonfunctional conformational variants that remain trapped inside the cell. XLRS disease has a broad heterogeneity in general, but subjects with the RS1 null-protein signal-sequence mutations are on the more severe end of the clinical phenotype. Results from the signal-sequence mutants are discussed in the context of the discoidin-domain mutations, clinical phenotypes, genotype–phenotype correlations, and implications for RS1 gene replacement therapy.
Keywords: retinoschisin, X-linked retinoschisis, XLRS, signal sequence, discoidin, splicing
Introduction
Young males with mutations in the X-chromosome retinoschisin gene (RS1; MIM# 312700) develop a bilateral, progressive retinal dystrophy known as X-linked retinoschisis (XLRS) (Sauer, et al., 1997). XLRS causes schisis (splitting) within the retinal layers, which impairs visual-signal processing and leads to progressive vision loss. When retinal function is assessed by clinical electroretinogram (ERG), XLRS patients display markedly reduced inner retinal b-wave amplitudes (“electronegative”) relative to the photoreceptor a-wave, which implicates abnormal synaptic signaling or neuronal processing (Sikkink, et al., 2007; Tantri, et al., 2004).
The human RS1 gene contains six separate exons interspaced by five introns (Fig. 1) (Sauer, et al., 1997). All five introns follow the “gt–ag rule” and vary greatly in size. The processed RNA transcript contains 3,040 nucleotides and translates into a 224 amino-acid cell-surface protein known as retinoschisin (RS1) (NP_000321.1). RS1 is prominently expressed by the retinal photoreceptor and bipolar cells and is also in the pineal gland (Molday, 2007; Takada, et al., 2006). RS1 encodes two functional sites of conserved sequence motifs: the N-terminus signal sequence (exons 1 and 2; aa 1–21/23) and a long and highly-conserved sequence motif termed the discoidin domain (exons 4–6; aa 64–219; Fig. 1). The signal sequence guides the translocation of RS1 from synthesis in the endoplasmic reticulum to the external leaflet of the cell plasma membrane (Molday, 2007).
Figure 1.

Schematic structure of the RS1 gene, mRNA, and protein products. RS1 is encoded on the minus strand of the X chromosome at Xp22.2–p22.1 covering 32.43 kb from 18600150 to 18567724. Exons are indicated by filled boxes, with numbers indicating the size of the exons and introns in nucleotides. The primary RNA transcript encoding both exons and introns undergoes post-transcriptional RNA splicing to remove introns and generate mRNA (NM_000330.3*) which is translated into a 224 amino-acid protein (NP_000321.1). The functional domains of RS1 are 1) a signal peptide (SP), 2) RS1, and 3) the discoidin domains. The signal sequence guides the translocation of nascent RS1 from the endoplasmic reticulum (the site of synthesis) to external leaflet of the plasma membrane, during which signal sequence is cleaved by signal peptidase to generate mature protein with characteristic RS1 and a highly conserved discoidin domain. The different subdomains of RS1 signal sequence are 1) the positively charged N region at the amino terminal end which mediates translocation, 2) the hydrophobic core (H) required for targeting and membrane insertion and 3) a polar “C” region that determines the site of recognition and cleavage by signal peptidase. The arrows indicate the sites at which the signal peptide is cleaved. *The numbering follows GenBank NCBI Reference Sequence: NM_000330.3. Nucleotide 1 is A of the ATG initiation codon (CDS 36–710).
In the biologically active conformation, RS1 is an octamer (Wu, et al., 2005). The discoidin domain apparently contributes to the adhesive function of RS1, which is essential to preserve the retinal cell architecture and to establish proper synaptic connectivity (Takada, et al., 2008). The L-type voltage-gated calcium channel (; Shi, et al., 2009;), Na/K ATPase-SARM1 complex (Molday, et al., 2007;), and phospholipids/Ca2+ (Vijayasarathy, et al., 2007) were identified as potential RS1 ligands, corroborating a role for RS1 in cell signaling events.
Many mutations, mostly missense, have been identified in XLRS patients in the RS1 discoidin-domain (exons 4–6) (The Retinoschisis Consortium: www.dmd.nl/rs/consortium.html). A structural model based on alignment of the RS1 discoidin domain with the C2 discoidin domain of coagulation factors FV and FVIII indicates that most of these mutations involve conserved amino-acid residues that are critical for the biologically active octomeric conformation of the RS1 protein (Sergeev, et al., 2010; Wu and Molday, 2003). Mutations in the conserved amino-acid residues in the discoidin domain lead to RS1 variants that have a secretion-incompetent, non-functional conformation that mislocalize within the cell (Wang, et al., 2002; Wu and Molday, 2003; Wu, et al., 2005). Less commonly, some discoidin-domain mutations lead to RS1 null phenotypes, as recently described for a frame shift due to an insertion-deletion-duplication (c.354del1–ins18) mutation in RS1exon 5 (Vijayasarathy, et al., 2009). In addition, point mutations, deletions, insertions, and splice-site alterations also have been identified at sites across the entire RS1 gene (The Retinoschisis Consortium).
XLRS patients display considerable clinical heterogeneity, and strict correlations between genetic mutations and clinical phenotype are elusive (Bradshaw, et al., 1999; Eksandh, et al., 2000; Hiriyanna, et al., 2001; Lesch, et al., 2008; Prenner, et al., 2006; Roesch, et al., 1998; Shinoda, et al., 2000; Sieving, et al., 1999), although scrutiny of the literature suggests a clinical difference between RS1 missense mutations and mutations expected to give a null-protein phenotype. We analyzed the biochemical basis of RS1 signal-sequence mutants c.1A>T (p.Met1Leu), c.35T>A (p.Leu12His), c.38T>C (p.Leu13Pro), and c.52G>A (Supp. Table S1). Results show a lack of mature RS1 protein involving several different mechanisms and a relatively more severe XLRS phenotype.
Materials and Methods
Clinical examinations
The study was conducted under the auspices of the National Institutes of Health (NIH) Intramural Research Program, and the Department of Ophthalmology, Peking Union Medical College, Chinese Academy of Medical Sciences, Beijing, China. Written informed consent consistent with the Helsinki Declaration was obtained from all participants. All study subjects underwent standard ophthalmological examinations: best corrected visual acuity by the Snellen charts, visual field measurements by Goldmann kinetic perimetry (Haag-Streit, Bern, Switzerland), slit-lamp biomicroscopy of the anterior segment, dilated indirect ophthalmoscopy of the posterior pole and the peripheral retina, and fundus photography. Retinal clinical structure was examined by optical coherence tomography (OCT) (3D OCT-2000 Spectral Domain; Topcon, Tokyo, Japan). ERG was performed (RetiPort ERG system, Roland Consult, Wiesbaden, Germany or Utas 2000, LKC Technologies, Gaithersburg, MD) with corneal “ERGjet” contact lens electrodes (Peking Union Medical College Hospital) and Burian-Allen bipolar corneal ERG electrodes (NIH), according to International Society for Clinical Electrophysiology of Vision (ISCEV) standards. Amplitudes and implicit times of scotopic and photopic full-field ERG responses were measured, and the ratio of standard combined b/a-wave amplitudes was calculated.
Molecular genetics
Genomic DNA was extracted from peripheral blood lymphocytes. RS1 coding regions (exons 1–6) and the flanking intronic sequences were amplified using primer sequences and PCR conditions listed in RetinoschisisDB© (www.dmd.nl/rs/). Amplicons were analyzed on 1.4% agarose gels, stained with ethidium bromide, and purified by the QIAEX II Gel Extraction Kit (Qiagen, Valencia, CA) and were used as sequencing templates. Sequencing was carried out in ABI3730XL analyzer with the Big Dye® 3.1 Sequencing Kit (Applied Biosystems, Foster City, CA, USA). Sequences were assembled and analyzed with Lasergene SeqMan software. The results were compared with the RS1 reference sequence.
RS1 expression plasmids: Cloning and mutagenesis
RS1 cDNA amplified from human retina total RNA (Zeng, et al., 2004) was subcloned in pCMV-Tag4A mammalian expression vector (Stratagene, Santa Clara, CA) with and without C-terminal Flag peptide tag. The primer sequences P13–P15 are listed in Supp. Table S1. The sense primer (P13) included the Kozak sequence (GCCACC) immediately upstream of the ATG start codon for efficient initiation of translation. Mutations were introduced by site-directed mutagenesis (primers P1-P12: see Supp. Table S1) using the QuickChange Kit (Stratagene). The effect of epitope tag on RS1 expression and localization was analyzed by immunoblotting of the secreted and cellular fractions from COS-7 cells expressing either wild-type RS1 or RS1-Flag (Supp. Fig. S1).
