

Abstract
The macrolactone archazolid is a novel, highly specific V-ATPase inhibitor with an IC50 value in the low nanomolar range. The binding site of archazolid is presumed to overlap with the binding site of the established plecomacrolide V-ATPase inhibitors bafilomycin and concanamycin in subunit c of the membrane-integral VO complex. Using a semi-synthetic derivative of archazolid for photoaffinity labeling of the V1VO holoenzyme we confirmed binding of archazolid to the VO subunit c. For the plecomacrolide binding site a model has been published based on mutagenesis studies of the c subunit of Neurospora crassa, revealing 11 amino acids that are part of the binding pocket at the interface of two adjacent c subunits (Bowman, B. J., McCall, M. E., Baertsch, R., and Bowman, E. J. (2006) J. Biol. Chem. 281, 31885–31893). To investigate the contribution of these amino acids to the binding of archazolid, we established in Saccharomyces cerevisiae mutations that in N. crassa had changed the IC50 value for bafilomycin 10-fold or more and showed that out of the amino acids forming the plecomacrolide binding pocket only one amino acid (tyrosine 142) contributes to the binding of archazolid. Using a fluorescent derivative of N,N′-dicyclohexylcarbodiimide, we found that the binding site for archazolid comprises the essential glutamate within helix 4 of subunit c. In conclusion the archazolid binding site resides within the equatorial region of the VO rotor subunit c. This hypothesis was supported by an additional subset of mutations within helix 4 that revealed that leucine 144 plays a role in archazolid binding.
Keywords: Antibiotics, Drug Action, Protein Drug Interactions, Vacuolar ATPase, Yeast, Manduca sexta, Saccharomyces cerevisiae, V-ATPase, Archazolid, Bafilomycin
Introduction
Vacuolar ATPases (V-ATPases)3 are heteromultimeric proteins that use the energy of ATP hydrolysis to translocate protons from the cytoplasm into intracellular compartments or across the plasma membrane of eukaryotic cells. This transport of protons is mediated by the membrane-integral VO complex, whereas the cleavage of ATP occurs at the cytoplasmatic V1 complex (1). The VO complex is composed of single copies of subunits a, d, and e, and the ring-forming proteolipid subunits c, c″, and in fungi subunit c′ also (2). Based on the crystal structure from the VO ring of K subunits, a homologue of the H+-translocating subunit c in the V-type Na+-ATPase from Enterococcus hirae, and a cryoelectron microscopy structure from the V-ATPase of Manduca sexta, an arrangement of 10 subunits is proposed for the VO ring (3, 4). The subunits c and c′ are predicted to have four transmembrane helices (TM 1 to 4), whereas subunit c″ contains an additional fifth transmembrane helix. All proteolipid subunits contain a conserved glutamate residue, subunits c and c′ in TM4 and subunit c″ in TM3, which are essential for proton transport across the membrane (2). This glutamate is a target for the covalent binding inhibitor N,N′-dicyclohexylcarbodiimide (DCCD) and its derivatives (5–8).
By regulating the pH homeostasis and membrane energization of cells, V-ATPases are involved in a variety of fundamental processes like vesicular trafficking or secondary transport. In addition, plasma membrane V-ATPases are responsible for extracellular acidification, e.g. in osteoclasts or metastasing tumor cells, and therefore play an important role in severe diseases such as osteoporosis or cancer (7). For these reasons the V-ATPase is a promising therapeutic target, and inhibitors of this enzyme are the focus of biomedical research. A variety of such compounds has been discovered of which the plecomacrolide inhibitors bafilomycin and concanamycin are the best studied examples (9). With IC50 values at low nanomolar concentrations these compounds are highly specific inhibitors of the V-ATPase (10). Throughout the past years the binding site and inhibition mechanism of the plecomacrolides has been studied in more detail. In 2002 Bowman et al. (11) identified via mutagenesis studies in Neurospora crassa amino acids in VO subunit c that contribute to the binding of bafilomycin. In the same year photoaffinity labeling studies with the radioactive concanamycin derivative 125I-concanolid A also resulted in the identification of subunit c as the binding partner for plecomacrolides (12). Further mutagenesis studies (13) and crystallization of the VO K-ring from E. hirae (4) finally resulted in a model of the plecomacrolide binding site within the V-ATPase of N. crassa in which the binding site is located at the interface of two adjacent c subunits in the cytosolic half of the membrane bilayer (14). It was suggested that the plecomacrolides inhibit V-ATPase function by blocking rotation of the c-ring relative to subunit a or by preventing internal torsion of the transmembrane helices within the c-ring (2, 13). An influence of amino acid exchanges on plecomacrolide binding was shown in Saccharomyces cerevisiae via site-directed mutagenesis also for subunit a (15).
