

Abstract
Interaction of SIRPα with its ligand, CD47, regulates leukocyte functions, including transmigration, phagocytosis, oxidative burst, and cytokine secretion. Recent progress has provided significant insights into the structural details of the distal IgV domain (D1) of SIRPα. However, the structural roles of proximal IgC domains (D2 and D3) have been largely unstudied. The high degree of conservation of D2 and D3 among members of the SIRP family as well as the propensity of known IgC domains to assemble in cis has led others to hypothesize that SIRPα forms higher order structures on the cell surface. Here we report that SIRPα forms noncovalently linked cis homodimers. Treatment of SIRPα-expressing cells with a membrane-impermeable cross-linker resulted in the formation of SDS-stable SIRPα dimers and oligomers. Biochemical analyses of soluble recombinant extracellular regions of SIRPα, including domain truncation mutants, revealed that each of the three extracellular immunoglobulin loops of SIRPα formed dimers in solution. Co-immunoprecipitation experiments using cells transfected with different affinity-tagged SIRPα molecules revealed that SIRPα forms cis dimers. Interestingly, in cells treated with tunicamycin, SIRPα dimerization but not CD47 binding was inhibited, suggesting that a SIRPα dimer is probably bivalent. Last, we demonstrate robust dimerization of SIRPa in adherent, stimulated human neutrophils. Collectively, these data are consistent with SIRPα being expressed on the cell surface as a functional cis-linked dimer.
Keywords: Cell-Cell Interaction, Ligand-binding Protein, Neutrophil, Protein-Protein Interactions, Site-directed Mutagenesis, CD47, IgC, SIRP, Homodimer
Introduction
Signal regulatory proteins (SIRPs)2 are immunoglobulin superfamily members and include SIRPα, SIRPβ, and SIRPγ (1, 2). Expression of SIRPs is largely restricted to leukocytes, where SIRPα and SIRPβ are expressed in myeloid lineages, and SIRPγ is expressed in lymphocytes (1, 2). Interestingly, SIRPα is also expressed in smooth muscle cells and neurons and has important signaling properties (3–5). The extracellular domains of SIRPs consist of a membrane-distal Ig variable-like (IgV) fold (D1), and two membrane-proximal Ig constant-like (IgC) folds (D2D3). The ectodomains of the various SIRP family members share a high degree of homology between their respective protein sequences. In contrast, the cytoplasmic signaling elements of different SIRPs differ greatly. For instance, the cytoplasmic tail of SIRPα contains four immunoreceptor tyrosine-based inhibitory motifs, which recruit Src homology 2 phosphatases upon phosphorylation (6). In contrast, SIRPβ and SIRPγ have short cytoplasmic tails that are only a few amino acids long. SIRPβ associates with the immunoreceptor tyrosine-based activating motif containing adaptor protein DAP12 through charge interactions in the transmembrane regions (7, 8). On the other hand, SIRPγ, lacking known signaling motifs, has been presumed to be a decoy receptor (9). However, a recent report suggests that SIRPγ on T cells may be able to transmit signals that can regulate transendothelial migration (10). The major cellular ligand of SIRPα and SIRPγ is CD47, a ubiquitously expressed transmembrane immunoglobulin superfamily protein, whereas the ligand of SIRPβ remains unknown (1, 2). CD47 consists of an extracellular IgV fold, a pentamembrane-spanning transmembrane domain, and a cytoplasmic tail (11). Binding interactions between SIRPα and CD47 have been implicated in regulating many aspects of leukocyte function.
SIRPα has emerged as an important inhibitory receptor that regulates a number of leukocyte functions. For example, SIRPα-CD47 interactions have been shown to regulate neutrophil migration across epithelial monolayers and monocyte migration across the blood brain barrier (12–14). These in vitro observations are consistent with in vivo experiments with CD47-deficient mice. In these experiments, CD47-deficient mice succumbed to acute bacterial peritonitis due to a delay in leukocyte recruitment (15). In other reports, activation of SIRPα has been shown to negatively regulate Toll-like-receptor signaling, inflammatory cytokine secretion, and leukocyte respiratory burst (16–19). SIRPα has also been shown to play a role in the regulation of phagocytosis in macrophages (20, 21). Specifically, activation of macrophage SIRPα results in inhibition of phagocytosis (20, 22–24). Recently, it was reported that the inhibition of phagocytosis by tissue-resident macrophages through SIRPα-CD47 interactions is essential for successful engraftment of hematopoietic stem cells (16, 25).
Given the multitude of functions attributed to SIRPα activation, understanding the molecular details of this interaction has become an important topic that might provide insights into novel therapeutic approaches. It has been shown that binding of SIRPα to CD47 is mediated through the membrane-distal IgV domain (D1) of SIRPα and the IgV domain of CD47. Recently, a series of mutagenesis studies using soluble, recombinant proteins and comparative analyses of the crystal structures of SIRPαD1, CD47, and SIRPαD1-CD47 complexes have provided important insights into the molecular mechanisms of the binding interaction between SIRPαD1 and CD47 (16, 26–29). However, little is known about the contributions of the membrane-proximal immunoglobulin domains of SIRPα. Previously published analyses of protein sequences indicated that proximal domains contained IgC motifs and are highly homologous among members of the SIRP family (6, 27–30). Given that many known IgC domains associate with other IgC domains, others have speculated that SIRPα may form dimers and higher order structures. Interestingly, we have previously shown that SIRPβ is present on the cell surface as a cis homodimer that is covalently linked through a disulfide bond on the most membrane-proximal domain (D3) (31). Although SIRPα lacks this particular cysteine, the marked similarities in protein sequences as well as the propensity for IgC domains to dimerize led us to hypothesize that SIPRα may form noncovalently linked cis homodimers on the cell surface.
Using full-length or truncated SIRPα proteins expressed on the cell surface and in solution, along with the use of a chemical cross-linker, we explored potential contributions of each of the three Ig folds in dimerization of SIRPα. Immunoprecipitation analyses of differentially tagged SIRPα molecules indicated that SIRPα dimerizes in cis. Furthermore, we report that dimerization of SIRPα is enhanced in stimulated adherent human PMN. These results have important implications in the understanding of SIRPα interactions with its ligands.
EXPERIMENTAL PROCEDURES
Antibodies
Rabbit polyclonal antibodies against the cytoplasmic tail of SIRPα (Prosci) and against green fluorescent protein (GFP) (Abcam) are commercially available. Rabbit polyclonal antiserum against recombinant SIRPα ectodomain as well as murine monoclonal antibodies against SIRPαD1 (SAF17.2) or SIRPαD3 (SAF4.2) were generated as described previously (28). A hybridoma that secretes a murine monoclonal antibody against Myc (9E10) was obtained from ATCC.
Cell Lines and Tissue Culture
A human promyelocytic leukemia cell line (HL60), human embryonic kidney cell line with T antigen (HEK293T), and wild type Chinese hamster ovary cell line (CHO) were purchased from ATCC. Cell culture and transfections of CHO and HEK293T cells using Lipofectamine (Invitrogen) were performed as described previously (28). Expression was assessed by immunoblotting as described previously (28) or flow cytometry as described below.
Chemical Cross-linking of SIRPα on the Cell Surface and in Solution
After washing with cold Hanks' balanced salt solution containing calcium and magnesium (HBSS+) (Sigma), confluent cultures of cells were cross-linked with 1 mm bis(sulfosuccinimidyl) suberate (BS3) (Pierce) in HBSS+ at 4 °C for 30 min. After cross-linking, cells were first washed with cold HBSS+, followed by quenching residual cross-linker with 40 mm Tris-HCl in HBSS+ at pH 8 for 20 min (4 °C) and washed again with HBSS+. The same procedure was used to cross-link cells in suspension. Purified soluble SIRPα proteins were cross-linked in the presence of 10 μm BS3 in PBS for 30 min (20 °C). The cross-linking reaction was quenched with 40 mm Tris-HCl at pH 8 (20 °C). Immunoblotting was performed as described previously (28).
Flow Cytometry
Cell suspensions were prepared from adherent cultures by trypsinization (trypsin/EDTA) (Sigma). After washing, cells were resuspended at 106/ml in serum-containing medium. Approximately 2 × 105 cells/staining condition were incubated with 5 μg/ml primary antibody for 1 h at 4 °C followed by washing and staining with 1 μg/ml secondary antibody conjugated with Alexa 488 dye (Invitrogen). After a final wash, cells were analyzed using a FACSCalibur (BD Biosciences) and FlowJo software (Treestar).