Construction of wild-type (WT) and c.52G>A mutant (Mut) RS1 minigenes with genomic intron-1 and intron-2 sequences
A cDNA-based minigene construct incorporating a Kozak sequence was prepared to guide the expression of WT and Mut (c.52G>A) minigenes in COS-7 cells. Because of the large intron-1 size (>14 kb in length), only end sequences (about 1271 bp) of intron 1 were cloned. To facilitate cloning, silent point mutations were created by site-directed mutagenesis in RS1 cDNA cloned in pCMVTag4 vector: C93T to create BamH1 site in exon 3 and G405A to silence BamH1 site in exon 5 (*RS1cDNA). Two cDNA fragments (0.56 kb Not1-EcoR1 and a second ∼1.7 kb EcoR1-BamH1. Supp. Table S1; primers18–21) were generated by PCR amplification of human genomic DNA from a normal control. The c.52G>A mutation was introduced by site-directed mutatgenesis in exon-1 region of Not1-EcoR1 fragment, and these two fragments were successively cloned in frame with exons 3–6 of *RS1cDNA at Not1-BamH1 sites. At each step, the authenticity of amplified DNA sequences and of sequences generated from the isolates was verified by direct sequencing. The schematic illustration of the minigene is depicted in the corresponding Results section.
Cell culture and immunoblot analysis
Transient expression analysis of WT and Mut RS1 plasmids was carried out in COS-7 cells using a lipid-based reagent (FuGENE® 6, Roche-Applied Science, Indianapolis, IN). Transfected cells were harvested after 72 h and the cellular and secreted fractions were assessed for RS1 expression by immunoblot analysis with anti-RS1 or anti-Flag antibody (Vijayasarathy, et al., 2009).
RT-PCR analysis
Total RNA was isolated with the TRIazol reagent (Invitrogen). Reverse transcription was carried out with 1 μg of RNA with Moloney Murine Leukaemia Virus (MMLV) based Reverse Transcriptase Kit (SuperScript One Step RT-PCR with Platinum®Taq, Invitrogen). All amplifications were primed by pairs of chemically synthesized 18–22-mer oligonucleotides designed with the RS1 transcript sequence (Supp. Table S1; primers 13–17). RT-PCR reactions were cloned into a Zero Blunt® TOPO vector (Invitrogen), and a minimum of 20 clones were sequenced for each.
Results
RS1 signal-sequence mutants: clinical presentation
XLRS disease clinical features include macular cystic cavities, peripheral schisis, and subnormal ERG b-wave amplitude relative to the a-wave (Figs. 2, 3). The signal-sequence mutants described in this study (p.Met1Leu, p.Leu12His, p. Leu13Pro, and c.52G>A; Supp. Table S1) result in null-protein products and appear to present a more severe disease phenotype by middle age, although two younger members of the p.Leu13Pro and c.52G>A mutation families displayed more modest clinical severity.
Figure 2.

Fundus photographs and non-invasive optical coherence tomograms (OCT) through a horizontal section of both eyes of 13-y/o III.4 and 45-y/o II.2, both with the c.52G>A mutation of the RS1 gene. III.4 has classical foveal schisis prototypical of XLRS, while II.2 shows abnormal RPE pigmentation and atrophic retinal changes in the macular area of the right eye and the periphery of the left eye. Atrophic macular thinning is quite evident by the OCT scans. II.2 also presents a large bullous peripheral schisis, as seen in patients with a severe XLRS phenotype.
Figure 3.
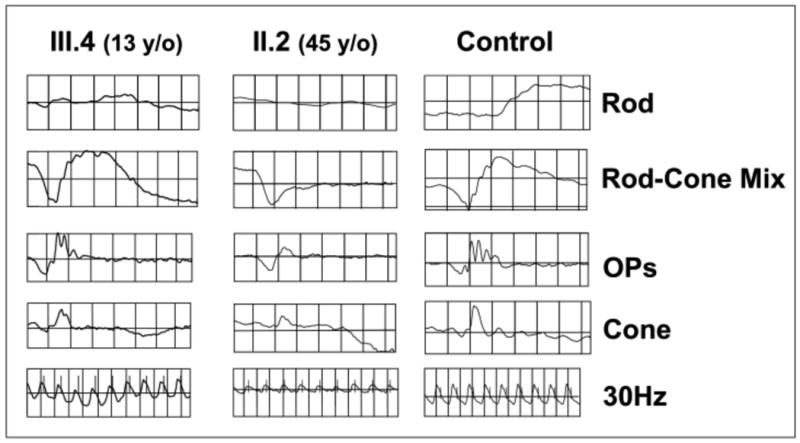
Electroretinogram depicting the rod- and cone-driven responses of the right eyes of two related males affected by XLRS (c.52G>A) and from a representative normal control subject. The rod-driven b-wave is relatively preserved in the 13-y/o III.4, but the 45-y/o-uncle II.2 shows remarkable reduction of b-wave amplitude, with the typical XLRS electronegative ERG waveform. The cone-mediated responses are overall normal in the young III.4 male, but are delayed and reduced in amplitude in the middle-aged II.2.
Three affected males in one family harboring the p.Met1Leu mutation reported that they had severe vision compromise since childhood that worsened with age (Kim, et al., 2006). One had vitreous hemorrhage at age 7; a second had severe progression of XLRS complicated by traumatic retinal detachment in one eye; and a third had poor vision in both eyes since childhood along with ocular trauma at age 8 and cataract extraction one year later. They presented extensive foveal pathology and large bullous peripheral retinoschisis. On our examination all three p.Met1Leu brothers showed vision loss ranging from 20/63 to “no light perception,” constricted Goldmann visual fields, and electronegative ERGs with reduced response amplitudes. One had only limited vision due to optic atrophy and neovascular glaucoma, and for this reason we could not perform kinetic perimetry testing nor the ERG recording.
The sole known affected person in the p.Leu13Pro family is 20 years old (y/o) and has visual acuity of 20/80 OD (right eye) and 20/70 OS (left eye), with refraction of + 6.50 sph OU. He had parafoveal cysts but without apparent peripheral involvement. The scotopic ERG b-wave amplitude, however, was reduced in both eyes leading to the electronegative ERG waveform, consistent with moderately severe XLRS disease for this age. This abnormal electrophysiological configuration, defined in different ways (Bradshaw, et al., 2004; Bradshaw, et al., 1999; Eksandh, et al., 2000; Eriksson, et al., 2004), is observed in approximately half of XLRS males (Renner, et al., 2008). The p.Leu12His mutation was described earlier (Wang, et al., 2002; Wang, et al., 2006), but pertinent clinical data are not available.
The c.52G>A males presented a history of clinical progression. The 13-y/o male (III.4 of the family pedigree, Supp. Fig. S2) had extensive central foveal cysts documented by OCT scan (Fig. 2) that impaired visual acuity (20/50 OD, 20/200 OS) but no peripheral schisis. The ERG b-wave amplitude was technically normal, but the b/a-wave ratio was subnormal (Fig. 3). The 45-y/o maternal uncle (II.2, Supp. Fig. S2) had more severe clinical XLRS features, with bilateral acuity loss (20/320 OU), macular atrophic thinning, peripheral retinal RPE changes in both eyes, and an extensive peripheral bullous schisis in the right eye. His ERG showed a markedly electronegative waveform, and even the cone-driven responses were reduced and delayed (Fig. 3).
RS1 signal-sequence mutants: molecular mechanisms
Mutations in ATG start codon (p.Met1Leu)
Protein synthesis is initiated universally with methionine (Met) (Kozak, 1999) decoded from the AUG initiating codon in eukaryotes. The initiator AUG codon is distinguished from non-initiator AUGs in mRNA by additional sequence information present at or near the initiation site and by the structural features of mRNA, such as 5′ m7G (5′) ppp (5′) N cap structure (Kozak, 1999; Kozak, 2005). These initiation factors maximize the accuracy of AUG codon and initiator anticodon selection (Antoun, et al., 2006). Examples of diseases caused by mutations in translation initiation codon or around the initiation codon have been described (Kozak, 2002).
