

Abstract
The recent investigation of a gene cluster encoding for a hybrid PKS-NRPS metabolite in the oral pathogen Streptococcus mutans UA159 yielded evidence that this natural product might play an important role regulating a range of stress tolerance factors. We have now characterized the major compound generated from this gene cluster, mutanobactin A, and demonstrated that this secondary metabolite is also capable of influencing the yeast-mycelium transition of Candida albicans.
A complex suite of cross-species interactions are anticipated to occur among the wide range of microorganisms that constitute the human microbiome.1–3 While a variety of compounds such as peptides, lipids, and acyl-homoserine lactones have emerged as key players in the multifarious exchanges among microbes and their hosts,4,5 the important contributions of many other families of secondary metabolites have been largely overlooked. We reported that the deletion of a gene cluster encoding for a putative hybrid polyketide synthase-nonribosomal peptide synthetase (PKS-NRPS) derived metabolite in the human oral pathogenic bacteria Streptococcus mutans UA159 resulted in a loss of several resistance traits associated with oxygen and hydrogen peroxide tolerance, as well as biofilm formation.6 Given that a variety of secondary metabolites are excreted into the extracellular environment, we suspected that the biosynthetic products of this gene cluster (which we have dubbed the mutanobactins6) may have additional yet undefined functions related to the interaction of S. mutans with other members of the oral microbiome. This hypothesis recently gained support based on co-culture studies in which mutanobactin-competent S. mutans and a mutanobactin deletion mutant strain (Δmub) were grown in the presence of Candida albicans. Whereas the mutanobactin producing strain of S. mutans was capable of maintaining C. albicans in a perpetual yeast morphological state (Fig. 1A), deletion of the mutanobactin cluster permitted C. albicans to shift into a mycelial growth pattern, which is believed to be the invasive form of the fungus (Fig. 1B). We now describe the distinctive structural features of the major hybrid PKS-NRPS-derived metabolite from S. mutans UA159 and demonstrate that this biomolecule is capable of suppressing the morphological transition of C. albicans from yeast to mycelium.
Fig. 1.
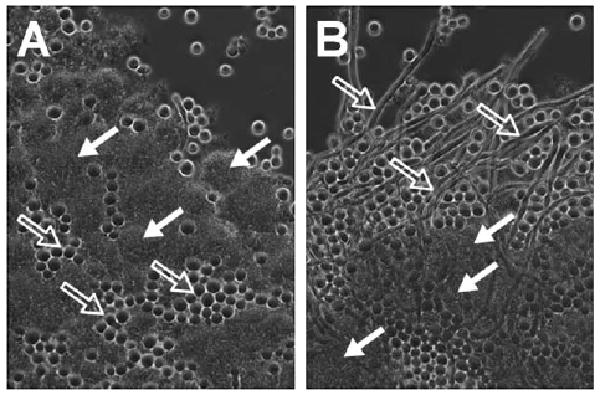
Phase contrast microscopy images showing co-cultures of S. mutans UA159 wild type (A) or Dmub strain (B) with C. albicans ATCC 10231. Solid arrows indicate S. mutans cells, while open arrows denote C. albicans yeast (A) or mycelia (B). S. mutans UA159 strains were grown anaerobically overnight at 37 °C in a semi-defined medium consisting of 50% saliva, 10% artificial saliva solution, 1% glucose, 0.5% sucrose, and 0.2% peptone in water. Cultures were diluted (1 : 20) in fresh medium immediately before testing. C. albicans was prepared separately as overnight cultures under aerobic conditions at 37 °C in yeast extract-peptone medium with 2% glucose. For experiments, aliquots of the C. albicans culture were diluted (1 : 50) in the S. mutans cultures and the mixtures of cells grown anaerobically for 16 h at 37 °C.
