

Abstract
α-Synuclein aggregation and accumulation in Lewy bodies are implicated in progressive loss of dopaminergic neurons in Parkinson disease and related disorders. In neurons, the Hsp70s and their Hsp40-like J-domain co-chaperones are the only known components of chaperone network that can use ATP to convert cytotoxic protein aggregates into harmless natively refolded polypeptides. Here we developed a protocol for preparing a homogeneous population of highly stable β-sheet enriched toroid-shaped α-Syn oligomers with a diameter typical of toxic pore-forming oligomers. These oligomers were partially resistant to in vitro unfolding by the bacterial Hsp70 chaperone system (DnaK, DnaJ, GrpE). Moreover, both bacterial and human Hsp70/Hsp40 unfolding/refolding activities of model chaperone substrates were strongly inhibited by the oligomers but, remarkably, not by unstructured α-Syn monomers even in large excess. The oligomers acted as a specific competitive inhibitor of the J-domain co-chaperones, indicating that J-domain co-chaperones may preferably bind to exposed bulky misfolded structures in misfolded proteins and, thus, complement Hsp70s that bind to extended segments. Together, our findings suggest that inhibition of the Hsp70/Hsp40 chaperone system by α-Syn oligomers may contribute to the disruption of protein homeostasis in dopaminergic neurons, leading to apoptosis and tissue loss in Parkinson disease and related neurodegenerative diseases.
Keywords: Amyloid, Atomic Force Microscopy, Chaperone Chaperonin, Neurodegeneration, Parkinson Disease, Protein Conformation
Introduction
A number of neurodegenerative disorders, such as Alzheimer, amyotrophic lateral sclerosis, Huntington, prion encephalopathy, and Parkinson diseases (PDs)2 are characterized by a loss of neurons associated with protein misfolding and the accumulation in and outside cells of stable protein aggregates composed of specific proteins, such as tau tangles, amyloid-β plaques, α-Syn fibrils, and Lewy bodies (1). Under ideal in vitro conditions, such as low protein concentrations and low temperatures, the primary amino acid sequence may suffice to dictate the spontaneous folding of polypeptides into discrete three-dimensional, active structures called the native state (2). Yet in the crowded environment of the cell (for review, see Ref. 3) and especially under stress conditions, such as heat shock, de novo synthesized or imported polypeptides and mutant or damaged proteins may undergo transient unfolding, thereby exposing hydrophobic segments that readily self-associate to form stable non-functional high molecular weight, β-sheet-enriched, oligomers and fibrillar assemblies, generally named “aggregates” and amyloids (4, 5).
PD is characterized by the selective degeneration of dopaminergic neurons in the substantia nigra of human brain alongside with the presence of cytoplasmic neuronal inclusions called Lewy bodies (LBs). LBs are proteinaceous inclusions mainly composed of aggregates and insoluble fibrillar α-Syn (6) associated with different members of the protein homeostasis machinery. LBs and other types of α-Syn inclusions are also found in cases of dementia in some types of Alzheimer disease, Down syndrome, and several other neurological diseases, collectively denominated synucleinopathies (for review, see Ref. 7).
α-Syn is a 14.5-kDa protein mainly expressed in the central and peripheral nervous system of vertebrates, from torpedo fish to humans (8). In humans, it is expressed mostly in presynaptic terminals (9), astrocytic and oligodendroglial cells (10). Under physiological conditions, native soluble α-Syn is involved in the differentiation of dopaminergic cells, where it functions as an activity-dependent negative regulator of dopamine neurotransmission (for review, see Ref. 11). Because in the purified state, human α-Syn is a monomer apparently devoid of stable secondary structures, it has been described as “natively” unfolded or intrinsically unstructured (12, 13). Yet, in the membrane-rich crowded environment of the cell, α-Syn may become structured and upon binding to membranes is thought to adopt partially an α-helical conformation (14). During PD pathogenesis or aging, loose α-helical or natively unfolded α-Syn monomers may spontaneously convert, by a mechanism still unclear, into highly stable β-sheet-enriched oligomers, some of which protofibrils (15) that can ultimately form more compact, protease-resistant and apparently less toxic fibrils in LBs (for review, see Ref. 16). Three specific point mutations in α-Syn have been linked to autosomal dominant inherited forms of PD and correlate with early onset of the disease (17–19). Interestingly, all three also accelerate the in vitro oligomerization and fibril formation of α-Syn and promote the spontaneous formation of toxic oligomers, including amyloid pores (20).
There is strong evidence that the soluble, low molecular weight forms of β-sheet-enriched oligomers are the primary toxic species in the disease (21), whereas fibril formation is already part of a detoxification mechanism whereby toxic oligomers become sequestered and, thus, incapacitated at participating in aberrant interactions with membranes and other proteins of the cell (for review, see Ref. 1). In solution and at high concentrations (>200 μm), α-Syn spontaneously forms annular pore-like structures (20, 21) that, when added externally to the culture medium, can cause toxic ion leakage in neuroblastoma cells (22).
Here, we developed a reproducible protocol for preparing and purifying a homogeneous population of highly stable β-sheet enriched, toroid-shaped α-Syn oligomers with a diameter typical of toxic protofibrils. We then compared the effect of purified native monomeric α-Syn to that of the purified oligomers on the in vitro ATP-dependent chaperone unfolding/refolding activity of bacterial (DnaK, DnaJ or CbpA, GrpE) or human (HSPA1A, DNAJA1) Hsp70/Hsp40 chaperones. We found that α-Syn oligomers can specifically inhibit the function of the Hsp70/Hsp40 chaperone systems, suggesting that α-Syn oligomer-induced toxicity may also result from the specific stalling of the cellular chaperone machinery, leading to the collapse of protein homeostasis and pathogenesis.
EXPERIMENTAL PROCEDURES
Purification and Characterization of Recombinant α-Syn
pT7 plasmid carrying the human wild-type (WT) α-Syn gene was expressed in BL21 (DE3) Escherichia coli cells. Cells were grown in 1.2 liters of Lysogeny (Luria) Broth medium containing 100 mg/liter ampicillin at 37 °C with shaking. At A0.6, 0.2 mm isopropyl-β-d-thiogalactopyranoside was added, and cells were further grown for 3 h. Centrifuged cells were sonicated in buffer A (50 mm Tris, pH 8.0, 50 mm KCl, 5 mm Mg(Ac)2, 0.1% Na azide), 0.2 mg/ml lysozyme, and the protease inhibitor mixture from Sigma (catalog no. P8465). After 15 min of centrifugation at 6000 rpm, the supernatant was heated at 73 °C for 4.5 min. After a second 15-min centrifugation at 10,000 rpm, the supernatant was precipitated in 40% (NH4)2SO4 and incubated overnight at 4 °C. After a 15-min centrifugation at 6000 rpm, the pellet was then resuspended in Buffer A, filtered through a 0.2-μm membrane, and loaded on a Superose 6 gel filtration column (Amersham Biosciences). Proteins were eluted in buffer A at a flow rate of 0.5 ml/min. For each eluted peak, fractions were pooled and concentrated by ultrafiltration in a 100-kDa cut-off membrane (Sartorius 13269E) and analyzed on a 15% SDS-PAGE or on a 5.5% native PAGE and stained with Coomassie Blue. The total protein concentration was determined at 560 nm with the bicinchoninic acid assay (BCA) (Sigma) using bovine serum albumin as a standard and then stored in buffer A supplemented with 20% glycerol at −80 °C.