ATG mutations have been reported in XLRS families p.Met1Leu (c.1A>T), p.Met1Val (c.1A>G), p.Met1Thr1 (c.2T>C) (Kim, et al., 2006; Mashima, et al., 1999). We examined the biochemical phenotype of one of these mutations (p.Met1Leu) for which we had clinical data. Immunoblot analysis of COS-7 cells transiently transfected with p.Met1Leu indicated that the mutant protein was expressed in the cells—albeit at a lower steady-state level (30–50%)—and secreted into the culture media (Fig. 4A, lane 6) similar to WT RS1 (Fig. 4A, lanes 4, 5). The cellular fractions appeared to retain little or no RS1 (Fig. 4A, lanes 1–3). These results suggest that RS1-protein synthesis is initiated at the UUG codon, albeit at reduced levels. Previous studies have shown that codons other than AUG can initiate protein synthesis to various degrees in vivo and in vitro: CUG (Leu), as an alternate initiation codon in a novel translation mechanism, which allowed stressed cells to display antigenic peptides (Schwab, et al., 2004); and AGG (Arg), GUC (Val), and CAG (Gln), as initiation codons in the presence of the corresponding anticodon sequence mutants of the eukaryotic initiator tRNA (Drabkin and RajBhandary, 1998). These findings indicate the potential of p.Met1Leu XLRS patients to synthesize a functional RS1 molecule. The result is surprising, since severe retinal clinical pathology is reported for p.Met1Leu patients (Kim, et al., 2006) raising the question whether the clinical phenotype is fully consistent with a reduction in RS1-protein levels.
Figure 4.


Effect of AUG alteration to UUG (p.Met1Leu) on RS1 mRNA and protein. (A) WT RS1 and p.Met1Leu mutants were transiently expressed in COS-7 cells. At 72 h post-transfection cultures were harvested and cellular and secreted fractions were analyzed for RS1 protein by immunoblotting using anti-RS1 rabbit polyclonal antibody raised against the N terminus amino-acid residues 24–37 of RS1. Both WT (AUG initiation codon) and Mut (UUG initiation codon) cells expressed RS1, which as expected, was mostly detected in the secreted fraction (lanes 4–6) as compared to the cellular bound RS1 (lanes 1–3). The results indicate that protein synthesis can be initiated at the UUG initiation codon. (B) RS1 expression in cells expressing plasmids encoding 5′ UTR immediately upstream of AUG (RS1*) or UUG (p.Met1Leu*) initiation codons. RS1 is profusely expressed in cells expressing the WT control plasmids RS1 (without 5′ UTR; lanes 1, 5) or RS1* (with 5′ UTR; lanes 3, 7). However, 5′ UTR immediately upstream of UUG initiation codon selectively abolished RS1 translation in cells expressing p.Met1Leu* (lanes 4, 8). (C) Detection of RS1 gene transcripts: total RNA isolated from cells expressing either WT or Mut expression plasmids without or with 5′ UTR was subjected to RT-PCR with RS1 cDNA specific primers and the products analyzed by agarose gel electrophoresis (semi-quantitative). Lane M is molecular size marker. The mRNA levels: lane 1, mouse retina; lane 3, the WT RS1* with 5′ UTR; lane 5, the mutant with 5′ UTR (p. Met1 Leu*); lane 7, p.Met1Leu without 5′ UTR. Parallel reactions were carried out in the absence of RT (even numbered lanes).
Exceptions to the universality of AUG as the initiator of protein synthesis in vertebrates should be understood in the context of sequence near the AUG codon (Kozak, 1999; Kozak, 2005). Most eukaryotic mRNA contains a short recognition sequence near the initiator AUG that facilitates the initial binding of mRNA to the small subunit of the ribosome. The consensus sequence for eukaryotic translation initiation sites derived substantially from vertebrate mRNA sequences is known as the Kozak sequence [(GCC)RCCATGG, where R is a purine (A or G)]. Many DNA constructs generated for protein-expression studies encode Kozak sequence immediately upstream to the ATG start codon to increase the efficiency of translation. We considered whether a favorable Kozak sequence might drive the initiation at UUG. Alternatively, because cells express only one initiator tRNA (Met-tRNAiMet), the use of non-AUG codon like UUG could represent “wobble” or cross reactivity when interacting with anticodon sequence of initiator tRNA. In such case, regardless of which AUG or non-AUG codon is used to initiate translation, it should always be decoded as methionine residue because the Met-tRNAiMet is loaded onto the 43S ribosome even before it binds to target mRNA.
To understand the in vivo mechanism involved in p.Met1Leu mutation, we generated WT and Mut cDNA constructs with a naturally occurring 18-bp untranslated region (UTR) just upstream from the ATG or TTG start codon (RS1*: 5′-gccgaggacgaggggaag ATG-3; p. Met1Leu*: 5′-gccgaggacgaggggaag TTG-3′). The cells transfected with the RS1*clone had high expression of 24-kDa RS1, and most of it was secreted and detected in the culture medium (Fig. 4B, lane 7). RS1* was also detected in the cellular fraction at reduced levels (Fig. 4B, lane 3). By contrast, the cells expressing the p.Met1Leu* clone failed to express the Mut RS1protein (Fig. 4B, lanes 4, 8). WT RS1 (lanes 1, 5) and RS1- Flag (lanes 2, 6) served as positive controls. To confirm that the absence of protein was not due to the lack of mRNA, RS1 mRNA levels were examined by RT-PCR using RS1 specific internal primers (Supp. Table S1), and gel electrophoresis of RT-PCR products demonstrate comparable levels of mRNA in cells expressing WT RS1* (Fig. 4C, lane 3), the Mut p.Met1Leu* (lane 5), and p.Met1Leu (lane 7) plasmids. Thus, the complete lack of RS1 protein in Mut p.Met1Leu* is due to a block in the initiation of translation at UUG codon. These findings imply that 5′ UTR mediates translational control by selectively initiating the translation at AUG but not at UUG. The potential of 5′ UTR (20–100 nucleotides) sequences and the structural features of mRNA to establish the fidelity of translation initiation were reviewed previously (Kozak, 1987; Kozak, 1991). The suboptimal translation initiation observed in the presence of statistically derived Kozak sequence (Fig. 4A, lane 6) is probably the outcome of artificially enhanced experimental condition. UUG was shown to initiate protein synthesis only in yeast strains that carried mutations in subunits of eukaryotic initiation factor (eIF), eIF2, or eIF5 (Donahue, et al., 1988; Huang, et al., 1997). In summary, these findings imply that an absolute deficiency of RS1 protein is the underlying cause of retinal pathology in p.Met1Leu patients with XLRS.
Mutations in hydrophobic segment of RS1 signal sequence: p.Leu12His and p.Leu13Pro
Mutations have been reported in the central portion of RS1 signal sequence that contains a segment of five leucines (L) [1-MSRKIEGFLLLLLFGYEATLGLSS-24] which are hydrophobic (lipophilic) (Hiriyanna, et al., 1999; Wang, et al., 2002). The structural features of the signal sequence are described in Fig. 1. This segment of hydrophobic amino acids is known to guide the growing polypeptide chain through the lipid membrane into the lumen of the endoplasmic reticulum (ER). We examined three mutations in this region, p.Leu12His, p.Leu13Pro, and pLeu13Phe for RS1-protein expression and localization by immunoblot analysis. No RS1 protein was detected in the cellular or secreted fractions derived from p.Leu12His and p.Leu13Pro mutants (Fig. 5A) indicating that disruption of the hydrophobic core of the signal sequence affects RS1 expression. By comparison, cells transfected with WT RS1 and p.Leu13Phe plasmids expressed RS1 that was readily secreted into the culture medium (lanes 5, 7). Since the mutation p.Leu13Phe replaced a hydrophobic Leu residue with another hydrophobic residue (Phe), it affected neither the synthesis nor secretion of RS1 and served as a positive control.
Figure 5.


Immunoblot analysis of RS1 expression in cellular and secreted fractions of COS-7 cells expressing p.Leu12His and p.Leu13Pro mutants. (A) The WT RS1 and the p.Leu13Phe mutant with intact signal sequence hydrophobic core both profusely expressed RS1, which was processed and secreted into the culture medium (lanes 5, 7) with little retained in the cellular fractions (lanes 1, 3). The p.Leu12His (lanes 2, 6) and p.Leu13Pro mutants (lanes 6, 8), with a disrupted hydrophobic core, showed virtual lack of RS1 expression. (B) Proteasomal inhibition stabilizes RS1 protein in the mutants. COS-7 cells were transfected with RS1 expression plasmids one day before MG132 (10 μM for 6h); or its vehicle dimethyl sulfoxide (DMSO) was added, and the cells were grown in serum-supplemented medium. The precursor form of RS1 (224 aa) stabilized in the presence of MG132 is seen in the cellular fractions of the signal sequence mutants (lanes 1–3). In p.Leu13Phe the precursor is processed into mature form of RS1 (201 aa) and secreted into the medium (lane 4) but not in the p.Leu12His and p.Leu13Pro mutants due to a processing defect.
