

Abstract
The Mycobacterium tuberculosis cytochrome P450 enzyme CYP142 is encoded in a large gene cluster involved in metabolism of host cholesterol. CYP142 was expressed and purified as a soluble, low spin P450 hemoprotein. CYP142 binds tightly to cholesterol and its oxidized derivative cholest-4-en-3-one, with extensive shift of the heme iron to the high spin state. High affinity for azole antibiotics was demonstrated, highlighting their therapeutic potential. CYP142 catalyzes either 27-hydroxylation of cholesterol/cholest-4-en-3-one or generates 5-cholestenoic acid/cholest-4-en-3-one-27-oic acid from these substrates by successive sterol oxidations, with the catalytic outcome dependent on the redox partner system used. The CYP142 crystal structure was solved to 1.6 Å, revealing a similar active site organization to the cholesterol-metabolizing M. tuberculosis CYP125, but having a near-identical organization of distal pocket residues to the branched fatty acid oxidizing M. tuberculosis CYP124. The cholesterol oxidizing activity of CYP142 provides an explanation for previous findings that ΔCYP125 strains of Mycobacterium bovis and M. bovis BCG cannot grow on cholesterol, because these strains have a defective CYP142 gene. CYP142 is revealed as a cholesterol 27-oxidase with likely roles in host response modulation and cholesterol metabolism.
Keywords: Cholesterol Metabolism, Crystal Structure, Cytochrome P450, Enzyme Catalysis, Hydroxylase, Azole Drugs, Drug Target, Hemoprotein, Mycobacterium tuberculosis, Redox Potentiometry
Introduction
Tuberculosis (TB)3 is a debilitating and frequently fatal disease caused by the bacterium Mycobacterium tuberculosis. The disease usually presents as a pulmonary condition but can also affect other parts of the body (1). M. tuberculosis can also remain in a dormant phase in the host (latent TB), and a proportion of individuals with latent TB will later develop active TB (2). The emergence and proliferation of multidrug-resistant and extensively drug-resistant M. tuberculosis strains have provided a major challenge for TB therapeutic development (3). Numerous M. tuberculosis clinical strains are now resistant to one or several front line and second line drugs, including antibiotics such as rifampicin, isoniazid, streptomycin, and ethambutol (4). TB and drug-resistant strains are prevalent in the less developed countries of Asia and Africa, and synergy between M. tuberculosis and HIV leads to high rates of TB infection and deaths in HIV-infected individuals (5). Consequently, there has been recent intensive research on the genetics and biochemistry of M. tuberculosis, as well as work to develop novel therapeutics and to identify new drug targets (6–8). Recent successes include the development of 1,3-benzothiazin-4-ones that inhibit the decaprenylphosphoryl-β-d-ribose 2′-epimerase enzyme and formation of cell wall arabinans, and clavulanate/β-lactam combinations inhibiting formation of cell wall peptidoglycan (9, 10).
The M. tuberculosis H37Rv and then the M. tuberculosis CDC1551 genome sequences provided key information on the M. tuberculosis proteome, highlighting the complexity of M. tuberculosis lipid metabolism as well as several genes previously found only in eukaryotes, e.g. the adrenodoxin reductase homolog FprA (11–13). The presence of 20 P450 (CYP) genes in M. tuberculosis pointed to their involvement in important biochemical processes. Of particular note was the identification of the first prokaryotic sterol demethylase (CYP51B1), which was shown to be particularly active in 14α-demethylation of the plant sterol obtusifoliol (14). However, the remaining M. tuberculosis P450s are dissimilar in sequence from other P450s of known structure/function, preventing simple functional assignment. However, such data have emerged for certain other M. tuberculosis P450s. CYP121 catalyzes oxidative coupling of tyrosyl side chains of the cyclic dipeptide cyclo-l-Tyr-l-Tyr in synthesis of a secondary metabolite of unknown function. CYP128 was predicted to catalyze terminal hydroxylation of respiratory dihydromenaquinone-9 to enable sulfation at the same position catalyzed by the product of its adjacent gene (stf3 or Rv2269c) (15, 16). Sulfated dihydromenaquinone-9 may have a role in M. tuberculosis virulence, as do other M. tuberculosis-sulfated lipids (16). M. tuberculosis CYP124 catalyzes ω-hydroxylation of phytanic acid and other methyl branched chain fatty acids (17).
More recently, studies on M. tuberculosis CYP125 have shown that this P450 binds cholesterol and cholest-4-en-3-one, transforming these substrates by 27-hydroxylation on their side chain (18, 19). CYP125 is located in a large gene regulon extensively conserved between Rhodococcus sp. strain RHA1 and M. tuberculosis, and is responsible for directing cholesterol import and catabolism (20). CYP125 (H37Rv gene Rv3545c) lies in a region known as igr (intracellular growth) (genes Rv3545c to Rv3540c). These genes are required for M. tuberculosis growth in macrophages and for virulence in mice, as well as for cholesterol metabolism (21, 22). CYP125 is predicted to be important in M. tuberculosis cholesterol metabolism that is crucial to infectivity and survival in the host, and the labile nature of its ferrous-NO complex was suggested to be advantageous in evasion of inhibition by host-generated NO (23, 24). Interestingly, both M. tuberculosis H37Rv and the TB vaccine strain Mycobacterium bovis BCG grow on cholesterol as a carbon source. However, the BCG CYP125 gene deletion strain did not grow on cholesterol (unless complemented with another copy of CYP125), although the M. tuberculosis CYP125 deletion strain grew well on cholesterol (19). In studies of CYP125 in the clinically relevant CDC1551 M. tuberculosis strain, cholest-4-en-3-one was shown to accumulate in cells of the ΔCYP125 strain, suggesting this cholesterol derivative is also a physiological substrate for CYP125 (25).
Data from Capyk et al. (19) suggested that there is redundancy in cholesterol oxidation capacity in M. tuberculosis H37Rv (but not in M. bovis BCG) and that a compensatory activity can overcome absence of CYP125 in this bacterium to enable growth on cholesterol. In this respect, it is notable that a further P450 gene (CYP142, Rv3518c) is located in the cholesterol regulon of M. tuberculosis. Here, we report expression, isolation, and characterization of CYP142. We demonstrate that CYP142 is a cholesterol 27-hydroxylase with intriguing relationships to the structurally resolved M. tuberculosis CYP125 and CYP124 proteins, pointing to a common evolutionary pathway.
EXPERIMENTAL PROCEDURES
Cloning and Expression of M. tuberculosis CYP142
The CYP142 gene (Rv3518c in the M. tuberculosis H37Rv genome) was amplified by PCR from an M. tuberculosis H37Rv chromosomal cosmid DNA library (supplied by Dr. Roland Brosch, Institut Pasteur, Paris). The BAC clone containing CYP142 (Rv416) was prepared by standard protocols and used as a template DNA for the PCR using Pfu turbo DNA polymerase (Agilent) and oligonucleotide primers designed from the M. tuberculosis genomic sequence as follows: upstream (CYP142-NdeF), 5′-GGAGGATCCATATGACTGAAGCTCCGGACGTGG-3′, and downstream (CYP142-BamHIR), 5′-CGTTCGGGATCCCTCAGCCCAGCGGCGTGAAC-3′. The letters underlined in the upstream primer indicate an engineered NdeI restriction-cloning site, including the initiation codon ATG (boldface). The underlined letters in the downstream primer indicate a BamHI restriction-cloning site, with the stop codon in boldface. Amplification conditions were 95 °C for 2 min, 30 cycles of 95 °C for 45 s, 62 °C for 30 s, and 72 °C for 1.5 min, followed by a final polymerization step of 72 °C for 10 min. CYP142 was cloned into pET15b (Merck) pre-digested with the same restriction enzymes, allowing the expression of the CYP142 gene from a T7lac promoter under isopropyl 1-thio-β-d-galactopyranoside induction and producing a recombinant P450 protein with an N-terminal His6 tag.
Purification of CYP142
Recombinant CYP142 was heterologously expressed in the Rosetta (DE3) Escherichia coli strain (Merck). Typically, 20 liters of Luria-Bertani (LB) medium was divided into 600-ml cultures in 2-liter conical flasks, and antibiotic plasmid selection was done using 50 μg ml−1 ampicillin and 34 μg ml−1 chloramphenicol. Cultures were started at 37 °C and inoculated with 10 ml from an overnight culture growth. Cultures were grown with agitation (220 rpm) for 1 h, and the temperature was then reduced to 30 °C. Cell culture was continued until an optical density (OD600) of 0.5. The temperature was then reduced to 22 °C and culture continued until an OD600 between 0.6 and 0.8 was reached. CYP142 expression was then induced by addition of 0.1 mm isopropyl 1-thio-β-d-galactopyranoside. After 24 h of culture post-induction at 22 °C, cells were harvested by centrifugation at 6,000 × g (15 min, 4 °C). Resulting pellets were pooled and resuspended in 50 mm KPi, 250 mm KCl, 10% glycerol, pH 8.0 (buffer A) and centrifuged as before. Cell pellets were either used immediately or stored at −20 °C until required.