Immunoblotting
Samples were separated on a 7.5% native PAGE or on a 12% SDS-PAGE (Fig. 1) and transferred by electroblotting to a nitrocellulose membrane (Bio-Rad) using standard protocols. Membranes were blocked in 1% skimmed milk and incubated overnight at 4 °C with a rabbit anti-α-Syn polyclonal antibody (Stressgen, Ann Arbor, MI, catalog no. 905-565) (1/1000, v/v) and then incubated with a HRP-labeled goat anti-rabbit antibody (Bio-Rad, catalog no. 170-5046) (1/15000, v/v). Immune complexes were visualized using the chemiluminescent ImmunstarTMKit (Bio-Rad) according to the manufacturer's instructions.
FIGURE 1.
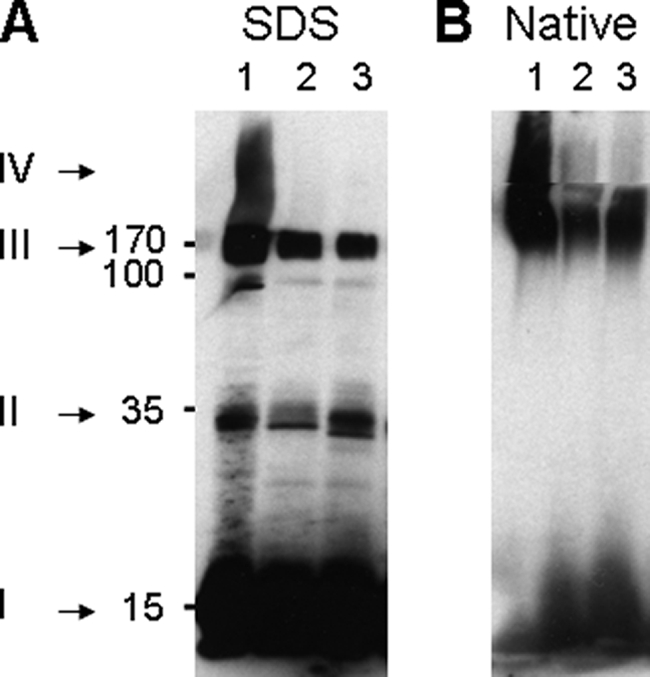
The initial stages in the purification of recombinant α-synuclein protofibrils. Shown is an immunoblot with an anti-α-synuclein antibody from a SDS gel (A) or a native gel (B) of cell extracts from BL21 E. coli cells expressing WT human α-synuclein. Lane 1, soluble initial crude extract. Lane 2, soluble extract after heat treatment. Lane 3, resolubilized ammonium sulfate pellet from the heat-treated soluble extract in lane 2. Numbers, estimated molecular masses from the SDS gel in kDa. Roman numbers with arrows: I, monomers; II, dimers; III, soluble oligomers; IV, large oligomers that do not enter the resolution gel.
Proteins
DnaK, DnaJ, and CbpA were purified according to Gur et al. (24); ClpB was purified according to Woo et al. (25). His-tagged Luciferase was expressed from the pT7lucC-His plasmid from A. S. Spirin (The Protein Research Institute, Moscow Region, Russia) and purified according to Svetlov et al. (26). GrpE was a gift from H.-J. Schönfeld, F. Hoffmann-La Roche, Basel, Switzerland. Glucose-6-phosphate Dehydrogenase (G6PDH) from Leuconostoc mesenteroides and BSA were from Sigma. Heat denaturation of the G6PDH was as described in Ben-Zvi et al. (27). Cold inactivation of His-tagged Luciferase was achieved by four consecutive cycles of freezing in liquid nitrogen and slow thawing at 4 °C (62).
Atomic Force Microscopy
10 μl of α-Syn monomers or oligomers (1 μm each, expressed in protomers) in buffer (50 mm Tris, 50 mm KCl, 10 mm MgCl2, pH 7.5) were deposited onto freshly cleaved mica at room temperature. After 1 min, the sample was gently rinsed with 1 ml of nanopure H2O and dried. Samples were imaged using a Nanoscope III (Digital Instrument, Santa Barbara, CA) in the tapping mode. The probes were NSC35 (NT-MDT, Troisk, Russia).
Reactivation of Heat-preaggregated G6PDH by Chaperones
Heat-preaggregated G6PDH was refolded by the DnaK chaperone system as described in Ben-Zvi et al. (27) with the following modifications; 0.75 μm heat-aggregated G6PDH (final concentration) was reactivated in the presence of 5 μm DnaK, 5 μm DnaJ, 0.5 μm GrpE (the full DnaK chaperone system) or 5 μm HSPA1A and 5 μm DNAJA1 (the minimal human Hsp70/40 system), and 4 mm ATP. G6PDH activity was measured at different times of chaperone-mediated refolding reaction at 30 °C.
Luciferase Assays
Luciferase activity was measured as described previously (26) using a Victor Light 1420 Luminescence Counter from PerkinElmer Life Sciences.
Inhibition of G6PDH Reactivation by α-Syn Species
Aliquots from each eluted fraction resulting from the gel filtration chromatography were diluted to a final concentration of 2.2 μm α-Syn and added to 0.75 μm heat-inactivated G6PDH and incubated in the presence of the full DnaK/DnaJ/GrpE bacterial system or the minimal Hsp70/Hsp40 human system at 30 °C for 1 h. G6PDH activity was measured at different times of chaperone-mediated refolding reaction.
Thioflavin T Fluorescence Assays
The β-sheet content of G6PDH and α-Syn were evaluated by measuring the fluorescence in a luminescence spectrometer, PerkinElmer Life Sciences LS 55 (excitation, 446 nm; emission, 480 nm), in a 500-μl cuvette containing 10 μm thioflavin T (Th-T) (final concentration) in the refolding buffer: 100 mm Tris-HCl, pH 7.5, 100 mm KCl, 10 mm MgAc, 10 mm DTT, 4 mm ATP, 4 mm PEP, and 0.02 mg/ml PK.