The mutations in the hydrophobic core of signal sequences affect protein secretion but not synthesis (Vijayasarathy, et al., 2006). In vivo, the uncleaved precursor protein molecule remains inserted in the ER. However, the failure to detect RS1 protein even in the cellular extracts of the mutants p.Leu12His and p.Leu13Pro led us to examine whether these mutants actually synthesize RS1 protein. Most mutant proteins, whether membrane or soluble but retained in ER, are degraded by the proteasome system (Goldberg, 2003). To test this possibility, we treated cells transfected with p.Leu12His, p.Leu13Pro, and p.Leu13Phe plasmids with proteasome-inhibitor MG-132 (10μM) for 6 h after which RS1 immunoreactivity was readily observed in the cellular fractions (Fig. 5B). The slowly migrating band observed in p.Leu12His, and p.Leu13Pro cellular extracts corresponded to the unprocessed RS1 precursor (224 aa). The results also demonstrate that the Mut p.Leu13Phe with an intact hydrophobic core in its signal sequence efficiently processes the 224-aa RS1 precursor (lane 1) into 201-aa mature form (lane 4), which is secreted into the medium. On the contrary, no mature protein was detected in the secreted fractions of p.Leu12His and p.Leu13Pro mutants, indicating that the unprocessed RS1 mutants undergo proteasome degradation. Therefore, p.Leu12His and p.Leu13Pro mutations in the signal peptide that disrupt the hydrophobic core result in loss of protein production.
Mutation in the terminal nucleotide of exon 1: c.52G>A
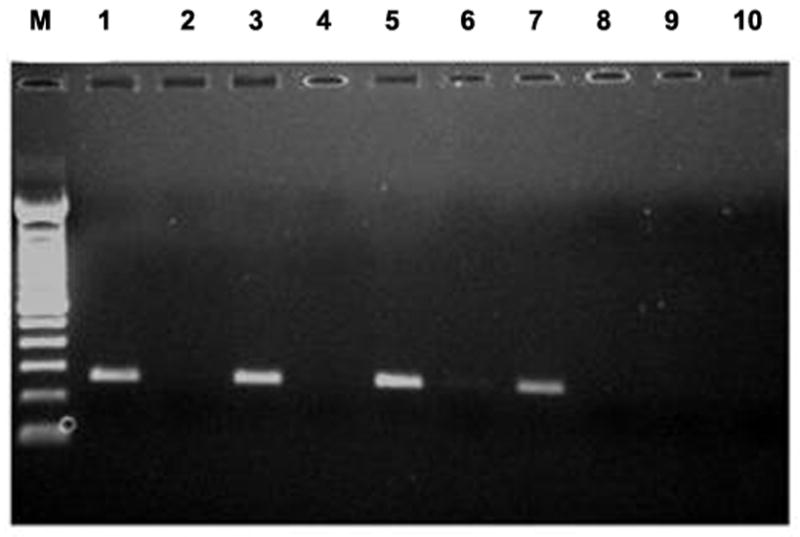
Sequence analysis in a Chinese family identified a novel XLRS mutation (c.52G>A) in exon 1 of RS1 gene. The c.52G residue is the terminal nucleotide of exon 1 and is within the 5′ donor splice site of intron 1. The 5′ donor and 3′ acceptor splice sites are the consensus sequences at the exon–intron (5′ splice site) and intron–exon junctions (3′ splice site) (Fig. 6) (Shapiro and Senapathy, 1987). The splicing factors recognize the splice sites and cleave the intron while joining the exons together (Faustino and Cooper, 2003; Kramer, 1996). We evaluated whether a 5′ splice-site mutation (c.52G>A) affected RNA processing. We first generated WT and Mut RS1minigenes with genomic intron-1 and intron-2 sequences in frame with exons 1–6 (Fig. 7A). Transient expression of the WT RS1 minigene in COS-7 cells gave the expected RS1 protein in the secreted fraction, which results from translation of an mRNA (Fig. 7B). However, the Mut c.52G>A minigene showed absolute lack of RS1 protein in both the cellular and secreted fractions (Fig. 7B). We then examined the possibility that RNA processing was defective. RNA derived from cells expressing either the WT or Mut minigene was subjected to RT-PCR for the analysis of RS1 transcripts. Various combinations of 5′ sense primers flanking the exon 1–6 sequences were used in combination with 3′ antisense primer flanking the termination codon. The total PCR products were cloned and sequenced from multiple reactions. Sequence analysis confirmed that the 700-bp band observed in the WT clones corresponded to normally spliced RNA species (Supp. Fig. S3A). The smaller (< 700-bp) band derived from the Mut c.52G>A clones lacked the 26-bp exon 2 (Supp. Fig. S3B). The 600-bp product also derived from mutant RNA clones corresponded to exons 3–6 of RS1 cDNA and lacked both exons 1 and 2 (Supp. Fig. S3C). In vitro data from COS-7 cells confirmed that the normal minigene could be spliced correctly, but the c.52G>A mutation that involves the 5′ donor splice site of intron 1 resulted in skipping of either exon 2 or both exons 1 and 2. Because the intron 1 interrupts the 18th codon (GCC) between the first and second nucleotides, exon-2 skipping would causes a frame shift at the exon-1/exon-3 junction upon the translation of the aberrantly spliced RS1 mRNA. The frame shift changes Ala 18 to Arg and creates a new reading frame ending in a UGA stop codon that terminates translation at position 19 (p.Ala18 Arg fsX2). The aberrant ∼600-bp mRNA generated due to skipping of both exons 1 and 2 retains the RS1 reading frame but lacks the initiating Met codon (AUG) required to generate a functional RS1 molecule.
Figure 6.
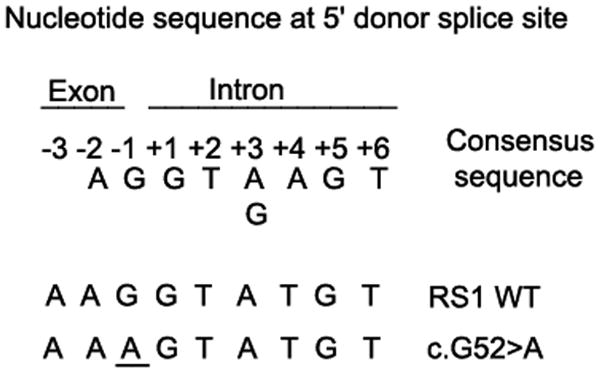
Sequence of eight highly conserved nucleotides at the boundary between an exon and an intron: the 5′ donor splice site (5′ ss) of eukaryotic mRNAs (Shapiro and Senapathy, 1987). Also shown are the sequences at the boundary between exon 1 and intron 1 in RS1 WT and c52 G>A mutant. Splicing is catalyzed by spliceosome, an RNA/protein complex consisting of small nuclear ribonucleoprotein particles (snRNPs) and SR family of splicing proteins (splicing factors with one or more RNA-recognition motif) through RNA–RNA and protein–protein interactions. snRNPs are composed of snRNAs (U1, U2, U4N6, and U5) each associated with 8 sm proteins. These snRNPs assemble on the conserved sequence motifs at three sites: U1 at exon–intron junction (5′ splice site), U2AF at intron–exon junction (3′ splice site) and U2 at branch point located 18–40 nucleotides upstream of the 3′ splice site.
Figure 7.


RS1 RNA and protein analysis in cells expressing the WT or c.52G>A Mut minigene. (A) Cartoon of the mutant c.52G>A minigene used. The minigene without splice-site mutation served as control WT. (B) Immunoblot analysis of RS1 protein with anti-RS1 antibody detected RS1 protein both in the cellular and secreted fractions of the cells expressing the WT minigene. RS1 protein was not detected in the fractions derived from cells expressing the c.52G>A mutant minigene.