Generation of Full-length and Truncated SIRP Plasmid Constructs
Constructs encoding SIRPα with deletions of specific Ig folds were generated as described previously (28). Specifically, the following wild type (WT) or truncated SIRPα constructs containing an intact ctyoplasmic tail and transmembrane domain were produced: SIRPαD1-Myc6, SIRPαD1D2, and SIRPαD2D3. Constructs containing WT or truncated soluble SIRPα ectodomains tagged with histidines were generated: solWTSIRPαHis, solSIRPαD1His, and solSIRPαD2D3His. In addition, constructs containing full-length WT SIRPα tagged with GFP or Myc6 were produced. Finally, CD47 cDNA was cloned into pcDNA3.1 containing a hygromycin resistance cassette (Invitrogen). All constructs were verified by sequencing.
Gel Filtration Chromatography and Matrix-assisted Laser Desorption/Ionization Time-of-flight (MALDI-TOF) Analyses
Purified protein samples were applied to a Sephacryl S100 (GE Healthcare) column on a Bio-Rad DuoFlo chromatography system. Gel filtration was performed at a flow rate of 1 ml/min using a buffer containing 20 mm Tris and 150 mm NaCl at pH 8. Protein standards consisting of blue dextran (3000 kDa), conalbumin (75 kDa), ovalbumin (45 kDa), chymotrypsinogen (25 kDa), and cytochrome c (12 kDa) were used. The logarithmic correlation of the elution peak volumes of the standard proteins and the molecular weights were calculated using MS Excel. The apparent molecular weights of the protein samples according to their peak elution volumes were calculated from the formula obtained, Mr = e([protein elusion volume] − 538.43)/−37.497 (R2 = 0.984). MALDI-TOF analyses of purified proteins were performed as described previously (32).
Inhibition of N-Linked Glycosylation by Tunicamycin and Kifunensine
To inhibit formation of N-linked glycans, confluent CHO cells were incubated with fresh medium containing 5 μg/ml tunicamycin (Calbiochem) for 18 h. To arrest maturation of N-linked glycans, subconfluent CHO cells were treated with fresh medium supplemented with 10 μg/ml kifunensine (Toronto Research). Cells were grown to confluence and subjected to cross-linking analyses and flow cytometric analyses using phytohemagglutinin type L conjugated with FITC (PHA-L-FITC; Vector Laboratories) to assess the reduction in N-linked glycosylation.
Binding Analysis of Soluble CD47 to SIRPα on the Cell Surface
Production and purification of soluble CD47 ectodomain fused with alkaline phosphatase (CD47AP) was as described previously (28, 33). Purified CD47AP was used to determine the concentrations of CD47AP in the cell culture supernatants. A series of dilutions of the CD47AP production supernatants was added to 96-well plates containing cultures of confluent CHO cells expressing SIRPα at 4 °C for 1 h. After washing with cold HBSS+, binding of CD47AP to CHO-SIRPα transfectants was assessed colorimetrically as described previously (28).
Stimulation of Human Blood PMNs
Human PMNs were freshly prepared using a dextran sedimentation method as described previously (34). After isolation, PMNs were resuspended in cold HBSS buffer free of calcium and magnesium (Sigma) at a density of 5 × 107/ml. Four million PMNs per well of HBSS+ were allowed to settle and adhere to the wells of 6-well tissue culture plates (Costar) at 37 °C for 30 min. The cells were then stimulated with 1 μg/ml LPS (Sigma) or 10−6 m fMLP (Sigma) for 20 min at 37 °C. To stimulate PMNs in suspension, 4 × 106 PMNs in 1 ml of HBSS+ were activated with 1 μg/ml LPS or 10−6 m fMLP for 20 min at 37 °C, followed by cross-linking and analysis as described above.
RESULTS
SIRPα Forms Noncovalently Linked Dimers on the Cell Surface
We have previously reported that SIRPβ but not SIRPα forms a covalent homodimer in cells through a disulfide bond at the membrane-proximal Ig domain (D3) (31). Because the ectodomains of SIRPα and SIRPβ exhibit a very high degree of homology in protein sequences (Fig. 1A) and the general belief that monomeric partners of a dimer must pair noncovalently in a thermodynamically favorable fashion in order to facilitate the formation of an intermolecular disulfide bond (35, 36), we hypothesized that SIRPα may form a noncovalently linked dimer on the cell surface. To address this possibility, we analyzed cell lysates of HL-60, a human promyelocytic leukemia cell line that is known to express SIRPα and SIRPβ (31, 37), by immunoblot for the presence of SIRPα dimers by SDS-PAGE. Under non-reducing conditions, covalent SIRPβ dimers but not SIRPα dimers were detected. Thus, if SIRPα forms dimers, they are probably non-covalently linked and unstable in the presence of SDS (Fig. 1B). To stabilize potential non-covalently linked SIRPα dimers, a membrane-non-permeable biochemical cross-linker, BS3, was used to covalently cross-link SIRPα on the surface of HL-60 cells. Subsequent immunoblot analyses under reducing conditions using a polyclonal anti-SIRPα antibody revealed three species of SIRPα protein complexes that were consistent with glycosylated monomers at molecular weights of 75,000–110,000, dimers at 220,000, and oligomers above 250,000 (Fig. 1C). Taken together, these results suggested that SIRPα may form noncovalently linked dimers at the cell surface.
FIGURE 1.

SIRPα is highly similar to SIRPβ and dimerizes after chemical cross-linking in HL60 cells. A, alignment of SIRPα and SIRPβ ectodomains revealing identical (stars) and conserved (dots) residues. The peptide sequences that represent each of the Ig folds as defined by Protein Family databases (PFAM) and the crystallization study are enclosed in gray boxes. The cysteine and tryptophan residues conserved in Ig folds are in boldface type. SIRPβ dimerization is mediated through a disulfide linkage at cysteine residue Cys299 (circled) as reported previously (31). B, immunoblots of HL-60 cell lysates under non-reduced (NR) and reduced (R) conditions with boiling (B) or without boiling (NB) were probed using rabbit antiserum against SIRPα cytoplasmic tail (Anti-SIRPαCT) and murine monoclonal antibody specific against SIRPβ (B4B6). Anti-SIRPβ antibody was unable to detect SIRPβ under reducing conditions (data not shown). C, immunoblot analysis using a rabbit polyclonal antibody against the SIRPα cytoplasmic tail detecting noncovalently linked SIRPα dimers after treatment of HL60 cells with a cell membrane-impermeable chemical cross-linker BS3. Results are representative of one of five independent experiments.
CD47 Is Not a Component of Cross-linked SIRPα Complexes
Because HL60 cells express CD47 (data not shown), it was important to determine whether the cross-linked SIRPα complexes contain CD47. Our anti-CD47 antibodies on hand did not detect denatured CD47 and inhibit binding to SIRPα (data not shown). Therefore, it was impossible to use these antibodies to assess the presence of CD47 in the cross-linked complexes. To address this issue, we used a recombinant protein expression system in CHO cells. Although CHO cells express CD47, it has been previously shown that hamster CD47 does not interact with human SIRPα (38). CHO cells stably expressing functional human SIRPα (CHO-SIRPα), CD47 (CHO-CD47), or both molecules (CHO-SIRPα/CD47) were generated. For CD47 to be a component of cross-linked complexes demonstrated in Fig. 1C, cross-linking CHO-SIRPα would yield a complex of a different molecular weight in the presence of CD47. However, immunoblot analyses under reducing conditions revealed no difference in mobility of the cross-linked species in CHO-SIRPα, CHO-SIRPα/CD47 double transfectants, and CHO-SIRPα cells co-cultured with CHO-CD47 (Fig. 2). High expression levels of both SIRPα and CD47 molecules in single and double transfectants were verified by flow cytometry (data not shown). Furthermore, sequence analyses of the protein bands corresponding to cross-linked complexes revealed the presence of SIRPα protein only (data not shown). Therefore, CD47 is not a component of the cross-linked SIRPα complex.
FIGURE 2.

The cross-linked SIRPα complex does not contain CD47. Shown is an immunoblot analysis of the stable CHO cell transfectants demonstrating that the apparent molecular weight of the cross-linked SIRPα complex does not change in the presence of human CD47. Results are representative of one of five independent experiments.