RESULTS
To address the possible effects of unstructured α-Syn monomers and structured oligomers on the in vitro activity of a typical Hsp70/Hsp40/NEF chaperone system (here, the E. coli DnaK/DnaJ/GrpE chaperone machinery), we first designed a protocol to purify a distinct population of stable α-Syn oligomers with reproducible characteristics. Immunoblots from SDS gels of soluble extracts from E. coli cells overexpressing plasmid-encoded human α-Syn revealed a predominant fraction of SDS-soluble 14.5-kDa α-Syn monomers (Fig. 1A, lane 1, I) and a minor fraction of SDS-resistant oligomers: some dimers (Fig. 1A, lane 1, II), high molecular weight species that migrated as 10–14-mers (Fig. 1A, lane 1, III), and very large insoluble oligomers that did not enter the resolution gel (Fig. 1A, lane 1, IV). To deplete this last insoluble fraction of very large SDS-resistant oligomers (fraction IV), the extract was heat-treated at 73 °C for 4.5 min and centrifuged (14,000 rpm, 15 min) (Fig. 1A, lane 2). After ammonium sulfate precipitation, we obtained a much reduced fraction, albeit of ∼95% pure α-Syn, that was devoid of large insoluble species, composed of fully soluble, discrete oligomers that resolved on native gel mainly as 10–14 mers (140–200 kDa) as well as some dimers and monomers (Fig. 1B, lane 3).
To better understand the relative size distribution of monomeric and oligomeric α-Syn complexes, this soluble fraction was further separated by gel filtration (Fig. 2A, blue line). As expected from the native gel (Fig. 1B), we observed two broad peaks; one corresponding to soluble high molecular weight (Mr) α-Syn oligomers ranging from 102 to 103 kDa (7.5–11 ml) with a very high specific affinity for the amyloid specific dye, Th-T, and a second that was resolved at the expected position of the 14.5-kDa α-Syn monomer (15–18 ml) with a very low specific affinity for Th-T (Fig. 2A, dark line). SDS and native gel electrophoresis confirmed that this low Mr fraction contained a uniform population of SDS-soluble 14.5-kDa monomers whose low affinity for Th-T is indicative of a lack of β-sheet structures, in agreement with the intrinsically unfolded state that is predominantly observed in the case of α-Syn monomers in vitro (13). In contrast, SDS and native gel electrophoresis confirmed that the high Mr, β-sheet-enriched fraction from the 7.5–11 ml peak was partially SDS-resistant (Fig. 2B) and formed on the native gel a remarkably discrete soluble α-Syn oligomer whose high stability was reflected by its ability to withstand extensive dilutions during gel filtration and native gel electrophoresis (Fig. 2C).
FIGURE 2.

Characterization of recombinant human α-synuclein (AS). A, shown is size exclusion chromatography of recombinant α-synuclein from the resolubilized amonium sulfate pellet of the heat-treated crude extract shown in lane 3 of Fig. 1, A and B. Elution profile from the Superose 6 column shows the protein concentration profile according to the absorbance at 280 nm (blue circles) and the Th-T fluorescence profile (black circles). Above the inset, Coomassie stain of the corresponding eluted fractions after separation on 12% SDS-PAGE is shown. B and C, two fractions were pooled from 15–18 ml as monomeric AS and from 7.5–11 ml as oligomeric AS, concentrated, and further separated by SDS-PAGE (B) or native-PAGE (C) and Coomassie-stained. Standard molecular masses (M) in kDa are indicated for the SDS-gel. D–F, shown are atomic force microscopy morphological studies of AS monomers and AS oligomers, as in B and C. Atomic force microscopy images (1 μm × 1 μm) of AS monomers (D) and AS oligomers (E) are shown. At higher magnitude, AS oligomers present a globular flattened-shape structure (F). G, shown is cumulative function of the diameter distribution of the AS oligomers with a mean size of 21.1 nm and a S.D. s = 2.4 nm. Inset, shown is s histogram of the distribution superimposed with a Gaussian (mean = 21.1 nm, s = 2.4 nm).
The discrete nature of this stable high Mr, yet soluble α-Syn oligomer was confirmed by atomic force microscopy (Fig. 2, D–G); whereas the low Mr fraction contained mostly α-Syn monomers, undetectable at this low resolution (Fig. 2D), the high Mr fraction showed a dense, remarkably homogeneous population of globular, mildly flattened oligomers (Fig. 2E), with a narrow size distribution (average diameter of 21 nm ± 3 nm) (Fig. 2G). The tapping procedure revealed a weaker region at the center of the particles, suggesting a less dense interior or a pore-like toroid (Fig. 2F) indistinguishable in size and shape from previously described toxic pore-forming α-Syn oligomers (20).
This protocol of production of stable α-Syn oligomers was highly reproducible. Yet, depending on the batches, α-Syn oligomers bound about 20 times more Th-T than monomeric α-Syn and about 8 times more than heat-aggregated G6PDH (Fig. 3A). G6PDH is a heat-generated misfolded protein substrate that is commonly used to test the unfolding/refolding activity of molecular chaperones (5, 27). The high stability of the β-sheet structures in the α-Syn oligomers was confirmed by their relative high resistance to prolonged (48 h) incubations after extensive dilutions; whereas prolonged incubations of heat-aggregated G6PDH caused a 33% loss of Th-T binding (Fig. 3B), only ∼8% loss of Th-T binding was observed in the case of α-Syn oligomers under identical conditions (Fig. 3C).
FIGURE 3.

AS oligomers are stable and β-sheet-enriched. A, the specific β-sheet content of various protein forms is shown. The relative Th-T fluorescence by a constant amount (1 μm, expressed in protomers) of AS oligomers was compared with AS monomers (black bars) to heat pre-aggregated and to native G6PDH (white bars). B and C, shown is an estimation of the relative stability of various oligomers. The relative Th-T fluorescence (normalized to 1 μm, expressed in protomers) of heat-pre-aggregated G6PDH (B) and AS oligomers (C) was measured either immediately after dilutions from 1 to 0.1 μm (open circles) or 48 h after dilutions (filled symbols). AU, absorbance units.
We next addressed the biochemical properties of α-Syn oligomers as a possible substrate of the full E. coli Hsp70 chaperone machinery. Confirming earlier observations (27), in the presence of DnaK, DnaJ, GrpE, and ATP, heat-preaggregated G6PDH lost 25% of its Th-T binding structures within 50 min (Fig. 4A, open squares) and correspondingly formed 25% of natively refolded G6PDH enzyme (Fig. 4A, filled circles). In contrast, the α-Syn oligomers lost less than 10% of their Th-T binding structures (Fig. 4A, filled squares), indicating that although interacting with the aggregates, the chaperones are less effective at unfolding the misfolded β-sheets structures in α-Syn oligomers than in heat-aggregated G6PDH substrate. Remarkably, immunoblots of native gels confirmed that the Hsp70 chaperone machinery can convert a significant fraction of the stable α-Syn oligomers into monomers in an ATP-dependent manner (Fig. 4B). This indicates that the chaperone machinery, although capable of interacting and causing the partial disassociation of the oligomers, are poorly effective at fully unfolding stably misfolded β-sheets structures into unstructured polypeptide segments.