Beside the established plecomacrolide antibiotics, new V-ATPase inhibitors have been identified in the past decade (9). In 2003 Sasse et al. (16) isolated the macrolactone archazolid from the myxobacteria Archangium gephyra and Cystobacter violaceus, which exhibited high cytotoxicity in mouse fibroblasts. Recently, the three-dimensional structure and stereochemistry was assigned by extensive high-field NMR studies in combination with molecular modeling and confirmed by total synthesis (17, 18). Archazolid was assumed to be a novel V-ATPase inhibitor because it induced the same morphological changes in mammalian cells as the approved plecomacrolide inhibitors bafilomycin and concanamycin (16, 19). Indeed, the new compound inhibited the V-ATPase from M. sexta half-maximally at a concentration of 20 nm, the ion translocating F- and P-type ATPases were not affected (19). These findings led to the conclusion that archazolid is a novel specific and highly efficient V-ATPase inhibitor. Although it exhibited only a low inhibitory effect against intact yeast cells (16), inhibition assays using isolated yeast vacuoles, which we present here, confirm that archazolid is also a highly potent inhibitor of the yeast V-ATPase.
Up to now information has been limited concerning the potential binding site of archazolid. In competition assays archazolid prevented, like bafilomycin, labeling of the V-ATPase subunit c with 125I-concanolid A, and therefore it was assumed that it shares at least part of its binding site in the V-ATPase with the plecomacrolide antibiotics (19). As this binding site had been characterized by mutagenesis studies (10, 11, 13), we supposed that the amino acids involved in plecomacrolide binding might also contribute to the binding of archazolid. In this regard, we chose those mutations in subunit c that had increased the IC50 value for bafilomycin 10-fold or more in N. crassa to carry out site-directed mutagenesis in S. cerevisiae. In a second approach, labeling experiments with NCD-4, which is a fluorescent derivative of DCCD, or with photoaffinity labeling derivatives of archazolid were performed to get more precise information about the interaction between archazolid and the V-ATPase. After the evaluation of these two sets of experiments we mutated additional amino acids within helix 4 of subunit c to improve the picture of how archazolid interacts with this subunit.
EXPERIMENTAL PROCEDURES
Strains and Culture Conditions
Yeast strains used in this study were BMA64-1B (MATα, ura3–52; trp1Δ2; leu2–3_112; his3–11; ade2-1; can1–100) and the corresponding diploid strain BMA64 (MATα/MATa; ura3–52/ura3–52; trp1Δ2/trp1Δ2; leu2–3_112/leu2–3_112; his3–11/his3–11; ade2-1/ade2-1; can1–100/can1–100) (Euroscarf, Frankfurt, Germany). Cells were usually grown on YPDA medium containing 1% yeast extract, 2% peptone, 2% glucose, and 0.02% adenine hemisulfate. The medium was buffered with 50 mm Na-MES/Na-MOPS and the pH was adjusted to either 5.5 or 7.5. For YPDA medium containing 0.1 m CaCl2, 10% CaCl2 was added from a sterile 1 m stock solution after autoclaving the medium. For YPDA plates 2% agar was added.
Selection was performed on plates containing 0.67% yeast nitrogen base without amino acids, 2% glucose, 1.5% agar, 10% drop-out solution (0.02% adenine hemisulfate, 0.02% l-arginine HCl, 0.02% l-histidine HCl, 0.03% l-isoleucine, 0.1% l-leucine, 0.03% l-lysine HCl, 0.02% l-methionine, 0.05% l-phenylalanine, 0.2% l-threonine, 0.02% l-tryptophan, 0.03% l-tyrosine, 0.02% uracil, 0.15% l-valine) lacking the respective amino acid. The medium was buffered with 50 mm Na-MES/Na-MOPS and pH was adjusted to 5.5.
