

Abstract
Von Willebrand Factor (vWF) is a plasma protein involved in thrombosis and hemostasis [1]. We examined whether common single nucleotide polymorphisms (SNPs) in the vWF gene were associated with vWF levels and platelet aggregation-related functional consequences in1230 Whites and 837 African Americans in a cross-sectional family based genetic study of platelet function. From a high-density scan, 28 SNPs with a minor allele frequency > 5% in both races were tested for association using age and sex adjusted variance components analysis in MERLIN. SNP rs216321, with the strongest association with vWF levels in biracial metaanalysis (p=9.5×10−6, Whites–p=8.1×10−4, African Americans–p=3.6×10−3), encoding a R852Q substitution in the D’D3 protein domain, demonstrated negative association with plasma vWF. The R852Q variant was recessively associated with 15.5% lower collagen-induced platelet aggregation adjusting for dose-response relationship (p=0.010, vWF-level adjusted p=0.003). Each copy of the R852Q variant was additively associated with 31% higher FVIII levels (p=0.039, vWF-adjusted p=0.033). In conclusion, this common missense polymorphism appears to have pleiotropic functional consequences.
The von Willebrand factor (vWF) is a large protein synthesized primarily in endothelial cells and blood platelets, stored in platelet alpha granules, and secreted into the plasma and subendothelial tissues [1]. vWF has important roles in hemostasis and thrombosis, serving both as a carrier for Factor VIII (FVIII), protecting it from degradation, and promoting platelet adhesion to vascular subendothelium by forming a bridge between platelet membrane glycoproteins (GP) Ib and IIb/IIIa and subendothelial collagen. Quantitative or qualitative reduction in vWF levels may result in bleeding disorders [1].
A number of DNA variants in the vWF gene that are associated with reduced vWF levels (Von Willebrand Disease Types 1 or 3) have been reported [2]. However, most of these known DNA variants are rare, implying that many common DNA variants are not well-known. We therefore examined whether any common polymorphisms included in a high density SNP scan of the vWF gene were associated with vWF levels and known vWF function, namely platelet tethering to collagen and transport of Factor VIII (FVIII).
Demographic characteristics are presented by race in Table 1. While racial differences existed in cardiovascular risk factors, there was no significant difference by race in vWF plasma protein levels.
Table 1.
Sample Characteristics (N= 2067)
| Whites n=1230 | African Americans n=837 | p | |
|---|---|---|---|
| Age, (years) | 42.0 ±11.7 | 41.2 ± 11.1 | 0.15 |
| Male sex (%) | 44.6 | 39.0 | 0.010 |
| vWF levels (percent) | 73 [46,112] | 74 [47,113] | 0.49 |
| BMI (kg/m2)* | 28.6± 6.3 | 31.2 ± 7.6 | <0.001 |
| LDL cholesterol mg/dl* | 125.0 ± 36.5 | 119.0 ± 38.3 | <0.001 |
| HDL cholesterol (mg/dl)* | 51.0 ± 14.8 | 55.1 ± 16.3 | <0.001 |
| Triglycerides (mg/dl)* | 122 [86,175] | 90 [61,132] | <0.001 |
| Fibrinogen mg/dl* | 352 [293,438] | 399 [313,495] | <0.001 |
| Hypertension (%)* | 26.6 | 40.1 | <0.001 |
| Current smoking (%)* | 23.5 | 30.0 | 0.002 |
| Diabetes (%)* | 6.0 | 12.2 | <0.001 |
Mean ± SD with t-test, or median [interquartile range] when rank sum test of the distribution is skewed;
risk factor data available N=1160 whites and 750 African-Americans
The association of log(vWF) levels with the SNPs is presented in Figure 1, Panel A, where the white, African American, and meta-analysis p-values are plotted against chromosomal position. Three SNPs in tight linkage disequilibrium (rs216321, rs216335 and rs216299) are associated with vWF levels above the Bonferroni corrected threshold of 0.0018 in the meta-analysis. Beta coefficients by race are presented in supporting information, Table 1. Of these, only one, rs216321 is a coding missense polymorphism.
Figure 1.

The strength of association of SNPs with log(VWF) positions plotted as log10(p-value) vs. SNP position on the chromosome (open circles: whites, closed circles: African-Americans, line: biracial metaanalysis). The insets below show the linkage disequilibrium plots for the SNPs in whites and African Americans.