For cell breakage, the cells were resuspended in ∼350 ml of buffer A (4 °C) containing phenylmethanesulfonyl fluoride (PMSF) and benzamidine hydrochloride (∼1 mm each) and 2 “complete” EDTA-free tablets (Roche Applied Science) as standard protease inhibitors. The cell suspension was initially treated with lysozyme and DNase (10 μm each) for 30 min with stirring at 4 °C and then sonicated on ice (∼10 cycles of 20 s, with 1-min rest periods between cycles) using a Bandelin Sonopuls sonicator, as described previously (26, 27). The lysate was centrifuged twice at 40,000 × g for 30 min at 4 °C to pellet insoluble material. Soluble supernatant was loaded onto a pre-equilibrated (buffer A) nickel-nitrilotriacetic acid column (Qiagen) and washed with 50 ml of buffer A plus 15 mm imidazole. Bound protein was eluted using buffer A plus 55 mm imidazole, and this fraction then dialyzed against 50 mm Tris, 1 mm EDTA, pH 7.2 (buffer B), also containing 50 mm KCl before being loaded on a Q-Sepharose column (GE Healthcare). The column was washed extensively with buffer B and then eluted using a linear gradient of KCl in buffer B between 50 and 500 mm. Red fractions were analyzed spectrally for CYP142 content, and the purest fractions (from A418/A280 ratio) were pooled. This sample was again dialyzed against buffer B, before concentration by ultrafiltration (Centriprep 30, Millipore) to <1 ml and final purification by gel filtration on a Sephacryl S-200 column. CYP142 purity was confirmed by a single band present on an SDS-polyacrylamide gel and by UV-visible spectroscopy, where an A418/A280 ratio ≥2 correlated with highly pure CYP142. CYP142 was stored for later use by dialysis into buffer B plus 10% glycerol before freezing at −20 °C. For crystallography, CYP142 was taken immediately from the S-200 purification step and buffer exchanged into 10 mm Tris, pH 7.5, using a 10 DG disposable chromatography column (Bio-Rad), then concentrated by ultrafiltration as before (typically to ∼15 mg/ml), and used directly for crystallogenesis.
Substrate and Ligand Binding Studies
UV-visible absorbance analysis of CYP142 was done on a Cary UV-50 UV-visible scanning spectrophotometer (Varian, UK) using a 1-cm path length quartz cuvette, recording spectra between 250 and 800 nm, and typically with CYP142 at ∼2–10 μm in buffer B. Binding of CO to ferrous CYP142 and nitric oxide (NO) to the ferric enzyme was done as described previously, with release of 5–6 bubbles of NO gas into a degassed CYP142 solution in the case of NO (28).
Optical titrations with potential substrates (including cholesterol and cholest-4-en-3-one) and triazole/imidazole drugs were done as described previously (26, 28) at 25 °C. Solutions of azole ligands (clotrimazole, econazole, fluconazole, itraconazole, ketoconazole, miconazole, and voriconazole) were made up in DMSO. 4-Phenylimidazole was made up in 50% ethanol. Steroid stocks (testosterone and androstenedione) were made up in ethanol. Fatty acids and other lipids (lauric acid, palmitic acid, 15-methylpalmitic acid, 13-methylmyristic acid, phytanic acid, geranylgeraniol, and 2-methylheptane) were made up in a 50% ethanol/methanol mixture. Volumes of ligand solutions added did not exceed 0.1% of the total assay volume. Titrations with cholesterol and cholest-4-en-3-one were performed similarly. However, 50 mm KPi buffer with 50 mm KCl, 5% glycerol, pH 7.0 (buffer C), was used, and these titrations were performed at 30 °C. Sterol solutions were made up in 45% 2-hydroxypropyl-β-cyclodextrin (in water). In all cases, Kd values were determined by plotting the induced optical change against ligand concentration and fitting using the Morrison equation (Equation 1), using Origin software (OriginLab, Northampton, MA). Equation 1 provides robust fitting of binding data for tight binding ligands, accounting for concentration of protein in cases where the Kd value is not substantially greater than the protein concentration, as described previously (26, 28, 29).
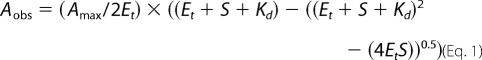 |
In Equation 1, Aobs is the observed absorbance change at ligand concentration S; Amax is the absorbance change at ligand saturation; Et is the CYP142 concentration, and Kd is the dissociation constant for the CYP142-ligand complex.
Hemoprotein Concentration Estimation
CYP142 hemoprotein concentration was estimated using the method of Omura and Sato (30) (Δϵ450–490 = 91 mm−1 cm−1 for the reduced/CO-bound minus reduced difference spectrum). The spectrum of the ferrous-CO [Fe(II)CO] complex of CYP142 was also analyzed in 50 mm KPi between pH 6 and 9 in both the ligand-free state and for the cholest-4-en-3-one-bound form. In view of variability of the P420 content in different buffer systems, the pyridine hemochromogen method was used to quantify CYP142 heme and to determine an extinction coefficient at the CYP142 Soret maximum. Analysis was done according to the method of Berry and Trumpower (31), as described in the supplemental material.
Redox Potentiometry
Redox potentiometry was performed anaerobically under a nitrogen environment in a Belle Technology glove box (Portesham, UK), as described previously (32, 33), using the method developed by Dutton (34). Full details are given in the supplemental material.
EPR Analysis of CYP142
Continuous wave EPR spectra for CYP142 were recorded at X-band (∼9.4 GHz) using a Bruker ELEXSYS E500/E580 EPR spectrometer (Bruker GmbH, Rheinstetten, Germany). Temperature was maintained at 10 ± 0.1 K using an Oxford Instruments ESR900 helium flow cryostat coupled to an ITC 503 controller from the same manufacturer. The microwave power was 0.5 milliwatt, the modulation frequency 100 KHz, and the modulation amplitude 5 G. EPR sample tubes were 4 mm Suprasil quartz supplied by Wilmad (Vineland, NJ). Sample volume was typically 200 μl, and CYP142 solutions were frozen in EPR tubes in liquid nitrogen prior to EPR analysis. CYP142 concentration was maintained at 200 μm for all samples, with econazole and cholest-4-en-3-one introduced at 500 μm for ligand-bound spectra.
Steady-state Kinetic Studies and Product Analysis
Steady-state kinetic studies were performed on a Cary UV-50 spectrophotometer. A heterologous electron transport system was set up using CYP142, spinach ferredoxin (spFDX), and ferredoxin reductase (spFDR), in the ratio 1:10:2 (200 nm CYP142, 2 μm FDX, and 400 nm FDR). At each concentration of substrate (0–50 μm cholest-4-en-3-one), 200 μm NADPH was added to initiate the reaction and the rate of change in absorbance at 340 nm monitored over 6 min to follow substrate-dependent NADPH oxidation. Reactions were performed in triplicate to produce a mean rate calculated as moles of NADPH oxidized/mol CYP142/min. These data were plotted versus the relevant cholest-4-en-3-one concentration and fitted using a standard (Michaelis-Menten) hyperbolic function with Origin software.
Product Analysis from CYP142 Incubations with Cholesterol and Cholest-4-en-3-one
To analyze the products by HPLC, enzyme activity assays were carried out with 0.5 μm CYP142, 10 μm spFDX or E. coli flavodoxin (FLD), 2.5 μm spFDR or E. coli flavodoxin reductase (FLDR), and 2 nm [3H]cholesterol to detect the product formation using a flow-through radioactive monitor attached to the HPLC system after the UV detector, or 1 μm cholest-4-en-3-one to detect the products by the UV absorbance. Reactions were initiated by the addition of 1 mm NADPH with a regenerating system (glucose 6-phosphate and glucose-6-phosphate dehydrogenase) in a reaction volume of 1 ml in 50 mm KPi, pH 7.5, and allowed to proceed at 37 °C. After a 30-min incubation time, reactions were terminated by the addition of 5 ml of dichloromethane. The organic phase was isolated following centrifugation, evaporated, and dissolved in acetonitrile and transferred to a vial for HPLC studies, as described previously (18, 35).
To characterize the products by gas chromatography-mass spectrometry (GC-MS), CYP142 assays were set up as described above, except [3H]cholesterol was omitted, and a higher concentration of substrate (10 μm cholesterol or cholest-4-en-3-one) was used. Following termination of duplicate incubations, the substrate and product(s) were extracted and converted into trimethylsilyl (TMS) ethers (either with or without prior methylation using diazomethane). Sterols were injected into a VF-35MS capillary column (60 m × 0.32 mm × 0.25 μm) in a splitless mode at an injection temperature of 270 °C with a helium flow rate of 1.1 ml/min. The initial oven temperature was kept at 200 °C for 1 min, then increased to 280 °C (20 °C/min), ramped up to 310 °C (3 °C/min), and was held for 14 min isothermally. The mass spectrometer (Agilent 5973N-MSD combined with an Agilent 6890 GC system) was operated in electron impact ionization mode (70 eV) at 230 °C. The retention time and mass spectrum for the TMS product of CYP142 oxidation of cholesterol with the E. coli FLDR/FLD system and for the methylated and TMS product of the cholesterol incubations with the spFDX/spFDR system were compared with the TMS ether derivative of authentic 27-hydroxycholesterol and the methyl ester-TMS ether derivative of authentic 5-cholestenoic acid (Steraloids, Newport RI).