Generation of a vma3 Deletion Mutant
The vma3 deletion mutant BMA64-1BΔVma3 (MATα, ura3–52, trp1Δ2, leu2–3_112, his3–11, ade2-1, can1–100, vma3::HIS3) used throughout this study was constructed by exchanging the native vma3 gene against the his3 gene via homologous recombination. A DNA fragment containing the his3 gene with the tef1 promoter and terminator flanked by 40 bp homologous to the regions upstream and downstream of the vma3 gene was amplified using the vector pFA6a-His3MX6, the forward primer, CAAAAAGACTAATCAATTAGAATAACAAAAGAAACATATACATATAGATCTGTTTAGCTTGCCTCGTCCCCG and the reverse primer, GTATACTCTATTCCTGCTTTAGTGATTCAGAAGCTGCCCTGGATGGCGGCGTTAGTATGAATC. The resulting fragment was transformed into the diploid strain BMA64 by electroporation using a Gene-Pulser (Bio-Rad). Cells were selected on S.D. medium without histidine and sporulated on potassium acetate plates (2% potassium acetate, 1.5% agar). The haploid spores were again selected on S.D. plates without histidine; exchange of the vma3 gene and mating type were verified by PCR on genomic DNA.
Site-directed Mutagenesis of the vma3 Gene
For mutagenesis of the yeast vma3 gene, flanked 300 bp upstream and downstream containing its native promoter and terminator, the gene was cloned into the yeast CEN vector pRS415. The mutagenesis was performed using the QuikChange II Site-directed Mutagenesis Kit (Stratagene). Amino acid exchanges were made by mutagenesis of each codon as indicated: T32A (act → gct), T32I (act → att), I39A (atc → gcc), I39F (atc → ttc), I54A (att → gct), I54F (att → ttt), G61A (ggt → gct), G61S (ggt → agt), Y66F (tac → ttc), Y66S (tac → tcc), F135A (ttt → gct), F135L (ttt → ctt), V138A (gtt → gca), V138T (gtt → act), Y142A (tac → gcc), Y142H (tac → cac), Y142N (tac → aac), L141F (cta → ttc), L141I (cta → ata), L144F (ttg → ttc), L144I (ttg → atc), I145F (att → ttt), I145L (att → ctt).
Primers (forward, TACTCACGGCCGTCTACGGCCTATTC, reverse, TACTCAGGATCCCTCATCGTACCCATTGTG) complementary to the endings of the insert were used to verify the resulting mutations. Sequencing was carried out at Seqlab (Sequence Laboratories GmbH, Göttingen, Germany). Plasmids were transformed into the yeast strain BMA64-1BΔVma3 via electroporation and cells were selected on S.D. plates without leucine. To confirm the V-ATPase function in these strains, spotting growth assays were performed. Yeast cells were grown in 5 ml of YPDA medium to early stationary phase and then diluted to 105 cells/ml in distilled water. Serial 10-fold dilutions from 104 cells/ml to 100 cells/ml were performed in water and 5 μl of each dilution was pipetted onto plates with different growth medium. Pictures were taken after 3 days of incubation at 30 °C.
Quinacrine Staining of Yeast Vacuoles
The fluorescent dye quinacrine was used to indicate acidification of the yeast vacuole (20). Cells were grown in YPDA (pH 5.5) to the exponential phase, 1 ml of the culture was then cooled on ice for 5 min and the cells were pelleted by brief centrifugation. The pellet was resuspended in 100 μl of YPDA (pH 7.5) containing 200 μm quinacrine (Sigma, Q3251) followed by incubation with shaking for 1 h at 30 °C. Afterward, the suspension was again cooled on ice, the cells were pelleted and washed twice with a solution containing 2% glucose and 50 mm Na-MES/Na-MOPS (pH 7.5). Finally, the cells were resuspended in 100 μl of 2% glucose (pH 7.5). Quinacrine staining was viewed within 15 min using inverted microscope system IX70 (Olympus) with 430/24 excitation and 535/30 emission filters.
Preparation of Yeast Vacuoles
Yeast vacuoles were basically purified according to Refs. 21 and 22. Glucose incubation after zymolyase treatment was performed for 30 min. After recovery, the spheroplasts were lysed in 15 ml of 10 mm Tris-MES (pH 6.9), 0.1 mm MgCl2, 12% Ficoll-400 containing 50 μl of protease inhibitor mixture (Calbiochem, number 535142). The purified vacuoles were stored in 1 ml of 10 mm Tris-MES (pH 6.9), 5 mm MgCl2, 25 mm KCl containing 10 μl of protease inhibitor mixture at −80 °C.
Inhibitors
Archazolid A, bafilomycin A1, concanamycin A, concanolide A, and [14C]concanolid A were isolated or synthesized according to published procedures (16, 23–25). Apicularen A was isolated by Brigitte Kunze according to Ref. 26. The synthesis of bis-[4-(3-trifluoromethyldiazirin-3-yl)benzoyl]-archazolid A (BD-archazolid) is described under the supplemental data.