While most of the mutant transcripts lacked either exons1–2 or exon 2, a less abundant and correctly spliced mRNA harboring the G-to-A mutation was also found in a mutant cDNA clone (Supp. Fig. S3D). This finding is consistent with the splice-junction consensus-sequences analysis that indicates that A is present as the last nucleotide (-1 position of any intron) in 11% of eukaryotic protein coding genes (compare to 78% G at the same position). The 5′ splice-site mutations disrupting the base pairing between the 5′ splice site and U1 snRNA could be expected to reduce the efficiency of normal splicing (Fig. 6). The matching scores for the WT 5′ AG splice site and the mutant 5′ AA splice sites of intron 1 in RS1 gene to the 5′ donor splice-site consensus sequences calculated by Shapiro and Senapathy formula (Shapiro and Senapathy, 1987) are 88.6% and 76.3%, respectively. Thus, the decreased interactions of a ribonucleoprotein particle at the mutant donor splice site influence its binding to the splice acceptor sites of preceding and/or succeeding introns resulting in a skip of intervening exon or exons. Mutations in the last nucleotide of an any exon underlie the pathology of some other genetic disorders: -1G>T in exon 3 of ATRX gene in ATR-X syndrome (Wada, et al., 2006); -1G>A in exon 5 of ß-hexosaminidase gene in a late infantile form of Tay-Sachs disease (Akli, et al., 1990); -1G>A in exon 6 of sterol 27-hydroxylase gene in cerebrotendinous xanthomatosis (Chen, et al., 1998); and -1G>T in exon 2 of purine nucleoside phosphorylase gene in severe immunodeficiency (Andrews and Markert, 1992). These results confirm that defective pre-RNA processing contributes to the etiopathogenesis of disease in persons harboring the c.52G>A mutation.
Point mutations in discoidin domain produce secretion-incompetent, misfolded RS1 variants
Previous studies have shown that point mutations that substitute conserved cysteine residues within the discoidin domain produced conformationally-defective, and secretion-incompetent RS1 protein (Wang, et al., 2002; Wu and Molday, 2003; Wu, et al., 2005). We evaluated the consequence of the substitution of amino acids other than cysteine in the discoidin domain, including p.Glu72Lys, p.Arg102Trp, p.Pro192Ser, p.Asn179Asp, p.Arg213Trp, p.Arg200cys, and p.Trp96cys (Supp. Table. S1). In COS-7 cells these mutants produced intracellularly retained misfolded RS1 protein (Fig. 8A). In non-reducing gels, the mutants appeared as dimers or as larger aggregates (not shown). Only the p.Glu72Lys mutant protein was secreted to a limited extent (Fig. 8A). We also tested whether a stable mutant RS1 protein exerts a trans-dominant-negative effect on WT RS1 and impairs its function. We co-transfected COS-7 cells with expression plasmids encoding WT RS1 and Flag epitope-tagged mutant RS1 to simulate heterozygous state. Presence of Flag tag facilitated the selective and specific identification of mutant variants of RS1 independent from WT RS1 (Supp. Fig. S1). Similar to WT, the epitope-tagged RS1 was secreted and served as a control. Immunoblot analysis of cellular and secreted fractions with anti-Flag antibody revealed none of the mutants in the secreted fraction, except p.Glu72Lys, (Fig. 8B). Similar results were obtained with both tagged and untagged RS1 variants, thus confirming that the epitope tag did not alter the molecular properties of the Mut RS1 proteins. Reprobing the blot with anti-RS1 antibody showed the presence of WT RS1 in all secreted fractions (Fig. 8B), confirming that co-expression of Mut RS1 with WT RS1 affected neither the synthesis nor the secretion of WT RS1.
Figure 8.

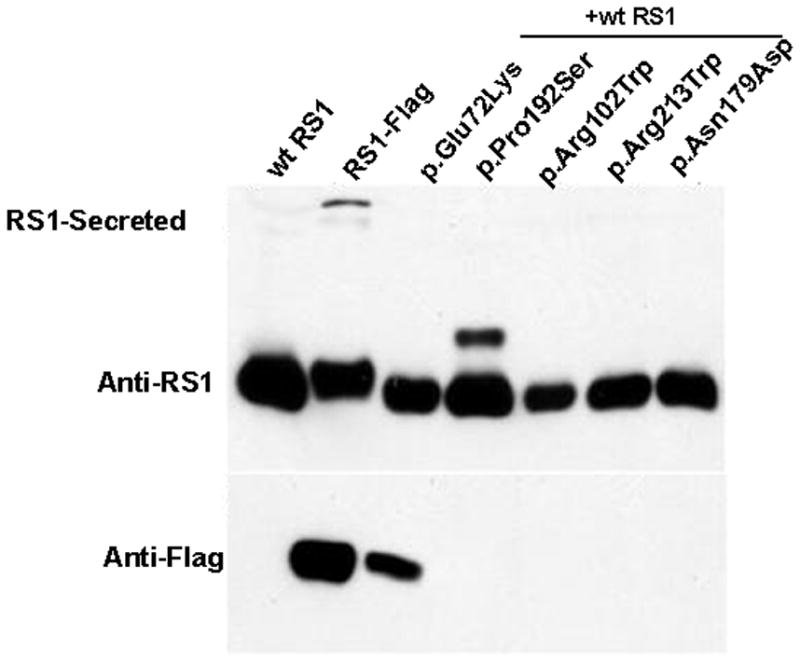
Effect of missense mutations in the discoidin domain on RS1-protein expression and localization. (A) The missense mutants were transiently expressed in COS-7 cells and analyzed for RS1 expression. The WT RS1 was predominantly detected in the secreted fraction as expected. The missense mutants which fail to be secreted are intracellularly retained. Only the p.Glu72Lys mutant was secreted. (B) Co-expression of Flag-epitope tagged RS1 mutant with WT RS1 to resemble heterozygous state. Anti-Flag antibody selectively identified epitope tagged RS1 mutants, and except for p.Glu72Lys none of the mutants were secreted. Reprobing the blot with anti-RS1 antibody revealed the presence of WT RS1 in all the secreted fractions. Flag-epitope tagged RS1 served as control. These results confirm that in a heterozygous state none of the subset of RS1 missense mutations studied here interfered with WT RS1 expression nor its secretion from the cell.
Discussion
Molecular phenotypes of XLRS mutations
This study describes how several point mutations in the RS1 signal sequence affect gene expression in XLRS patients. The molecular consequences affect one of the many steps in RS1 biosynthesis, namely post-transcriptional pre-RNA splicing (c.52G>A), translation initiation (p.Met1Leu), or the ability to mature in the secretory pathway (p.Leu12His; p.Leu13Pro). These signal-sequence mutants result in no RS1 protein either by an absolute lack of functional protein production (p.Met1Leu) or by rapid ER-associated proteasomal degradation of immature (p.Leu12His; p.Leu13Pro) or truncated (c.52G>A) RS1 variants.
The c.52G>A mutation disrupts base pairing between the splice site and small nuclear RNA and reduces normal splicing efficiency (Shapiro and Senapathy, 1987). We observed a mixture of three spliced RNA species (exon 1 skipped; exons 1 and 2 skipped; or normally spliced RNA). Exon skipping precludes synthesis of functional RS1 protein. Other point mutations in the exonic and intronic donor- or acceptor-site splice junctions have been reported in XLRS patients (www.dmd.nl/rs/consortium.html). The most common outcome of mutations affecting splice sites is exon skipping resulting in deletion of one or more exons. Other XLRS mutations include nonsense deletions, and/or insertions of one or several nucleotides (Vijayasarathy, et al., 2009) leading to frame shifts that may introduce premature termination codons. Such truncated proteins may be harmful to cells, and cellular quality-control mechanisms ensure that the anomalous mRNAs are rapidly degraded by nonsense-mediated mRNA decay (NMD) or eliminated by the proteasomal pathway. Both mechanisms result in null-protein phenotype.
By contrast to RS1 null phenotypes, missense mutations involving conserved amino-acid residues in the discoidin domain generally cause protein misfolding, as we and others have demonstrated (Wang, et al., 2002; Wu and Molday, 2003; Wu, et al., 2005). The misfolded discoidin-domain mutants (Glu72Lys, p. Arg102Trp, p.Pro192Ser, p.Asn179Asp, p.Arg213Trp, p.Arg200cys, and p.Trp96cys) are stabilized in the cell and form high molecular mass aggregates. Accumulation of misfolded mutant proteins result in ER stress that induces an unfolded protein response (UPR) (Nakatsukasa and Brodsky, 2008). With excessive or prolonged ER stress, cells undergo apoptosis. The extent to which ER stress modulates the XLRS clinical phenotype is not clear. A recent study (Gleghorn, et al., 2010) demonstrated that UPR in cells expressing pCys110Tyr mutant RS1 may actually promote cell viability. Apoptosis of photoreceptor cells would have drastic consequences for vision. How photoreceptors dispose of misfolded proteins while preserving cell viability remains unexplained.