CYP142 Crystallization and Structural Analysis
CYP142 was crystallized using highly purified protein concentrated to 15 mg ml−1 in 10 mm Tris, pH 7.5. Diffraction quality crystals of bipyramidal morphology (∼0.05 × 0.1 mm) were obtained using 0.1 m sodium acetate at a pH range of 5.0–6.0 with 100 mm potassium thiocyanate, 10% PEG 200 (v/v), and 10% PEG 550MME (v/v). Sitting drops were made by mixing equal volumes of protein solution and mother liquor and incubating at 4 °C. Crystals appeared within 2–3 days and reached final size within 2 weeks. Single crystals were flash-cooled by plunge freezing into liquid nitrogen with the PEG 200 acting as a cryoprotectant. Diffraction data were collected from a single cryofrozen crystal at the Diamond Light Source, beamline (IO2). The data were scaled and integrated using the XDS package (36) and subsequently handled using the CCP4 suite (37). The CYP142 structure was solved following molecular replacement (38) with the CYP124 structure as a search model (PDB code 2WM5) (17). The CYP142 model was built using COOT (39) in conjunction with MOLPROBITY (40) and refined using Phenix (41) to a resolution of 1.60 Å. Data and final refinement statistics are presented in Table 1.
TABLE 1.
X-ray data collection and refinement statistics for M. tuberculosis CYP142
| CYP142 | PDB code 2XKR |
|---|---|
| Space group | P212121 |
| Resolution | 40.0 to 1.6 Å (1.66 to 1.60 Å) |
| Unit cell a, b, c | 55.3, 65.5, 129.5 Å |
| Redundancy | 6.9 (6.3) |
| Reflections total | 430,506 |
| Unique | 62,547 |
| Completeness | 99.7% (99.0%) |
| Rsym | 5.6% (46.0%) |
| 〈I/σI〉 | 22.7 (4.1) |
| Rwork | 16.8% |
| Rfree | 19.5% |
| Overall B-factor | 19.9 Å2 |
| Protein | 18.1 Å2 |
| Main chain | 16.6 Å2 |
| Side chain | 19.7 Å2 |
| Solvent | 31.3 Å2 |
| Heme | 11.9 Å2 |
| PEG | 29.2 Å2 |
| Ramachandran plot | |
| Favorable | 98% |
| Allowed | 100% |
| Outliers | 0% |
| No. of atoms | |
| Protein | 3020 |
| Solvent | 510 |
| Heme | 43 |
| PEG | 7 |
| Root mean square deviation from ideal geometry | |
| Bond lengths | 0.006 Å |
| Bond angles | 1.032° |
Modeling of a Cholesterol-CYP142 Complex
Molecular modeling of the interaction of cholesterol with CYP142 was based on a soft-restrained molecular dynamics approach previously described for P450s (42) and successfully applied in our previous studies with M. tuberculosis CYP125 (18). All molecular dynamics simulations, energy minimization experiments, and stabilization energy calculations were performed as described previously (18).
Materials
Bacterial growth media (tryptone and yeast extract) were from Melford Laboratories (Ipswich, Suffolk, UK). Azole drugs were from MP Biomedicals. All other reagents were from Sigma and were of the highest grade available.
RESULTS
Isolation and Spectral Features of CYP142
CYP142 was isolated from an E. coli Rosetta (DE3) expression system using affinity for the N-terminal His6 tag, followed by ion exchange chromatography and size exclusion chromatography to produce a pure protein (by SDS-PAGE, Fig. 1) with a Rheinheitzahl (Rz) value (A418/A280 ratio) of ∼2.0. Previous studies on CYP125, the other P450 in the cholesterol regulon of M. tuberculosis, showed that this enzyme bound heme in an extensively high spin (HS) ferric form, regardless of whether substrate was associated, and with its major heme absorbance band (Soret, γ) at 393 nm (18). In contrast, purified CYP142 has an electronic spectrum indicative of an extensively low spin (LS) ferric P450 enzyme, with the Soret band at 418 nm, and the smaller α and β bands at 566 and 535 nm, respectively (Fig. 1). CYP142 undergoes heme optical shifts typical of the P450s on binding the heme-coordinating inhibitor econazole (Soret shift to 423 nm) and NO (shift to 433.5 nm with development of features in the Q-band region at 542.5 nm and 573.5 nm).
FIGURE 1.

Purification and spectral features of CYP142. Spectra in the main figure show the UV-visible absorption features of pure, substrate-free ferric M. tuberculosis CYP142 (3.7 μm) (thick solid line) and for CYP142 bound to econazole (13 μm, thin solid line), nitric oxide (dotted line), and in the Fe2+-CO form bound to cholest-4-en-3-one (10 μm, dashed line). The major (Soret) absorption band is centered at 418, 423, 433.5, and 448 nm, respectively. The inset shows an SDS-polyacrylamide gel with molecular weight markers of indicated mass in the 1st lane and purified CYP142 (0.5 and 2.5 μg) as a single band in the remaining lanes.
CYP142 Binds Cholesterol and Cholest-4-en-3-one
In view of the genetic location of CYP142 and the possibility of its involvement in metabolism of host-generated cholesterol, we examined spectral changes on binding of cholesterol and cholest-4-en-3-one. For both these molecules, substantial shifts in the ferric heme iron spin-state equilibrium were induced, with CYP142 becoming extensively HS in both cases. Fig. 2A shows an optical titration of CYP142 with cholest-4-en-3-one, displaying a Soret shift from 418 to 393 nm as cholest-4-en-3-one binds, and a clear LS/HS isosbestic point at 406 nm. Spectral perturbations are also evident in the Q-band region, with the development of a charge-transfer band at ∼650 nm also consistent with substrate binding and the P450 HS shift. Fig. 2B shows an overlaid set of difference spectra derived from the cholest-4-en-3-one titration of CYP142 and a plot of the induced spectral change versus the steroid concentration, fitted to generate a Kd value of 0.36 ± 0.04 μm. Cholesterol induced similar absorption shifts and bound with an apparent Kd of 0.34 ± 0.20 μm (supplemental Fig. S9). In contrast, no spectral perturbations were observed on titration of CYP142 with the steroids testosterone and androstenedione, with a range of long straight and branched chain molecules, or with the terpene geranylgeraniol. The latter molecule and various branched chain fatty acids were shown previously to be good substrates for the M. tuberculosis CYP124 enzyme (17). In addition, no spectral binding to CYP142 was detected for 2-methyl heptane, which mimics the side chain of cholesterol. Thus, cholesterol binding affinity likely originates mainly from the sterol ring structure.
FIGURE 2.

Spectral binding of cholest-4-en-3-one to CYP142. A shows absolute spectra recorded during titration of CYP142 (4.8 μm) with cholest-4-en-3-one. The Soret band shifts from 418 to 393 nm as the HS ferric heme iron form accumulates. The development of a charge-transfer species at 649 nm is further confirmatory of the substrate-like nature of the binding event. B shows overlaid difference spectra from the titration shown in A, and cholest-4-en-3-one-induced absorption change plotted versus cholest-4-en-3-one concentration, with data fitted using Equation 1 to produce a Kd value of 0.36 ± 0.04 μm.
CYP142 Heme Coordination by Carbon Monoxide and Azole Drugs
Consistent with a number of the M. tuberculosis P450 enzymes investigated to date, CYP142 binds to a range of imidazole and triazole antifungals, inducing a type II (Soret red shift) spectral shift to ∼423 nm (see Fig. 1 for example of econazole). Among the panel tested, highest affinity was observed for clotrimazole, econazole, and miconazole (Kd values of 3.8 ± 0.9, 4.6 ± 0.2, and 4.0 ± 0.5 μm, respectively). Negligible binding was observed for the more polar voriconazole and the bulky itraconazole (Table 2).
TABLE 2.