Based on the NLM databases [3], the negative strand sequence around the rs216321 variant (by mRNA position) is as follows:
(2789) AAC ACT TGT GTC TGT C[G/A]G GAC CGG AAG TGG AAC (2821)
and the amino acid sequence around the corresponding R852Q variant (by residue number in pre-pro-vWF) is as follows:
(847) N T C V C [R/Q] D R K W N (857)
This variant is in exon 20 of the gene, and the D’-D3 domain of the vWF protein. The percentages of individuals by genotype were: GG 85.3%, GA 14.3%, and AA, 0.5%. The medians and interquartile ranges of vWF levels by genotype were: GG 74.9% [47.8 to 114.2], GA 59.8% [41.4 to 93.2], AA 48.7% [26.1 to 84.7].
In all study participants, we examined the association of collagen-induced platelet function with this functional polymorphism using growth curve models. Accounting for collagen dose-response in individuals, there was no significant difference in the collagen-induced aggregation between those who were heterozygous versus the homozygous wildtype (0.0 ± 1.2%, p=0.96). However, collagen-induced aggregation was 15.5 ± 6.0% (p=0.010) lower in persons who were homozygous for the R852Q polymorphism as compared to those who were homozygous for the major allele. This recessive association remained significant even after adjusting for vWF protein levels (p=0.003).
Figure 2 shows the FVIII levels in the 5 families with at least one individual with the minor allele homozygote, and at least one first-degree relative with a different genotype (N=14). Within each family, the number of R852Q alleles is associated with a 31% higher level of FVIII(p=0.039, vWF-adjusted p=0.033).
Figure 2.
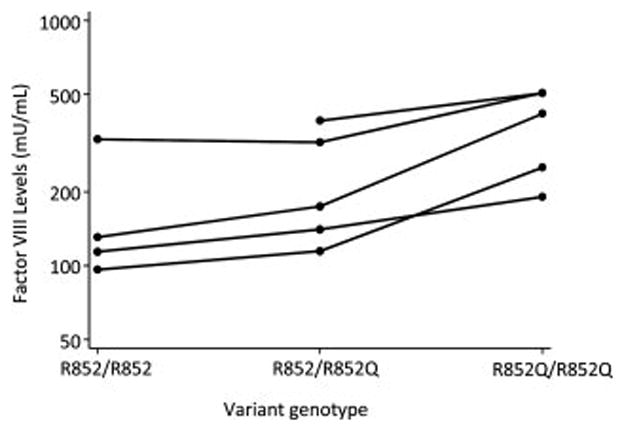
FVIII levels (semi-log scale plot) in the 5 families with at least one individual with the minor allele homozygote, and at least one first-degree relative with a different genotype. Persons of the same family are connected with solid lines.
We have identified a common missense polymorphism that is associated with lower vWF protein levels, and pleiotropic association with the platelet functional aspects of vWF. This polymorphism is in the D′D3 region of the protein and is associated with a R852Q substitution. Interestingly, some rare mutations identified in this same region are associated with a reduced levels of vWF, and targeted deletion of the D′D3 region results in increased clearance of the vWF protein from the blood [4, 5]. A study of the clearance rate of variously truncated vWF protein molecules revealed that molecules lacking the D′D3 domain were more rapidly cleared from the circulation, while those lacking the A1-CK domains were cleared more slowly [6]. Further single amino acid mutations in the D′D3 domain could mimic the rapid clearance phenotype, suggesting that the D′D3 domain could play a regulatory role in shielding vWF from the receptors involved in its cellular uptake, while domains more towards the carboxyl terminal would be more involved with the uptake of the protein by the clearance mechanism [6]. This suggests that the D′D3 region is involved in the recognition of the protein for catabolism. Thus our finding that the R852Q is associated with lower levels of the vWF protein is consistent with the involvement of the D′D3 domain in vWF protein clearance.
The better known function of the D′D3 domain is FVIII binding, and many rare mutations in the D′D3 region have been shown to be associated with altered FVIII levels in the blood [1]. Recent work suggests that the D′D3 domain also shields the A1 domain of the vWF protein [7]. Thus it is possible that changes in the D′D3 domain may affect the interaction of the A1 domain with the platelet GpIb membrane protein.
