

Abstract
We found that adenosine 5′-monophosphate-activated protein kinase (AMPK)—which is considered the “fuel sensor” of mammalian cells because it directly responds to the depletion of the fuel molecule ATP—is strongly activated by tumor-like hypoxia and glucose deprivation. We also observed abundant AMPK activity in tumor cells in vivo, using subcutaneous tumor xenografts prepared from cells transformed with oncogenic H-Ras. Such rapidly growing transplants of tumor cells, however, represent fully developed tumors that naturally contain energetically stressed microenvironments that can activate AMPK. Therefore, to investigate the induction of AMPK activity during experimental tumorigenesis, we used an established model of brain tumor (glioma) development in the offspring of rats exposed prenatally to the mutagen N-ethyl-N-nitrosourea (ENU). We observed that immunostaining for a specific readout of AMPK activity (AMPK-dependent phosphorylation of acetyl-CoA carboxylase) was prominent during ENU-initiated neurocarcinogenesis, from the occurrence of early hyperplasia (microtumors) to the emergence of large gliomas. Moreover, we observed that immunostaining for activating phosphorylation of AMPK correlated with the same stages of glioma development, notably in mitotic tumor cells where the signal showed punctate as well as cytoplasmic patterns associated with spindle formation. Based on these observations, we propose that neurocarcinogenesis requires AMPK-dependent regulation of cellular energy metabolism.
Keywords: AMPK, ENU model, rat glioma, hypoxia, tumor microenvironment, centrosomes, C6
INTRODUCTION
Adenosine 5′-monophosphate-activated protein kinase (AMPK) functions as a direct sensor of cellular ATP status—in response to ATP depletion, AMPK acts to restore energy homeostasis by inhibiting ATP-consuming processes and stimulating ATP-generating processes (e.g., reviewed in [1–4]). These general physiological consequences of AMPK activation suggest that AMPK could be important for the survival of tumor cells exposed to metabolic (energy) stress in the pathophysiological microenvironments present in solid tumors, such as low oxygen and glucose conditions (hypoxia and hypoglycemia) (5, 6). In previous work (7) we used wild type (WT) mouse embryo fibroblasts (MEFs) and control MEFs genetically manipulated to lack expression of AMPK catalytic α subunits (AMPK null cells) to demonstrate that AMPK is activated by tumor-like hypoxic and hypoglycemic conditions. To determine whether AMPK activation also occurs in authentic tumor microenvironments, in the same study we prepared tumor xenografts from H-Ras G12V-transformed derivatives of the same WT and AMPK null cells, as well as from identically transformed MEFs lacking expression of hypoxia-inducible factor-1 (HIF-1 null tumors). Immunohistochemical analysis of these tumors indicated that AMPK activity was widely distributed in both the WT and HIF-1 null tumors, especially in viable areas near necrosis. We also determined that the growth of the AMPK null tumors was strongly suppressed compared with the WT tumors (7), which suggested that AMPK can contribute to tumor cell survival, at least in a transplanted tumor model such as mouse fibrosarcoma xenograft.
There are reports of elevated AMPK expression, activation, or activity in human tumors (e.g., [8–11]); however, it has not been established when AMPK is induced during tumorigenesis using an experimental model. Therefore, we used an established model of primary brain tumor (glioma) development in the offspring of rats exposed prenatally to the mutagen N-ethyl-N-nitrosourea (ENU) (12, 13) to determine when the induction of AMPK activity can be detected during a carcinogenic process. ENU-initiated gliomas contain hypoxic and other stressful microenvironments that activate AMPK, similar to microenvironments found in malignant human gliomas (e.g., reviewed in [14–16]). We observed that AMPK activity is induced at the earliest stages of hyperplasia in developing rat gliomas, suggesting that metabolic perturbations associated with proliferation occur early in the glioma process. The early and sustained elevation of AMPK activity during glioma development is an important correlative observation, particularly in the context of mechanistic models in which activation of AMPK is considered to have a tumor suppressive function (17).
MATERIALS AND METHODS
Materials
The following antibodies were obtained from Cell Signaling Technology (Danvers, IL): rabbit polyclonal anti-phospho-acetyl-CoA carboxylase (Ser79) antibody (CST, P-ACC Ser79, Cat. No. 3661; P-ACC hereafter); rabbit polyclonal anti-ACC antibody (acetyl-CoA carboxylase antibody, Cat. No. 3662); and rabbit monoclonal anti-phospho-AMPKα (Thr172) antibody (Phospho-AMPKα [Thr172] [40H9], Cat. No. 2535; P-AMPK hereafter). ACC and P-AMPK blocking peptides were also obtained from Cell Signaling Technology (Acetyl-CoA Carboxylase Blocking Peptide, Cat. No. 1062; P-AMPK Blocking Peptide, SKU 21200). The ACC antibody was raised against ACC1, and does not appear to clearly detect ACC2 (our findings; also, see reference [18]). A rabbit polyclonal anti-phospho-ACC2 (Ser219) antibody was obtained from Santa Cruz Biotechnology, Inc. (p-ACCβ [Ser 219/Ser 221]-R, Cat. No. sc-30446-R; ACCβ is ACC2).
Cell Culture
C6 rat glioma cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA); C6 cells were cultured in DMEM containing 25 mM HEPES buffer (pH 7.4, DMEMH; Invitrogen GIBCO) and 10% fetal bovine serum (FBS, JRH Biosciences, Lenexa, KS) at 37°C in 5% CO2–air. The origin of the ENU3.2 rat cell line is described in (19). ENU3.2 cells were cultured in Neurobasal-A Medium containing N-2 supplement (Invitrogen GIBCO) and 20 ng/ml EGF (Collaborative Research) at 37°C in 5% CO2–air. Synchronized cultures of C6 cells were prepared by a double thymidine block procedure. Briefly, cells were placed on 22-mm2 glass cover slips in 35-mm-diameter tissue culture dishes (5×104 cells/dish) and incubated overnight at 37°C. Cells were washed with PBS and incubated at 37°C in DMEMH–FBS containing 2 mM thymidine for 24 h (first block). Cells were washed with PBS, incubated at 37°C in fresh DMEMH–FBS for 9 h, and then incubated at 37°C in DMEMH–FBS containing 2 mM thymidine at for 19 h (second block). Cells were washed with PBS, fresh DMEMH–FBS was added, and incubation was continued at 37°C for 36 h; cells were fixed for immunofluorescence at various times up to 36 h, as described below. To expose C6 cells to hypoxia (7), cells were incubated in a 5% CO2–air atmosphere at 37°C overnight and then placed in an oxygen-regulated chamber (Invivo2 400 Hypoxia Work Station, Ruskinn Corp., Bridgend. UK) maintained at 37°C and 1% O2–5% CO2–N2. All manipulations of hypoxic cells were performed in the chamber.