FIGURE 4.

AS oligomers are partially resistant to the Hsp70 chaperone machinery. A, AS oligomers are more resistant to the unfolding chaperones than G6PDH aggregates. Time-dependent loss of Th-T fluorescence (squares) of 1 μm heat pre-aggregated G6PDH (open squares) or 1 μm (protomers) AS oligomers (filled squares) in the presence of 5 μm DnaK, 0.75 μm DnaJ, and 1 μm GrpE (KJE) and ATP and the corresponding time-dependent reactivation of native G6PDH (filled circles) is shown. B, AS oligomers are partially dissociated by the ATP-fueled DnaK/DnaJ/GrpE chaperone system. AS oligomers were incubated as in A but for 3 h at 30 °C with or without ATP and chaperones, as specified. Samples were separated on native gel, and α-synuclein was detected by Western blot analysis as in Fig. 1.
Thus, we next addressed the possibility that the oligomers may stall the Hsp70/40 chaperone machinery. The in vitro chaperone-mediated ATP-dependent reactivation of G6PDH was tested in the presence of increasing amounts of monomeric or oligomeric α-Syn (Fig. 5A). We found that the refolding rate of heat-pre-aggregated G6PDH by an excess of chaperones (5.0 μm DnaK, 1 μm DnaJ, and 0.5 μm GrpE) was strongly inhibited by α-Syn oligomers but not by the monomers. Half-inhibition of the chaperone reaction was observed in the presence of 2 μm α-Syn oligomers (expressed in protomers) (Fig. 5B). Remarkably, up to a 10-fold molar excess of monomeric α-Syn did not inhibit and was even slightly beneficial to the chaperone reaction (Fig. 5A, open circles). Chaperone inhibition was specific to α-Syn oligomers and not to other types of protein aggregates, as equimolar amounts of heat-aggregated MDH or of cold-inactivated luciferase did not compete with the chaperone-mediated reactivation of heat-denatured G6PDH (supplemental Fig. S1). Moreover, the oligomers inhibited the chaperone-mediated refolding of cold-inactivated Luciferase to the same extent as they did in the case of heat-denatured G6PDH (supplemental Fig. S2), indicating that inhibition is not caused by the competition between the different substrates for a limited amount of chaperones but rather by a non-competitive action mediated by a specific interaction of the oligomers with a component of the chaperone system. This specific chaperone inhibitory effect was further tested on different α-Syn oligomers with increasing apparent molecular masses (and/or shapes, as elongated proteins may elute with larger than real apparent molecular masses) obtained by gel filtration fractionation. Remarkably, the size/shape-dependent profile of the specific chaperone inhibitory activity (Fig. 5C) matched that of the specific Th-T binding (Figs. 2A and 5C), suggesting a strong correlation between the β-sheet or amyloid-like content of the α-Syn oligomers and their ability to inhibit the chaperone activity.
FIGURE 5.

AS oligomers inhibit the unfolding/refolding activity of the Hsp70 chaperone system. A, time-dependent reactivation of heat-pre-aggregated G6PDH by the full DnaK chaperone system is shown. The heat pre-aggregated G6PDH (0,75 μm) was first incubated with increasing concentrations of AS oligomers (0- 20 μm protomers) (filled symbols) or 20 μm AS monomers (open symbols, asterisk) and then incubated with 5 μm DnaK, 1 μm DnaJ, 0.5 μm GrpE, and ATP. B, optimal G6PDH refolding rates from A as a function of increasing concentrations of AS monomers (open triangles) or oligomers (filled triangles) are shown. C, the specific chaperone inhibitory activity profile (open circles) compared with the Th-T binding profile (filled circles) of AS species in the different fractions from the size exclusion chromatography in Fig. 2B is shown.
The specific in vitro inhibitory effect of the α-Syn oligomers was confirmed with HSPA1A and DNAJA1, which represent a central hub of the stress-induced chaperone network and are among the most strongly expressed Hsp70 chaperone and Hsp40 co-chaperones of the cytoplasm of human cells, including PD neurons prone to α-Syn aggregation (61). We found that, similar to E. coli Dnak/DnaJ/GrpE chaperones, α-Syn oligomers, but not monomers, strongly inhibited the human HSPA1A/DNAJA1 chaperone machinery (supplemental Fig. S3A). Moreover, in the presence of 1.5 μm DNAJA1, inhibition was half, and in the presence of 3 μm DNAJA1, inhibition was fully alleviated. This confirms that, as with bacterial chaperones, the inhibition of the human chaperone system by the α-Syn oligomers, but not by the monomers, is also predominantly caused by the incapacitation of the Hsp40 co-chaperone (supplemental Fig. S3B).
Both chaperone and co-chaperone can bind to unfolded or misfolded proteins, albeit in different manners (28). Whereas in its ADP-liganded and substrate-bound state, DnaK may tightly bind only to protruding unfolded hydrophobic polypeptide segments by tightly enclosing them, DnaJ may directly bind with much less steric limitations to bulky hydrophobic surfaces on misfolded structures on the substrate (28–30). Therefore, we next considered the possibility that β-sheet-enriched oligomers expose binding sites with a particular affinity for DnaJ, thereby potentially stalling the chaperone reaction. This was addressed by measuring the rates of ATP- and chaperone-mediated refolding without or with a fixed amount of oligomeric α-Syn in the presence of increasing concentrations of DnaK (Fig. 6A) or of DnaJ (or the DnaJ-like co-chaperone CbpA) (Fig. 6, B and C) or of chaperone substrate (Fig. 6D). The half inhibitory effect (IC50), which was observed at 5 μm DnaK, was not alleviated by higher DnaK concentrations of up to 13 μm (Fig. 6A), suggesting that inhibition is not because DnaK becomes sequestered by the oligomers. As expected, excess GrpE did not alleviate inhibition (data not shown), consistent with the fact that GrpE is a mere nucleotide exchange factor that exclusively interacts with DnaK. In contrast, increasing concentrations of the J-domain co-chaperones were found to gradually alleviate chaperone inhibition by oligomers (Fig. 6, B and C); inhibition was strongly diminished above 2.5 μm DnaJ and was negligible at 5 μm DnaJ (Fig. 6B). This was confirmed using CbpA, which can substitute for DnaJ in our type of in vitro DnaK-mediated protein disaggregation assays (24); the inhibition, which was initially set to 50%, remained so below 1 μm CbpA. The inhibition gradually decreased above 1 μm CbpA and in the presence of 5 μm CbpA was reduced to 10% (Fig. 6C). This suggests that α-Syn oligomers may inhibit the chaperone reaction by way of specifically sequestering the J-domain co-chaperones.
FIGURE 6.