Consistent with the above two functions or protein interactions of the D′D3 domain, we have found truly pleiotropic functional effects of the R852Q polymorphism with collagen-induced platelet activation, and FVIII levels in the blood. These associations are not epiphenomena related to reduced vWF levels, as they remain significant after adjustment for levels of the protein. Further underlining the pleiotropy is the fact that associations of the R852Q polymorphism with collagen-induced platelet aggregation and FVIII levels follow different genetic models: the association with platelet aggregation is recessive, while the association with FVIII levels is additive. It is reasonable that a missense polymorphism in the D′D3 region, which shields the A1 domain[7], may partially hinder the binding of the A1 domain to bind collagen. We posit that enough platelet tethering to collagen can take place in the heterozygous state, and because platelet activation may proceed through other positive feedback mechanisms, there is no functional change in the heterozygous state. However, in the homozygous R852Q condition, platelet activation due to collagen is reduced.
The association of FVIII with the R852Q polymorphism resulted enhancing of the protein-carrying function. Because the binding of FVIII with the D′D3 domain is stoichiometric, the additive association we found is as expected. It is possible that this polymorphism preserves or enhances the coagulation function due to FVIII carriage, though there is the partial loss of hemostatic function related to collagen-induced activation.
Although the FVIII association is statistically significant, the level of FVIII protein in all genotypes is in the clinically normal range and unlikely to be clinically significant. Rather, these differences elucidate the chemistry of protein interactions around these amino acid residues. Interestingly, a mutation with the substitution of the nearby arginine 854 residue with glutamine has been reported as associated with von Willebrand disease type 2N, i.e., with reduced FVIII [2, 8]. For the variant that we have characterized, replacement of arginine 852 by glutamine is associated with slightly higher FVIII levels, suggesting that this location in the polypeptide chain has a complex association with FVIII binding, not fully explained by the average local balance of acidic and basic amino acid side chains. These residues are located in a region rich with cysteine residues, and very close to an N-linked glycosylation site at N857 [9]. All cysteines in the vWF protein are presumed to take part in disulphide bond formation, because no free sulphydryl groups can be detected in native vWF [10, 11]. We conjecture that a change in the acid-base balance of different residues in this region may result in subtly different local polypeptide chain conformations in this region that is rich in post-translational modifications.
A major strength of our study is the large study population of both whites and African Americans, and the capacity to replicate findings in one race with another. The presence of family structures allow for the determination of small but consistent associations in spite of great interfamily variance that would have overwhelmed the associations in a sample of unrelated individuals, and reduces bias due to population stratification. A limitation of the study is its observational nature, in the sense that it cannot be definitely said whether the polymorphism is directly associated or only linked with a functional polymorphism that was not genotyped. However, it is likely that we have studied the functional polymorphism because there are no large linkage disequilibrium blocks around this locus in the population studied. Furthermore, the demonstration of hypothesized associations with three different phenotypes lends biological plausibility to the functionality of the variant. Another limitation is that we have measured vWF antigen level and collagen-aggregation levels and not ristocetin cofactor levels that more directly measure the platelet adhesion function of vWF. However, this does not change the interpretation of the function association with the amount of vWF protein or with the other measured phenotypes.
Thus, our findings support the discovery of a missense polymorphism R852Q in the D′D3 region that is associated with reduced vWF plasma protein levels. Consistent with the multiple functions of this domain, the polymorphism is associated with lower collagen-induced platelet activation, but enhanced ability to carry FVIII. Future research is needed to determine if this polymorphism and protein region can be a target for developing an anti-hemostatic drug.
Methods
Population and Study Design
This study constituted a part of the Genetic Study of Aspirin Responsiveness (GeneSTAR) and focuses on aspirin-free native platelet and vWF measures. The sample size was planned to have 80% statistical power to detect a locus with heritability of 0.4 as significant at the 10−4 level. The population consisted of 2069 subjects (1230 white, 839 African American) identified from probands with documented CAD events before 60 years of age, including siblings (N=763), adult offspring of patients and siblings ≥ 21 years (N=1097), and the co-parent of the adult offspring (N=209). The probands were identified from Baltimore area hospitals and families were recruited and examinations conducted over the 2003–2006 period. Participants had no clinical CAD, peripheral vascular disease, vascular thrombotic events, bleeding disorders, hemorrhagic events, or autoimmune diseases. Participants were also ineligible if they were taking anticoagulants, antiplatelet agents, or nonsteroidal anti-inflammatory drugs. Dietary supplements (e.g. vitamin E) and foods (coffee, grapes, red wine, chocolate) known to influence platelet function were proscribed for the week preceding platelet measurement.