Tumor Models
The induction of rat gliomas/astrocytomas (called gliomas hereafter) following the transplacental administration of ENU is detailed in (13). Briefly, timed pregnant Sprague-Dawley rats (Taconic Farms, Hudson, NY) were placed in a restrainer and injected ip at gestational days 17–18 with 50 mg/kg ENU (Sigma, Cat No. N3385) using a 26-gauge needle. During the experiments, at least one control rat received an identical injection with the saline vehicle. Pups from ENU-exposed mothers (usually born on embryonic day 23) appeared normal and healthy. After weaning, rats with the same sex were housed two per cage and observed weekly for signs of illness. ENU-exposed rats remained healthy until they were at least 100 days of age (designated as P100); they began to die from complications of neural tumors starting at about 120 days of age (P120). No benign ENU-initiated tumors (neurinomas) were observed in the present study. Whole brains were removed from P90 and P140 rats to evaluate early- and late-stage ENU-initiated gliomas, respectively (13), as described below. The establishment of C6 gliomas by intracranial inoculation of Sprague-Dawley rats was performed essentially as described in (20). All procedures were approved by Stanford University’s IACUC.
Immunohistochemistry
Rats were transcardially perfused with 4% paraformaldehyde in PBS; then brains were removed and immersed in the same fixative overnight before infiltrating through 10 and 30% sucrose + PBS and freezing in OCT (Optimal Cutting Temperate media). Fifty-μm-thick coronal sections were cut from the anterior commissure through the posterior hippocampus using an Erma type sliding microtome equipped with a freezing device (Physitemp Instruments, Clifton, NJ). Immunohistochemistry (IHC) was performed as described in (13). The following antibodies and dilutions were used for IHC: P-ACC (1:500), P-AMPK (1:500). The specificity of immunostaining with the P-ACC and P-AMPK antibodies was demonstrated by exposing an adjacent or proximal section of a glioma with antibody that had been pre-adsorbed with the ACC or P-AMPK blocking peptide—the specific P-ACC or P-AMPK signal was strongly diminished in the tissue (Figures 1D and 2D). Our IHC protocol (e.g., with the P-ACC antibody) was as follows, with all incubation performed on a platform shaker: histological sections were washed with Tris-buffered saline (TBS) and treated with 0.5% hydrogen peroxide in TBS for 20–30 min. Sections were washed with TBS and blocked with TBS containing 10% goat serum (GS; Vector Laboratories, Burlingame, CA) and 0.1% Triton X-100 for 1 hour, and then incubated with the primary antibody in TBS containing 2% GS and 0.1% Triton X-100 (incubation buffer). After washing, sections were incubated with a species-specific biotinylated secondary antibody in incubation buffer and then ABC reagent (Vector Laboratories) was added. 3, 3′-Diaminobenzidine (DAB, 2 min treatment; Vector Laboratories) was used to visualize P-ACC immunoreactivity on the sections.
Figure 1. AMPK-dependent phosphorylation of ACC (P-AAC) in a C6 rat glioma and in ENU-initiated rat brain tumors.

A P-ACC immunostaining in an implanted C6 glioma in a rat brain; tissue was isolated for immunostaining (DAB) seven days after implantation and counterstained with hematoxylin. The inserts show (a/top, b/middle) cytoplasmic P-ACC immunostaining of tumor cells; (c/bottom) P-ACC immunostaining of a mitotic tumor cell. B P-ACC immunostaining in a fully developed ENU-initiated macrotumor in the forceps major of the corpus callosum of the brain of a 140-day-old (P140) rat. The insert shows tumor cells with strong cytoplasmic immunostaining for P-ACC. C P-ACC immunostaining in an ENU-initiated microtumor in the gray matter (caudate putamen) of the brain of 90-day-old rat (P90). The insert shows P-ACC immunostaining in groups of tumor cells. D A contiguous section with that in panel B showing a control for antibody specificity (the primary P-ACC antibody was pre-adsorbed with an ACC peptide antigen; see Materials and Methods for details. Nonspecific immunostaining was detected only in necrotic regions (e.g., indicated by an arrow in panel D).
Figure 2. Activating phosphorylation of AMPK (P-AMPK) in a C6 rat glioma and in ENU-initiated rat brain tumors.

A P-AMPK immunostaining (DAB) in an implanted C6 rat glioma in a rat brain; tissue was counterstained with hemtoxylin. The inserts show diffuse and punctate P-AMPK immunostaining in interphase/anaphase and mitotic tumor cells. B P-AMPK immunostaining in an ENU-initiated macrotumor (glioma) at the border of the cortex and hippocampus in the brain of a P140 rat. The inserts show (a/top) diffuse cytoplasmic P-AMPK immunostaining in a group of tumor cells; (b/middle) diffuse and punctate cytoplasmic P-AMPK immunostaining in mitotic tumor cells; and (c/bottom) diffuse and punctate cytoplasmic P-AMPK immunostaining in interphase/anaphase tumor cells. C P-AMPK immunostaining in an ENU-initiated microtumor in the white matter of the brain of a P90 rat. The insert shows a punctate pattern of P-AMPK immunostaining in the cytoplasm of tumor cells. D A control for antibody specificity in which the primary P-AMPK antibody was pre-adsorbed with a P-AMPK peptide antigen; see Materials and Methods for details. The section in the upper panel, which was from a C6 rat glioma, can be compared to the P-AMPK immunostaining in a proximal section in the lower panel.
Immunofluorescence (IF)
C6 cells at ~50% confluency were washed twice with PBS, fixed for 20 min in freshly prepared 4% paraformaldehyde (PFA) in 0.1 M phosphate buffer, washed three times in PBS, and permeabilized with PBS containing 0.1% Triton X-100 (30 min). Cells were blocked in 10% normal donkey serum in bovine serum albumin (BSA) buffer (2% BSA, 0.1 M phosphate buffer, pH 7.4) for 30 min, aspirated before incubating with a rabbit monoclonal anti-phospho-AMPKα [Thr172] antibody (diluted at 2μg/ml in BSA buffer) for 60 min at room temperature. A rabbit IgG isotype control antibody at the same concentration was included in the study. Cells were washed three times with PBS (10 min each time), and then labeled with a Cy3-conjugated donkey anti-rabbit F(ab′)2 fragment antibody (minimally cross-reactive IgG [H+L]; Jackson ImmunoResearch, West Grove, PA) at 5 μg/ml in BSA buffer for 60 min at room temperature. Cells were rinsed two times with PBS (10 min each), counterstained with diamidinophenylindole dihydrochloride (DAPI; ProLong Gold, Molecular Probes Invitrogen), and rinsed with 0.1 M Tris–HCl, pH 8.5, for 5 min. The cover slips were mounted on microscope slides under an anti-fade medium and sealed before imaging. Images were obtained using a Leica DM 5500B microscope (Bannockburn, IL; e.g., at 63x magnification) equipped with a Retiga-SRV camera (Q-imaging, Surrey, BC) and Image Pro Plus acquisition software (Bethesda, MD).