AS protofibrils interact with J-domain chaperones. The reactivation of heat-pre-aggregated G6PDH was performed under the same conditions as in Fig. 4A with constant GrpE (0.5 μm) and increasing DnaK, DnaJ, or CbpA or luciferase substrate concentrations without (open circles) or with AS oligomers (4 μm protomers) (filled circles). A, shown are optimal rates of G6PDH reactivation by constant (1 μm) DnaJ and increasing DnaK concentrations. B, shown are optimal rates of G6PDH reactivation by constant (5 μm) DnaK and increasing DnaJ concentrations. C, procedures are as in B but with increasing CbpA concentrations instead of DnaJ. D, shown is optimal rates of luciferase reactivation by constant (0.8 μm) DnaK, (0.4 μm) DnaJ, and (0.8 μm) GrpE and increasing concentrations of inactive luciferase substrate without (open circles) or with AS oligomers (2 μm protomers) (filled circles).
Because the same quality of G6PDH aggregates cannot be obtained when they have been heat-denatured at different concentrations (31), we chose to use freeze-inactivated luciferase, which shows no differences of chaperone reactivity once inactivated at different concentrations (62) to test the effect of increasing concentrations of a chaperone substrate. Here again, the same amount of oligomers, which inhibited by half the chaperone-mediated refolding of 125 nm luciferase, kept inhibiting by more than half the refolding of five times more luciferase (650 nm) substrate (Fig. 6D). Thus, inhibition cannot be attributed to the sequestering of the chaperone substrate by α-Syn oligomers.
DnaJ and α-Syn being devoid of Trp residues, we could follow the changes in the steady-state intrinsic fluorescence of the unique Trp residue, which is in the nucleotide binding domain of DnaK, in the presence of ATP and increasing amounts of monomeric or oligomeric α-Syn with or without DnaJ. Fluorescence changes (supplemental Fig. S4) showed that in the absence of DnaJ, DnaK had the same low apparent affinity for monomeric α-Syn as for oligomeric α-Syn. In contrast, in the presence of DnaJ, increasing amounts of oligomeric α-Syn caused a dramatic change in the DnaK fluorescence, whereas increasing amounts of monomeric α-Syn caused similar minor fluorescence changes, as without DnaJ. This confirms that it is the DnaJ co-chaperone rather than the DnaK chaperone that is able to specifically distinguish between oligomeric and monomeric α-Syn. Moreover, DnaJ acts as if it instructed DnaK to interact with the oligomers and specifically act upon them as a disaggregating/unfolding chaperone.
DISCUSSION
Recombinant α-Syn Oligomers Recapitulate Biochemical Properties of Oligomers from PD Tissues
The physiological non-toxic species of α-Syn is presumably a soluble monomer that in healthy neurons may loosely bind to membranes as amphiphilic α-helices (14). In the purified state the full-length α-Syn polypeptide is found in a so-called natively unfolded or unstructured state, apparently devoid of secondary structures. At high concentrations, β-sheet-enriched oligomers may spontaneously form by a yet unclear mechanism and undergo subsequent growth and/or self-assembly to form the mature fibrils found in the Lewy bodies of PD brains (32). Soluble oligomers, typically composed of about 12–24 subunits, were isolated from the cerebral cortex of PD and dementia with LBs patients and from α-Syn-transgenic mesencephalic neuronal (MES) cells and mouse brains (33). Because oligomers have a higher surface/volume ratio as compared with large fibrils and amyloids, they can be very toxic, and their apparition in cells correlates with a general failure of the protein homeostasis (proteostasis) machinery, the onset of apoptosis, tissue loss, and neurodegeneration (34, 35).
Both artificially formed and in vivo formed α-Syn oligomers are partially resistant to detergents such as Triton and SDS (36) and have a typical high affinity for the amyloid-specific dyes like Congo red or Th-T (37). Upon reconstitution in synthetic membranes, they can spontaneously form discrete spheroid pore-like structures that cause ion leakage (38). α-Syn oligomers have been previously isolated from cold-induced dissociation of amyloid fibrils (39). Here, we described a protocol to isolate recombinant human α-Syn oligomers that recapitulate a number of important properties of the toxic α-Syn oligomers found in PD neuronal tissues; they were very stable soluble oligomeric species that were partially resistant to SDS. They could withstand extensive dilutions and native gel electrophoresis and bound much more Th-T (here about 20 times) than the monomeric form. Moreover, the α-Syn oligomers were a remarkably uniform population of discrete spheroid-like flattened structures of the same diameter as previously described for the toxic species shown to spontaneously form pores in artificial membranes and cause ion leakage in cell cultures.
Yet, in contrast to classic protofibrils, we found that these oligomers remained ineffective as seeds of spontaneous α-Syn fibril formation in vitro (supplemental Fig. S5). This suggests that our specific α-Syn oligomers are off-fibrillation pathway species whose negative effects on the chaperone network and possibly on the cell cannot be neutralized by the spontaneous or aggresome-mediated formation of less toxic larger fibrils and amyloids.
The α-Syn Oligomers Feebly but Effectively Interact with the Hsp70 Chaperone Machinery
Although Th-T fluorescence showed that, as compared with denatured G6PDH or luciferase, the α-Syn oligomers were relatively resistant, Western blot analysis of native gels revealed that oligomers were relatively efficiently disaggregated by the bacterial ATP-dependent Hsp70 chaperone machinery. This is consistent with earlier finding (40) showing that human Hsp70 can efficiently inhibit α-Syn fibril formation in vitro and further suggests that partial ATP-fuelled, chaperone-mediated unfolding can lead to the partial disaggregation of α-Syn oligomers. It should be noted, however, that incomplete chaperone-mediated unfolding of more compact and, therefore, less toxic aggregates could lead to a transient increase of protein toxicity when more active species are formed but are not further converted into nontoxic monomers. The concomitant binding of more than one Hsp70 molecule to the same misfolded polypeptide has been shown to be essential for the effective cooperative unfolding action of the chaperone on relatively resistant protein aggregates such as heat-denatured G6PDH species (27, 41). According to the algorithm of Rüdiger et al. (29), the α-Syn sequence has only one good potential Hsp70 binding site (instead of the three-four sites expected on an average polypeptide of the same size). The lack of another chaperone binding site would strongly limit the ability of the single chaperone molecule to efficiently pull and unfold the stable β-sheets and could explain the apparent resistance of misfolded α-Syn conformers to the protein quality control machineries in neurons affected by PD (for review, see Ref. 4).
α-Syn Oligomers Inhibit the Unfolding Activity of the Hsp70/40 System
Consistent with the observed co-localization of human Hsp40 proteins (and Hsp70) with α-Syn in Lewy Bodies (34), our α-Syn oligomers behaved in vitro as a competitive inhibitor of the human and bacterial (E. coli) Hsp70/40 chaperone machineries by interacting with J-domain co-chaperones. Noticeably, we found that DnaJ preincubated either with α-Syn monomers or with oligomers did not show differences of elution profiles on gel filtration and, moreover, that DnaJ did not co-immunoprecipitate with α-Syn oligomers better than with monomers (data not shown). We conclude that the specific strong inhibitory effect that is exerted by the α-Syn oligomers on the J-domain co-chaperones must be mediated by rather weak interactions. This is not entirely surprising given that a very ubiquitous “generalist” chaperone, such as Hsp70/Hsp40 could hardly afford having very strong affinities for hundreds of different substrate proteins in various misfolded and aggregated states in the same cell.