Analytical-scale HPLC comparison of the crude extracts generated from wild-type S. mutans UA159 and Δmub strains grown on brain-heart-infusion agar plates enabled us to identify two metabolites in the elution profile that were present only in the wild-type organism. Scale-up fermentation of the wild-type strain was performed in a bioreactor with 15 L of brain-heart-infusion broth under microaerobic conditions at 37 °C to enable the isolation and structure characterization of the major metabolite. After 48 h, the cells and broth were partitioned against ethyl acetate and the solvent removed in vacuo. The resulting extract was resuspended in methanol and the organic soluble material was defatted with hexane. The methanol layer was mixed with an equal volume of water and subjected to partitioning (3×) against dichloromethane. Solvent from the dichloromethane layer was removed under vacuum yielding 1.5 g of a dark brown crude extract. The extract was subjected to preparative-scale C18 flash chromatography under gradient elution conditions (mobile phase: 20% to 100% methanol in water) to generate a single fraction (240 mg) containing the mutanobactins. The major mutanobactin metabolite was targeted for isolation by gradient preparative-scale C18 HPLC (mobile phase: 50% to 100% methanol in water) followed by isocratic semi-preparative C18 HPLC (mobile phase 88% methanol in water), which yielded 8 mg of pure mutanobactin A (1) (Fig. 2 - refer to ESI for a summary of the physical and chemical data for 1).†
Fig. 2.
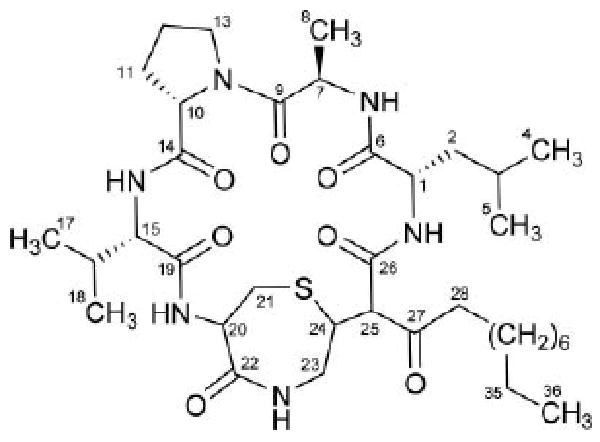
Structure of mutanobactin A (1), the major metabolite generated from the mutanobactin gene cluster in S. mutans UA159.
Inspection of the 1H NMR data collected in DMSO-d6 for 1 (Fig. S3†) confirmed our prior hypothesis6 concerning the partial peptidic nature of the mutanobactins (five exchangeable amide resonances were observed at δH 7.23, 7.77, 7.90, 8.05, and 8.59) (Table S1†). However, we were surprised by the presence of several overlapping methylene resonances centered at δH 1.23 that integrated for 12 protons since our previous inspection of the mutanobactin gene cluster did not provide evidence for this feature.6 Turning to the FT-ICR-MS data generated for 1, we observed a pseudomolecular ion with a m/z of 719.41713 corresponding to a molecular formula of C36H59N6O7S ([M − H]−, calcd 719.41714, −0.01 mmu error) (Figure S1†). These data reinforced our suspicion that in addition to the predicted amino acid residues in 1, the compound also contained a substantial number of non-NRPS-derived atoms.
Considering the disparity between the expected and observed structural data for 1, we set about investigating the metabolite's structural features using a combination of 1H and 13C NMR, 1H–1H COSY, 1H–1H TOCSY, 1H–1H NOESY, 1H–13C HSQC, 1H–13C HMBC, and 1H–15N HMBC experiments (Fig. S3–10†). This approach yielded three distinct substructures (fragments A–C) that formed the backbone of 1 (Fig. 3). We quickly deduced that fragment A consisted of a tetrapeptide (Val-Pro-Ala-Leu) (Fig. 3) based on analysis of two-dimensional NMR correlation data (Fig. S5–10†). We found the 1H–1H TOCSY data were particularly useful for defining the spin systems originating from each of the alpha-protons to the hydrogens embedded in their respective amino acid side chains (Fig. 3). Marfey's analysis7 of the proposed amino acids was carried out by treating 1 with 6 M HCl at 120 °C for 18 h and derivatizing the hydrolysate with 1-fluoro-2,4-dinitrophenyl-5-l-alanineamide. Analytical HPLC analysis of the resultant mixture established the absolute configuration of four amino acids as l-valine (15S), l-proline (10S), d-alanine (7R), and l-leucine (1S). The other hydrolysis fragments generated from 1 were not examined due to their limited sample sizes.
Fig. 3.