Immunoblotting
Immunoblotting protocols have been described in detail in (7). Briefly, cells were placed on ice in the hypoxia chamber, and the medium was removed. Cells were lysed by adding 200 μl of lysis buffer (50 mM Tris-HCl, pH 7.4, 0.5% NP-40, 250 mM NaCl, 1 mM DTT, 50 mM NaF, 15 mM Na4P2O7, 25 mM β-glycerophosphate, 1 mM Na3VO4, 100 nM okadaic acid, 1× Protease Inhibitor Cocktail III, PIC III, Calbiochem, Carlsbad, CA). After spinning at 9,000 × g for 5 min at 4°C, the protein concentrations of the supernatants were determined by using a bicinchoninic acid assay (Pierce Biotechnology, Rockford, IL). Equal protein samples (typically 10–15 μg) were resolved in 4–12% SDS-polyacrylamide gels (Nu-PAGE Bis-Tris Gels, Invitrogen Corp.) and electroblotted onto Immobilon-FL membranes (Millipore, Billerica, MA). Blots were blocked in a 1:1 mixture of Odyssey Blocking Buffer (LI-COR Biosciences, Lincoln, NE) and PBS (pH 7.4) at 4°C overnight. For protein detection, blots were incubated overnight at 4°C with a primary antibody typically diluted 1:1,000 in Odyssey Blocking Buffer containing 0.1% Tween-20. Blots were then incubated for 1 h at room temperature containing a species-specific IRDye 800-conjugated IgG antibody (LI-COR Biosciences) diluted in Odyssey Blocking Buffer containing 0.1%Tween-20 and 0.01%SDS (e.g., 1:10,000). Primary antibody binding was detected and visualized by using an Odyssey Infrared Imaging System (LI-COR Biosciences) according to the supplier’s instructions. SeeBlue Plus 2 Prestained or HiMark Prestained HMW standards (Invitrogen) were used to calibrate the immunoblots.
Statistical Analysis
Statistical calculations were performed using a GraphPad Prism 4 package (GraphPad Software, La Jolla, CA). Data (mean ± SD) were analyzed using an unpaired t test (P<0.05 was considered a significant difference).
RESULTS
Elevated AMPK-Dependent Phosphorylation of Acetyl-CoA Carboxylase In Experimental Rat Brain Tumors Compared with Surrounding Normal Tissue
AMPK activity is commonly monitored by detecting specific phosphorylation of the substrate acetyl-CoA carboxylase (called ACC hereafter), which collectively defines ubiquitous rate-limiting enzymes for long-chain fatty acid (FA) synthesis (ACC1) and oxidation (ACC2) (1, 21). In particular, AMPK phosphorylates ACC1 on Ser79 and ACC2 on Ser219 in rat (21–23). Previously, we used Wt and AMPK null cells to confirm that immunological detection of ACC1 Ser79 phosphorylation (P-ACC1) with a commercially available antibody provides a surrogate marker for AMPK activity (7). Here we used immunostaining to investigate AMPK-dependent phosphorylation—AMPK activity—in histological sections of brains from rats exposed prenatally to ENU to initiate neurocarcinogenesis. The antibody used for detecting P-ACC1 is considered to recognize the equivalent AMPK phosphorylation site on ACC2; here we designate both phosphorylated forms of ACC as P-ACC. In principle, P-ACC immunostaining represents diverse AMPK-regulated cellular processes that could influence multiple physiological or pathophysiological states (1, 3).
We used the C6 rat glioma cell line, which was originally derived from an N-methyl-N-nitrosourea (MNU)-initiated glioma (24), to investigate P-ACC immunostaining in a common in vivo model of an experimental rat glioma. Figure 1A shows that P-ACC immunostaining in a representative brain section containing a C6 rat glioma was substantially higher than in surrounding normal tissue (parenchyma). Because P-ACC is a specific readout for AMPK activity, this finding indicates that AMPK activity was pervasively induced in an example of an implanted malignant rat glioma. To determine the earliest time at which AMPK activity could be detected during ENU-initiated glioma development, we investigated P-ACC immunostaining in examples of early and fully developed tumors. Figures 1B and 1C show representative brain sections, respectively, from a 140-day-old rat (P140; see [13]) with a fully developed ENU-initiated glioma, and from a 90-day-old rat (P90) with an ENU-initiated microtumor (early glioma). ENU-initiated gliomas are readily distinguished from normal brain parenchyma with conventional histological staining (e.g., hematoxylin); histologically, the tumors are circumscribed and consist mainly of cells having characteristics of astrocytes or oligodendrocytes (12, 25). In the ENU model, the term microtumor is used to describe the smallest pathologic lesions (<300 μm in diameter) in the brains of ENU-exposed rats because serial histological studies have shown that these tumors, which exhibit mitotic cells, appear in the same locations as advanced tumors (macrotumors, >1 mm in diameter) (e.g., see references [12, 26–28]).
The results shown in Figures 1B and 1C demonstrate that both fully developed macrotumors (gliomas) and early microtumors had high levels of P-ACC immunostaining compared with surrounding normal tissue, similar to the immunostaining result for C6 gliomas (Figure 1A). We did not detect significant P-ACC immunostaining in brain sections from ENU-exposed rats between ages P60 and P90 in which there was no evidence of hyperplasia (result not shown). In terms of the distribution of P-ACC immunostaining in tumor beds, P-ACC signals were predominantly, if not entirely, cytoplasmic (Figures 1A and 1B) and present in a heterogeneous pattern of cells throughout areas of microtumors (Figure 1C). The cytoplasmic location of P-ACC agrees with the reported subcellular location of ACC1 and −2 (29). This distribution of P-ACC immunostaining closely resembles the P-ACC immunostaining results reported previously for mouse fibrosarcoma xenografts prepared from H-Ras G12V-transformed MEFs (7). In summary, we found complete concordance between elevated P-ACC immunostaining and the presence of ENU-initiated microtumors and fully developed gliomas—these findings indicate that the induction of AMPK activity is an early event in the neurocarcinogenic process, which coincides with the appearance of a proliferative tumor cell phenotype.