Dissociation constants for the binding of selected azole drugs and substrates to M. tuberculosis CYP142
Kd values were determined as described under “Experimental Procedures.” Comparative data for the binding of azoles to the M. tuberculosis CYP144 enzyme are from Driscoll et al. (60). Azole binding data for the M. tuberculosis CYP125 cholesterol 27-hydroxylase are from McLean et al. (18), and CYP125 data for cholesterol/cholest-4-en-3-one binding are from Ouellet et al. (25). ND indicates that a Kd value could not be determined due to lack of any significant heme spectral perturbation induced on binding of the relevant azole or steroid to the particular M. tuberculosis P450.
| Inhibitor/substrate |
Kd value |
||
|---|---|---|---|
| CYP144 | CYP125 | CYP142 | |
| μm | μm | μm | |
| Econazole | 0.78 ± 0.29 | 11.7 ± 0.7 | 4.6 ± 0.2 |
| Clotrimazole | 0.37 ± 0.08 | 5.3 ± 0.6 | 3.8 ± 0.9 |
| Miconazole | 0.98 ± 0.22 | 4.6 ± 0.4 | 4.0 ± 0.5 |
| Ketoconazole | 134 ± 5 | 27.1 ± 0.9 | 21 ± 4 |
| Fluconazole | ND | 43.1 ± 3.8 | 860 ± 108 |
| Voriconazole | 174 ± 14 | ND | ND |
| Itraconazole | ND | 30.2 ± 4.3 | ND |
| 4-Phenylimidazole | 280 ± 18 | 216 ± 5 | 12.0 ± 1.5 |
| 4-Cholesten-3-one | ND | 1.2 ± 0.1 | 0.36 ± 0.04 |
| Cholesterol | ND | 0.11 ± 0.06 | 0.34 ± 0.20 |
The binding of CO to P450 enzymes is a diagnostic test whereby these enzymes are characterized based on a Soret shift to ∼450 nm in the ferrous-CO (Fe2+-CO) complex (30). This shift is indicative of the retention of the thiolate proximal ligand to the heme iron (Cys-339 in CYP142), and a distinctive Soret shift to ∼420 nm instead usually indicates a Fe2+-CO “P420” form in which thiolate protonation has occurred and the cysteine thiol becomes the proximal ligand (28, 43, 44). Fig. 3A shows the spectra collected for substrate-free CYP142 at the pH values 7–9. At pH 6, the enzyme was unstable and aggregated. Of the remaining conditions tested, the P450 form of CYP142 is most stable at pH 7, and larger proportions of the P420 species are formed at the higher pH values, with near-complete P420 formation at pH 9. At pH 8, the spectrum for the Fe2+-CO form is notably unstable, and the P450 species progressively “collapses” over time with P420 accumulation, a phenomenon that was also observed for the M. tuberculosis CYP51B1 enzyme in its substrate-free form (45, 46). Fig. 3A, inset, shows the time-dependent collapse of the P450 form to P420 at pH 8, leading to the near-complete accumulation of the cysteine thiol-coordinated form once equilibrium is reached. Fig. 3B shows the stabilizing effect of substrate binding on the thiolate-coordinated CYP142, to the extent that the P420 form of CYP142 can be converted almost completely to the P450 state on binding of cholest-4-en-3-one at pH 8.0. The stabilization of the P450 species is also evident from the spectrum for the CYP142 Fe2+-CO complex shown in Fig. 1, where CYP142 pre-bound to cholest-4-en-3-one was reduced and complexed with CO, leading to an almost complete spectral conversion to the thiolate-coordinated P450 form.
FIGURE 3.

Carbon monoxide binding to CYP142. A, influence of pH on the CYP142 P450/P420 equilibrium. Figure shows spectra for ligand-free CYP142 (5.1 μm) at pH 7 (dotted line), pH 8 (dashed line), and pH 9 (solid line). The spectral maxima for the P450 and P420 Fe2+-CO species of CYP142 are at ∼449 and ∼421 nm, respectively. Spectra were recorded immediately after introduction of CO to the ferrous enzyme and mixing. Inset shows the progressive spectral conversion of the CYP142 P450 species to the P420 form at pH 8. The black solid line with maximal absorbance at ∼450 nm was recorded immediately following formation of the CYP142 Fe2+-CO species. Subsequent gray lines are examples of spectra recorded at 30-min intervals and represent the gradual collapse of the spectrum to a near-complete P420 form (black solid line with maximal absorbance at ∼420 nm collected 3.5 h after formation of the Fe2+-CO species). Arrows show direction of absorbance change with time. B, regeneration of the P450 spectrum on binding cholest-4-en-3-one. Figure shows spectrum for the CYP142 Fe2+-CO species (5 μm at pH 8) in the absence of steroid (black solid line with maximal absorbance at ∼420 nm) and selected subsequent spectra (gray solid lines) from a set collected following addition of cholest-4-en-3-one (50 μm) and recorded at ∼15-min intervals over a period of 510 min. The final spectrum (black solid line with maximal absorbance at ∼450 nm) was collected at 510 min and has converted to a mainly P450 form with heme thiolate coordination. The inset shows the time course of A448 increase and A422 decrease following cholest-4-en-3-one addition to CYP142.
In previous studies of the P450 EpoK from the bacterium Sorangium cellulosum, Ogura et al. (47) demonstrated “substrate-mediated rescue” of the enzyme, whereby addition of the substrate epothilone D effected a gradual conversion of the EpoK Fe2+-CO complex at 422 nm (P420) to 446 nm (P450), indicating a substrate binding-dependent structural rearrangement leading to deprotonation of the proximal cysteine thiol ligand. In our studies of the M. tuberculosis CYP51B1 enzyme, binding of the substrate analog estriol decreased the rate of the P450-to-P420 “collapse” in this enzyme, but it could not prevent altogether a time-dependent Cys-394 thiolate protonation in this enzyme (46). However, our data here indicate that substrate-mediated rescue of the M. tuberculosis CYP142 P420 form can be achieved by binding of the substrate cholest-4-en-3-one.
In view of the variability of the P450 complex with pH, we determined the extinction coefficient of the LS, ligand-free CYP142 using the pyridine hemochromogen method of Berry and Trumpower (31). This was established as ϵ418 = 140 mm−1 cm−1 (supplemental Fig. S1).
EPR Spectroscopy of CYP142
To further characterize the heme coordination in CYP142 and to examine the effects of binding of substrate- and inhibitor-like molecules, we collected EPR spectra for native CYP142 and for the enzyme bound to cholest-4-en-3-one and the inhibitor econazole. Fig. 4 shows overlaid X-band EPR spectra for these species. A characteristic P450 rhombic spectrum was observed in each case. For the native and econazole-bound forms of CYP142, spectra were indicative of almost completely LS heme iron. The ligand-free CYP142 has g values at gz = 2.40, gy = 2.23, and gx = 1.92, consistent with a LS thiolate-coordinated P450 enzyme and similar to spectra previously reported for the heme domain of the well studied flavocytochrome P450 BM3 fatty acid hydroxylase enzyme from Bacillus megaterium (2.42, 2.26, and 1.92) and the M. tuberculosis cholesterol 27-hydroxylase CYP125 (2.40, 2.25, and 1.94) (18, 48), for example. In the CYP142 complex with econazole, the P450 is mainly converted to two novel species with g values consistent with changes to the 6th ligand environment of the heme iron. These species have sets of g values at 2.53, 2.24, and 1.86 (minor) and at 2.45, 2.24, and 1.89 (major). The former (gz = 2.53) is consistent with direct coordination by the imidazole nitrogen from the drug. The other species (gz = 2.45) could reflect direct ligation with the imidazole ring in a different orientation or possibly retention of the 6th aqua ligand with g values influenced by interactions between the bound water and the imidazole drug.
FIGURE 4.

EPR analysis of CYP142. Upper spectrum, econazole-bound CYP142 showing formation of two novel ligand-bound species with sets of g values at 2.53, 2.24, and 1.86 (major) and 2.45, 2.24, and 1.89 (minor), different from those for the ligand-free enzyme (2.40, 2.23, and 1.92) as seen in the middle panel. The lower panel shows the cholest-4-en-3-one-bound CYP142. Optical spectra at room temperature show predominantly HS character, and HS features are retained at 10 K with g values at 7.78, 3.65, and 1.71, although the P450 is mainly LS at this temperature. The derivative at g = 3.65 is not shown in the lower panel for cholest-4-en-3-one-bound CYP142 to enable scaling for direct comparisons of other features between the three spectra. The full spectrum for cholest-4-en-3-one bound CYP142 is shown as supplemental Fig. S2.
In the cholest-4-en-3-one-bound CYP142 sample (Fig. 4 and supplemental Fig. S2), the EPR spectrum is dominated by LS species with g values identical to that for the substrate-free CYP142. However, there is also clearly a set of g values associated with a smaller proportion of HS CYP142 at gz = 7.87, gy = 3.65, and gx = 1.71, confirming the ability of cholest-4-en-3-one to induce formation of CYP142 HS heme iron and demonstrating that some of the HS form is retained even at a temperature as low as 10 K.