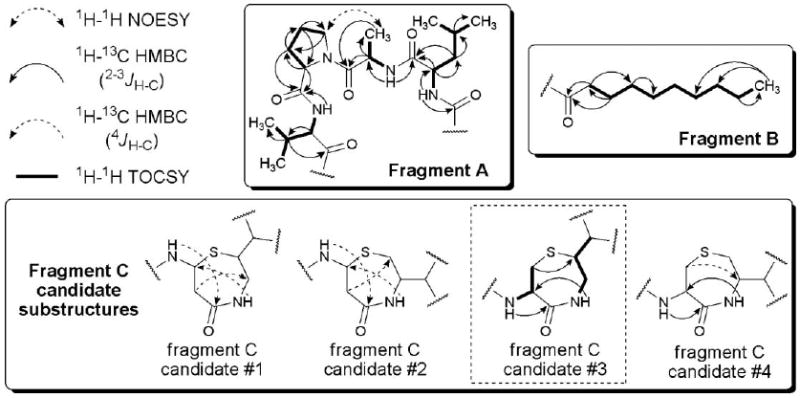
Substructure fragments and selected two-dimensional NMR correlation data used to construct the planar structure of compound 1. Fragment C candidates #1, #2, and #4 were ultimately rejected due to the inclusion of several incongruous 4JH–C couplings, as well as inconsistencies with 1H–1H TOCSY data.
Fragment B was established using a combination of NMR (i.e., 1H–13C HMBC) (Fig. 3) and MS approaches. While many of the methylenes extending from both the ketone (C-27) and methyl (C-36) groups were readily assigned using 1H–13C HMBC correlation data, the significant overlap among a portion of the resonances required us to probe the metabolite using a different analytical method. Turning to ESI-MS/MS, we observed a prominent fragment ion at m/z 155 that was consistent with alpha-cleavage between C-27 and C-25 (Fig. S2†). This fragmentation accounted for a loss of C10H19O, which we interpreted as corresponding to a hydrocarbon chain consisting of eight methylenes and one methyl group.
The remaining atoms constituting fragment C consisted of C6H9N2OS. Considering that this fragment must be covalently bonded with the two unassigned termini of fragment A and the single unassigned terminus of fragment B, we concluded that fragment C required two units of unsaturation. One unit of unsaturation was assigned as an amide carbonyl based on the appearance of a distinctive 13C NMR resonance at δC 170.4. The second unit of unsaturation was attributed to a ring since no additional double bonds could be accounted for in the 1H and 13C NMR data. The remaining non-hydrogen atoms in fragment C were rationalized to constitute a thiazepanone system, but five variations of this system could be envisioned. Fortunately, we could immediately eliminate 1,3-thiazepan-2-one and 1,2-thiazepan-3-one as possibilities since their predicted chemical shifts and/or questionable chemical stability were inconsistent with our data. In addition, 1,4-thiazepan-3-one and 1,3-thiazepan-4-one were also rejected since both ring systems necessitated the inclusion of isolated methylenes, which were incompatible with the observed 1H–1H TOCSY data. Therefore, we deduced that fragment C was based on a 1,4-thiazepan-5-one ring system.
Noting that H-25 and C-20-NH were part of two separate spin systems (based on1H–1H TOCSY data), we restricted our investigation of fragment C to consider four possible substructure candidates (Fig. 3). Analysis of the 1H–13C HMBC (optimized for JH–C = 8 Hz) revealed that candidates #1 and #2 were unlikely possibilities since both of these substructures required at least three H–C couplings across four bonds. Similarly, candidate #4 also required at least one H–C coupling across four bonds and it too was removed from further consideration. In contrast, candidate #3 was strongly supported by the observed 1H–13C HMBC data with all of the H–C couplings occurring within the target range of two to three bonds. Therefore, this fragment candidate was selected as the only plausible planar substructure for 1. Unfortunately, extensive overlap among several critical 1H NMR resonances within this fragment resulted in the generation of inconclusive 1H–1H NOESY data, which prevented us from confidently assigning the relative configuration of fragment C. We are currently exploring whether this problem may be exacerbated by C-25 epimerization occurring as a consequence of tautomerization involving the C-27 ketone and C-26 amide carbonyl groups.