Soluble Ectodomains of SIRPα Spontaneously Dimerize in Solution
To determine whether SIRPα dimerizes through its extracellular domains, purified histidine-tagged soluble SIRPα containing all three extracellular Ig domains SIRPαD1D2D3His (predicted molecular weight of ∼40,000) were expressed in HEK293T followed by purification and analyzed using gel filtration chromatography on an S100 column. Interestingly, the majority of SIRPαD1D2D3His eluted at a peak volume corresponding to an apparent molecular weight of 70,000, almost twice the predicted molecular weight (Fig. 3A). The eluted sample was further analyzed by MALDI-TOF, and the molecular weight was determined to be 40,000 (Fig. 3B). As revealed by immunoblot analysis with anti-SIRPα mAb SAF17.2, non-cross-linked SIRPαD1D2D3His had a broad apparent molecular weight in the range of 37,000–70,000, typical of glycosylation effects. Consistent with the gel filtration analysis, dimers (Mr 100,000) and oligomers (Mr >200,000) were observed after cross-linking (Fig. 3C). These data strongly suggest that, under native conditions in solution, SIRPα dimerizes through its extracellular Ig domains.
FIGURE 3.

The IgC folds of SIRPα ectodomains are critical for dimerization of SIRPα. Soluble full-length and truncated ectodomains tagged with 10 histidines were produced in HEK293T cells and purified using nickel-NTA columns. A, gel filtration chromatography analysis demonstrating that full-length SIRPα ectodomain (SIRPαD1D2D3His) with a predicted molecular weight of 38,000 eluted as a single peak with an apparent molecular weight of 70,000. SIRPαD1His (membrane-distal IgV fold) has a predicted molecular weight of 16,000 but eluted as two major peaks with apparent molecular weights of 25,000 and 18,000. SIRPαD2D3His (membrane-proximal IgC folds) has a predicted molecular weight of 26,000 but eluted as a single peak with an apparent molecular weight of 49,000. Void volume is indicated by the gray-shaded area. B, MALDI-TOF analysis of the eluted recombinant SIRP proteins. As can be seen, SIRPαD1D2D3His consisted of monomers at 42 kDa and dimers at 85 kDa, and SIRPαD2D3His consisted of monomers at 30 kDa and dimers at 60 kDa. In contrast, SIRPαD1His consisted of only monomers at 16 kDa. C, immunoblot analysis of purified SIRPα proteins using rabbit antiserum against the full-length SIRPα ectodomain, confirming the presence of noncovalently linked dimers after cross-linking with BS3. Results are representative of one of five independent experiments. C, control; X, cross-linked.
Interpretation of the recently solved crystal structures of soluble SIRPαD1 suggested the formation of transdimers (26, 27, 29). However, subsequent analyses failed to demonstrate dimerization of soluble SIRPαD1 under native conditions (27). Given our evidence that the full-length ectodomain of SIRPα may dimerize, we tested whether SIRPαD1 also dimerizes under similar conditions. Purified histidine-tagged soluble SIRPα membrane-distal domain SIRPαD1His with a predicted molecular weight of ∼19,000 was analyzed using gel filtration chromatography. Chromatography analyses of SIRPαD1His revealed four protein peaks. Two of the peaks were obtained at or near the void volume and contained mostly co-purifying contaminants, whereas two other peaks at 158 ml (Mr 25,000) and 169 ml (Mr 19,000) contained the majority of SIRPαD1His (Fig. 3A). Because we could not separate the two peaks completely, they were pooled for subsequent analyses. However, it is likely that the Mr 18,000 peak represented monomeric SIRPαD1His, and the Mr 25,000 peak may represent dimers. In support of this prediction, MALDI-TOF analysis of Mr 18,000/25,000 peaks revealed a single species with a molecular mass of 14 kDa (Fig. 3B). On immunoblot analysis, the apparent molecular weight of SIRPαD1His ranged from 15,000 to 25,000. After cross-linking, a dimer product was apparent at Mr 37,000, and oligomers were apparent at Mr 55,000 (Fig. 3C). Collectively, these data suggest that SIRPαD1His forms weak dimers in solution.
We hypothesized that weak dimerization mediated through D1 was not sufficient to account for robust dimerization observed with the full-length ectodomain SIRPαD1D2D3His. This would require significant contributions of SIRPαD2D3 in addition to SIRPαD1. Purified SIRPαD2D3His was thus analyzed using gel filtration chromatography. SIRPαD2D3His eluted as a single peak on gel filtration chromatography with an apparent molecular weight of 49,000, which is roughly twice the predicted molecular mass (26 kDa; Fig. 3A) and suggests that SIRPαD2D3His is a dimer in solution. In addition, MALDI-TOF analysis revealed a 30-kDa monomeric species and a 60-kDa dimeric species (Fig. 3B). Furthermore, a dimeric species was also detected by immunoblot after cross-linking (Fig. 3C). These data indicate that the D2 and D3 membrane-proximal domains of SIRPα also play an important role in mediating dimerization of soluble SIRPα.
Extracellular Domains Contribute to the Dimerization of SIRPα on the Cell Surface
Because our findings indicated that soluble ectodomains of SIRPα formed dimers in solution, we performed experiments to verify whether dimerization of full and truncated SIRPα ectodomains could also occur on the cell surface. Expression of truncated SIRPα on the surface of transiently transfected CHO cells was verified and followed by cross-linking experiments. Following transfection, surface expression of WTSIRPα, SIRPαD1D2, SIRPαD1, SIRPαD2D3, and SIRPαD3 was assessed by flow cytometry using anti-SIRPαD1 antibody (SAF17.2) and an anti-SIRPαD3 antibody (SAF4.2). All truncated SIRPα proteins were expressed on the cell surface similarly except for reduced amounts of SIRPαD1-Myc6 (Fig. 4A). Immunoblot analyses of cross-linked WTSIRPα, SIRPαD1D2, SIRPαD1-Myc6, SIRPαD2D3, and SIRPαD3 on the cell surface revealed dimerization and oligomerization (Fig. 3B). Although high amounts of SIRPαD1-Myc6 were observed in Western blots of cell lysates, we only observed a low level of cell surface expression by flow cytometric analyses of non-permeabilized cells. An explanation for this difference became apparent in confocal immunofluorescence analyses of permeabilized cells transfected with SIRPαD1-Myc6 that revealed abundant intracellular staining distributed in a reticular pattern typical of endoplasmic reticulum staining (not shown). However, given that the cross-linker we used does not cross cell membranes, the SIRPαD1-Myc6 dimers detected by Western blot after cross-linking represent cell surface protein only. These data suggest that all three Ig domains of SIRPα mediate dimerization, and the collective contribution of individual domains may be important. Furthermore, we hypothesized that if all three domains participate in dimerization, it is likely that the dimers are formed in cis.
FIGURE 4.

All three Ig folds of SIRPα ectodomain participate in dimerization on the cell surface. Membrane-bound SIRPα with full-length or truncated ectodomains was expressed transiently in CHO cells. A, flow cytometric analysis of surface expression. Each transfectant was probed with murine monoclonal anti-SIRPαD1 (SAF17.2; dashed line), anti-SIRPαD3 (SAF4.2; solid line), and an isotype control (dotted line). B, immunoblot analysis with rabbit antiserum against the full-length SIRPα ectodomain demonstrating that membrane-bound WTSIRPα, SIRPαD1-Myc6, and SIRPαD2D3 dimerize on the cell surface after cross-linking with BS3. Results are representative of one of five independent experiments. C, control; X, cross-linked.