Correlation of Activating Phosphorylation of AMPK with P-ACC Immunostaining In Experimental Rat Brain Tumors
AMP binding to the γ subunit of AMPK promotes activation of the enzyme by enabling or retaining specific phosphorylation within the activation loop of the catalytic domain of the α subunit (Thr172 in rat) by an AMPK kinase (AMPKK) (1, 30). Therefore, immunological detection of AMPKα Thr172 phosphorylation with a specific antibody (P-AMPK; see Materials and Methods) provides a surrogate marker for AMPK activation (30). To determine the relationship between AMPK-dependent phosphorylation (P-ACC immunostaining) and AMPK activation in rat gliomas, we investigated P-AMPK immunostaining (Figure 2) in brain sections from an implanted C6 glioma and from ENU-initiated gliomas, equivalent to the tumor stages used for investigating P-ACC immunostaining. We found that P-AMPK immunostaining in the C6 glioma (Figure 2A) and in examples of macro- (Figure 2B) and microtumors (Figure 2C) in ENU-initiated gliomas resembled the heterogeneous distribution of P-ACC immunostaining shown in Figure 1, but also showed a unique subcellular pattern: specifically, P-AMPK immunostaining showed a punctate pattern in cells in tumor tissue (inserts in Figure 2). Moreover, P-AMPK immunostaining was prominent in proliferating or mitotic cells (Figures 2A and 2B), whereas P-ACC immunostaining was distributed more uniformly among cell populations, as mentioned above.
Recent reports concluding that P-AMPK immunostaining is enhanced in mitotic tumor cells (10, 31) confirm our finding of strong P-AMPK immunostaining in mitotic cells in C6 and ENU-initiated gliomas. One report demonstrated that P-AMPK immunostaining in mitotic cells is particularly intense on two major subcellular structures: centrosomes attached to the spindle (punctate P-AMPK immunostaining) and the cleavage furrow that forms during cytokinesis in telophase (31). Because these structures can be difficult to resolve in histological sections, we performed P-AMPK immunofluorescence with synchronized cultures of C6 glioma cells. In agreement with the observations reported in reference (31), Figure 3 shows that P-AMPK immunostaining was localized to centrosomes (e.g., in metaphase C6 cells; Figure 3B) and the cleavage furrow in anaphase and telophase C6 cells (Figures 3C and 3D). Figure 3E shows relative P-ACC and P-AMPK levels in total protein lysates prepared from asynchronous cultures of normoxic and hypoxic C6 cells: P-ACC levels were constitutive under normoxia, but also inducible by hypoxia (1% O2), which is a physiological stimulus for AMPK activation (7, 32). C6 cells also showed constitutive levels of AMPK phosphorylated in the α subunit activation loop (P-AMPK), which is considered a necessary state for the induction of AMPK activity by stimuli such as metabolic stress (e.g., reviewed in [1, 33]). Overall, the immunoblotting results shown in Figure 3E demonstrate that AMPK activity and activation were constitutive in monolayer cultures of proliferating C6 cells, but that AMPK also retained a normal physiological response to energy stress. Thus, the relatively high levels of AMPK activity found in C6 gliomas (Figures 1A and 2A) could represent both constitutive activity and activity induced in response to energy stress within tumor microenvironments.
Figure 3. Activated AMPK (P-AMPK) associates with centrosomes and the cleavage furrow in mitotic C6 rat glioma cells.
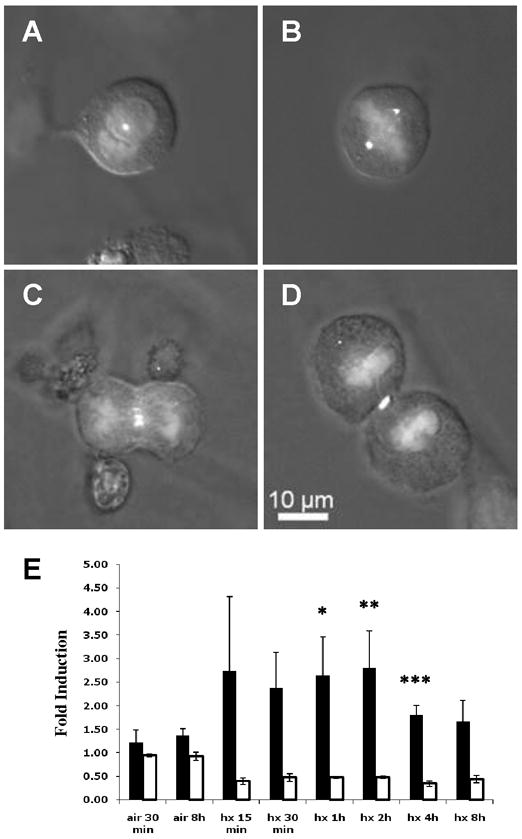
Examples of mitotic C6 cells showing P-AMPK (Cy3; white) and nuclear DNA (DAPI; light grey) immunofluorescence (IF): A prophase; B metaphase; C anaphase; D telophase. A–D, composite IF and DIC (differential image contrast) images; 63x. E Histogram showing relative P-ACC (black) and P-AMPK (white) levels in hypoxic (hx, 1% O2) compared with normoxic (air, 21% O2) C6 cell monolayers. Replicate immunoblots of total cellular protein were probed for the levels of P-ACC, total ACC, P-AMPK, or total AMPK. P-ACC and P-AMPK levels were normalized to the corresponding levels of total ACC and AMPK protein, and these ratios were normalized to that for the normoxic control (air, 30 min; air, 8 h was used to normalize hx, 8 h). Error bars represent standard deviations (n=3). Statistical comparisons were made between normoxic (air, 30 min; air, 8 h was paired with hx, 8 h) and hypoxic cells for each condition shown (only the differences indicated by one or more asterisks are statistically significant). *P=0.04, hx, 1 h;**P=0.03, hx, 2 h; ***P=0.04, hx 4 h.
In summary, in parallel with the relatively high levels of AMPK activity (P-ACC immunostaining) detected in C6 gliomas, there was also substantial AMPK activation in these tumors (P-AMPK immunostaining) at all stages of ENU-initiated gliomagenesis. A proportion of the P-AMPK immunostaining was associated with mitotic tumor cells.
Prominent P-ACC and P-AMPK Immunostaining In the Rat Subventricular Zone and Hippocampus
While investigating P-ACC immunostaining in C6 and ENU-initiated gliomas, we determined whether significant P-ACC immunostaining could be detected in regions of normal rat brain (Figure 4). There are several reports of physiological AMPK activity in rodent brain (e.g., reviewed in [34, 35]), particularly in the hypothalamus, where alteration of intracellular malonyl-CoA levels—the product of ACC activity (21)—contributes to the regulation of feeding behavior (35, 36). In the present study, we detected significant immunostaining of P-ACC and P-AMPK in two areas of adult rodent brain characterized by continued neurogenesis—the subventricular zone (SVZ) and hippocampus (recently reviewed in [37]). Figures 4A and 4C show strong P-ACC immunostaining in the subventricular zone (SVZ) and hippocampus of normal rat brain: just as with subcellular P-ACC immunostaining in tumor tissue, the P-ACC signal in the SVZ and hippocampus is cytoplasmic. Figures 4B and 4D show that P-AMPK immunostaining in the SVZ and hippocampus had a punctate cytoplasmic pattern that resembled that found for P-AMPK immunostaining in the C6 and ENU-initiated gliomas (Figure 2).