Correlation between clinical severity and associated gene mutations in XLRS
XLRS patients exhibit considerable heterogeneity in clinical severity. The common denominator is the presence of bilateral foveo-macular cystic cavities (III.4 in Fig. 2) involving the inner and intermediate retinal layers (Gregori, et al., 2009) that impairs vision in most XLRS males (George, et al., 1996). Half of affected males also show peripheral retinoschisis (II.2, Fig. 2) with inner holes, outer retinal holes, and vitreo-retinal tractions (George, et al., 1996), leading to loss of visual-field sensitivity (Li, et al., 2007), vitreous veils and/or later-stage RPE degeneration. Foveal involvement in XLRS generally causes acuity reduction by school age or earlier (George, et al., 1996). Greater reduction of the ERG b-wave compared to the a-wave gives the classic XLRS electronegative waveform (Fig. 3), and implicates deficient synaptic function or bipolar-cell action (Khan, et al., 2001).
These discussions invariably raise the question of whether null mutations lead to more severe XLRS disease than do missense mutations, as some have reported (Hiriyanna, et al., 2001). Such correlations have been indicated for other retinal diseases (Lorenz, et al., 2008). One notes in the XLRS clinical literature that insertions, deletions, and splice-site mutations expected to produce no RS1 protein generally have an aggressive clinical phenotype with subnormal or undetectable ERG b-wave and reduced a-wave, poor visual acuity, extensive macular lesions, and even retinal detachment (Bradshaw, et al., 1999; Shinoda, et al., 2000; Shinoda, et al., 2004). Even three XLRS homozygous females in a family with an RS1 frame-shift causing deletion displayed severe clinical phenotypes (Mendoza-Londono, et al., 1999). By contrast, most patients carrying some missense mutations (e.g., p.Arg200Cys or p.Arg102Trp) more commonly have only modest b-wave loss, present generally a good or modest blunting of the macular reflex, and generally have benign peripheral retinal change (Bradshaw, et al., 1999).
Unfortunately, the methods to grade clinical severity of XLRS disease are not standardized and vary considerably (Bradshaw, et al., 1999; Eksandh, et al., 2000; Lesch, et al., 2008; Prenner, et al., 2006; Roesch, et al., 1998; Shinoda, et al., 2000; Sieving, et al., 1999) which makes it difficult to establish definitive genotype–phenotype correlations across different reports. Genetic factors affecting severity and disease penetrance have been mapped in mouse models of XLRS (Johnson, et al., 2006) but have not been identified for human XLRS.
The clinical severity of the disease in these four families with RS1 signal-sequence mutations ranged from severe in the middle-aged affected men with p.Met1Leu or c.52G>A to moderately severe in the boys (given the young ages) with c.52G>A and p.Leu13Pro. We and others have reported XLRS variability and progression of severity with age (Lesch, et al., 2008; Sergeev, et al., 2010; Vijayasarathy, et al., 2009). The less severe clinical presentation of the 13-y/o boy with a c.52G>A mutation may reflect younger age or could reflect the 5–10% normally spliced mRNA that we found biochemically, which would promote RS1 synthesis albeit at quite reduced levels. Since in vivo kinetics of mRNA splicing could be modulated by subtle cell specific variations (Chen, et al., 1998), these variables are difficult to predict from cell culture. That reduced normal RS1 expression does not fully rescue these subjects is consistent with the hypothesis that an absolute lack of RS1 protein is the underlying cause of XLRS in c.52G>A. The graded severity of disease from young age to middle-age affected men raises questions about possible cumulative pathology with age that is beyond the restorative capacity of the retina.
Implications for gene therapy
Experiments in RS1 knockout-mouse models have demonstrated the feasibility of AAV-mediated RS1 gene delivery/transfer as an effective treatment approach (Janssen, et al., 2008; Kjellstrom, et al., 2007; Park, et al., 2009; Zeng, et al., 2004). The knockout-mouse models completely lack RS1 protein, which mimics our human patients harboring the p.Met1Leu, p.Leu12His, p.Leu13Pro, c.52G>A, and c.354del1–ins18 mutations leading to null-protein phenotype. By comparison, introducing the RS1 gene into XLRS individuals harboring conformationally-defective misfolded RS1 could result in expression of both the native Mut protein and also the WT transduced protein. Fortunately, most missense mutations that have been examined in heterozygous form by co-transfection of WT- and Mut-RS1 cDNA plasmids in cultured cells did not impair the synthesis or localization of WT RS1 (Fig. 8B). Only a small number of secreted mutants (p.Glu72Lys, p.Cys110Tyr) were found to associate with WT RS1 and possibly to interfere with the functional competency of the assembled RS1 complex (Gleghorn, et al., 2010). Thus, in the absence of a dominant-negative effect of mutant RS1 protein, RS1 gene-transfer therapy is likely to confer therapeutic benefit. Results of the present study and others (Dyka and Molday, 2007) validate the general principles of a therapeutic potential of RS1 gene therapy to treat a majority of XLRS patients across a spectrum of gene mutations.
Supplementary Material
Acknowledgments
We thank Drs. Brian Brooks and Michael Redmond of NEI/NIH for useful discussions; Sean Finnegan for editorial assistance with manuscript preparation, and Doug Kesselring for help with illustrations. The author CV thanks Yogesh Sharma, B.E., M.S., of Patni Computer Systems, Mumbai, India. This research was supported by the Intramural Research Programs of the NIH. Dr. Ruifang Sui was a recipient of the Foundation Fighting Blindness (FFB Grant No. CD-CL-0808-0470-PUMCH). Dr. Richard Lewis is a Senior Scientific Investigator for Research to Prevent Blindness, New York. We thank the individuals and families reported here for their generous and continuing commitment to these programs.
Footnotes
Supporting Information for this preprint is available from the Human Mutation editorial office upon request (humu@wiley.com)
References
- Akli S, Chelly J, Mezard C, Gandy S, Kahn A, Poenaru L. A “G” to “A” mutation at position -1 of a 5′ splice site in a late infantile form of Tay-Sachs disease. J Biol Chem. 1990;265(13):7324–30. [PubMed] [Google Scholar]
- Andrews LG, Markert ML. Exon skipping in purine nucleoside phosphorylase mRNA processing leading to severe immunodeficiency. J Biol Chem. 1992;267(11):7834–8. [PubMed] [Google Scholar]
- Antoun A, Pavlov MY, Lovmar M, Ehrenberg M. How initiation factors maximize the accuracy of tRNA selection in initiation of bacterial protein synthesis. Mol Cell. 2006;23(2):183–93. doi: 10.1016/j.molcel.2006.05.030. [DOI] [PubMed] [Google Scholar]
- Bradshaw K, Allen L, Trump D, Hardcastle A, George N, Moore A. A comparison of ERG abnormalities in XLRS and XLCSNB. Doc Ophthalmol. 2004;108(2):135–45. doi: 10.1023/b:doop.0000036786.22179.44. [DOI] [PubMed] [Google Scholar]
- Bradshaw K, George N, Moore A, Trump D. Mutations of the XLRS1 gene cause abnormalities of photoreceptor as well as inner retinal responses of the ERG. Doc Ophthalmol. 1999;98(2):153–73. doi: 10.1023/a:1002432919073. [DOI] [PubMed] [Google Scholar]