We found that tryptophan fluorescence independently supported the evidence from the chaperone assays and confirmed that the J-domain co-chaperone is the primary component of the Hsp70 chaperone system that can specifically distinguish between the oligomeric and the monomeric form of α-Syn and, moreover, direct Hsp70 to interact with α-Syn oligomers in a limited attempt to disaggregate them.
The binding of de novo-forming oligomers to Hsp40 co-chaperones would partially sequester and, therefore, neutralize the whole Hsp70/Hsp40 chaperone system, adding to the direct deleterious effects of oligomers on membrane integrity (38) and the indirect effects on the stability of other labile and mutant proteins in the cell, as observed in the case of polyQ aggregates in thermo-sensitive mutants of Caenorhabditis elegans (42). In addition, because Hsp70/Hsp40 activity can repress IkB and, therefore, repress NF-κB-mediated apoptosis (43), the blockage of Hsp70/Hsp40 by α-Syn oligomers could promote neuroinflammation, cell death, and tissue loss in PD.
Moreover, the toxicity of α-Syn oligomers may not only result from the inhibition of the Hsp70/40 chaperone machinery. α-Syn oligomers have also been shown to strongly inhibit in vitro protein degradation mediated by the 26 S proteasome (44). Likewise, PC12 cells expressing the pathogenic A53T and/or A30P α-Syn diseases-associated mutants showed an altered lysosomal activity as compared with WT α-Syn (45). This fits in vivo observations of altered proteasomal and/or lysosomal pathways in dopamine neurons of the substantia nigra from PD patients (35). Together, this suggests that α-Syn oligomers may impair their own degradation along with that of other damaged proteins in the cell, leading to a vicious cycle of proteostasis imbalance, causing cell death (for review, see Ref. 46).
The Hsp70/Hsp40 Is the Main Chaperone That Can Unfold Toxic Protein Conformers in Neurons
Previous studies have demonstrated the positive effect of molecular chaperones, more particularly of the Hsp70/40 chaperone system, on the cytotoxicity of α-Syn oligomers. Hsp70 overexpression in flies expressing α-Syn prevents dopaminergic neuronal loss caused by α-Syn oligomers. It also reduces α-Syn oligomerization both in mice and in H4 cells expressing α-Syn. In the mouse model Hsp70 overexpression leads to a significant reduction both in high molecular weight and detergent-insoluble α-Syn species (47). Moreover, purified Hsp70 effectively inhibits α-Syn fibril formation in vitro and suppresses the permeabilization of synthetic vesicles induced by prefibrillar α-Syn (40). Chaperones other than Hsp70/Hsp40 may also contribute to the reduction of toxic oligomers in the cytoplasm of mammalian cells; the expression of Hsp27 or of the yeast Hsp104 can reduce toxicity associated with α-Syn oligomers in human H4 neuroglioma cells or rat models of PD, respectively (48, 49). However, the contribution of sHSPs is expected to be limited to the passive prevention of protein aggregation as, unlike Hsp70/Hsp40, they cannot use the energy of ATP hydrolysis to unfold misfolded proteins. It can, however, stabilize and deliver misfolded proteins to the Hsp70/Hsp40 chaperone system (50).
True functional homologues of ClpB acting together with Hsp70/Hsp40/NEF in the disaggregation of large compact aggregates (51) have been identified only in bacteria in the organelles and the cytoplasm of plants, yeast, and fungi (for review, see Ref. 5). In animal cells, among the many cytoplasmic AAA+ proteins with conserved Walker motives, none has been convincingly shown thus far to function as true ClpB/Hsp104 homologues. Our immunoblot analysis showed that, unlike with a classic heat-aggregated chaperone substrate, α-Syn oligomers remained resistant to the unfolding action of ClpB, which remained unable to improve the limited disaggregating activity of DnaK/DnaJ/GrpE alone (data not shown). Hence, the Hsp70/40/NEF remains thus the only known chaperone machinery in the cytoplasm of mammalian cells that can use the energy of ATP hydrolysis to unfold toxic misfolded protein conformers, such as α-Syn oligomers, into non-toxic natively refoldable or protease-degradable species (5, 27).
Recruiting Hsp70/Hsp40/NEF to Reduce PD Is a Promising Therapeutic Approach
An age-dependent progressive failure of the protein clearance machinery, including the Hsp70/Hsp40 chaperone system, has been shown in aging organisms (for review, see Ref. 52). Interestingly, in agreement with our finding that J-domain co-chaperones play a central role in aggregate recognition and processing, a systematic screen in human cell cultures expressing disease-associated polyglutamine proteins (polyQ) revealed that two human DnaJ homologues, DNAJB6b and DNAJB8, are effective suppressors of polyQ aggregation and toxicity (53). This suggests new avenues of therapeutic approaches using chaperone-inducing drugs, such as Hsp90 inhibitors (54) or non-steroidal anti-inflammatory drugs (55). Alternatively, vectors could mediate the specific expression of particular effective J-domain co-chaperones in aging or diseased neurons, thus, targeting more effectively the unfoldase activity of the cytoplasmic Hsp70/Hsp40s to the cytotoxic α-Syn oligomers in PD.
Mechanistic Implications; J-domain Co-chaperones May Preferably Bind Misfolded Structures
Although the native unstructured monomeric form of human α-Syn apparently optimally exposes a typical high affinity binding motive, 32KTKEGVLYVGSKTR45, for the potential binding and locking of Hsp70 (29) (see supplemental Fig. S6), we found that even in large molar excess, it did not inhibit the ATP- and DnaK/DnaJ/GrpE-mediated unfolding/refolding reaction of classic amenable chaperone substrates. This is consistent with NMR and gel filtration studies that show no interaction of Hsp70 with monomeric α-Syn (56, 57). Our work further shows that DnaJ too has no apparent affinity for the unstructured monomeric form of α-Syn. In contrast, we found that the three J-domain co-chaperones, DnaJ, CbpA, and DNAJA1, have an apparent specific affinity for the structured oligomeric form of the same protein. This is consistent with earlier findings showing that the yeast Sis1 and Ydj1 Hsp40s co-chaperones have a stronger affinity for Sup35 prionogenic oligomers than for the soluble monomers (58). Thus, unlike Hsp70, which in the ADP-liganded state may only “lock” around extended loops extruding from the aggregate, J-domain co-chaperones may directly bind surfaces of bulky misfolded structures made of hydrophobic residues joined in trans, which are otherwise individually scattered in the primary sequence. This could explain why in the unfolded α-Syn monomer, for lack of a DnaJ binding site, DnaK lacks the ability to lock onto the DnaK binding site and moreover could justify why J-domain co-chaperones do not necessarily compete with Hsp70 for substrate binding and why chaperone and co-chaperone best collaborate when they are bound alongside rather than instead of each other on the same native (59) or unfolded polypeptide substrate (23, 28). Moreover, this would explain for the first time how, subsequent to their initial binding to the substrate, J-domain co-chaperones tend to dissociate from the product of the Hsp70-mediated ATP-fueled unfolding reaction (60); with the co-chaperones preferentially binding the composite trans-assembled structures, the unfolding action of substrate-bound Hsp70 (41) would result in the progressive destruction of the co-chaperone-binding sites on substrate.