Figure 4. P-ACC and P-AMPK immunostaining in the subventricular zone (SVZ) and hippocampus of normal rat brain.

A P-ACC immunostaining in the SVZ of a control rat. LV, lateral ventricle. B P-AMPK immunostaining in the SVZ of a control rat. Punctate cytoplasmic P-AMPK immunostaining (e.g., arrows) is prominent in the image. C P-ACC immunostaining in the hippocampus of a control rat. D P-AMPK immunostaining in the hippocampus of a control rat, showing punctate cytoplasmic immunostaining (e.g., arrows). Sections shown in A–D were counterstained with hematoxylin.
DISCUSSION
The reliable development of gliomas in rats exposed prenatally to ENU offers the unique opportunity to investigate the induction of AMPK activity in a defined model of neurocarcinogenesis. As background, when ENU-exposed rats are examined serially over time there is a characteristic pattern of primary brain tumor development that closely correlates with age (13, 25, 38). Prior work from our and other groups using the ENU model identified areas of early hyperplasia or microtumors (at approximately sixty days of age, P60) as the first abnormality that can be visualized using conventional histological techniques (12, 26–28); these microtumors, which can also be detected using magnetic resonance imaging (38), become fully developed gliomas (13). An important conclusion of the present study is that AMPK activity is elevated at the stage of early microtumors and remains elevated throughout subsequent stages of glioma development in the ENU model. This conclusion is based on our key finding that P-ACC immunostaining—a specific readout for endogenous AMPK activity at the cell or tissue level (7) was prominent in all stages of ENU-initiated tumor development from the microtumor stage (Figure 1). As indicated in the Results, we note that there was no significant P-ACC immunostaining before the microtumor stage—thus, P-ACC immunostaining was not associated with the “nest” stage of early glioma development that is characterized by expression of the neural progenitor protein nestin (13, 25). Finally, we routinely compared AMPK activity (P-ACC) and activation (P-AMPK) in tumor and normal tissue in the ENU model, and observed that both P-ACC and P-AMPK immunostaining were relatively high in neurogenic regions of rat brain (Figure 4).
While the present study does not establish a causal role for AMPK in the development of ENU-initiated gliomas, the physiological function of AMPK as an energy sensor suggests a plausible hypothesis to explain the elevated AMPK activity observed within these tumors. AMPK provides metabolic support by directing cells to generate ATP and thus restore or maintain energy homeostasis when challenged by increased energy demand. Indeed, AMPK has substrates that control diverse energy-intensive cellular processes, including protein synthesis, mitochondrial genesis, control of cell size, and cell-cycle regulation (1, 39–42). From the perspective of cellular energy demand, a straightforward mechanistic explanation for the elevated AMPK activity detected in both C6 and developing ENU-initiated gliomas is the presence of proliferating cells, which consume substantial amounts of ATP as they double in mass during the cell cycle (43). In agreement with this explanation, we found that both AMPK activity (P-ACC) and activation (P-AMPK) were prominent in mitotic cells in tumor beds (Figures 1 and 2). Recent reports that activated AMPK may be considered a mitotic kinase (10, 31) provide confirmation of our findings. In particular, our observations of P-AMPK immunostaining in mitotic centrosomes in C6 cells (Figure 3) and punctate patterns of P-AMPK immunostaining in tumor beds (Figure 2) are consistent with the conclusion that activated AMPK is associated with centrosomes in mitotic human tumor cells (31). Evidence for a centrosomal location for activated AMPK may also explain the punctate pattern of P-AMPK immunostaining found in neurogenic regions of rat brain (Figure 4). It is noteworthy, however, that the SVZ in particular has been reported to contain basal bodies, which have a different function from mitotic centrosomes (44).
The widespread nature of P-ACC immunostaining in tumor tissue and the constitutive and inducible P-ACC levels found in cultured C6 cells (Figure 3), suggest that AMPK’s role during ENU-initiated neurocarcinogenesis is not restricted to regulating ATP levels during proliferation. The physiological model of AMPK activation, in which AMPK functions as a direct sensor for energy homeostasis (1), predicts that AMPK activity is generally induced by ATP-depleting stresses—such as the hypoxic, hypoglycemic, and oxidative conditions common in solid tumors (5, 6). In a developing glioma, the canonical model thus predicts that AMPK would be activated within emergent pathophysiological microenvironments distributed heterogeneously in tumor tissue, as part of a physiological adaptive or pro-survival response to cellular ATP depletion. For example, in energetically stressed tumor cells that are not severely hypoxic, AMPK could generate ATP from long-chain FAs by phosphorylating ACC2 and thus stimulating FA catabolism in mitochondria, while suppressing FA synthesis by phosphorylating ACC1 (1, 45). Figure 5 shows immunoblotting results for AMPK-dependent phosphorylation of ACC2 on Ser219 (P-Ser219) in total protein lysates of C6 and ENU 3.2 cells; ENU3.2 cells were derived from neurospheres prepared from ENU-exposed rats (19). The P-Ser219 antibody appeared to recognize both ACC isoforms (here designated as ACC1/2); this result is expected considering that Ser219 in rat ACC2 (~280 kD) is biochemically equivalent to the Ser79 AMPK phosphorylation site in rat ACC1 (~260 kD kD) (21, 45). Both cell lines showed bands for P-ACC1/2, which indicates that both AMPK substrates were present in at least some of the C6 and ENU-initiated gliomas investigated in the present study. In this context, expression of both ACC1 and −2 has been reported in rat brain (Sprague-Dawley rats; e.g., see reference [46]). In hypoxic tumor cells, inducible AMPK activity could also have a pro-survival role analogous to that proposed for hypoxia-inducible factors (HIFs) during tumorigenesis (e.g., reviewed in reference [47]). Relatively high levels of AMPKα2 expression have been detected in human glioma specimens in pseudopalisading regions, which could be severely hypoxic and hypoglycemic (48). The overall patterns of P-ACC immunostaining detected in ENU-initiated gliomas, as well as in mouse fibrosarcoma xenografts described elsewhere (7), resemble the heterogeneous patterns of hypoxia detected within experimental and clinical tumors (e.g., [49, 50]). It should be emphasized that regions of tumor hypoxia and associated nutrient stress do not appear as gradients extending from peripheral to interior regions of solid tumors. Reports indicating that AMPK is an important regulator of macroautophagy (autophagy) in tumor cells exposed to energy stress (32, 41) are also consistent with a pro-survival role for AMPK activation in solid tumors, assuming that transient autophagy is a physiological mechanism for cell survival (51).