Stock solutions (10 mm) of the inhibitors in dimethyl sulfoxide were stored at −80 °C. For activity assays serial dilutions were prepared in dimethyl sulfoxide. Stock solutions (10 mm) of DCCD (Sigma, D80002) and NCD-4 (Invitrogen, C428) in ethanol were stored at −20 °C.
Activity Assays with Purified Yeast Vacuoles
Activity assays at pH 6.9 were performed in triplicate using 3 μg of yeast vacuolar protein in a total volume of 160 μl of 50 mm Tris-MES with 3.75 mm MgCl2, 0.1 mm sodium orthovanadate, 20 mm KCl, 0.5 mm sodium azide, 5 mm Tris-HCl, 2 mm ATP, and 6.25% dimethyl sulfoxide, with or without V-ATPase inhibitor. The samples were preincubated for 10 min at 30 °C, and the reaction was started by the addition of ATP. After incubation for 20 min at 30 °C the reaction was stopped by freezing the samples in liquid nitrogen. Inorganic phosphate was determined according to Ref. 27.
Radioactive Labeling
Thirty μg of M. sexta V-ATPase were incubated with 0.6 mm [14C]BD-archazolid (Fig. 4) or 1 μm 23-O-(3-trifluoromethyldiazirin-3-yl)-[1-14C]benzoyl-21-desoxyconcanolide A ([14C]concanolid A) in a total volume of 40 μl for 1 h at 25 °C. Afterward ATP was added to a final concentration of 1.5 mm and the samples were irradiated for 1 min with UV light (366 nm) on ice, followed by SDS-PAGE and phosphoscreen analysis according to Ref. 12. In addition, the subunits of interest were cut out of the gel and subjected to scintillation counting (10,000 counts; LS 6500, Beckman Coulter). For competition assays the V-ATPase was incubated with 50 μm concanamycin A for 1 h at 25 °C before proceeding as described above.
FIGURE 4.

Labeling of subunit c with a radioactive derivative of archazolid A. Purification of the M. sexta V-ATPase was carried out as described under “Experimental Procedures.” A, the V-ATPase was preincubated with 50 μm concanamycin A or with the solvent dimethyl sulfoxide. Afterward [14C]BD-archazolid was added and the cross-linking reaction was induced by exposure to UV light (366 nm) for 1 min (+). Controls were without UV exposure (−). Note that due to the lower affinity of the V-ATPase for BD-archazolid the concentration of [14C]BD for labeling has to be very high (Fig. 3). This leads to an unavoidable high background in the gel. B, line scan of the autoradiography shown in A. C, the V-ATPase was incubated with the radioactive derivatives of either archazolid ([14C]-BD) or concanamycin A ([14C]-Ccl) and the cross-linking reaction was induced as described above. Controls without UV exposure are not shown. The arrow indicates subunit c. DMSO, dimethyl sulfoxide.
NCD-4 Labeling
For labeling 100 μg of the purified M. sexta V1VO holoenzyme or 20 μg of the VO complex was preincubated for 10 min at 25 °C in 100 μl of solution containing 50 μm archazolid A or 50 μm bafilomycin A1, 20 mm Tris-HCl (pH 8.1), 50 mm NaCl, 9.6 mm 2-mercaptoethanol, 0.01% C12E10, followed by the addition of NCD-4 to a final concentration of 0.1 mm. After incubation for 3 h at 25 °C, 20 μl of each sample were subjected to SDS-PAGE, and the gels were then fixed in 10% acetic acid, 40% ethanol for 30 min. The NCD-4 label was documented using the Versa Doc system (Bio-Rad) with UV transillumination and 520LP filter. Additional experiments were performed also using 100 μg of the purified M. sexta V-ATPase in 100 μl of buffer (see above), which was preincubated for 1 h at 25 °C with 500 μm bafilomycin A1, concanamycin A, concanolid A, or apicularen A, respectively. Next, the samples were incubated in the presence of 50 μm archazolid A or dimethyl sulfoxide as described above. Then, NCD-4 was added to a final concentration of 0.1 mm. After a third incubation for 1 h at 25 °C, the samples were treated as described above.