Supplementary Material
Acknowledgments
We thank Paolo De Los Rios for discussions, Eliora Ron and Eyal Gur for CbpA and DjlA plasmids, and Sandeep Sharma and Valerie Grimminger for technical assistance.
This work was supported by Swiss National Science Foundation Grant 3100A0-109290 and by a Zwahlen grant from the Faculty of Biology and Medicine of Lausanne University.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S6.
- PD
- Parkinson disease
- LB
- Lewy bodies
- G6PDH
- glucose-6-phosphate dehydrogenase
- Th-T
- thioflavin T
- AS
- α-synuclein
- NEF
- Nucleotide Exchange Factor
- PEP
- phosphoenolpyruvate
- PK
- pyruvate kinase.
REFERENCES
- 1.Muchowski P. J. (2002) Neuron 35, 9–12 [DOI] [PubMed] [Google Scholar]
- 2.Anfinsen C. B. (1973) Science 181, 223–230 [DOI] [PubMed] [Google Scholar]
- 3.Hartl F. U., Hayer-Hartl M. (2009) Nat. Struct. Mol. Biol. 16, 574–581 [DOI] [PubMed] [Google Scholar]
- 4.Hinault M. P., Ben-Zvi A., Goloubinoff P. (2006) J. Mol. Neurosci. 30, 249–265 [DOI] [PubMed] [Google Scholar]
- 5.Sharma S. K., Christen P., Goloubinoff P. (2009) Curr. Protein Pept. Sci. 10, 432–446 [DOI] [PubMed] [Google Scholar]
- 6.Mezey E., Dehejia A. M., Harta G., Tresser N., Suchy S. F., Nussbaum R. L., Brownstein M. J., Polymeropoulos M. H. (1998) Mol. Psychiatry 3, 493–499 [DOI] [PubMed] [Google Scholar]
- 7.Spillantini M. G., Goedert M. (2000) Ann. N.Y. Acad. Sci. 920, 16–27 [DOI] [PubMed] [Google Scholar]
- 8.Lavedan C. (1998) Genome Res. 8, 871–880 [DOI] [PubMed] [Google Scholar]
- 9.Murphy D. D., Rueter S. M., Trojanowski J. Q., Lee V. M. (2000) J. Neurosci. 20, 3214–3220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wakabayashi K., Hayashi S., Yoshimoto M., Kudo H., Takahashi H. (2000) Acta Neuropathol. 99, 14–20 [DOI] [PubMed] [Google Scholar]
- 11.Tofaris G. K., Spillantini M. G. (2007) Cell. Mol. Life Sci. 64, 2194–2201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weinreb P. H., Zhen W., Poon A. W., Conway K. A., Lansbury P. T., Jr. (1996) Biochemistry 35, 13709–13715 [DOI] [PubMed] [Google Scholar]
- 13.Uversky V. N. (2003) J. Biomol. Struct. Dyn. 21, 211–234 [DOI] [PubMed] [Google Scholar]
- 14.Davidson W. S., Jonas A., Clayton D. F., George J. M. (1998) J. Biol. Chem. 273, 9443–9449 [DOI] [PubMed] [Google Scholar]
- 15.Goldberg M. S., Lansbury P. T., Jr. (2000) Nat. Cell Biol. 2, E115–E119 [DOI] [PubMed] [Google Scholar]
- 16.Volles M. J., Lansbury P. T., Jr. (2003) Biochemistry 42, 7871–7878 [DOI] [PubMed] [Google Scholar]
- 17.Polymeropoulos M. H., Lavedan C., Leroy E., Ide S. E., Dehejia A., Dutra A., Pike B., Root H., Rubenstein J., Boyer R., Stenroos E. S., Chandrasekharappa S., Athanassiadou A., Papapetropoulos T., Johnson W. G., Lazzarini A. M., Duvoisin R. C., Di Iorio G., Golbe L. I., Nussbaum R. L. (1997) Science 276, 2045–2047 [DOI] [PubMed] [Google Scholar]
- 18.Krüger R., Kuhn W., Müller T., Woitalla D., Graeber M., Kösel S., Przuntek H., Epplen J. T., Schöls L., Riess O. (1998) Nat. Genet. 18, 106–108 [DOI] [PubMed] [Google Scholar]
- 19.Zarranz J. J., Alegre J., Gómez-Esteban J. C., Lezcano E., Ros R., Ampuero I., Vidal L., Hoenicka J., Rodriguez O., Atarés B., Llorens V., Gomez Tortosa E., del Ser T., Muñoz D. G., de Yebenes J. G. (2004) Ann. Neurol. 55, 164–173 [DOI] [PubMed] [Google Scholar]
- 20.Lashuel H. A., Petre B. M., Wall J., Simon M., Nowak R. J., Walz T., Lansbury P. T., Jr. (2002) J. Mol. Biol. 322, 1089–1102 [DOI] [PubMed] [Google Scholar]
- 21.Volles M. J., Lansbury P. T., Jr. (2002) Biochemistry 41, 4595–4602 [DOI] [PubMed] [Google Scholar]
- 22.Furukawa K., Matsuzaki-Kobayashi M., Hasegawa T., Kikuchi A., Sugeno N., Itoyama Y., Wang Y., Yao P. J., Bushlin I., Takeda A. (2006) J. Neurochem. 97, 1071–1077 [DOI] [PubMed] [Google Scholar]
- 23.Laufen T., Mayer M. P., Beisel C., Klostermeier D., Mogk A., Reinstein J., Bukau B. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 5452–5457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gur E., Biran D., Shechter N., Genevaux P., Georgopoulos C., Ron E. Z. (2004) J. Bacteriol. 186, 7236–7242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Woo K. M., Kim K. I., Goldberg A. L., Ha D. B., Chung C. H. (1992) J. Biol. Chem. 267, 20429–20434 [PubMed] [Google Scholar]
- 26.Svetlov M. S., Kolb V. A., Spirin A. S. (2007) Mol. Biol. 41, 86–92 [PubMed] [Google Scholar]
- 27.Ben-Zvi A., De Los Rios P., Dietler G., Goloubinoff P. (2004) J. Biol. Chem. 279, 37298–37303 [DOI] [PubMed] [Google Scholar]
- 28.Han W., Christen P. (2004) FEBS Lett. 563, 146–150 [DOI] [PubMed] [Google Scholar]
- 29.Rüdiger S., Germeroth L., Schneider-Mergener J., Bukau B. (1997) EMBO J. 16, 1501–1507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rüdiger S., Schneider-Mergener J., Bukau B. (2001) EMBO J. 20, 1042–1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Diamant S., Ben-Zvi A. P., Bukau B., Goloubinoff P. (2000) J. Biol. Chem. 275, 21107–21113 [DOI] [PubMed] [Google Scholar]