- Chen W, Kubota S, Seyama Y. Alternative pre-mRNA splicing of the sterol 27-hydroxylase gene (CYP 27) caused by a G to A mutation at the last nucleotide of exon 6 in a patient with cerebrotendinous xanthomatosis (CTX) J Lipid Res. 1998;39(3):509–17. [PubMed] [Google Scholar]
- Donahue TF, Cigan AM, Pabich EK, Valavicius BC. Mutations at a Zn(II) finger motif in the yeast eIF-2 beta gene alter ribosomal start-site selection during the scanning process. Cell. 1988;54(5):621–32. doi: 10.1016/s0092-8674(88)80006-0. [DOI] [PubMed] [Google Scholar]
- Drabkin HJ, RajBhandary UL. Initiation of protein synthesis in mammalian cells with codons other than AUG and amino acids other than methionine. Mol Cell Biol. 1998;18(9):5140–7. doi: 10.1128/mcb.18.9.5140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyka FM, Molday RS. Coexpression and interaction of wild-type and missense RS1 mutants associated with X-linked retinoschisis: its relevance to gene therapy. Invest Ophthalmol Vis Sci. 2007;48(6):2491–7. doi: 10.1167/iovs.06-1465. [DOI] [PubMed] [Google Scholar]
- Eksandh LC, Ponjavic V, Ayyagari R, Bingham EL, Hiriyanna KT, Andreasson S, Ehinger B, Sieving PA. Phenotypic expression of juvenile X-linked retinoschisis in Swedish families with different mutations in the XLRS1 gene. Arch Ophthalmol. 2000;118(8):1098–104. doi: 10.1001/archopht.118.8.1098. [DOI] [PubMed] [Google Scholar]
- Eriksson U, Larsson E, Holmstrom G. Optical coherence tomography in the diagnosis of juvenile X-linked retinoschisis. Acta Ophthalmol Scand. 2004;82(2):218–23. doi: 10.1111/j.1600-0420.2004.00227.x. [DOI] [PubMed] [Google Scholar]
- Faustino NA, Cooper TA. Pre-mRNA splicing and human disease. Genes Dev. 2003;17(4):419–37. doi: 10.1101/gad.1048803. [DOI] [PubMed] [Google Scholar]
- George ND, Yates JR, Moore AT. Clinical features in affected males with X-linked retinoschisis. Arch Ophthalmol. 1996;114(3):274–80. doi: 10.1001/archopht.1996.01100130270007. [DOI] [PubMed] [Google Scholar]
- Gleghorn LJ, Trump D, Bulleid NJ. Wild-type and missense mutants of retinoschisin co-assemble resulting in either intracellular retention or incorrect assembly of the functionally active octamer. Biochem J. 2010;425(1):275–83. doi: 10.1042/BJ20091179. [DOI] [PubMed] [Google Scholar]
- Goldberg AL. Protein degradation and protection against misfolded or damaged proteins. Nature. 2003;426(6968):895–9. doi: 10.1038/nature02263. [DOI] [PubMed] [Google Scholar]
- Gregori NZ, Berrocal AM, Gregori G, Murray TG, Knighton RW, Flynn HW, Jr, Dubovy S, Puliafito CA, Rosenfeld PJ. Macular spectral-domain optical coherence tomography in patients with X linked retinoschisis. Br J Ophthalmol. 2009;93(3):373–8. doi: 10.1136/bjo.2007.136127. [DOI] [PubMed] [Google Scholar]
- Hiriyanna KT, Bingham EL, Yashar BM, Ayyagari R, Fishman G, Small KW, Weinberg DV, Weleber RG, Lewis RA, Andreasson S, et al. Novel mutations in XLRS1 causing retinoschisis, including first evidence of putative leader sequence change. Hum Mutat. 1999;14(5):423–7. doi: 10.1002/(SICI)1098-1004(199911)14:5<423::AID-HUMU8>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Hiriyanna KT, Singh-Parikshak R, Bingham EL, Kemp JA, Ayyagari R, Yashar BM, Sieving PA. In: Searching for genotype-phenotype correlations in X-linked juvenile retinoschisis. Anderson RE, LV M, Hollyfield JG, editors. New York: Plenum Publishers; 2001. pp. 45–53. [Google Scholar]
- Huang HK, Yoon H, Hannig EM, Donahue TF. GTP hydrolysis controls stringent selection of the AUG start codon during translation initiation in Saccharomyces cerevisiae. Genes Dev. 1997;11(18):2396–413. doi: 10.1101/gad.11.18.2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen A, Min SH, Molday LL, Tanimoto N, Seeliger MW, Hauswirth WW, Molday RS, Weber BH. Effect of late-stage therapy on disease progression in AAV-mediated rescue of photoreceptor cells in the retinoschisin-deficient mouse. Mol Ther. 2008;16(6):1010–7. doi: 10.1038/mt.2008.57. [DOI] [PubMed] [Google Scholar]
- Johnson BA, Ikeda S, Pinto LH, Ikeda A. Reduced synaptic vesicle density and aberrant synaptic localization caused by a splice site mutation in the Rs1h gene. Vis Neurosci. 2006;23(6):887–98. doi: 10.1017/S0952523806230244. [DOI] [PubMed] [Google Scholar]
- Khan NW, Jamison JA, Kemp JA, Sieving PA. Analysis of photoreceptor function and inner retinal activity in juvenile X-linked retinoschisis. Vision Res. 2001;41(28):3931–3942. doi: 10.1016/s0042-6989(01)00188-2. [DOI] [PubMed] [Google Scholar]
- Kim DY, Neely KA, Sassani JW, Vrabec TR, Tantri A, Frost A, Donoso LA. X-linked retinoschisis: novel mutation in the initiation codon of the XLRS1 gene in a large family. Retina. 2006;26(8):940–6. doi: 10.1097/01.iae.0000224321.93502.a3. [DOI] [PubMed] [Google Scholar]
- Kjellstrom S, Bush RA, Zeng Y, Takada Y, Sieving PA. Retinoschisin gene therapy and natural history in the Rs1h-KO mouse: long-term rescue from retinal degeneration. Invest Ophthalmol Vis Sci. 2007;48(8):3837–45. doi: 10.1167/iovs.07-0203. [DOI] [PubMed] [Google Scholar]
- Kozak M. At least six nucleotides preceding the AUG initiator codon enhance translation in mammalian cells. J Mol Biol. 1987;196(4):947–50. doi: 10.1016/0022-2836(87)90418-9. [DOI] [PubMed] [Google Scholar]
- Kozak M. Structural features in eukaryotic mRNAs that modulate the initiation of translation. J Biol Chem. 1991;266(30):19867–70. [PubMed] [Google Scholar]
- Kozak M. Initiation of translation in prokaryotes and eukaryotes. Gene. 1999;234(2):187–208. doi: 10.1016/s0378-1119(99)00210-3. [DOI] [PubMed] [Google Scholar]
- Kozak M. Emerging links between initiation of translation and human diseases. Mamm Genome. 2002;13(8):401–10. doi: 10.1007/s00335-002-4002-5. [DOI] [PubMed] [Google Scholar]
- Kozak M. Regulation of translation via mRNA structure in prokaryotes and eukaryotes. Gene. 2005;361:13–37. doi: 10.1016/j.gene.2005.06.037. [DOI] [PubMed] [Google Scholar]
- Kramer A. The structure and function of proteins involved in mammalian pre-mRNA splicing. Annu Rev Biochem. 1996;65:367–409. doi: 10.1146/annurev.bi.65.070196.002055. [DOI] [PubMed] [Google Scholar]
- Lesch B, Szabo V, Kanya M, Somfai GM, Vamos R, Varsanyi B, Pamer Z, Knezy K, Salacz G, Janaky M, et al. Clinical and genetic findings in Hungarian patients with X-linked juvenile retinoschisis. Mol Vis. 2008;14:2321–32. [PMC free article] [PubMed] [Google Scholar]
- Li X, Ma X, Tao Y. Clinical features of X linked juvenile retinoschisis in Chinese families associated with novel mutations in the RS1 gene. Mol Vis. 2007;13:804–12. [PMC free article] [PubMed] [Google Scholar]
- Lorenz B, Poliakov E, Schambeck M, Friedburg C, Preising MN, Redmond TM. A comprehensive clinical and biochemical functional study of a novel RPE65 hypomorphic mutation. Invest Ophthalmol Vis Sci. 2008;49(12):5235–42. doi: 10.1167/iovs.07-1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashima Y, Shinoda K, Ishida S, Ozawa Y, Kudoh J, Iwata T, Oguchi Y, Shimizu N. Identification of four novel mutations of the XLRS1 gene in Japanese patients with X-linked juvenile retinoschisis. Mutation in brief no. 234. Online. Hum Mutat. 1999;13(4):338. doi: 10.1002/(SICI)1098-1004(1999)13:4<338::AID-HUMU16>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Mendoza-Londono R, Hiriyanna KT, Bingham EL, Rodriguez F, Shastry BS, Rodriguez A, Sieving PA, Tamayo ML. A Colombian family with X-linked juvenile retinoschisis with three affected females finding of a frameshift mutation. Ophthalmic Genet. 1999;20(1):37–43. doi: 10.1076/opge.20.1.37.2299. [DOI] [PubMed] [Google Scholar]