Modeling a Vma3-ring
To map the approximate positions of the key residues involved in binding archazolid and bafilomycin, a model of the S. cerevisiae c-ring was generated. This model is based on the crystal structure of the E. hirae K-ring structure, which shares 24% sequence identity and 68% similarity to the Saccharomyces c-ring and as such is believed to share a very similar fold. In addition to replacing the residues in the E. hirae crystal structure with their equivalents in the Saccharomyces c-ring, loop modeling was also carried out using the Phyre program (28). The resulting model provides a structure with high confidence in the main chain position of those residues that form the 4-helix bundle, a conserved motif of the V-ATPase c subunit.
Other Methods
SDS-PAGE and Western blotting were carried out as described earlier (12). For immunodetection the monoclonal antibody A6422 (Invitrogen) against Vma1p and the secondary antibody A3682 (Sigma) were used. The V1VO holoenzyme and the VO complex from M. sexta were purified according to established protocols (27). Activity assays for the V1VO holoenzyme and determination of the produced inorganic phosphate were performed according to Refs. 12 and 27. Protein content was determined with Amido Black (27).
RESULTS
Site-directed Mutagenesis of the vma3 Gene
To create a defined genetic background for our mutagenesis experiments, a Vma3 deletion mutant was engineered in yeast strain BMA64. The resulting strain BMA64-1BΔVma3 exhibited the typical Vma− phenotype (29), which could be completely restored via transformation with the plasmid pRS415_Vma3 bearing the wild type Vma3 gene (Table 1). This strain also showed wild type growth at pH 7.5 in the presence of 0.1 m CaCl2, whereas transformation with the empty vector pRS415 had no effect on the Vma− phenotype (data not shown).
TABLE 1.
In vivo assays of yeast strains expressing mutated versions of the V-ATPase subunit Vma3p
For growth assays, the cells were dropped on YPDA plates (pH 5.5) with or without calcium chloride and on YPDA buffered to pH 7.5 (+++ indicates wild type growth; ++ indicates slightly impaired growth; + indicates strongly impaired growth; −, indicates no growth). Quinacrine staining was used to visualize accumulation of protons in the vacuole of living yeast cells as indication for an intact V-ATPase (see above).
| Mutation in Vma3p | YPDA (pH 5.5) | YPDA (pH 5.5), 100 mm CaCl2 | YPDA (pH 7.5) | Quinacrine fluorescence |
|---|---|---|---|---|
| Controls | ||||
| BMA64-1B | +++ | +++ | +++ | Positive |
| ΔVma3 | ++ | − | − | Negative |
| pRS415-Vma3 | +++ | +++ | +++ | Positive |
| Helix 1 | ||||
| T32I | +++ | +++ | ++ | Positive |
| I39F | +++ | +++ | +++ | Positive |
| Helix 2 | ||||
| I54F | +++ | +++ | + | Positive |
| G61S | +++ | +++ | +++ | Positive |
| Y66F | +++ | +++ | + | Positive |
| Helix 4 | ||||
| F135L | ++ | ++ | + | Positive |
| V138A | +++ | +++ | +++ | Positive |
| V138T | +++ | ++ | − | Negative |
| L141F | +++ | ++ | − | Negative |
| L141I | +++ | +++ | + | Negative |
| Y142H | ++ | ++ | − | Negative |
| Y142N | ++ | ++ | + | Negative |
| L144F | +++ | +++ | + | Positive |
| L144I | +++ | + | + | Positive |
| I145 | +++ | +++ | +++ | Positive |
| I145L | +++ | +++ | ++ | Positive |
Because previous results had indicated that archazolid and bafilomycin interact with the same region of the VO subunit c (19), we wanted to find out whether mutations in subunit c, which have an effect on the binding of bafilomycin, also influence archazolid binding. All the mutations selected for this work (T32I, I39F, I54F, G61S, F136L, Y143H, and Y143N) had been shown to increase the resistance of the V-ATPase from N. crassa to bafilomycin A1 at least 10-fold (14). In the following these mutations are referred to as “bafilomycin” mutations. So far a similar effect for only two of the corresponding mutations in S. cerevisiae (T32I and I54F) was described (13). We established the mutations via site-directed mutagenesis on plasmid pRS415-Vma3, followed by expression of the altered versions of Vma3p in yeast strain BMA64-1BΔVma3. In addition we mutated amino acids that we, in regard to the results described later, presumed to interact with archazolid (Tyr-66, Val-138, Leu-141, Leu-144, Ile-145). Each of these amino acids was replaced by similar and structurally different amino acids, respectively.