The trial was approved by the Institutional Review Board of the Johns Hopkins University School of Medicine and was monitored by a Data Safety and Monitoring Board appointed by the National Heart, Lung and Blood Institute. Written informed consent was obtained from all participants.
Covariable Risk Factor Measurements
Age and race were self-reported. Blood pressure was measured at rest and hypertension was considered present if the average of 4 blood pressure measurements was ≥ 140/90 mm Hg and/or the participant was taking an antihypertensive medication. Current cigarette smoking was defined as any self-reported smoking within the past 30 days, verified by exhaled carbon monoxide levels. All blood tests were performed after participants had fasted for 12 hours overnight. Serum glucose, total cholesterol, high-density lipoprotein cholesterol (HDL-C), and triglyceride levels were measured directly. Low-density lipoprotein cholesterol (LDL-C) level was estimated using the Friedewald formula [12]. Diabetes was defined as having a glucose level of ≥ 126 mg/dL and/or use of an antidiabetic agent. Height and weight were measured and body mass index (BMI) was calculated as weight in kilograms divided by height in meters squared.
Platelet Function and vWF Testing
Blood was obtained by venipuncture and collected into tubes containing either EDTA (for blood cell counts) or 3.2% sodium citrate (for platelet function testing and fibrinogen levels). Genotyping, platelet aggregometry, and vWF antigen were measured in all 2069 participants. Optical aggregometry in platelet rich plasma (PRP, 200 000 platelets per microliter) was performed in a 4-channel PAP-4 Aggregometer (Chronolog Corporation, Havertown, PA) after stimulating samples with collagen (0.25, 0.5, 1, 2, 5, 10 and 20 micrograms/mL). Levels of vWF were measured using ELISA at the University of Maryland Cytokine Laboratory.
FVIII was assayed in a subset of participants. This subset included all participants who were minor allele homozygotes for the SNP to be further studied and at least one person of the other genotypes from the same families as the homozygotes (total n=14 from 5 families). Plasma samples from these individuals were sent to the University of Maryland Cytokine Laboratory for FVIII assessment using ELISA. The coefficient of variation for FVIII measurements was 5.3%.
Genotyping
Genotyping of SNPs was performed by the Johns Hopkins Genetics Core using the Illumina BeadArray platform. SNPs within the vWF gene were selected to achieve high density coverage using a modification of the “cherry picker method” [13], described by Herrera-Galeano et al [14]. Briefly, SNPs were prioritized if (a) they tagged haplotypes in the HapMap database (www.hapmap.org), (b) were reported to have higher quality score by the genotyping platform manufacturer Illumina (proprietary), (c) had greater heterozygosity and success rate in the dbGAP database (http://www.ncbi.nlm.nih.gov/projects/SNP/). The chosen SNPs were spaced to achieve as even a density of coverage as possible.
Of the 91 typed SNPs, 28 had minor allele frequencies exceeding 5% in the sample in each of the racial groups, and only these were used for association analysis.
Missing Data
Phenotyping and genotyping data were considered to be missing at random, and for any genotype-phenotype combination, observations with missing data were not used.
Statistical methods
Distributions of all variables were examined by race. Linkage disequilibrium maps were generated using the Haploview software [15], and blocks were identified using criteria suggested by Gabriel et al [16].
Analyses for additive associations of SNPs with log-transformed vWF levels were performed separately in whites and Blacks using the MERLIN software package [17], which performs a maximum likelihood based variance components analysis in familial data.
The p-values thus obtained among African Americans and whites were meta-analyzed using Fisher’s chi-square. Any associations that bypassed the multiple testing threshold (<0.05/28= 0.0018) were examined further for candidacy as functional variants, including the protein sequence. Only candidate functional polymorphisms were further analyzed for platelet and vWF functional associations.
Growth curve models for aggregation due to collagen
Aggregation due to various doses of collagen was modeled as a third degree polynomial growth curve versus log(collagen dose). A multilevel mixed model with the individual growth curves nested within families was estimated. The association of genotypes with collagen aggregation was tested in models adjusting for age, sex, race and log(vWF) levels.
Mixed model analysis of FVIII levels in families
Individuals were selected from families with at least one individual with the minor allele homozygote, and at least one first-degree relative with a different genotype (n=14 from 5 families). The association of FVIII levels with genotype was modeled using family as the random effect, adjusting for age, sex and race, and log(vWF) levels.