Figure 5. AMPK-dependent phosphorylation of ACC2 on Ser219 (P-Ser219) in C6 rat glioma cells and ENU3.2 neurosphere-derived cells from ENU-exposed rats.
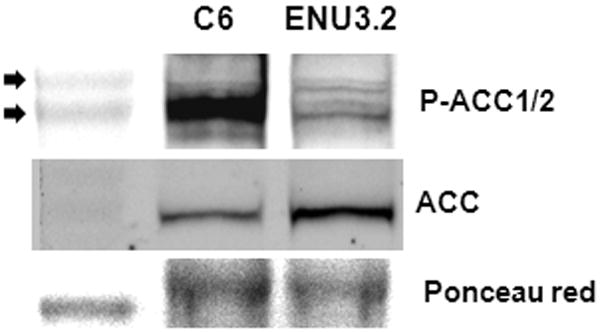
Immunoblots of total cellular protein from C6 and ENU3.2 cells. Replicate blots were probed for the relative levels of the Ser219 AMPK phosphorylation site in rat ACC2, which is biochemically equivalent to the Ser79 AMPK phosphorylation site in rat ACC1 (P-ACC1/2), and for total ACC (see Materials and Methods for details). The replicate P-ACC1/2 immunoblot was stained with Ponceau red as a loading control for the total lysates. Top arrow, 290 kD molecular size standard; bottom arrow, 240 kD molecular size standard.
In summary, the widespread AMPK activity detected in ENU-initiated gliomas—using P-ACC immunostaining as a readout for AMPK activity—could be explained in part by two major pathophysiological mechanisms: AMPK activation in response to microenvironmental energy stress within pathophysiological tumor microenvironments (e.g., hypoxia [5, 6]), and increased energy consumption in proliferating tumor cells. Therefore, AMPK activity in response to energy stress could be elevated within both quiescent or slowly proliferating and proliferating compartments of an ENU-initiated glioma. The evidence presented here that AMPK activity is induced early and remains sustained within developing gliomas contributes to our understanding of the role of AMPK in mammalian tumor development (e.g., reviewed in [40, 52]). In this case, it is difficult to reconcile persistently elevated AMPK activity with mechanistic models in which the activation of AMPK is considered to antagonize tumor development or progression. There is increasing evidence to support the hypothesis that AMPK activity in solid tumors has potential therapeutic value (e.g., [9, 41, 48, 53–56]). It will be important to determine the effect of both activation and inhibition of AMPK on experimental tumor growth, particularly using human tumor models. Future studies using selective inhibitors of AMPK activity (e.g., small molecule protein kinase inhibitors, RNAi reagents) could be used to begin to address this fundamental issue in solid tumor microenvironments.
Acknowledgments
This work was supported by grant CA73807 from the NIH/NCI (KL). To Susan.
References
- 1.Hardie DG. AMP-activated/SNF1 protein kinases: Conserved guardians of cellular energy. Nat Rev Mol Cell Biol. 2007;8:774–785. doi: 10.1038/nrm2249. [DOI] [PubMed] [Google Scholar]
- 2.Carling D, Sanders MJ, Woods A. The regulation of AMP-activated protein kinase by upstream kinases. Int J Obes (Lond) 2008;32 (Suppl 4):S55–59. doi: 10.1038/ijo.2008.124. [DOI] [PubMed] [Google Scholar]
- 3.Lage R, Dieguez C, Vidal-Puig A, Lopez M. AMPK: A metabolic gauge regulating whole-body energy homeostasis. Trends Mol Med. 2008;14:539–549. doi: 10.1016/j.molmed.2008.09.007. [DOI] [PubMed] [Google Scholar]
- 4.Viollet B, Athea Y, Mounier R, Guigas B, Zarrinpashneh E, Horman S, Lantier L, Hebrard S, Devin-Leclerc J, Beauloye C, Foretz M, Andreelli F, et al. AMPK: Lessons from transgenic and knockout animals. Front Biosci. 2009;14:19–44. doi: 10.2741/3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cairns R, Papandreou I, Denko N. Overcoming physiologic barriers to cancer treatment by molecularly targeting the tumor microenvironment. Mol Cancer Res. 2006;4:61–70. doi: 10.1158/1541-7786.MCR-06-0002. [DOI] [PubMed] [Google Scholar]
- 6.Bristow RG, Hill RP. Hypoxia and metabolism. Hypoxia, DNA repair and genetic instability. Nat Rev Cancer. 2008;8:180–192. doi: 10.1038/nrc2344. [DOI] [PubMed] [Google Scholar]
- 7.Laderoute KR, Amin K, Calaoagan JM, Knapp M, Le T, Orduna J, Foretz M, Viollet B. 5′-amp-activated protein kinase (AMPK) is induced by low-oxygen and glucose deprivation conditions found in solid-tumor microenvironments. Mol Cell Biol. 2006;26:5336–5347. doi: 10.1128/MCB.00166-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang FY, Chiu PM, Tam KF, Kwok YK, Lau ET, Tang MH, Ng TY, Liu VW, Cheung AN, Ngan HY. Semi-quantitative fluorescent PCR analysis identifies PRKAA1 on chromosome 5 as a potential candidate cancer gene of cervical cancer. Gynecol Oncol. 2006;103:219–225. doi: 10.1016/j.ygyno.2006.02.028. [DOI] [PubMed] [Google Scholar]
- 9.Park HU, Suy S, Danner M, Dailey V, Zhang Y, Li H, Hyduke DR, Collins BT, Gagnon G, Kallakury B, Kumar D, Brown ML, et al. AMP-activated protein kinase promotes human prostate cancer cell growth and survival. Mol Cancer Ther. 2009;8:733–741. doi: 10.1158/1535-7163.MCT-08-0631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vazquez-Martin A, Lopez-Bonet E, Oliveras-Ferraros C, Perez-Martinez MC, Bernado L, Menendez JA. Mitotic kinase dynamics of the active form of AMPK (phospho-AMPKalphaThr172) in human cancer cells. Cell Cycle. 2009;8:788–791. doi: 10.4161/cc.8.5.7787. [DOI] [PubMed] [Google Scholar]
- 11.Hadad SM, Baker L, Quinlan PR, Robertson KE, Bray SE, Thomson G, Kellock D, Jordan LB, Purdie CA, Hardie DG, Fleming S, Thompson AM. Histological evaluation of AMPK signalling in primary breast cancer. BMC Cancer. 2009;9:307. doi: 10.1186/1471-2407-9-307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zook BC, Simmens SJ, Jones RV. Evaluation of ENU-induced gliomas in rats: Nomenclature, immunochemistry, and malignancy. Toxicol Pathol. 2000;28:193–201. doi: 10.1177/019262330002800124. [DOI] [PubMed] [Google Scholar]