- 32.Conway K. A., Harper J. D., Lansbury P. T. (1998) Nat. Med. 4, 1318–1320 [DOI] [PubMed] [Google Scholar]
- 33.Sharon R., Bar-Joseph I., Frosch M. P., Walsh D. M., Hamilton J. A., Selkoe D. J. (2003) Neuron 37, 583–595 [DOI] [PubMed] [Google Scholar]
- 34.Auluck P. K., Chan H. Y., Trojanowski J. Q., Lee V. M., Bonini N. M. (2002) Science 295, 865–868 [DOI] [PubMed] [Google Scholar]
- 35.Chu Y., Dodiya H., Aebischer P., Olanow C. W., Kordower J. H. (2009) Neurobiol. Dis. 35, 385–398 [DOI] [PubMed] [Google Scholar]
- 36.Campbell B. C., McLean C. A., Culvenor J. G., Gai W. P., Blumbergs P. C., Jäkälä P., Beyreuther K., Masters C. L., Li Q. X. (2001) J. Neurochem. 76, 87–96 [DOI] [PubMed] [Google Scholar]
- 37.Conway K. A., Harper J. D., Lansbury P. T., Jr. (2000) Biochemistry 39, 2552–2563 [DOI] [PubMed] [Google Scholar]
- 38.Volles M. J., Lee S. J., Rochet J. C., Shtilerman M. D., Ding T. T., Kessler J. C., Lansbury P. T., Jr. (2001) Biochemistry 40, 7812–7819 [DOI] [PubMed] [Google Scholar]
- 39.Kim H. Y., Cho M. K., Kumar A., Maier E., Siebenhaar C., Becker S., Fernandez C. O., Lashuel H. A., Benz R., Lange A., Zweckstetter M. (2009) J. Am. Chem. Soc. 131, 17482–17489 [DOI] [PubMed] [Google Scholar]
- 40.Huang C., Cheng H., Hao S., Zhou H., Zhang X., Gao J., Sun Q. H., Hu H., Wang C. C. (2006) J. Mol. Biol. 364, 323–336 [DOI] [PubMed] [Google Scholar]
- 41.De Los Rios P., Ben-Zvi A., Slutsky O., Azem A., Goloubinoff P. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 6166–6171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gidalevitz T., Ben-Zvi A., Ho K. H., Brignull H. R., Morimoto R. I. (2006) Science 311, 1471–1474 [DOI] [PubMed] [Google Scholar]
- 43.Weiss Y. G., Bromberg Z., Raj N., Raphael J., Goloubinoff P., Ben-Neriah Y., Deutschman C. S. (2007) Crit. Care Med. 35, 2128–2138 [DOI] [PubMed] [Google Scholar]
- 44.Zhang N. Y., Tang Z., Liu C. W. (2008) J. Biol. Chem. 283, 20288–20298 [DOI] [PubMed] [Google Scholar]
- 45.Cuervo A. M., Stefanis L., Fredenburg R., Lansbury P. T., Sulzer D. (2004) Science 305, 1292–1295 [DOI] [PubMed] [Google Scholar]
- 46.Balch W. E., Morimoto R. I., Dillin A., Kelly J. W. (2008) Science 319, 916–919 [DOI] [PubMed] [Google Scholar]
- 47.Klucken J., Shin Y., Masliah E., Hyman B. T., McLean P. J. (2004) J. Biol. Chem. 279, 25497–25502 [DOI] [PubMed] [Google Scholar]
- 48.Outeiro T. F., Klucken J., Strathearn K. E., Liu F., Nguyen P., Rochet J. C., Hyman B. T., McLean P. J. (2006) Biochem. Biophys. Res. Commun. 351, 631–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lo Bianco C., Shorter J., Régulier E., Lashuel H., Iwatsubo T., Lindquist S., Aebischer P. (2008) J. Clin. Invest. 118, 3087–3097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Veinger L., Diamant S., Buchner J., Goloubinoff P. (1998) J. Biol. Chem. 273, 11032–11037 [DOI] [PubMed] [Google Scholar]
- 51.Goloubinoff P., Mogk A., Zvi A. P., Tomoyasu T., Bukau B. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 13732–13737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Koga H., Kaushik S., Cuervo A. M. (2010) Ageing Res. Rev., doi: 10.1016/j.arr.2010.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hageman J., Rujano M. A., van Waarde M. A., Kakkar V., Dirks R. P., Govorukhina N., Oosterveld-Hut H. M., Lubsen N. H., Kampinga H. H. (2010) Mol. Cell 37, 355–369 [DOI] [PubMed] [Google Scholar]
- 54.Putcha P., Danzer K. M., Kranich L. R., Scott A., Silinski M., Mabbett S., Hicks C. D., Veal J. M., Steed P. M., Hyman B. T., McLean P. J. (2010) J. Pharmacol. Exp. Ther. 332, 849–857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hirohata M., Ono K., Morinaga A., Yamada M. (2008) Neuropharmacology 54, 620–627 [DOI] [PubMed] [Google Scholar]
- 56.Dedmon M. M., Christodoulou J., Wilson M. R., Dobson C. M. (2005) J. Biol. Chem. 280, 14733–14740 [DOI] [PubMed] [Google Scholar]
- 57.Ahmad A. (2010) Int. J. Biol. Macromol. 46, 275–279 [DOI] [PubMed] [Google Scholar]
- 58.Shorter J., Lindquist S. (2008) EMBO J. 27, 2712–2724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rodriguez F., Arsène-Ploetze F., Rist W., Rüdiger S., Schneider-Mergener J., Mayer M. P., Bukau B. (2008) Mol. Cell 32, 347–358 [DOI] [PubMed] [Google Scholar]
- 60.Summers D. W., Douglas P. M., Ramos C. H., Cyr D. M. (2009) Trends Biochem. Sci. 34, 230–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Finka A., Mattoo R. U., Goloubinoff P. (2010) Cell Stress Chaperon., doi:10.1007/s12192-010-0216-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sharma S. K., De Los Rios P., Christen P., Lustig A., Goloubinoff P. (2010) Nat. Chem. Biol., doi:10.1038/nchembio.455 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.