- Molday LL, Wu WW, Molday RS. Retinoschisin (RS1), the protein encoded by the X-linked retinoschisis gene, is anchored to the surface of retinal photoreceptor and bipolar cells through its interactions with a Na/K ATPase-SARM1 complex. J Biol Chem. 2007;282(45):32792–801. doi: 10.1074/jbc.M706321200. [DOI] [PubMed] [Google Scholar]
- Molday RS. Focus on molecules: retinoschisin (RS1) Exp Eye Res. 2007;84(2):227–8. doi: 10.1016/j.exer.2005.12.013. [DOI] [PubMed] [Google Scholar]
- Nakatsukasa K, Brodsky JL. The recognition and retrotranslocation of misfolded proteins from the endoplasmic reticulum. Traffic. 2008;9(6):861–70. doi: 10.1111/j.1600-0854.2008.00729.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park TK, Wu Z, Kjellstrom S, Zeng Y, Bush RA, Sieving PA, Colosi P. Intravitreal delivery of AAV8 retinoschisin results in cell type-specific gene expression and retinal rescue in the Rs1-KO mouse. Gene Ther. 2009;16(7):916–26. doi: 10.1038/gt.2009.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prenner JL, Capone A, Jr, Ciaccia S, Takada Y, Sieving PA, Trese MT. Congenital X-linked retinoschisis classification system. Retina. 2006;26(7 Suppl):S61–4. doi: 10.1097/01.iae.0000244290.09499.c1. [DOI] [PubMed] [Google Scholar]
- Renner AB, Kellner U, Fiebig B, Cropp E, Foerster MH, Weber BH. ERG variability in X-linked congenital retinoschisis patients with mutations in the RS1 gene and the diagnostic importance of fundus autofluorescence and OCT. Doc Ophthalmol. 2008;116(2):97–109. doi: 10.1007/s10633-007-9094-5. [DOI] [PubMed] [Google Scholar]
- Roesch MT, Ewing CC, Gibson AE, Weber BH. The natural history of X-linked retinoschisis. Can J Ophthalmol. 1998;33(3):149–58. [PubMed] [Google Scholar]
- Sauer CG, Gehrig A, Warneke-Wittstock R, Marquardt A, Ewing CC, Gibson A, Lorenz B, Jurklies B, Weber BH. Positional cloning of the gene associated with X-linked juvenile retinoschisis. Nat Genet. 1997;17(2):164–70. doi: 10.1038/ng1097-164. [DOI] [PubMed] [Google Scholar]
- Schwab SR, Shugart JA, Horng T, Malarkannan S, Shastri N. Unanticipated antigens: translation initiation at CUG with leucine. PLoS Biol. 2004;2(11):e366. doi: 10.1371/journal.pbio.0020366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergeev YV, Caruso RC, Meltzer MR, Smaoui N, Macdonald IM, Sieving PA. Molecular modeling of retinoschisin with functional analysis of pathogenic mutations from human X-linked retinoschisis. Hum Mol Genet. 2010;27:27. doi: 10.1093/hmg/ddq006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro MB, Senapathy P. RNA splice junctions of different classes of eukaryotes: sequence statistics and functional implications in gene expression. Nucleic Acids Res. 1987;15(17):7155–74. doi: 10.1093/nar/15.17.7155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Jian K, Ko ML, Trump D, Ko GY. Retinoschisin, a new binding partner for L-type voltage-gated calcium channels in the retina. J Biol Chem. 2009;284(6):3966–75. doi: 10.1074/jbc.M806333200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinoda K, Ishida S, Oguchi Y, Mashima Y. Clinical characteristics of 14 japanese patients with X-linked juvenile retinoschisis associated with XLRS1 mutation. Ophthalmic Genet. 2000;21(3):171–80. [PubMed] [Google Scholar]
- Shinoda K, Ohde H, Ishida S, Inoue M, Oguchi Y, Mashima Y. Novel 473-bp deletion in XLRS1 gene in a Japanese family with X-linked juvenile retinoschisis. Graefes Arch Clin Exp Ophthalmol. 2004;242(7):561–5. doi: 10.1007/s00417-004-0878-y. [DOI] [PubMed] [Google Scholar]
- Sieving PA, Yashar BM, Ayyagari R. Juvenile retinoschisis: a model for molecular diagnostic testing of X-linked ophthalmic disease. Trans Am Ophthalmol Soc. 1999;97:451–64. 464–9. discussion. [PMC free article] [PubMed] [Google Scholar]
- Sikkink SK, Biswas S, Parry NR, Stanga PE, Trump D. X-linked retinoschisis: an update. J Med Genet. 2007;44(4):225–32. doi: 10.1136/jmg.2006.047340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takada Y, Fariss RN, Muller M, Bush RA, Rushing EJ, Sieving PA. Retinoschisin expression and localization in rodent and human pineal and consequences of mouse RS1 gene knockout. Mol Vis. 2006;12:1108–16. [PubMed] [Google Scholar]
- Takada Y, Vijayasarathy C, Zeng Y, Kjellstrom S, Bush RA, Sieving PA. Synaptic pathology in retinoschisis knockout (Rs1-/y) mouse retina and modification by rAAV-Rs1 gene delivery. Invest Ophthalmol Vis Sci. 2008;49(8):3677–86. doi: 10.1167/iovs.07-1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tantri A, Vrabec TR, Cu-Unjieng A, Frost A, Annesley WH, Jr, Donoso LA. X-linked retinoschisis: a clinical and molecular genetic review. Surv Ophthalmol. 2004;49(2):214–30. doi: 10.1016/j.survophthal.2003.12.007. [DOI] [PubMed] [Google Scholar]
- Vijayasarathy C, Gawinowicz MA, Zeng Y, Takada Y, Bush RA, Sieving PA. Identification and characterization of two mature isoforms of retinoschisin in murine retina. Biochem Biophys Res Commun. 2006;349(1):99–105. doi: 10.1016/j.bbrc.2006.07.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijayasarathy C, Takada Y, Zeng Y, Bush RA, Sieving PA. Retinoschisin is a peripheral membrane protein with affinity for anionic phospholipids and affected by divalent cations. Invest Ophthalmol Vis Sci. 2007;48(3):991–1000. doi: 10.1167/iovs.06-0915. [DOI] [PubMed] [Google Scholar]
- Vijayasarathy C, Ziccardi L, Zeng Y, Smaoui N, Caruso RC, Sieving PA. Null retinoschisin-protein expression from an RS1 c354del1-ins18 mutation causing progressive and severe XLRS in a cross-sectional family study. Invest Ophthalmol Vis Sci. 2009;50(11):5375–83. doi: 10.1167/iovs.09-3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada T, Sakakibara M, Fukushima Y, Saitoh S. A novel splicing mutation of the ATRX gene in ATR-X syndrome. Brain Dev. 2006;28(5):322–5. doi: 10.1016/j.braindev.2005.09.005. [DOI] [PubMed] [Google Scholar]
- Wang T, Waters CT, Rothman AM, Jakins TJ, Romisch K, Trump D. Intracellular retention of mutant retinoschisin is the pathological mechanism underlying X-linked retinoschisis. Hum Mol Genet. 2002;11(24):3097–105. doi: 10.1093/hmg/11.24.3097. [DOI] [PubMed] [Google Scholar]
- Wang T, Zhou A, Waters CT, O'Connor E, Read RJ, Trump D. Molecular pathology of X linked retinoschisis: mutations interfere with retinoschisin secretion and oligomerisation. Br J Ophthalmol. 2006;90(1):81–6. doi: 10.1136/bjo.2005.078048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu WW, Molday RS. Defective discoidin domain structure, subunit assembly, and endoplasmic reticulum processing of retinoschisin are primary mechanisms responsible for X-linked retinoschisis. J Biol Chem. 2003;278(30):28139–46. doi: 10.1074/jbc.M302464200. [DOI] [PubMed] [Google Scholar]
- Wu WW, Wong JP, Kast J, Molday RS. RS1, a discoidin domain-containing retinal cell adhesion protein associated with X-linked retinoschisis, exists as a novel disulfide-linked octamer. J Biol Chem. 2005;280(11):10721–30. doi: 10.1074/jbc.M413117200. [DOI] [PubMed] [Google Scholar]
- Zeng Y, Takada Y, Kjellstrom S, Hiriyanna K, Tanikawa A, Wawrousek E, Smaoui N, Caruso R, Bush RA, Sieving PA. RS-1 Gene Delivery to an Adult Rs1h Knockout Mouse Model Restores ERG b-Wave with Reversal of the Electronegative Waveform of X-Linked Retinoschisis. Invest Ophthalmol Vis Sci. 2004;45(9):3279–85. doi: 10.1167/iovs.04-0576. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.