SIRPα Dimerizes in cis on the Cell Surface
Analyses of the crystal structure have suggested that SIRPαD1 may form dimers in trans; however, no studies thus far have confirmed or ruled out this possibility (26, 27, 29). On the other hand, our data on cross-linked SIRPα support dimerization in cis as a more plausible model. Using tagged fusion SIRPα proteins, including SIRPα-GFP (predicted molecular weight of ∼80,000) and SIRPα-Myc6 (predicted weight of ∼72,000), a strategy was devised to test whether dimerization of SIRPα can occur in trans. In these experiments, populations of CHO cells expressing individual tagged SIRPα were mixed and cocultured, treated with cross-linker, solubilized, and subjected to immunoprecipitation analyses for the presence or absence of individual tagged proteins. In Fig. 5A, it is shown that each of the tags that were introduced do not interfere with dimerization in CHO cells that co-express both proteins or in cocultures of cells that express each protein separately. In assessing the results of these experiments, some biochemical considerations are worth noting. In particular, monomeric forms of the tagged proteins have higher apparent molecular weight than native SIRPα protein due to contributions from added tags. Furthermore, predicted molecular weights of highly glycosylated proteins, such as SIRPα often yield higher than predicted Mr values. Based on the mass of monomers, the predicted mass of SIRPα dimers should range from 140,000 to 240,000. However, tagged dimers in Fig. 5A had apparent molecular weight ranging from 250,000 to 310,000. Although the difference between the observed apparent and the predicted molecular weight values only roughly accounts for the weight of an additional tagged monomer, separation of proteins by SDS-PAGE can also be affected by protein conformation (as in apparent Stokes radii), thereby resulting in deviation of observed molecular weight from predicted molecular weight. Given our other data, we think it is reasonable to assume that the slight discrepancy in apparent molecular mass observed on SDS-PAGE is secondary to the increased glycosylation, the addition of tags as well as the presence of the flexible cross-linker. As a positive control, immunoprecipitation was performed with anti-GFP from lysates of CHO cells coexpressing SIRPα-GFP and SIRPα-Myc6. Both SIRPα-GFP and SIRPα-Myc6 were precipitated with anti-GFP antibody with and without cross-linking, suggesting that the SIRPα dimer is stable in the presence of non-denaturing detergent (Fig. 5B). Under this scenario, dimers containing both tags could potentially form through either cis or trans interactions (Fig. 5B). On the other hand, immunoprecipitation with anti-GFP from lysates of co-cultures of CHO cells that separately express each of the tagged proteins precipitated SIRPα-GFP but not SIRPα-Myc6, even after cross-linking (Fig. 5B). The lack of tagged dimers containing both GFP and Myc6 in this case argues against the possibility of trans interaction. These results strongly support the notion of cis dimerization of SIRPα on the cell surface.
FIGURE 5.
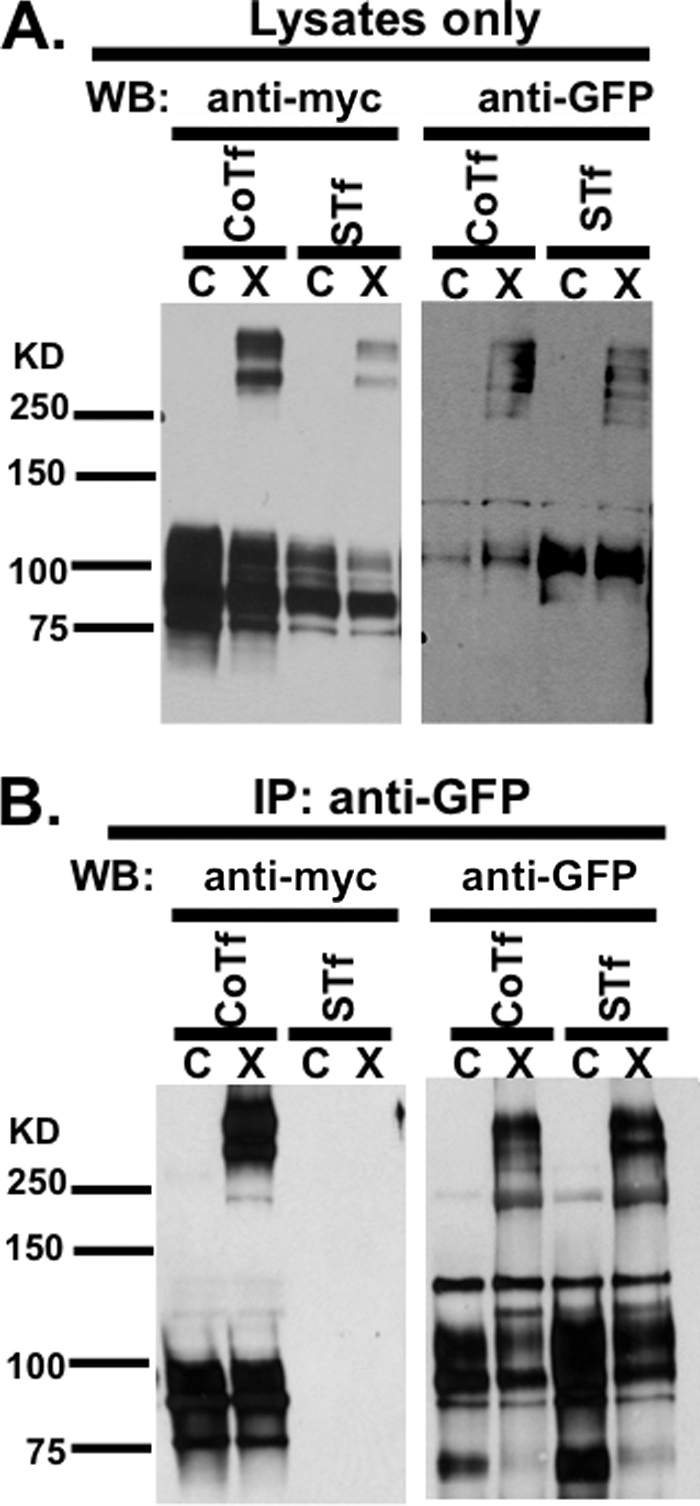
SIRPα forms homodimers in cis on the cell surface. Cross-linked SIRPα dimers from a mixture of CHO cells that separately expressed SIRPα-EGFP and SIRPα-Myc6 (separate transfection; STf) were compared with dimers from a culture of CHO cells that were co-expressing SIRPα-EGFP and SIRPα-Myc6 (co-transfection; CoTf). A, immunoblots of separately transfected and co-transfected cell lysates were probed with a murine monoclonal anti-Myc antibody (9E10; left blot) and with a rabbit polyclonal anti-EGFP antibody (right blot) after cross-linking, demonstrating that the EGFP and Myc tags did not interfere with SIRPα dimerization. B, immunoprecipitation (IP) of non-treated (C) and cross-linked (X) cell lysates of separately transfected and co-transfected cells with polyclonal rabbit anti-EGFP antibody and probe (WB) with anti-Myc antibody in order to determine the presence of SIRPα dimers that contain both Myc6 and EGFP tags (left blot). As a positive control for immunoprecipitation, proteins precipitated by the rabbit polyclonal anti-GFP antibody were also probed with the same anti-GFP antibody (right blot). Results are representative of one of three independent experiments.
N-Glycans Facilitate SIRPα Dimerization
Previous studies that failed to detect SIRPα dimers utilized only SIRPαD1 and not the full-length ectodomain (26, 27, 29). As shown in Fig. 3, B and C, SIRPαD1 by itself exhibited weak dimerization. However, SIRPαD2D3 formed stronger dimers, as evidenced by gel filtration and MALDI-TOF data in Fig. 3B. It is important to note that, in one study, SIRPαD1 was produced in Escherichia coli, which lacked post-translational modifications, such as N-linked glycosylation (27). In this study, recombinant proteins were produced in HEK293T cells and retained proper N-linked glycosylation. Thus, we speculated that the discrepancies in SIRPα dimerization may stem from the differences in N-linked glycosylation.
To examine this possibility, SIRPα dimerization was assessed in CHO-SIRPα treated with tunicamycin and kifunensine. Treatment with tunicamycin prevents the addition of any polysaccharide onto asparagine by inhibiting GlcNAc phosphotransferase (39, 40). On the other hand, treatment with kifunensine arrests the N-glycation at the high mannose stage without the addition of terminal structures by inhibiting endoplasmic reticulum mannosidase (41). To assess the efficacy of the inhibitors, the degree of N-glycation was measured with PHA-L-FITC by flow cytometry (42). In parallel, surface expression of SIRPα was assessed by flow cytometry and to allow for normalization of protein loading on SDS-polyacrylamide gels for Western blots. In SIRPα-expressing CHO cells treated with tunicamycin, the total amount of N-linked glycation was reduced by ∼80%, as shown by PHA-L-FITC staining, whereas the expression of cell surface SIRPα was maintained at ∼70% of the levels observed in untreated cells (Fig. 6A). Interestingly, after cross-linking and immunoblot, SIRPα dimerization was greatly inhibited (Fig. 6A). These observations suggest that N-glycans on SIRPα may play an important role in facilitating dimerization.
FIGURE 6.