In light of the previously noted 1H–13C HMBC correlations joining fragment C to fragments A and B (vide supra), we propose that 1 (Fig. 2) represents the structure of the major mutanobactin produced by S. mutans UA159. Further consideration of the mutanobactin biosynthetic gene cluster revealed that although most of the component building blocks predicted to appear in 1 are present, many unexpected features are also incorporated. One of the surprising components is the integration of an extended hydrocarbon chain. We speculate that regions of high homology to fatty-acid biosynthesis genes in the vicinity of the mutanobactin gene cluster may be involved in the generation of this structural component. In addition, we had predicted that the mutanobactins would contain an amino acid sequence consisting of Asp, Leu, Ser, Pro, Val, Cys, and Gly. Instead, we observed Leu, Ala, Pro, Val, Cys, and Gly (the latter two amino acids residues appear to have undergone further cyclization to generate the 1,4-thiazepan-5-one ring system). At this time, the fate of the Asp residue is unclear; however, we predict that it may have contributed to the generation of the C-26 amide carbonyl contained in the mutanobactin macrocycle.
Having already observed that a functional mutanobactin gene cluster was correlated with the ability of S. mutans to control the morphological transition of C. albicans from yeast to mycelium under co-culture conditions (Fig. 1), we next addressed whether purified 1 was capable of eliciting the same biological effect. Whereas control cultures of C. albicans readily formed tangled mats of intertwined mycelia in vitro (Fig. 4A), cells treated with 1 exhibited a remarkable preservation of yeast morphological characteristics (Fig. 4B), but did not show signs of decreased cell division. Based on our data, compound 1 joins a select group of two other S. mutans-derived biomolecules (i.e., competence-stimulating peptide8 and trans-2-decenoic acid9) that are capable of re-directing the yeast-mycelium dimorphism of C. albicans.
Fig. 4.
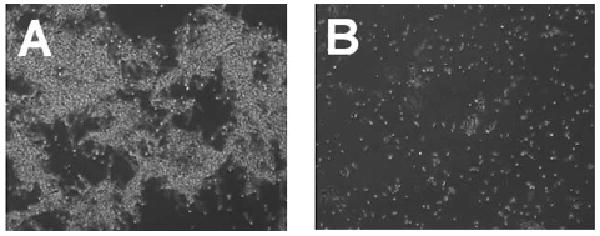
Phase contrast microscopy images illustrating the morphology of C. albicans ATCC 10231 cultures grown with vehicle only (A) or treated with compound 1 (B). Whereas the mycelia morphology of the control C. albicans cells appeared as mats of tangled filaments, mycelia were not apparent in cells treated with 1. For the experiment, overnight shake cultures of C. albicans were prepared under aerobic conditions in yeast extract-peptone medium with 2% glucose. The cells were diluted in fresh medium (1 : 100) and 100 μL aliquots added to the wells of a flat-bottom 96-well plate. Cells were treated with DMSO only (vehicle control) or compound 1 (2 μL of compound prepared in DMSO at 5 mg mL−1).
The human microbiome is certain to yield scores of chemically unique and biologically intriguing secondary metabolites. For example, it was recently reported that Staphylococcus aureus biosynthesizes the NRPS-derived metabolites phevalin and au-reusimine A as part of a virulence regulatory pathway.10 As new research tools emerge that further enhance the natural product research community's ability to deeply probe microbial secondary metabolite diversity,11,12 we expect that many new and complex intra- and inter-species relationships will come to light.13–15
Conclusion
The discovery of 1 is noteworthy since a very limited number of other signaling molecules are known to non-lethally influence the interactions between bacteria and fungi despite the expectation that these types of chemical exchanges are quite prevalent.16,17 Whereas the ability to generate compound 1 has been shown to be an important stress-resistance factor in S. mutans,6 we have now demonstrated that the mutanobactin metabolite is also capable of regulating cross-kingdom interactions with C. albicans. Furthermore, our data indicate that 1, which contains a structurally rare 1,4-thiazepan-5-one ring system, represents a new and unusual addition to the emerging list of natural products biosynthesized by microbial species that are part of the human microbiome.