- 13.Jang T, Savarese T, Low HP, Kim S, Vogel H, Lapointe D, Duong T, Litofsky NS, Weimann JM, Ross AH, Recht L. Osteopontin expression in intratumoral astrocytes marks tumor progression in gliomas induced by prenatal exposure to N-ethyl-N-nitrosourea. Am J Pathol. 2006;168:1676–1685. doi: 10.2353/ajpath.2006.050400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jensen RL. Hypoxia in the tumorigenesis of gliomas and as a potential target for therapeutic measures. Neurosurg Focus. 2006;20:E24. doi: 10.3171/foc.2006.20.4.16. [DOI] [PubMed] [Google Scholar]
- 15.Rong Y, Durden DL, Van Meir EG, Brat DJ. 'Pseudopalisading' necrosis in glioblastoma: A familiar morphologic feature that links vascular pathology, hypoxia, and angiogenesis. J Neuropathol Exp Neurol. 2006;65:529–539. doi: 10.1097/00005072-200606000-00001. [DOI] [PubMed] [Google Scholar]
- 16.Wong ET, Brem S. Antiangiogenesis treatment for glioblastoma multiforme: Challenges and opportunities. J Natl Compr Canc Netw. 2008;6:515–522. doi: 10.6004/jnccn.2008.0039. [DOI] [PubMed] [Google Scholar]
- 17.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gao S, Kinzig KP, Aja S, Scott KA, Keung W, Kelly S, Strynadka K, Chohnan S, Smith WW, Tamashiro KL, Ladenheim EE, Ronnett GV, et al. Leptin activates hypothalamic acetyl-CoA carboxylase to inhibit food intake. Proc Natl Acad Sci U S A. 2007;104:17358–17363. doi: 10.1073/pnas.0708385104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Savarese TM, Jang T, Low HP, Salmonsen R, Litofsky NS, Matuasevic Z, Ross AH, Recht LD. Isolation of immortalized, INK4a/ARF-deficient cells from the subventricular zone after in utero N-ethyl-N-nitrosourea exposure. J Neurosurg. 2005;102:98–108. doi: 10.3171/jns.2005.102.1.0098. [DOI] [PubMed] [Google Scholar]
- 20.Lachyankar MB, Ross AH, Litofsky NS, Condon PJ, Quesenberry PJ, Recht LD. TrkA expression decreases the in vivo aggressiveness of C6 glioma cells. Cancer Res. 1997;57:532–536. [PubMed] [Google Scholar]
- 21.Hardie DG, Pan DA. Regulation of fatty acid synthesis and oxidation by the AMP-activated protein kinase. Biochem Soc Trans. 2002;30:1064–1070. doi: 10.1042/bst0301064. [DOI] [PubMed] [Google Scholar]
- 22.Ha J, Daniel S, Broyles SS, Kim KH. Critical phosphorylation sites for acetyl-CoA carboxylase activity. J Biol Chem. 1994;269:22162–22168. [PubMed] [Google Scholar]
- 23.Abu-Elheiga L, Almarza-Ortega DB, Baldini A, Wakil SJ. Human acetyl-CoA carboxylase 2. Molecular cloning, characterization, chromosomal mapping, and evidence for two isoforms. J Biol Chem. 1997;272:10669–10677. doi: 10.1074/jbc.272.16.10669. [DOI] [PubMed] [Google Scholar]
- 24.Benda P, Lightbody J, Sato G, Levine L, Sweet W. Differentiated rat glial cell strain in tissue culture. Science. 1968;161:370–371. doi: 10.1126/science.161.3839.370. [DOI] [PubMed] [Google Scholar]
- 25.Jang T, Litofsky NS, Smith TW, Ross AH, Recht LD. Aberrant nestin expression during ethylnitrosourea-(ENU)-induced neurocarcinogenesis. Neurobiol Dis. 2004;15:544–552. doi: 10.1016/j.nbd.2003.11.016. [DOI] [PubMed] [Google Scholar]
- 26.Lantos PL, Cox DJ. The origin of experimental brain tumours: A sequential study. Experientia. 1976;32:1467–1468. doi: 10.1007/BF01937439. [DOI] [PubMed] [Google Scholar]
- 27.Schiffer D, Giordana MT, Mauro A, Racagni G, Bruno F, Pezzotta S, Paoletti P. Experimental brain tumors by transplacental ENU. Multifactorial study of the latency period. Acta Neuropathol. 1980;49:117–122. doi: 10.1007/BF00690751. [DOI] [PubMed] [Google Scholar]
- 28.Ikeda T, Mashimoto H, Iwasaki K, Shimokawa I, Matsuo T. A sequential ultrastructural and histoautoradiographic study of early neoplastic lesions in ethylnitrosourea-induced rat glioma. Acta Pathol Jpn. 1989;39:487–495. doi: 10.1111/j.1440-1827.1989.tb01514.x. [DOI] [PubMed] [Google Scholar]
- 29.Abu-Elheiga L, Brinkley WR, Zhong L, Chirala SS, Woldegiorgis G, Wakil SJ. The subcellular localization of acetyl-CoA carboxylase 2. Proc Natl Acad Sci U S A. 2000;97:1444–1449. doi: 10.1073/pnas.97.4.1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sanders MJ, Grondin PO, Hegarty BD, Snowden MA, Carling D. Investigating the mechanism for AMP activation of the AMP-activated protein kinase cascade. Biochem J. 2007;403:139–148. doi: 10.1042/BJ20061520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vazquez-Martin A, Oliveras-Ferraros C, Menendez JA. The active form of the metabolic sensor: AMP-activated protein kinase (AMPK) directly binds the mitotic apparatus and travels from centrosomes to the spindle midzone during mitosis and cytokinesis. Cell Cycle. 2009;8:2385–2398. doi: 10.4161/cc.8.15.9082. [DOI] [PubMed] [Google Scholar]
- 32.Papandreou I, Lim AL, Laderoute K, Denko NC. Hypoxia signals autophagy in tumor cells via AMPK activity, independent of HIF-1, BNIP3, and BNIP3L. Cell Death Differ. 2008;15:1572–1581. doi: 10.1038/cdd.2008.84. [DOI] [PubMed] [Google Scholar]
- 33.Oakhill JS, Scott JW, Kemp BE. Structure and function of AMP-activated protein kinase. Acta Physiol (Oxf) 2009;196:3–14. doi: 10.1111/j.1748-1716.2009.01977.x. [DOI] [PubMed] [Google Scholar]
- 34.Ramamurthy S, Ronnett GV. Developing a head for energy sensing: AMP-activated protein kinase as a multifunctional metabolic sensor in the brain. J Physiol. 2006;574:85–93. doi: 10.1113/jphysiol.2006.110122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Minokoshi Y, Shiuchi T, Lee S, Suzuki A, Okamoto S. Role of hypothalamic AMP-kinase in food intake regulation. Nutrition. 2008;24:786–790. doi: 10.1016/j.nut.2008.06.002. [DOI] [PubMed] [Google Scholar]