SIRPα dimerization is disrupted after treatment with tunicamycin. A, flow cytometric analysis of tunicamycin-treated (solid line) and untreated (dashed line) CHO-SIRPα transfectants labeled with PHA-L-FITC (top left panel) to measure the degree of deglycosylation and with anti-SIRPαD1 (SAF17.2; bottom left panel) to assess the level of SIRPα expression. After cross-linking, treated cells were analyzed by immunoblots for the presence of SIRPα dimers. B, immunoblot after cross-linking, demonstrating that SIRPα on CHO cells still dimerized in the presence of 100 mm α-methyl-d-mannoside or 100 mm lactose, which were added to competitively inhibit potential dimerization by multivalent mannose or galatose binding lectins. C, flow cytometric analysis of kifunensine-treated CHO-SIRPα transfectants labeled with PHA-L-FITC (top left panel) to measure the degree of deglycosylation and with anti-SIRPαD1 (SAF17.2; bottom left panel) to assess the level of SIRPα expression. After cross-linking, treated cells were analyzed by immunoblots for the presence of SIRPα dimers. D, immunoblot after cross-linking demonstrating that SIRPα expressed in mutant CHO cells lacking enzymes necessary for terminal modification of N-glycans, such as Lec1 (mannose-rich), Lec2 (sialic acid-deficient), and Lec8 (galactose-deficient), can still dimerize. Results are representative of one of four independent experiments. C, control; X, cross-linked.
Several studies recently demonstrated that complex N-glycan interactions with lectins may facilitate the formation of higher order molecular complexes (43). Therefore, it is possible that lectin interactions with SIRPα may be in part responsible for higher order oligomers in the fully glycosylated protein. To test this, we incubated cells with inhibitors of common cell surface carbohydrate-lectin interactions: α-methyl-d-mannoside, which inhibits mannose recognizing C type lectins, and lactose, which effectively blocks galectin interactions (32, 44). However, incubation of CHO cells with either 100 mm α-methyl-d-mannoside or 100 mm lactose failed to alter SIRPα dimerization (Fig. 6B). Furthermore, incubation of cells with kifunensine, which specifically inhibits the formation of many N-glycan modifications required for lectin interactions (41, 45), also failed to alter surface expression or dimerization of SIRPα (Fig. 6C).
Although kifunensine significantly inhibited total cell surface complex N glycan formation, it was not possible to determine whether specific inhibition of complex N-glycans on SIRPα also occurred. To further examine the potential role of N-glycans in dimerization, we expressed SIRPα in mutant CHO cells that lack enzymes necessary for terminal modification of N-glycans. Specifically, CHO-Lec1 (mannose-rich), CHO-Lec2 (sialic acid-deficient), and CHO-Lec8 (galactose-deficient) were used (46). However, consistent with kifunensine-treated cells, the dimerization of SIRPα in these CHO cell mutants remained unaffected (Fig. 6D). These results suggest that terminal modifications of N-glycans do not facilitate dimerization although complete inhibition of the formation of N-glycans appears to play an important role in SIRPα dimerization.
SIRPα Dimerization Is Not Necessary for Binding to Soluble CD47
From these data, we speculated that dimerization of SIRPα may be functionally important in the interaction with CD47. Comparison of the binding affinities of soluble CD47 to monomeric and dimeric SIRPα on the cell surface would allow for determination of whether dimeric SIRPα is necessary for CD47 binding. As shown in Fig. 6A, treatment of CHO-SIRPα with tunicamycin resulted in a major reduction in dimerized SIRPα on the cell surface. Therefore, the majority of SIRPα on the cell surface after tunicamycin treatment is in a monomeric state. Binding analyses were then performed on CHO-SIRPα cells grown in the presence or the absence of tunicamycin. Consistent with flow cytometry analyses (Fig. 6A), the number of SIRPα molecules on the surface of treated CHO-SIRPα was reduced as indicated by the lower Bmax (Fig. 7). More importantly, the binding affinity (Kd) of SIRPα in treated cells was not significantly altered (Fig. 7). Thus, dimerization of SIRPα is not necessary for CD47 binding and does not change the affinity of SIRPα for CD47. Furthermore, consistent with the recently published crystal structure of SIRPαD1 complexed with CD47, the stoichiometry of CD47 binding to SIRPα is 1:1 (26). These observations suggest that dimerization of SIRPα is not necessary for binding CD47 and that SIRPα acts as a bivalent homodimer.
FIGURE 7.

Dimerization of SIRPα is not required for binding to CD47. Serial dilutions of cell supernatants containing known concentrations of soluble CD47 fused with alkaline phosphatase (CD47AP) were assessed in binding assays to cell surface-expressed SIRPα in tunicamycin-treated (Tunica.) or non-treated control (No Tx) CHO-SIRPα cells grown in 96-well tissue culture plates. Analysis of binding curves for affinity constant (Kd) and maximal binding value (Bmax) was performed with PRISM software. Data are representative of a quadruplicate set of determinations from an experiment that is representative of one of three independent experiments.
Evidence of SIRPα Dimerization in Stimulated Adherent Human PMN
Because stimulation of PMN with LPS on fMLP has previously been shown to alter SIRPα signaling and trafficking (12, 47), we sought to verify whether SIRPα exists as dimers on the cell surface of human PMN that naturally express this protein. Human PMNs isolated from normal donors were stimulated with LPS and fMLP, either in suspension or after adhesion to plastic. We assessed SIRPα dimerization by immunoblot from PMN after cross-linking. Interestingly, only low levels of dimers were detected on the cell surface of suspended PMNs even after stimulation with LPS or fMLP (data not shown). However, significant dimerization was detected after cross-linking treatment of stimulated adherent PMNs (Fig. 8). Although these data may suggest that dimerization of SIRPα is a regulated physiological event, we have previously shown that there are significant intracellular pools of SIRPa in human PMN that are up-regulated to the cell surface after stimulation of adherent cells. Thus, it is also likely that the increase observed in Fig. 8 is a reflection of increased cell surface expression of SIRPa dimers. Nevertheless, these findings verify the existence of SIRPa dimers in normal human PMNs.
FIGURE 8.
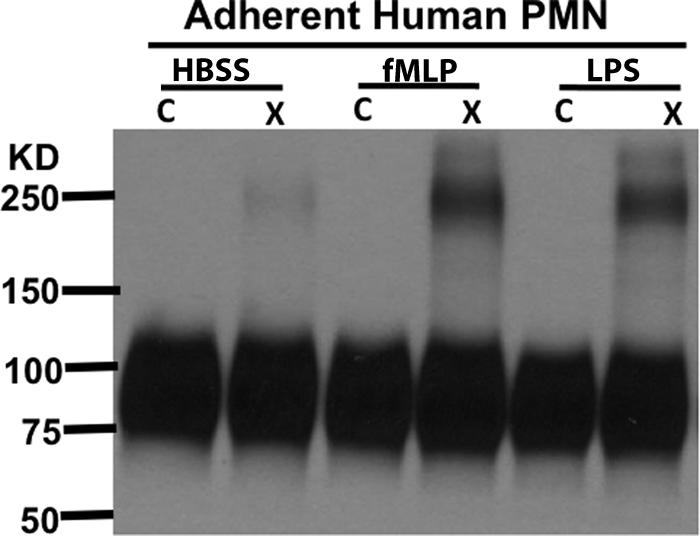
SIRPα dimerization is enhanced in stimulated adherent human PMNs. Immunoblot of cell lysates from non-cross-linked and cross-linked human blood PMNs stimulated with vehicle, 10−6 m fMLP, or 1 μg/ml of LPS for 20 min at 37 °C after adhesion to plastic tissue culture wells. The cross-linked dimers were detected with polyclonal rabbit antiserum against the SIRPα cytoplasmic tail. Results are representative of one of four independent experiments. C, control; X, cross-linked.
DISCUSSION
Recently, a series of structural and mutagenesis studies reported on molecular mechanisms involved in binding interactions of soluble SIRPαD1 to CD47 (16, 25–29). However, despite these advances, little is known about the structural role of the proximal IgC domains. It was previously shown that a closely related family member, SIRPβ, which expresses on the cell surface, is a dimer that is covalently linked through a disulfide bond in the most membrane-proximal IgC domain (31). Due to the very high degree of homology between the ectodomains of SIRPα and SIRPβ, we investigated the possibility that SIRPα also dimerizes. Analysis of the Ig domains of SIRPα indicates that SIRPαD1 is an IgV domain, whereas SIRPαD2 and SIRPαD3 are categorized by the Protein Family database (PFAM) as IgC1 domains by virtue of their conservation in protein sequences with other known IgC domains (27, 30, 48, 49). There is only a modest degree of similarity between SIRPαD2 and SIRPαD3, each having only 57% homology. However, SIRPαD2 and SIRPαD3 exhibit high degrees of homology to the corresponding domains in SIRPβ and SIRPγ (Fig. 1A). A notable difference in SIRPβD3 is the presence of an extra cysteine residue that forms an intermolecular disulfide bond linking two SIRPβ monomers (31). The formation of such intermolecular disulfide bonds is dependent on intimate noncovalent association between monomers before bond formation. In immunoglobulins, for example, noncovalent association of light chains and heavy chains has been shown to occur before interchain covalent linkage (35, 36, 50). The high degree of homology between SIRPα and SIRPβ led us to hypothesize that SIRPα may form noncovalently linked dimers.