Redox Potentiometry of CYP142
Spectroelectrochemistry was used to determine the midpoint reduction potential for the Fe3+/Fe2+ transition of the CYP142 heme iron. In the substrate-free enzyme, the CYP142 Soret signal is blue-shifted from 418 to ∼413 nm at maximal reduction with dithionite, and spectral fitting using the Nernst function indicates the potential to be −416 ± 6 mV versus normal hydrogen electrode. For the cholest-4-en-3-one-bound CYP142, the Soret shifts from 393 (HS) to 410 nm, suggesting a more complete conversion to the ferrous form in the substrate-bound enzyme. Consistent with this conclusion, the potential is substantially increased to −192 ± 7 mV (supplemental Fig. S3). The shift in potential of >200 mV is rather greater than observed in previous studies of e.g. P450 BM3 and the M. tuberculosis CYP51B1 sterol demethylase, where binding of a substrate or a substrate analog (arachidonic acid and estriol, respectively) induced positive shifts in heme iron potential of ∼130 and ∼150 mV, respectively, which are considered typical for a substrate-mediated conversion of a P450 heme iron from a predominantly LS to a mainly HS form (32, 46). The origin of the unexpectedly large difference in potential for substrate-free and cholest-4-en-3-one-bound CYP142 remains uncertain, but it is also of note that hysteretical behavior was observed during redox titration experiments in which cholest-4-en-3-one-bound CYP142 was successively reduced (using dithionite) and oxidized (using ferricyanide) in the presence of mediators (supplemental Fig. S4). Although the reductive phase exhibited a midpoint potential at approximately −190 mV, the oxidative phase consistently occurred at a more negative potential (approximately −295 mV). We hypothesize that CYP142 may occupy different conformational states in the substrate-bound form (and possibly also in the substrate-free state) and that such states could influence the heme environment differently, and thus the redox potential for its Fe3+/Fe2+ couple.
The Soret features for the reduced forms of the substrate-free and cholest-4-en-3-one-bound CYP142 are at 413 and 410 nm, respectively (reduction by dithionite is not complete for the substrate-free form, and thus a complete Soret shift to ∼410 nm is not observed), and thus retain cysteine thiolate coordination in both cases. This is rather different from the behavior of the CO-bound, substrate-free enzyme at neutral pH, which has an equilibrium mixture of the P450 and P420 forms. This likely indicates that heme thiolate protonation in the substrate-free CYP142 Fe2+-CO complex is a consequence of CO ligation to the heme iron and not the iron reduction per se. This contrasts with the properties of CYP51B1, where cysteine thiolate protonation is independent of CO binding as the 6th ligand to the heme iron and where there is a red shift in the Soret band (from 419 to 423 nm) and development of strong features in the Q-band region indicative of thiol coordination of the CYP51B1 ferrous heme iron (46).
Steady-state Kinetics and Product Analysis from CYP142 Assays with Cholesterol and Cholest-4-en-3-one
In light of the substrate-like binding properties of cholesterol and cholest-4-en-3-one to CYP142, we attempted first to analyze the rate of turnover by reconstituting the P450 with spFDR and spFDX and monitoring substrate-dependent NADPH oxidation on addition of the sterols. This redox system was shown recently to support branched chain fatty acid hydroxylation by M. tuberculosis CYP124 and cholesterol/cholest-4-en-3-one oxidation by M. tuberculosis CYP125 (17, 18). Fig. 5 shows a hyperbolic dependence of NADPH oxidation rate on cholest-4-en-3-one concentration for a 1:10:2 P450/spFDX/spFDR reaction mixture, with an apparent Km of 3.2 ± 0.8 μm for cholest-4-en-3-one and a kcat of 0.0062 ± 0.0004 min−1 following accounting for the background rate of NADPH oxidation in the absence of substrate. In the case of cholesterol, where some stimulation of NADPH oxidation was also observed for this compound, its lack of solubility in aqueous buffer prevented accurate determination of catalytic parameters.
FIGURE 5.

Steady-state analysis of cholest-4-en-3-one turnover by CYP142. A CYP142 cholest-4-en-3-one-oxidizing system was reconstituted using the spinach ferredoxin reductase/ferredoxin proteins and NADPH, as described under “Experimental Procedures.” The system was effective in driving 27-hydroxylation of both cholest-4-en-3-one and cholesterol, and data for substrate-dependent NADPH oxidation versus cholest-4-en-3-one concentration were fitted using the Michaelis-Menten equation to provide parameters of kcat = 0.0062 ± 0.0004 min−1 and Km = 3.2 ± 0.8 μm.
To establish whether the sterol-stimulated NADPH oxidation was coupled to substrate oxidation, organic products derived from extended reactions were analyzed first by HPLC separation and then by GC-MS. The HPLC profile (Fig. 6A) shows that under the experimental conditions used, a single product was generated from cholesterol when the E. coli FLDR/FLD redox partner system was used. Therefore, the enzyme assay was repeated and analyzed by GC-MS. A comparison of the fragmentation pattern of the TMS product with the TMS ether of authentic 27-hydroxycholesterol indicates that the product is 27-hydroxycholesterol (Fig. 6C and supplemental Figs. S5B and S6B). This is the same product as observed in our previous studies of the enzymatic transformation of cholesterol by M. tuberculosis CYP125 (18). In contrast, use of the spFDX/spFDR redox partner system under identical conditions produced a major cholesterol metabolite distinct from 27-hydroxycholesterol that was assigned as 5-cholestenoic acid (3β-hydroxy-5-cholesten-27-oic acid, the C27 carboxylic acid derivative of cholesterol), based on HPLC and GC-MS analyses and comparison with the methyl ester-TMS ether of the authentic 5-cholestenoic acid (Fig. 6B and supplemental Fig. S5A and Fig. S6A). A similar result was obtained with cholest-4-en-3-one as the substrate, with the majority of the substrate converted to a different single product dependent on whether the E. coli FLDR/FLD or spinach spFDX/spFDR redox partner systems were used. These were assigned to 27-hydroxycholest-4-en-3-one and cholest-4-en-3-one-27-oic acid (3-keto-4-cholestene-27-oic acid), respectively, with reference to the similarities in product profiles and the relevant retention times in comparison with the HPLC analysis of cholesterol oxidation products (Fig. 6 and supplemental Fig. S7). There was no evidence for formation of an aldehyde intermediate formed in our studies of cholesterol or cholest-4-en-3-one conversion by CYP142 using spFDX/spFD redox partners. We thus favor a concerted three-step oxidation of these substrates to the 27-acids, consistent with data for CYP125 supported by these partners (25).
FIGURE 6.

CYP142 oxidation of cholesterol. A, HPLC separations of the extracts from the CYP142 incubations with 2 nm [3H]cholesterol (room temperature for 17.21 min) and spinach (spFDX/spFDR, solid line) or E. coli (FLDR/FLD, dashed line) redox partner systems. The products were almost exclusively 5-cholestenoic acid (room temperature for 8.34 min, solid line) and 27-hydroxycholesterol (room temperature for 9.59 min, dashed line), respectively. B, mass spectrum of the major product from the incubation with the spinach redox system following GC separation, showing key peaks at m/z values = 373, 412, and 502 that are diagnostic of the methyl ester-TMS ether of the 5-cholestenoic acid (27-COOH) product. C, mass spectrum of the major product from the incubation with the E. coli redox system following GC separation, showing key peaks at m/z values = 417, 456, and 546 that are diagnostic of the TMS ether of the 27-hydroxycholesterol product.
Crystal Structure of Substrate-free CYP142
The crystal structure of substrate-free CYP142 was obtained to a resolution of 1.6 Å using molecular replacement with M. tuberculosis CYP124 as a search model (PDB code 2XKR, Table 1) (17). The final CYP142 model reveals a fold typical of the P450 enzyme, but it displays its own unique conformation of the various structural elements involved in active site formation. The overall fold of CYP142 is similar to that of M. tuberculosis CYP124 as well as to the cholesterol-metabolizing CYP125 (18), and the CYP142 structure can be superimposed with a root mean square deviation of 1.74 Å over 308 Cα atoms on CYP124 (PDB code 2WM5) and a root mean square deviation of 1.85 Å over 296 Cα atoms on CYP125 (PDB code 3IVY) (Fig. 7A) (17, 25). Extensive differences occur in the conformation of the FG-loop, the orientation of the F- and G-helices, as well as the extended loop region that connects the B- and C-helices. CYP142 furthermore lacks the extended loop region connecting the β1 and β2 strands in CYP124/125 and that interacts with other active site loops from the latter enzymes.
FIGURE 7.

Structural features of CYP142. A, overlay of CYP142 with CYP124 (left panel) and with CYP125 (right panel). The β1-β2-loop, BC-loop, and the FG-helices are colored blue (CYP142), red (CYP124), and green (CYP125), respectively. B, solvent-accessible surface of CYP124 (left), CYP142 (middle), and CYP125 (right), with color coding as in A. The arrow indicates the access site entry for CYP124, which is not directly visible in this orientation. In case of CYP142 and CYP125, the access channel can be readily identified by the direct view onto the heme cofactor. C, overlay of the CYP142 distal heme pocket (residues in blue) with CYP124 (left panel; in red) and CYP125 (right panel; in green). CYP124 and CYP125 substrates (phytanic acid and cholest-4-en-3-one) are also shown in the respective overlays.