Supplementary Material
Acknowledgments
Financial support for this project was provided in part through a grant from the National Institutes of Health (1R01AI085161-01) and funds from the University of Oklahoma College of Arts and Sciences and the Department of Chemistry and Biochemistry.
Footnotes
Electronic supplementary information (ESI) available: General methods, physical data, MS and NMR (1H, 13C, 1H–1H COSY, 1H–1H TOCSY, 1H–1H NOESY, 1H–13C HSQC, 1H–13C HMBC, and 1H–15N HMBC) for 1. See DOI: 10.1039/c0ob00579g
References
- 1.Peleg AY, Hogan DA, Mylonakis E. Nat Rev Microbiol. 2010;8:340–349. doi: 10.1038/nrmicro2313. [DOI] [PubMed] [Google Scholar]
- 2.Hughes DT, Terekhova DA, Liou L, Hovde CJ, Sahl JW, Patankar AV, Gonzalez JE, Edrington TS, Rasko DA, Sperandio V. Proc Natl Acad Sci U S A. 2010;107:9831–9836. doi: 10.1073/pnas.1002551107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Antunes LCM, Ferreira RBR. Crit Rev Microbiol. 2009;35:69–80. doi: 10.1080/10408410902733946. [DOI] [PubMed] [Google Scholar]
- 4.Pacheco AR, Sperandio V. Curr Opin Microbiol. 2009;12:192–198. doi: 10.1016/j.mib.2009.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hsiao WWL, Metz C, Singh DP, Roth J. Endocrinol Metab Clin North Am. 2008;37:857–871. doi: 10.1016/j.ecl.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu C, Cichewicz R, Li Y, Liu J, Roe B, Ferretti J, Merritt J, Qi F. Appl Environ Microbiol. 2010;76:5815–5826. doi: 10.1128/AEM.03079-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bhushan R, Brückner H. Amino Acids. 2004;27:231–247. doi: 10.1007/s00726-004-0118-0. [DOI] [PubMed] [Google Scholar]
- 8.Jarosz LM, Deng DM, Van Der Mei HC, Crielaard W, Krom BP. Eukaryotic Cell. 2009;8:1658–1664. doi: 10.1128/EC.00070-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vílchez R, Lemme A, Ballhausen B, Thiel V, Schulz S, Jansen R, Sztajer H, Wagner-Döbler I. ChemBioChem. 2010;11:1552–1562. doi: 10.1002/cbic.201000086. [DOI] [PubMed] [Google Scholar]
- 10.Wyatt MA, Wang W, Roux CM, Beasley FC, Heinrichs DE, Dunman PM, Magarvey NA. Science. 2010;329:294–296. doi: 10.1126/science.1188888. [DOI] [PubMed] [Google Scholar]
- 11.Cichewicz RH, Henrikson JC, Wang X, Branscum KM. In: Manual of Industrial Microbiology and Biotechnology. 3rd. Baltz RH, Demain AL, Davies JE, editors. ch. 7. ASM Press; Washington, D. C.: 2010. pp. 78–95. [Google Scholar]
- 12.Zerikly M, Challis GL. ChemBioChem. 2009;10:625–633. doi: 10.1002/cbic.200800389. [DOI] [PubMed] [Google Scholar]
- 13.Yang YL, Xu Y, Straight P, Dorrestein PC. Nat Chem Biol. 2009;5:885–887. doi: 10.1038/nchembio.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schroeckh V, Scherlach K, Nützmann HW, Shelest E, Schmidt-Heck W, Schuemann J, Martin K, Hertweck C, Brakhage AA. Proc Natl Acad Sci U S A. 2009;106:14558–14563. doi: 10.1073/pnas.0901870106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Partida-Martinez LP, Monajembashi S, Greulich KO, Hertweck C. Curr Biol. 2007;17:773–777. doi: 10.1016/j.cub.2007.03.039. [DOI] [PubMed] [Google Scholar]
- 16.Piel J. Nat Prod Rep. 2009;26:338–362. doi: 10.1039/b703499g. [DOI] [PubMed] [Google Scholar]
- 17.Tarkka M, Sarniguet A, Frey-Klett P. Curr Genet. 2009;55:233–243. doi: 10.1007/s00294-009-0241-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.