Given that conventional immunoblot analyses require denaturation of proteins in detergents, noncovalently linked protein complexes most often dissociate, and thus detection of the dimers is not possible. To circumvent this problem, chemical cross-linkers capable of forming stable covalent linkages between closely associated partners are often used to stabilize dimers under denaturing conditions. BS3, a membrane-impermeable homobifunctional cross-linker that reacts specifically with primary amines, has been used extensively for this purpose (51). After cross-linking with BS3, we demonstrate that SIRPα exists as a dimer on the cell surface. We excluded the possibility of CD47 as a component of the cross-linked SIRPα complex by demonstrating that the presence or lack of CD47 does not alter the size of cross-linked SIRPα dimers (Fig. 2). Furthermore, mass spectrometry of cross-linked protein did not yield evidence of CD47 in the complex. Our attempts at cross-linking SIRPα-CD47 complexes that were formed either by binding soluble CD47AP (data not shown) to WT SIRPα expressed on CHO cell surface, by mixing cultures of cells expressing separately cell surface CD47 and SIRPα, or coexpressing both molecules in the same cell (Fig. 2) were not successful. These results strongly suggest that the spatial configurations of free amine groups on CD47 and SIRPα are not permissible for cross-linking. Given that binding of CD47 to SIRPα does not inhibit SIRPα dimerization, it is reasonable to infer that the contact sites in the SIRPα-SIRPα homodimer are distinct from contact sites in the SIRPα-CD47 complex. Therefore, free amine groups of each molecule in the SIRPα-CD47 complex are most likely distributed at distances greater than the 11-Å cross-linking requirement with BS3. In support of this, the apparent molecular weight of soluble SIRPαD1D2D3 ectodomain estimated by gel filtration was approximately twice the predicted molecular weight. Also, cross-linked dimers of soluble SIRPαD1D2D3 ectodomains were readily detected on immunoblots after SDS-PAGE (Fig. 3). These results strongly suggest that dimerization of SIRPα occurs through the ectodomain under native conditions.
To identify which Ig folds in the ectodomain mediate dimerization, soluble truncated and fully glycosylated ectodomains produced in eukaryotic cells were analyzed by gel filtration chromatography and immunoblots (Fig. 3). Soluble SIRPαD2D3His eluted from gel filtration columns as a single peak with an apparent molecular weight that corresponded to a dimeric species. The same was true when recombinant soluble SIRPαD1D2-His was purified by gel filtration (data not shown). The presence of dimeric SIRPαD2D3 was then verified by immunoblot analysis after cross-linking. Corresponding results were obtained in cross-linked membrane-bound SIRPαD1, SIRPαD1D2, and SIRPαD2D3 (Fig. 4) as well as D3 (data not shown), suggesting that all three domains of SIRPα participate in dimerization. Interestingly, soluble SIRPαD1His eluted as two overlapping peaks by gel filtration. The species in the lower molecular weight peak approximated the predicted molecular weight, whereas the higher molecular weight species was ∼10,000 short of the predicted weight of the dimer. Analysis of the pooled protein fractions by MALDI-TOF revealed only the smaller species of SIRPαD1, suggesting that the higher molecular weight species is most likely a weak dimer. Cross-linking studies confirmed the presence of D1 dimers. However, the fact that SIRPαD1 eluted as both monomer and dimer suggests that dimerization of SIRPαD1 is weak and may account for the inability of other investigators to detect this interaction (27, 29). In our hands, this weak D1 interaction was difficult to detect without stabilization by chemical cross-linkers. In addition, it is worth noting that the preparation of SIRPαD1 used in other studies was not produced in mammalian cells, and thus differences in glycosylation may have also affected the degree of dimerization.
Our findings that both the IgV and the IgC loops contribute to the dimerization of SIRPα led us to propose a working model of SIRPα cis homodimers on the cell surface. This model would predict that in cells co-expressing two differently tagged SIRPα, SIRPα dimers containing both tags should be detectable after cross-linking. On the other hand, in a mixture of two cultures that each express a unique type of tagged SIRPα, cross-linked SIRPα dimers would contain only one type of tag. Indeed, as revealed by our immunoprecipitation analysis, SIRPα dimers with two different tags are only detectable in cells that co-express both types of tagged SIRPα and not in a mixture of cells that each express a single type of tagged SIRPα (Fig. 5) (52). This latter result was obtained even in the absence of any cross-linking and also indicates that dimerization does not occur in trans. Therefore, these results strongly support a model of cis dimerization.
In our working model, dimerization of SIRPα on the cell surface is primarily mediated through interactions between the IgC folds. A notable precedent that illustrates many modes of cis dimerization between IgC folds comes from structural studies of immunoglobulins. For example, in IgG there are four pairs of IgC folds, with two identical pairs in the two arms of the Fab2 region and two distinct pairs in the Fc region. The IgC folds in the Fab region pair through noncovalent and covalent linkages. In the Fc region, the residues of one pair of IgC folds (Ch1) do not contact at all. Here, stabilization occurs through weak interactions of glycans and amino acid residues. In contrast, the other pair (Ch2) dimerize through noncovalent interactions between the residues (53, 54).
It is also possible that the addition of N-linked glycans can directly affect the conformation of Ig folds and in turn change the propensity to dimerize (55, 56). Using pharmacological inhibitors and CHO cell glycosylation mutants, we found that SIRPα dimerization was inhibited only when all components of N-linked glycosylation were absent (Fig. 6). On the other hand, when maturation of N-linked glycans was arrested in a high mannose stage or terminal modification remained incomplete, dimerization was not affected. These results suggest that terminal modification of N-linked glycosylation is not likely to facilitate dimerization. In addition, the results of our competitive inhibition assays with soluble disaccharides excluded the possibility that SIRPα dimers are mediated through cross-linking interactions by a third party lectin. Therefore, the most likely role of N-linked glycosylation is to maintain a conformation that is favorable for dimerization.
The presence of SIRPα as cis dimers on the cell surface adds a new dimension of complexity in considering interactions with its ligand CD47. Three mutually exclusive models are possible. First, cis dimerization of SIRPα may be independent of CD47 binding. Second, cis dimerization of SIRPα may be necessary to form a proper binding site for CD47. Third, cis dimerization of SIRPα may block the CD47 binding site, thereby inhibiting interaction with CD47. Although the first and second models include dimerized SIRPα binding to CD47, interactions on the cell surface would be considerably different in the first model due to changes in avidity secondary to dimerization. In particular, the first model would require one SIRPα to bind to one CD47, whereas the second model would require two SIRPα to bind one CD47. There are several observations from this study and others that support the first model. First, the CD47 binding region and those required for cis dimerization are structurally distinct. Previous studies have already established that the membrane-distal IgV fold of SIRPα is the only site necessary and sufficient in binding CD47 (12, 26, 27, 29, 37, 57). In this study, we demonstrate that the membrane-proximal IgC folds of SIRPα, although not important for binding to CD47, are important in cis dimerization. Furthermore, we observed that cis dimerization of full-length SIRPα occurs despite extensive mutagenesis of contacting residues of a SIRPαD1 putative transdimer predicted in the crystal structure, as well as after numerous mutations of regions that have been shown to affect CD47 binding (data not shown). Second, SIRPα interactions with CD47 and cis dimerization are functionally independent. Previous studies have already shown that N-linked glycosylation is not important in SIRPα CD47 binding interactions (28, 29, 38, 58, 59). In this report, we show that SIRPα expressed at the cell surface after treatment with tunicamycin is capable of binding CD47 to an extent comparable with controls, although dimerization was greatly diminished. In other words, the CD47 binding site of membrane-bound SIRPα monomers and dimers both appear to exhibit equivalent affinity for CD47. However, it is worth noting that the measured affinity in our system between soluble CD47AP and SIRPα is higher than that reported for soluble SIRPα-CD47 proteins. It is possible that AP-tagged fusion proteins can dimerize through AP (60). Therefore, in this system, it is possible that CD47AP may act as a bivalent receptor, thereby accounting for the observed increase in affinity. Despite this possibility, the dimerization of SIRPα we observe is clearly independent of CD47AP binding. In particular, we show that SIRPα expressed on CHO cells bound to full-length CD47 (Fig. 2) or soluble CD47AP (data not shown) is easily cross-linked, thereby demonstrating that CD47 binding has no significant effect on SIRPα dimerization. Last, recently published crystal structures of SIRPαD1-CD47 complexes reveal that one SIRPα binds to one CD47 (26). Therefore, it is likely that SIRPα binding to CD47 and cis dimerization are mutually independent events.