Despite the differences in active site loop conformations, CYP142 and CYP125 share a similar active site topology, with a similar letterbox-shaped entry-exit channel formed by the FG-loop, the BC-loop and the I-helix N-terminal region (Fig. 7B). This channel, which is lined by predominantly hydrophobic residues, curves upwards away from the heme. In contrast, the CYP124 entry-exit channel is located virtually perpendicular to the heme plane and is formed by FG- and BC-loops in addition to the β1-β2-loop region. However, the CYP142 distal heme pocket is highly similar to that of CYP124, with most residues involved in formation of this part of the active site being conserved in identity and position. A similar overlay with CYP125 clearly reveals a much lower degree of similarity between these P450s in the distal pocket region, despite their apparently identical catalytic activities (Fig. 7C). Of particular note in the structural overlay of CYP142 with CYP124 is the close positioning of the conserved residues Leu-226 (CYP142)/Leu-263 (CYP124), Ile-76/Ile-111, Ile-65/Ile-94, Met-280/Met-318, Val-277/Val-315, and Phe-380/Phe-416. With the exception of the Val-277 (CYP142)/Val-313 (CYP125) residue pair, those CYP142 amino acids closely overlaid with their counterparts in CYP124 are either replaced by different amino acids in CYP125 or display altered conformations of side chains where the amino acid is conserved (Fig. 7C). However, in both overlays, the retention of a conserved threonine (Thr-234 in CYP142) is clear. This I-helix residue is likely important in active site water organization and/or protonation of, or hydrogen bonding to, an iron-hydroperoxy intermediate in the P450 cycle (compound 0) to enable dioxygen scission and catalysis (44–46).
The CYP142 active site contains a narrow and continuous stretch of electron density that cannot be accounted for by protein or solvent atoms and could be observed in all crystals. The density appears to correspond to a long and narrow molecule, whose terminus approaches close to the heme iron and occupies space overlapping that for the water distal ligand. We have therefore modeled this molecule as a partially ordered PEG derived from the mother liquor, with the oxygen of its terminal alcohol group close to the 6th ligand position on the heme iron (supplemental Fig. S8).
Cholesterol was docked using soft restrained dynamics docking (42) into the CYP142 active site, using the funnel formed by the FG- and BC-loops as the access channel. The substrate was docked with the cholesterol alkyl chain pointing to the heme, in accordance with the observed enzyme activity, and similar to data for CYP125 (18). During molecular dynamics, it was necessary to allow a reorientation of the backbone CYP142 coordinates from the conformation observed in the crystal structure to allow the docking to converge. This resulted in a minor reorientation of the FG-loop to avoid a clash of the Leu-163 side chain with the cholesterol steroid moiety. Unlike with CYP125, two (rather than one) conformations of the docked cholesterol were obtained, both equal in stabilization energy (−56.3 and −57.6 kcal/mol, respectively). In both cases, the cholesterol is deeply buried in the CYP142 active site, surrounded by several hydrophobic residues, identified in Fig. 8. The main difference between these conformations is the relative position that the tetracyclic portion of the cholesterol lies between the FG- and BC-loop regions. In contrast to the large difference in cholesterol conformation, the CYP142 structures are very similar for both the cholesterol-CYP142 complexes. Regardless of the exact position of the tetracyclic ring, both structures reveal the close proximity of C27 to the heme iron (as observed with CYP125).
FIGURE 8.

Models of the CYP142:cholesterol complex. An overlay of the active site of the CYP142 crystal structure (in gray) with both the modeled cholesterol-CYP142 complexes (green). The images in the left and right panels show the two different docked conformations of cholesterol defined in the modeling studies. Key residues contacting the cholesterol tetracyclic ring are shown in atom colored sticks, with the docked cholesterol molecule depicted in cyan. The largest differences in protein structure occur in the FG-loop region. Both models indicate that the terminal part of the cholesterol side chain approaches the heme iron, consistent with the observed cholesterol 27-oxidation reactions observed for CYP142.
DISCUSSION
Studies by van der Geize et al. (20) highlighted a large gene cluster in Rhodococcus sp. strain RHA1 involved in cholesterol uptake/metabolism and reported that 51 of these genes that are specifically expressed during RHA1 growth on cholesterol are also conserved in an 82 gene cluster found in both M. tuberculosis H37Rv and M. bovis BCG. M. tuberculosis H37Rv and CDC1551 strains were then both shown to grow using cholesterol as a carbon source, as was M. bovis BCG (19, 25). The CYP125 gene lies in this cluster and also within the M. tuberculosis H37Rv igr locus required for bacterial virulence and growth in the macrophage. Our group and others characterized the CYP125 P450, showing it to be a 27-hydroxylase of both cholesterol and cholest-4-en-3-one (18, 19, 25) and likely enabling an early and obligate step in cholesterol metabolism in mycobacteria and Rhodococcus strains that precedes cholesterol ring oxidation (49). Capyk et al. (19) showed that although M. bovis BCG grew on cholesterol, the BCG CYP125 deletion strain did not, unless complemented by another CYP125 copy. However, the M. tuberculosis H37Rv ΔCYP125 strain still grew on cholesterol, suggesting a compensatory cholesterol 27-hydroxylase activity in this strain. The BCG ΔCYP125 strain also failed to grow on cholest-4-en-3-one, and this molecule inhibited the growth of the strain, suggesting this molecule is also a potential substrate in vivo and likely accumulates in this strain due to action of the 3β-hydroxysteroid dehydrogenase and/or cholesterol oxidase enzymes on cholesterol (19).
Recently, Ouellet et al. (25) examined the genetics of the M. tuberculosis CDC1551 strain, reporting that the ΔCYP125 strain accumulated cholest-4-en-3-one that was toxic to the cells. They concluded that the M. tuberculosis CDC1551 CYP125 was involved in detoxification of cholest-4-en-3-one by 27-hydroxylation, leading to its breakdown. An increase in mass of the virulence factor phthiocerol dimycocerosate in WT M. tuberculosis CDC1551 cells (but not in the ΔCYP125 strain) grown on cholesterol was consistent with the role of CYP125 in breakdown of the cholesterol side chain and its incorporation into phthiocerol dimycocerosate by an increased metabolic flux of methylmalonyl-CoA via propionyl-CoA (25, 50).
In searching for an alternative cholesterol oxidase to explain the inability of the M. bovis BCG ΔCYP125 strain to grow on cholesterol (although ΔCYP125 M. tuberculosis strains can grow), we noted that M. tuberculosis H37Rv has a further CYP gene outside the igr region. We considered that CYP142 (the Rv3518c gene product) might also oxidize cholesterol, cholest-4-en-3-one, or breakdown products from these molecules. This led us to clone and express the enzyme for biochemical and structural analysis. Further data supporting the hypothesis also came from comparative analysis of the genome sequences of M. tuberculosis H37Rv/CDC1551 and M. bovis/M. bovis BCG. In M. tuberculosis strains, the CYP142 gene is intact, encoding a 397-amino acid protein of 44,267-Da predicted mass. In M. bovis strains, a single nucleotide deletion (C591) results in a premature stop codon prior to the heme-binding region, resulting in an inactive 205-amino acid CYP142 apoprotein.
Data presented in this paper are consistent with our hypothesis, identifying CYP142 as a second cholesterol 27-hydroxylase and a likely compensatory enzyme in absence of CYP125 function. CYP142 is distinct from CYP125 in being a LS heme iron P450, rather than being extensively HS (due to the conformation of the Val-267 side chain in CYP125 and its influence on the occupancy of the distal water ligand) (18). Binding of cholesterol and cholest-4-en-3-one to CYP142 is thus straightforward to observe by UV-visible spectroscopy (see Fig. 2) based on a substantial Soret LS to HS shift from 418 to 393 nm, concomitant with a large increase in heme iron Fe3+/Fe2+ potential that favors electron transfer from redox partners.