- 36.Andrews ZB, Liu ZW, Walllingford N, Erion DM, Borok E, Friedman JM, Tschop MH, Shanabrough M, Cline G, Shulman GI, Coppola A, Gao XB, et al. UCP2 mediates ghrelin's action on NPY/AgRP neurons by lowering free radicals. Nature. 2008;454:846–851. doi: 10.1038/nature07181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhao C, Deng W, Gage FH. Mechanisms and functional implications of adult neurogenesis. Cell. 2008;132:645–660. doi: 10.1016/j.cell.2008.01.033. [DOI] [PubMed] [Google Scholar]
- 38.Jang T, Sathy B, Hsu YH, Merchant M, Recht B, Chang C, Recht L. A distinct phenotypic change in gliomas at the time of magnetic resonance imaging detection. J Neurosurg. 2008;108:782–790. doi: 10.3171/JNS/2008/108/4/0782. [DOI] [PubMed] [Google Scholar]
- 39.Richter EA, Ruderman NB. AMPK and the biochemistry of exercise: Implications for human health and disease. Biochem J. 2009;418:261–275. doi: 10.1042/BJ20082055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Motoshima H, Goldstein BJ, Igata M, Araki E. AMPK and cell proliferation--AMPK as a therapeutic target for atherosclerosis and cancer. J Physiol. 2006;574:63–71. doi: 10.1113/jphysiol.2006.108324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liang J, Shao SH, Xu ZX, Hennessy B, Ding Z, Larrea M, Kondo S, Dumont DJ, Gutterman JU, Walker CL, Slingerland JM, Mills GB. The energy sensing LKB1-AMPK pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or apoptosis. Nat Cell Biol. 2007;9:218–224. doi: 10.1038/ncb1537. [DOI] [PubMed] [Google Scholar]
- 42.Okoshi R, Ozaki T, Yamamoto H, Ando K, Koida N, Ono S, Koda T, Kamijo T, Nakagawara A, Kizaki H. Activation of AMP-activated protein kinase induces p53-dependent apoptotic cell death in response to energetic stress. J Biol Chem. 2008;283:3979–3987. doi: 10.1074/jbc.M705232200. [DOI] [PubMed] [Google Scholar]
- 43.DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: Metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7:11–20. doi: 10.1016/j.cmet.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 44.Mirzadeh Z, Merkle FT, Soriano-Navarro M, Garcia-Verdugo JM, Alvarez-Buylla A. Neural stem cells confer unique pinwheel architecture to the ventricular surface in neurogenic regions of the adult brain. Cell Stem Cell. 2008;3:265–278. doi: 10.1016/j.stem.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brownsey RW, Boone AN, Elliott JE, Kulpa JE, Lee WM. Regulation of acetyl-CoA carboxylase. Biochem Soc Trans. 2006;34:223–227. doi: 10.1042/BST20060223. [DOI] [PubMed] [Google Scholar]
- 46.Puglianiello A, Germani D, Antignani S, Tomba GS, Cianfarani S. Changes in the expression of hypothalamic lipid sensing genes in rat model of intrauterine growth retardation (IUGR) Pediatr Res. 2007;61:433–437. doi: 10.1203/pdr.0b013e3180332d4e. [DOI] [PubMed] [Google Scholar]
- 47.Rankin EB, Giaccia AJ. The role of hypoxia-inducible factors in tumorigenesis. Cell Death Differ. 2008;15:678–685. doi: 10.1038/cdd.2008.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Neurath KM, Keough MP, Mikkelsen T, Claffey KP. AMP-dependent protein kinase alpha 2 isoform promotes hypoxia-induced VEGF expression in human glioblastoma. Glia. 2006;53:733–743. doi: 10.1002/glia.20326. [DOI] [PubMed] [Google Scholar]
- 49.Koch CJ. Measurement of absolute oxygen levels in cells and tissues using oxygen sensors and 2-nitroimidazole EF5. Methods Enzymol. 2002;352:3–31. doi: 10.1016/s0076-6879(02)52003-6. [DOI] [PubMed] [Google Scholar]
- 50.Vaupel P, Mayer A. Hypoxia in cancer: Significance and impact on clinical outcome. Cancer Metastasis Rev. 2007;26:225–239. doi: 10.1007/s10555-007-9055-1. [DOI] [PubMed] [Google Scholar]
- 51.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fay JR, Steele V, Crowell JA. Energy homeostasis and cancer prevention: The AMP-activated protein kinase. Cancer Prev Res (Phila Pa) 2009;2:301–309. doi: 10.1158/1940-6207.CAPR-08-0166. [DOI] [PubMed] [Google Scholar]
- 53.Kato K, Ogura T, Kishimoto A, Minegishi Y, Nakajima N, Miyazaki M, Esumi H. Critical roles of AMP-activated protein kinase in constitutive tolerance of cancer cells to nutrient deprivation and tumor formation. Oncogene. 2002;21:6082–6090. doi: 10.1038/sj.onc.1205737. [DOI] [PubMed] [Google Scholar]
- 54.Suzuki A, Kusakai GI, Shimojo Y, Chen J, Ogura T, Kobayashi M, Esumi H. Involvement of TGF-beta1 signaling in hypoxia-induced tolerance to glucose starvation. J Biol Chem. 2005;280:31557–31563. doi: 10.1074/jbc.M503714200. [DOI] [PubMed] [Google Scholar]
- 55.Kim HS, Hwang JT, Yun H, Chi SG, Lee SJ, Kang I, Yoon KS, Choe WJ, Kim SS, Ha J. Inhibition of AMP-activated protein kinase sensitizes cancer cells to cisplatin-induced apoptosis via hyper-induction of p53. J Biol Chem. 2008;283:3731–3742. doi: 10.1074/jbc.M704432200. [DOI] [PubMed] [Google Scholar]
- 56.Natsuizaka M, Ozasa M, Darmanin S, Miyamoto M, Kondo S, Kamada S, Shindoh M, Higashino F, Suhara W, Koide H, Aita K, Nakagawa K, et al. Synergistic up-regulation of hexokinase-2, glucose transporters and angiogenic factors in pancreatic cancer cells by glucose deprivation and hypoxia. Exp Cell Res. 2007;313:3337–3348. doi: 10.1016/j.yexcr.2007.06.013. [DOI] [PubMed] [Google Scholar]