The crystal structure of the full-length extracellular region of SIRPα was recently reported by Hatherley et al. (30). Deglycosylated SIRPα was used to facilitate crystallization and yielded a monomeric configuration of the SIRPα extracellular region. This result is consistent with our findings that demonstrate a great reduction in dimerization of deglycosylated SIRPα (Fig. 6). Furthermore, their structural studies showed that there is a constant (C-1 set) domain in each of the D2 and D3 domains of SIRPα. The C-1 set domains are restricted to proteins involved in vertebrate antigen recognition, such as immunoglobulins, T cell receptors, MHC molecules class I and class II, and tapasin and β2-microglobulin. It is well known that all of these molecules form either homodimers or heterodimers with strong avidity.
In primary leukocytes that natively express SIRPα, we observed dimerization of SIRPα. Specifically, SIRPα dimerization was enhanced in stimulated adherent human PMNs (Fig. 8). In PMNs, SIRPα is present both on the cell surface and in the secondary granule fraction and is up-regulated to the cell surface after stimulation (12). Given these observations, although the data in Fig. 8 may suggest that dimerization of SIRPα is a regulated physiological event, it is also likely that the increase observed in Fig. 8 is a reflection of increased cell surface expression of SIRPα dimers. Nevertheless, these findings verify the existence of SIRPα dimers in normal human PMNs.
Interestingly, it has been proposed that SIRPα is restricted within certain membrane microdomains and is tethered to certain cytoskeletal structures because cross-linking with antibody resulted in the formation of microclusters (58). Therefore, mechanisms such as membrane partitioning or cytoskeletal reorganization may also have important roles in regulating SIRPα dimerization.
To our knowledge, this is the first report that demonstrates SIRPα forms cis dimers. Previous studies utilized purified deglycosylated SIRPαD1 fragments that were either treated with kifunensine and EndoHf or produced in bacterial systems and thus did not observe SIRPα dimers (26, 27, 29, 30). Our data also show that the D1 domains of SIRPα form weak dimers. Results from this study may have significant implications for our understanding on how SIRPα interacts with CD47. It is probable that SIRPα dimers on the cell surface acting as a bivalent receptor can bind two CD47 molecules in trans in a manner reminiscent of the interaction between B7–1/2 and CD28/CTLA4, in which the interaction of ligand dimers with receptor dimers is enhanced by increased avidity (61). Furthermore, this model is consistent with reported observations of “synapse-like” structures between SIRPα-expressing macrophages and CD47-expressing red blood cells that inhibited phagocytosis (49, 62, 63).
In conclusion, we demonstrate that full-length, cell surface-expressed SIRPα and soluble SIRPα ectodomains form noncovalently linked dimers. Generation of reagents that can manipulate SIRPα dimerization will advance understanding of the functional roles of SIRPα in cells and may facilitate development of novel immunoregulatory therapies.
This work was supported, in whole or in part, by National Institutes of Health Grants DK079392 and DK72564 (to C. A. P.) and Digestive Diseases Minicenter Grant DK064399 (epithelial cell culture core and microscopy core support).
- SIRP
- signal regulatory protein
- BS3
- bis(sulfosuccinimidyl) suberate
- fMLF
- formyl-methionyl-leucyl-phenylalanine
- PHA-L-FITC
- phytohemagglutinin type L conjugated with FITC
- PMN
- polymorphonuclear neutrophil
- EGFP
- enhanced green fluorescent protein.
REFERENCES
- 1.van Beek E. M., Cochrane F., Barclay A. N., van den Berg T. K. (2005) J. Immunol. 175, 7781–7787 [DOI] [PubMed] [Google Scholar]
- 2.Barclay A. N., Brown M. H. (2006) Nat. Rev. Immunol. 6, 457–464 [DOI] [PubMed] [Google Scholar]
- 3.Maile L. A., Capps B. E., Miller E. C., Aday A. W., Clemmons D. R. (2008) Diabetes 57, 2637–2643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maile L. A., Clemmons D. R. (2002) J. Biol. Chem. 277, 8955–8960 [DOI] [PubMed] [Google Scholar]
- 5.Ling Y., Maile L. A., Lieskovska J., Badley-Clarke J., Clemmons D. R. (2005) Mol. Biol. Cell 16, 3353–3364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kharitonenkov A., Chen Z., Sures I., Wang H., Schilling J., Ullrich A. (1997) Nature 386, 181–186 [DOI] [PubMed] [Google Scholar]
- 7.Dietrich J., Cella M., Seiffert M., Bühring H. J., Colonna M. (2000) J. Immunol. 164, 9–12 [DOI] [PubMed] [Google Scholar]
- 8.Tomasello E., Cant C., Bühring H. J., Vély F., André P., Seiffert M., Ullrich A., Vivier E. (2000) Eur. J. Immunol. 30, 2147–2156 [DOI] [PubMed] [Google Scholar]
- 9.Brooke G., Holbrook J. D., Brown M. H., Barclay A. N. (2004) J. Immunol. 173, 2562–2570 [DOI] [PubMed] [Google Scholar]
- 10.Stefanidakis M., Newton G., Lee W. Y., Parkos C. A., Luscinskas F. W. (2008) Blood 112, 1280–1289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brown E. J., Frazier W. A. (2001) Trends Cell Biol. 11, 130–135 [DOI] [PubMed] [Google Scholar]
- 12.de Vries H. E., Hendriks J. J., Honing H., De Lavalette C. R., van der Pol S. M., Hooijberg E., Dijkstra C. D., van den Berg T. K. (2002) J. Immunol. 168, 5832–5839 [DOI] [PubMed] [Google Scholar]
- 13.Liu Y., Merlin D., Burst S. L., Pochet M., Madara J. L., Parkos C. A. (2001) J. Biol. Chem. 276, 40156–40166 [DOI] [PubMed] [Google Scholar]
- 14.Parkos C. A., Colgan S. P., Liang T. W., Nusrat A., Bacarra A. E., Carnes D. K., Madara J. L. (1996) J. Cell Biol. 132, 437–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lindberg F. P., Bullard D. C., Caver T. E., Gresham H. D., Beaudet A. L., Brown E. J. (1996) Science 274, 795–798 [DOI] [PubMed] [Google Scholar]
- 16.Ide K., Wang H., Tahara H., Liu J., Wang X., Asahara T., Sykes M., Yang Y. G., Ohdan H. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 5062–5066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miyake A., Murata Y., Okazawa H., Ikeda H., Niwayama Y., Ohnishi H., Hirata Y., Matozaki T. (2008) Genes Cells 13, 209–219 [DOI] [PubMed] [Google Scholar]
- 18.Smith R. E., Patel V., Seatter S. D., Deehan M. R., Brown M. H., Brooke G. P., Goodridge H. S., Howard C. J., Rigley K. P., Harnett W., Harnett M. M. (2003) Blood 102, 2532–2540 [DOI] [PubMed] [Google Scholar]
- 19.Dong L. W., Kong X. N., Yan H. X., Yu L. X., Chen L., Yang W., Liu Q., Huang D. D., Wu M. C., Wang H. Y. (2008) Mol. Immunol. 45, 3025–3035 [DOI] [PubMed] [Google Scholar]