CYP142 is driven by heterologous redox partners from spinach (spFDX/spFDR) to produce 5-cholestenoic acid from cholesterol, whereas E. coli FLDR/FLD partners generate 27-hydroxycholesterol (Fig. 6 and supplemental Fig. S5). The same outcome occurs with cholest-4-en-3-one as the substrate (supplemental Fig. S7). In previous studies of CYP125, the 27-alcohol was formed exclusively using either FLDR/FLD or M. tuberculosis KshB (a ketosteroid 9α-hydroxylase reductase) as redox partners in cholesterol and cholest-4-en-3-one transformations (18, 51), but spFDR/spFDX partners enabled cholest-4-en-3-one-27-oic acid formation from cholest-4-en-3-one (25). The differences in CYP142 product formation may be explained (at least in part) by the greater driving force for electron transfer to the CYP142 heme from the spFDX, the standard potential of which has been measured at −428 mV, pH 7.0, and at −420 mV, pH 8.4 (52, 53). Although electron transfer from the E. coli FLD hydroquinone may also be strongly favored (E′ = −433 mV), under in vitro assay conditions the electron transfer may be mainly from the FLD semiquinone, a weaker reductant (E′ = −254 mV) (54). Although 5-cholestenoic acid is a likely product of successive oxidations of cholesterol by CYP142/CYP125 to facilitate cholesterol breakdown, it is important to consider that the 27-hydroxycholesterol product has functions such as regulating macrophage differentiation and cholesterol homeostasis, modulating estrogen receptor structure (with positive control over gene transcription and cellular proliferation in breast cancer cell models), and regulating cholesterol uptake and metabolism (19, 55–58). In the latter case, liver X receptors are activated by oxysterol ligands to regulate expression of genes involved in fatty acid and cholesterol metabolism (57). Thus, there are clearly possibilities for modulation of host metabolism and immune response of M. tuberculosis through generation of 27-hydroxycholesterol by CYP142/CYP125. This is in addition to energy generation from cholesterol that likely initiates by formation of the 5-cholestenoic acid product. Potentially, formation of 5-cholestenoic acid or 27-hydroxycholesterol products might be favored by different M. tuberculosis redox partner systems, and we are currently investigating interactions of various putative M. tuberculosis redox partners with CYP142 and other M. tuberculosis P450s.
CYP142 has little apparent affinity for various lipids and fatty acids, including ones that bind to CYP124 (17). Lauric acid does not induce a reverse type I optical change in HS cholest-4-en-3-one-bound CYP142, suggesting low affinity of the fatty acid for this P450 (data not shown). Despite this, the CYP142 crystal structure reveals certain structural aspects that are more akin to those of the M. tuberculosis branched chain fatty-acid oxidase CYP124, although others are similar to those observed in the CYP125 crystal structure (Fig. 7) (17, 18). Because the substrate specificity and reaction product of CYP142 are similar to that for CYP125, it is interesting to note that the active site topology of both enzymes bears significant resemblance. Despite this, the CYP142 region directly involved in controlling the P450 oxidation chemistry, i.e. in the distal heme pocket, is remarkably similarly to that of CYP124 (Fig. 7C). CYP124 oxidizes terminally methylated fatty acids, and an overlay of the substrate-bound CYP124 structure with CYP142 reveals near-identical active site conformations. Cholesterol has a terminally methylated alkyl portion similar to methylated fatty acid substrates of CYP124. It thus seems likely that CYP142 catalyzes a similar oxidation to CYP124, albeit on a different substrate. The difference in substrate specificity between the CYP142/CYP124 P450s may be a consequence of the altered shape of their respective active sites. An overlay of CYP142 with the cholest-4-en-3-one-CYP125 complex (PDB code 2X5W) (25) reveals that the CYP142 active site can easily accommodate a cholesterol-type substrate in a similar orientation. However, from available CYP124 structures, it seems unlikely cholesterol can bind in this particular confirmation, due to the noncomplementary shape of the entry channel to the active site (contrary to observations made for cholesterol binding in the CYP142 model and the CYP125 complex structure). However, this does not rule out an induced-fit reorganization of CYP124 upon cholesterol binding. Such a reorganization does not occur to any significant extent in CYP125, where the active site is largely preformed. In addition, our modeling (Fig. 8) suggests only a minor structural rearrangement is required to accommodate cholesterol in CYP142. However, on the basis of biphasic kinetics of binding of NO to CYP142, the presence of different conformational states of CYP142 was predicted (24).
These comparisons point to evolutionary relationships between the three M. tuberculosis P450s, and pairwise alignments of the protein sequences using the ClustalW program indicate higher similarity between CYP124/CYP125 (40.7% identity over 428 residues) than between CYP124/CYP142 (35.5% identity over 392 residues) or CYP125/CYP142 (28.0% identity over 397 residues). The closer relationship between CYP124/CYP125 is consistent with our previous work showing phylogenetic relationships within the M. tuberculosis P450 enzyme group (59). Thus, structural analysis (at both the amino acid sequence and tertiary structure levels) provides further hints toward commonality in function and evolutionary origin among these M. tuberculosis P450s.
In conclusion, we provide structural and biochemical characterization of M. tuberculosis CYP142, a 27-oxidase of cholesterol and cholest-4-en-3-one with likely roles in cholesterol catabolism and modulation of host responses and cholesterol homeostasis, according to whether CYP142 catalyzes the three-step oxidative transformation to the 27-acid (via 27-alcohol and 27-alkanal) or 27-hydroxylation in a single P450 turnover. The product outcome is sensitive to the redox partner, raising the possibility of another level of regulation of CYP142 activity according to cellular requirements. CYP142 activities provide an explanation for the inability of ΔCYP125 strains of M. bovis and M. bovis BCG strains to grow on cholesterol, given that the CYP142 gene is mutated in these strains. Structural data point to intriguing (and likely ancient) evolutionary relationships between CYP142, CYP125, and CYP124 and also provide further key data in unraveling the roles of these P450s in the complex biology of the human pathogen M. tuberculosis.
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grant GM062882 (to I. A. P.). This work was also supported by European Union FPVI Project NM4TB.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S9.
The atomic coordinates and structure factors (code 2XKR) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
- TB
- tuberculosis
- TMS
- trimethylsilyl
- spFDX
- spinach ferredoxin
- spFDR
- spinach ferredoxin reductase
- FLD
- flavodoxin
- FLDR
- flavodoxin reductase
- LS
- low spin
- HS
- high spin
- PDB
- Protein Data Bank.
REFERENCES
- 1.Barry C. E., 3rd, Boshoff H. I., Dartois V., Dick T., Ehrt S., Flynn J., Schnappinger D., Wilkinson R. J., Young D. (2009) Nat. Rev. Microbiol. 7, 845–855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Campbell I. A., Bah-Sow O. (2006) Br. Med. J. 332, 1194–1197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shenoi S., Friedland G. (2009) Annu. Rev. Med. 60, 307–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Riccardi G., Pasca M. R., Buroni S. (2009) Future Microbiol. 4, 597–614 [DOI] [PubMed] [Google Scholar]
- 5.Shenoi S., Heysell S., Moll A., Friedland G. (2009) Curr. Opin. Infect. Dis. 22, 11–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Singh R., Manjunatha U., Boshoff H. I., Ha Y. H., Niyomrattanakit P., Ledwidge R., Dowd C. S., Lee I. Y., Kim P., Zhang L., Kang S., Keller T. H., Jiricek J., Barry C. E., 3rd (2008) Science 322, 1392–1395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Andries K., Verhasselt P., Guillemont J., Göhlmann H. W., Neefs J. M., Winkler H., Van Gestel J., Timmerman P., Zhu M., Lee E., Williams P., de Chaffoy D., Huitric E., Hoffner S., Cambau E., Truffot-Pernot C., Lounis N., Jarlier V. (2005) Science 307, 223–227 [DOI] [PubMed] [Google Scholar]
- 8.Rivers E. C., Mancera R. L. (2008) Drug Discov. Today 13, 1090–1098 [DOI] [PubMed] [Google Scholar]