Supplementary Material
Acknowledgments
This research was supported by the NIH Grants NR02241, R01 HL49762, R01 HL59684, and the Johns Hopkins University School of Medicine Institute for Clinical and Translational Research, UL1 RR 025005 (National Institutes of Health, Bethesda, MD, USA).
Footnotes
Disclosures: The authors have no conflicts to disclose.
References
- 1.Sadler JE. Biochemistry and genetics of von Willebrand factor. Annu Rev Biochem. 1998;67:395–424. doi: 10.1146/annurev.biochem.67.1.395. [DOI] [PubMed] [Google Scholar]
- 2.ISTH-SSC-VWF-Database. [Accessed September 17, 2009];Sheffield University vWF Homepage. http://www.vwf.group.shef.ac.uk/index.html.
- 3.NCBI-NLM. SNP rs216321. National Library of Medicine; [Accessed September 17, 2009]. http://www.ncbi.nlm.nih.gov/projects/SNP/snp_ref.cgi?rs=216321. [Google Scholar]
- 4.Casonato A, Pontara E, Sartorello F, et al. Reduced von Willebrand factor survival in type icenza von Willebrand disease. Blood. 2002;99:180–4. doi: 10.1182/blood.v99.1.180. [DOI] [PubMed] [Google Scholar]
- 5.Schooten CJ, Tjernberg P, Westein E, et al. Cysteine-mutations in von Willebrand factor associated with increased clearance. J Thromb Haemost. 2005;3:2228–37. doi: 10.1111/j.1538-7836.2005.01571.x. [DOI] [PubMed] [Google Scholar]
- 6.Lenting PJ, Westein E, Terraube V, et al. An experimental model to study the in vivo survival of von Willebrand factor. Basic aspects and application to the R1205H mutation. J Biol Chem. 2004;279:12102–9. doi: 10.1074/jbc.M310436200. [DOI] [PubMed] [Google Scholar]
- 7.Ulrichts H, Udvardy M, Lenting PJ, et al. Shielding of the A1 domain by the D′D3 domains of von Willebrand factor modulates its interaction with platelet glycoprotein Ib-IX-V. J Biol Chem. 2006;281:4699–707. doi: 10.1074/jbc.M513314200. [DOI] [PubMed] [Google Scholar]
- 8.Bowen DJ, Standen GR, Mazurier C, et al. Type 2N von Willebrand disease: rapid genetic diagnosis of G2811A (R854Q), C2696T (R816W), T2701A (H817Q) and G2823T (C858F)--detection of a novel candidate type 2N mutation: C2810T (R854W) Thromb Haemost. 1998;80:32–6. [PubMed] [Google Scholar]
- 9.Purvis AR, Gross J, Dang LT, et al. Two Cys residues essential for von Willebrand factor multimer assembly in the Golgi. Proc Natl Acad Sci U S A. 2007;104:15647–52. doi: 10.1073/pnas.0705175104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Counts RB, Paskell SL, Elgee SK. Disulfide bonds and the quaternary structure of factor VIII/von Willebrand factor. J Clin Invest. 1978;62:702–9. doi: 10.1172/JCI109178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hessel B, Jornvall H, Thorell L, et al. Structure-function relationships of human factor VIII complex studied by thioredoxin dependent disulfide reduction. Thromb Res. 1984;35:637–51. doi: 10.1016/0049-3848(84)90267-6. [DOI] [PubMed] [Google Scholar]
- 12.Friedewald WT, Levy RI, Fredrickson DS. Estimation of the concentration of low-density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clin Chem. 1972;18:499–502. [PubMed] [Google Scholar]
- 13.Harris M, Martin JM, Peden JF, Rawlings CJ. SNP cherry picker: maximizing the chance of finding an association with a disease SNP. Bioinformatics. 2003;19:2141–3. doi: 10.1093/bioinformatics/btg258. [DOI] [PubMed] [Google Scholar]
- 14.Herrera-Galeano JE, Becker DM, Wilson AF, et al. A novel variant in the platelet endothelial aggregation receptor-1 gene is associated with increased platelet aggregability. Arterioscler Thromb Vasc Biol. 2008;28:1484–90. doi: 10.1161/ATVBAHA.108.168971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 16.Gabriel SB, Schaffner SF, Nguyen H, et al. The structure of haplotype blocks in the human genome. Science. 2002;296:2225. doi: 10.1126/science.1069424. [DOI] [PubMed] [Google Scholar]
- 17.Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin--rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet. 2002;30:97–01. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.