- 20.Ishikawa-Sekigami T., Kaneko Y., Okazawa H., Tomizawa T., Okajo J., Saito Y., Okuzawa C., Sugawara-Yokoo M., Nishiyama U., Ohnishi H., Matozaki T., Nojima Y. (2006) Blood 107, 341–348 [DOI] [PubMed] [Google Scholar]
- 21.Oldenborg P. A., Gresham H. D., Lindberg F. P. (2001) J. Exp. Med. 193, 855–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oldenborg P. A., Zheleznyak A., Fang Y. F., Lagenaur C. F., Gresham H. D., Lindberg F. P. (2000) Science 288, 2051–2054 [DOI] [PubMed] [Google Scholar]
- 23.Gardai S. J., McPhillips K. A., Frasch S. C., Janssen W. J., Starefeldt A., Murphy-Ullrich J. E., Bratton D. L., Oldenborg P. A., Michalak M., Henson P. M. (2005) Cell 123, 321–334 [DOI] [PubMed] [Google Scholar]
- 24.Gardai S. J., Xiao Y. Q., Dickinson M., Nick J. A., Voelker D. R., Greene K. E., Henson P. M. (2003) Cell 115, 13–23 [DOI] [PubMed] [Google Scholar]
- 25.Takenaka K., Prasolava T. K., Wang J. C., Mortin-Toth S. M., Khalouei S., Gan O. I., Dick J. E., Danska J. S. (2007) Nat. Immunol. 8, 1313–1323 [DOI] [PubMed] [Google Scholar]
- 26.Hatherley D., Graham S. C., Turner J., Harlos K., Stuart D. I., Barclay A. N. (2008) Mol. Cell 31, 266–277 [DOI] [PubMed] [Google Scholar]
- 27.Nakaishi A., Hirose M., Yoshimura M., Oneyama C., Saito K., Kuki N., Matsuda M., Honma N., Ohnishi H., Matozaki T., Okada M., Nakagawa A. (2008) J. Mol. Biol. 375, 650–660 [DOI] [PubMed] [Google Scholar]
- 28.Lee W. Y., Weber D. A., Laur O., Severson E. A., McCall I., Jen R. P., Chin A. C., Wu T., Gernert K. M., Parkos C. A. (2007) J. Immunol. 179, 7741–7750 [DOI] [PubMed] [Google Scholar]
- 29.Hatherley D., Harlos K., Dunlop D. C., Stuart D. I., Barclay A. N. (2007) J. Biol. Chem. 282, 14567–14575 [DOI] [PubMed] [Google Scholar]
- 30.Hatherley D., Graham S. C., Harlos K., Stuart D. I., Barclay A. N. (2009) J. Biol. Chem. 284, 26613–26619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu Y., Soto I., Tong Q., Chin A., Bühring H. J., Wu T., Zen K., Parkos C. A. (2005) J. Biol. Chem. 280, 36132–36140 [DOI] [PubMed] [Google Scholar]
- 32.Stowell S. R., Arthur C. M., Slanina K. A., Horton J. R., Smith D. F., Cummings R. D. (2008) J. Biol. Chem. 283, 20547–20559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu Y., Bühring H. J., Zen K., Burst S. L., Schnell F. J., Williams I. R., Parkos C. A. (2002) J. Biol. Chem. 277, 10028–10036 [DOI] [PubMed] [Google Scholar]
- 34.Lee W. Y., Chin A. C., Voss S., Parkos C. A. (2006) Methods Mol. Biol. 341, 205–215 [DOI] [PubMed] [Google Scholar]
- 35.Elkabetz Y., Ofir A., Argon Y., Bar-Nun S. (2008) Mol. Immunol. 46, 97–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leitzgen K., Knittler M. R., Haas I. G. (1997) J. Biol. Chem. 272, 3117–3123 [DOI] [PubMed] [Google Scholar]
- 37.Seiffert M., Brossart P., Cant C., Cella M., Colonna M., Brugger W., Kanz L., Ullrich A., Bühring H. J. (2001) Blood 97, 2741–2749 [DOI] [PubMed] [Google Scholar]
- 38.Subramanian S., Boder E. T., Discher D. E. (2007) J. Biol. Chem. 282, 1805–1818 [DOI] [PubMed] [Google Scholar]
- 39.Mahoney W. C., Duksin D. (1979) J. Biol. Chem. 254, 6572–6576 [PubMed] [Google Scholar]
- 40.Dorner A. J., Bole D. G., Kaufman R. J. (1987) J. Cell Biol. 105, 2665–2674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Elbein A. D., Tropea J. E., Mitchell M., Kaushal G. P. (1990) J. Biol. Chem. 265, 15599–15605 [PubMed] [Google Scholar]
- 42.Cummings R. D., Trowbridge I. S., Kornfeld S. (1982) J. Biol. Chem. 257, 13421–13427 [PubMed] [Google Scholar]
- 43.Demetriou M., Granovsky M., Quaggin S., Dennis J. W. (2001) Nature 409, 733–739 [DOI] [PubMed] [Google Scholar]
- 44.Feinberg H., Park-Snyder S., Kolatkar A. R., Heise C. T., Taylor M. E., Weis W. I. (2000) J. Biol. Chem. 275, 21539–21548 [DOI] [PubMed] [Google Scholar]
- 45.Demotte N., Stroobant V., Courtoy P. J., Van Der Smissen P., Colau D., Luescher I. F., Hivroz C., Nicaise J., Squifflet J. L., Mourad M., Godelaine D., Boon T., van der Bruggen P. (2008) Immunity 28, 414–424 [DOI] [PubMed] [Google Scholar]
- 46.Nagayama Y., Namba H., Yokoyama N., Yamashita S., Niwa M. (1998) J. Biol. Chem. 273, 33423–33428 [DOI] [PubMed] [Google Scholar]
- 47.Kong X. N., Yan H. X., Chen L., Dong L. W., Yang W., Liu Q., Yu L. X., Huang D. D., Liu S. Q., Liu H., Wu M. C., Wang H. Y. (2007) J. Exp. Med. 204, 2719–2731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Finn R. D., Tate J., Mistry J., Coggill P. C., Sammut S. J., Hotz H. R., Ceric G., Forslund K., Eddy S. R., Sonnhammer E. L., Bateman A. (2008) Nucleic Acids Res. 36, D281–D288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tsai R. K., Discher D. E. (2008) J. Cell Biol. 180, 989–1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chothia C., Novotný J., Bruccoleri R., Karplus M. (1985) J. Mol. Biol. 186, 651–663 [DOI] [PubMed] [Google Scholar]
- 51.Lopez M., Aoubala M., Jordier F., Isnardon D., Gomez S., Dubreuil P. (1998) Blood 92, 4602–4611 [PubMed] [Google Scholar]
- 52.Jain R. K., Joyce P. B., Molinete M., Halban P. A., Gorr S. U. (2001) Biochem. J. 360, 645–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Padlan E. A. (1994) Mol. Immunol. 31, 169–217 [DOI] [PubMed] [Google Scholar]
- 54.Alzari P. M., Lascombe M. B., Poljak R. J. (1988) Annu. Rev. Immunol. 6, 555–580 [DOI] [PubMed] [Google Scholar]
- 55.Jiménez D., Roda-Navarro P., Springer T. A., Casasnovas J. M. (2005) J. Biol. Chem. 280, 5854–5861 [DOI] [PubMed] [Google Scholar]
- 56.Wyss D. F., Choi J. S., Li J., Knoppers M. H., Willis K. J., Arulanandam A. R., Smolyar A., Reinherz E. L., Wagner G. (1995) Science 269, 1273–1278 [DOI] [PubMed] [Google Scholar]
- 57.Vernon-Wilson E. F., Kee W. J., Willis A. C., Barclay A. N., Simmons D. L., Brown M. H. (2000) Eur. J. Immunol. 30, 2130–2137 [DOI] [PubMed] [Google Scholar]
- 58.Subramanian S., Parthasarathy R., Sen S., Boder E. T., Discher D. E. (2006) Blood 107, 2548–2556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ogura T., Noguchi T., Murai-Takebe R., Hosooka T., Honma N., Kasuga M. (2004) J. Biol. Chem. 279, 13711–13720 [DOI] [PubMed] [Google Scholar]
- 60.Flanagan J. G., Cheng H. J. (2000) Methods Enzymol. 327, 198–210 [DOI] [PubMed] [Google Scholar]
- 61.Sharpe A. H., Freeman G. J. (2002) Nat. Rev. Immunol. 2, 116–126 [DOI] [PubMed] [Google Scholar]
- 62.Ohnishi H., Kobayashi H., Okazawa H., Ohe Y., Tomizawa K., Sato R., Matozaki T. (2004) J. Biol. Chem. 279, 27878–27887 [DOI] [PubMed] [Google Scholar]
- 63.Umemori H., Sanes J. R. (2008) J. Biol. Chem. 283, 34053–34061 [DOI] [PMC free article] [PubMed] [Google Scholar]