- 9.Makarov V., Manina G., Mikusova K., Möllmann U., Ryabova O., Saint-Joanis B., Dhar N., Pasca M. R., Buroni S., Lucarelli A. P., Milano A., De Rossi E., Belanova M., Bobovska A., Dianiskova P., Kordulakova J., Sala C., Fullam E., Schneider P., McKinney J. D., Brodin P., Christophe T., Waddell S., Butcher P., Albrethsen J., Rosenkrands I., Brosch R., Nandi V., Bharath S., Gaonkar S., Shandil R. K., Balasubramanian V., Balganesh T., Tyagi S., Grosset J., Riccardi G., Cole S. T. (2009) Science 324, 801–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hugonnet J. E., Tremblay L. W., Boshoff H. I., Barry C. E., 3rd, Blanchard J. S. (2009) Science 323, 1215–1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cole S. T., Brosch R., Parkhill J., Garnier T., Churcher C., Harris D., Gordon S. V., Eiglmeier K., Gas S., Barry C. E., 3rd, Tekaia F., Badcock K., Basham D., Brown D., Chillingworth T., Connor R., Davies R., Devlin K., Feltwell T., Gentles S., Hamlin N., Holroyd S., Hornsby T., Jagels K., Krogh A., McLean J., Moule S., Murphy L., Oliver K., Osborne J., Quail M. A., Rajandream M. A., Rogers J., Rutter S., Seeger K., Skelton J., Squares R., Squares S., Sulston J. E., Taylor K., Whitehead S., Barrell B. G. (1998) Nature 393, 537–544 [DOI] [PubMed] [Google Scholar]
- 12.Fleischmann R. D., Alland D., Eisen J. A., Carpenter L., White O., Peterson J., DeBoy R., Dodson R., Gwinn M., Haft D., Hickey E., Kolonay J. F., Nelson W. C., Umayam L. A., Ermolaeva M., Salzberg S. L., Delcher A., Utterback T., Weidman J., Khouri H., Gill J., Mikula A., Bishai W., Jacobs W. R., Jr., Venter J. C., Fraser C. M. (2002) J. Bacteriol. 184, 5479–5490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McLean K. J., Scrutton N. S., Munro A. W. (2003) Biochem. J. 372, 317–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bellamine A., Mangla A. T., Nes W. D., Waterman M. R. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 8937–8942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Belin P., Le Du M. H., Fielding A., Lequin O., Jacquet M., Charbonnier J. B., Lecoq A., Thai R., Courçon M., Masson C., Dugave C., Genet R., Pernodet J. L., Gondry M. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 7426–7431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Holsclaw C. M., Sogi K. M., Gilmore S. A., Schelle M. W., Leavell M. D., Bertozzi C. R., Leary J. A. (2008) ACS Chem. Biol. 3, 619–624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johnston J. B., Kells P. M., Podust L. M., Ortiz de Montellano P. R. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 20687–20692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McLean K. J., Lafite P., Levy C., Cheesman M. R., Mast N., Pikuleva I. A., Leys D., Munro A. W. (2009) J. Biol. Chem. 284, 35524–35533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Capyk J. K., Kalscheuer R., Stewart G. R., Liu J., Kwon H., Zhao R., Okamoto S., Jacobs W. R., Jr., Eltis L. D., Mohn W. W. (2009) J. Biol. Chem. 284, 35534–35542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Van der Geize R., Yam K., Heuser T., Wilbrink M. H., Hara H., Anderton M. C., Sim E., Dijkhuizen L., Davies J. E., Mohn W. W., Eltis L. D. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 1947–1952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chang J. C., Harik N. S., Liao R. P., Sherman D. R. (2007) J. Infect. Dis. 196, 788–795 [DOI] [PubMed] [Google Scholar]
- 22.Chang J. C., Miner M. D., Pandey A. K., Gill W. P., Harik N. S., Sassetti C. M., Sherman D. R. (2009) J. Bacteriol. 191, 5232–5239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McLean K. J., Belcher J., Driscoll M. D., Fernandez C., Le Van D., Bui S., Golovanova M., Munro A. W. (2010) Future Med. Chem. 2, 1339–1353 [DOI] [PubMed] [Google Scholar]
- 24.Ouellet H., Lang J., Couture M., Ortiz de Montellano P. R. (2009) Biochemistry 48, 863–872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ouellet H., Guan S., Johnston J. B., Chow E. D., Kells P. M., Burlingame A. L., Cox J. S., Podust L. M., de Montellano P. R. (2010) Mol. Microbiol. 77, 730–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McLean K. J., Cheesman M. R., Rivers S. L., Richmond A., Leys D., Chapman S. K., Reid G. A., Price N. C., Kelly S. M., Clarkson J., Smith W. E., Munro A. W. (2002) J. Inorg. Biochem. 91, 527–541 [DOI] [PubMed] [Google Scholar]
- 27.Leys D., Mowat C. G., McLean K. J., Richmond A., Chapman S. K., Walkinshaw M. D., Munro A. W. (2003) J. Biol. Chem. 278, 5141–5147 [DOI] [PubMed] [Google Scholar]
- 28.McLean K. J., Carroll P., Lewis D. G., Dunford A. J., Seward H. E., Neeli R., Cheesman M. R., Marsollier L., Douglas P., Smith W. E., Rosenkrands I., Cole S. T., Leys D., Parish T., Munro A. W. (2008) J. Biol. Chem. 283, 33406–33416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morrison J. F. (1969) Biochim. Biophys. Acta 185, 269–286 [DOI] [PubMed] [Google Scholar]
- 30.Omura T., Sato R. (1964) J. Biol. Chem. 239, 2370–2378 [PubMed] [Google Scholar]
- 31.Berry E. A., Trumpower B. L. (1987) Anal. Biochem. 161, 1–15 [DOI] [PubMed] [Google Scholar]
- 32.Daff S. N., Chapman S. K., Turner K. L., Holt R. A., Govindaraj S., Poulos T. L., Munro A. W. (1997) Biochemistry 36, 13816–13823 [DOI] [PubMed] [Google Scholar]
- 33.Girvan H. M., Marshall K. R., Lawson R. J., Leys D., Joyce M. G., Clarkson J., Smith W. E., Cheesman M. R., Munro A. W. (2004) J. Biol. Chem. 279, 23274–23286 [DOI] [PubMed] [Google Scholar]
- 34.Dutton P. L. (1978) Methods Enzymol. 54, 411–435 [DOI] [PubMed] [Google Scholar]
- 35.Mast N., Norcross R., Andersson U., Shou M., Nakayama K., Bjorkhem I., Pikuleva I. A. (2003) Biochemistry 42, 14284–14292 [DOI] [PubMed] [Google Scholar]
- 36.Kabsch W. (1993) J. Appl. Crystallogr. 26, 795–800 [Google Scholar]
- 37.Collaborative Computation Project No. 4 (1994) Acta Crystallogr. D Biol. Crystallogr. 50, 760–76315299374 [Google Scholar]
- 38.McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Emsley P., Cowtan K. (2004) Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 40.Davis I. W., Leaver-Fay A., Chen V. B., Block J. N., Kapral G. J., Wang X., Murray L. W., Arendall W. B., 3rd, Snoeyink J., Richardson J. S., Richardson D. C. (2007) Nucleic Acids Res. 35, W375–W383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) Acta Crystallogr. D. Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lafite P., André F., Zeldin D. C., Dansette P. M., Mansuy D. (2007) Biochemistry 46, 10237–10247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Perera R., Sono M., Sigman J. A., Pfister T. D., Lu Y., Dawson J. H. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 3641–3646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dunford A. J., McLean K. J., Sabri M., Seward H. E., Heyes D. J., Scrutton N. S., Munro A. W. (2007) J. Biol. Chem. 282, 24816–24824 [DOI] [PubMed] [Google Scholar]
- 45.Aoyama Y., Horiuchi T., Gotoh O., Noshiro M., Yoshida Y. (1998) J. Biochem. 124, 694–696 [DOI] [PubMed] [Google Scholar]
- 46.McLean K. J., Warman A. J., Seward H. E., Marshall K. R., Girvan H. M., Cheesman M. R., Waterman M. R., Munro A. W. (2006) Biochemistry 45, 8427–8443 [DOI] [PubMed] [Google Scholar]
- 47.Ogura H., Nishida C. R., Hoch U. R., Perera R., Dawson J. H., Ortiz de Montellano P. R. (2004) Biochemistry 43, 14712–14721 [DOI] [PubMed] [Google Scholar]
- 48.Girvan H. M., Levy C. W., Williams P., Fisher K., Cheesman M. R., Rigby S. E., Leys D., Munro A. W. (2010) Biochem. J. 427, 455–466 [DOI] [PubMed] [Google Scholar]
- 49.Rosłoniec K. Z., Wilbrink M. H., Capyk J. K., Mohn W. W., Ostendorf M., van der Geize R., Dijkhuizen L., Eltis L. D. (2009) Mol. Microbiol. 74, 1031–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jain M., Petzold C. J., Schelle M. W., Leavell M. D., Mougous J. D., Bertozzi C. R., Leary J. A., Cox J. S. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 5133–5138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Capyk J. K., D'Angelo I., Strynadka N. C., Eltis L. D. (2009) J. Biol. Chem. 284, 9937–9946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stombaugh N. A., Sundquist J. E., Burris R. H., Orme-Johnson W. H. (1976) Biochemistry 15, 2633–2641 [DOI] [PubMed] [Google Scholar]
- 53.Batie C. J., Kamin H. (1981) J. Biol. Chem. 256, 7756–7763 [PubMed] [Google Scholar]
- 54.McIver L., Leadbeater C., Campopiano D. J., Baxter R. L., Daff S. N., Chapman S. K., Munro A. W. (1998) Eur. J. Biochem. 257, 577–585 [DOI] [PubMed] [Google Scholar]
- 55.DuSell C. D., Umetani M., Shaul P. W., Mangelsdorf D. J., McDonnell D. P. (2008) Mol. Endocrinol. 22, 65–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hansson M., Ellis E., Hunt M. C., Schmitz G., Babiker A. (2003) Biochim. Biophys. Acta 1593, 283–289 [DOI] [PubMed] [Google Scholar]
- 57.Chen W., Chen G., Head D. L., Mangelsdorf D. J., Russell D. W. (2007) Cell Metab. 5, 73–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Javitt N. B. (2002) J. Lipid Res. 43, 665–670 [PubMed] [Google Scholar]
- 59.McLean K. J., Clift D., Lewis D. G., Sabri M., Balding P. R., Sutcliffe M. J., Leys D., Munro A. W. (2006) Trends Microbiol. 14, 220–228 [DOI] [PubMed] [Google Scholar]
- 60.Driscoll M. D., McLean K. J., Cheesman M. R., Jowitt T. A., Howard M., Carroll P., Parish T., Munro A. W. (2010) Biochim. Biophys. Acta, in press [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.