Effect of Mutations in Vma3p on the V-ATPase
To check the V-ATPase function in vivo, all strains were tested for growth on medium with pH 7.5 or on medium containing 0.1 m CaCl2 at pH 5.5 (Table 1). In most cases, transformation of BMA64-1BΔVma3 with the mutated versions of pRS415-Vma3 resulted in total complementation of the Vma− phenotype on medium containing 0.1 m CaCl2 at pH 5.5. Strains I39F, G61S, V138A, and I145F exhibited wild type growth, whereas T32I, I54F, Y66F, L141I, L144F, and I145L grew more slowly at pH 7.5. Mutations F135L, V138T, L141F, Y142H, Y142N, and L144I resulted in reduced growth, particularly at pH 7.5, suggesting that these mutations in helix 4 are less tolerable. Y66S exhibited a typical Vma− phenotype (data not shown). Nevertheless, the results from the growth assays pointed to the assembly of an, at least partially, functional V-ATPase in all yeast strains bearing a mutation in Vma3p used in this study. This outcome was supported by the results from vacuolar staining using the fluorescent dye quinacrine. With the exception of V138T, L141F, L141I, and Y142H, and Y142N, a clear staining of the vacuolar lumen could be observed in all strains (Table 1).
The results from the in vivo assays that indicated a functional V-ATPase were strengthened by Western blot analysis of isolated vacuoles from the mutant strains. To prove the attachment of the V1 complex to the VO complex at the vacuolar membrane, an antibody against Vma1p was used (30). As shown in Fig. 1, for all strains a clear signal of Vma1p could be detected at the purified vacuolar membranes, confirming that none of the mutations impeded assembly of the V-ATPase at the membrane. The reduced amount of Vma1p detected in I54F, L144F, and L144I points to a smaller amount of assembled V-ATPase in these strains. Nevertheless, the in vivo function of the enzyme was not drastically reduced as all three strains did not exhibit a Vma− phenotype.
FIGURE 1.

Assembly of the V-ATPase in yeast strains expressing the mutated Vma3 proteins. The assembly of the mutated versions of Vma3p into V-ATPase complexes was verified by Western blot analysis of isolated vacuolar membranes. Using a monoclonal antibody against subunit A (Vma1p, A6422, Invitrogen), the attachment of the V1 complex to the vacuolar membrane was interpreted as a proof for the correct assembly of the V1VO holoenzyme (30). WT, BMA64-1B; M, BMA64-1BΔVma3, strains expressing the Vma3p with the indicated mutations.
Functionality of the mutated V-ATPases was also tested by measurement of the ATPase activity using isolated yeast vacuoles (Table 2). Most of the strains containing V-ATPase with mutated Vma3p showed bafilomycin-sensitive activity of more than 50% up to 100% of the wild type activity. Only mutation Y142N led to a greater reduction of the V-ATPase activity with only 0.1 μmol/min/mg, which was, however, still 30% of wild type activity.
TABLE 2.
Bafilomycin A1 and archazolid A sensitive activity of vacuolar membranes isolated from yeast strains expressing either the wild type or a mutated version of the Vm3p
Vacuolar membrane preparations and ATPase activity assays of the wild type (BMA64-1B) and the mutant strains were carried out as described under “Experimental Procedures.” The activities were corrected for the residual activity at the point of maximal inhibition by bafilomycin A1 and archazolid A, respectively. Values are the averages obtained from two or three independent vacuolar preparations with S.D. as indicated.
| Mutation in Vma3p | ATPase activity sensitive to bafilomycin A1 | ATPase activity sensitive to archazolid A |
|---|---|---|
| μmol/min/mg | ||
| Control | ||
| BMA64-1B | 0.30 ± 0.06 | 0.29 ± 0.11 |
| Helix 1 | ||
| T32I | 0.27 ± 0.08 | 0.25 ± 0.04 |
| I39F | 0.32 ± 0.11 | 0.36 ± 0.18 |
| Helix 2 | ||
| I54F | 0.18 ± 0.02 | 0.19 ± 0.01 |
| G61S | 0.18 ± 0.05 | 0.23 ± 0.02 |
| Y66F | 0.31 ± 0.05 | 0.34 ± 0.06 |
| Helix 4 | ||
| F135L | 0.16 ± 0.03 | 0.15 ± 0.01 |
| V138A | 0.28 ± 0.02 | 0.27 ± 0.06 |
| V138T | 0.25 ± 0.10 | 0.26 ± 0.10 |
| L141F | 0.14 ± 0.01 | 0.10 ± 0.01 |
| L141I | 0.28 ± 0.11 | 0.26 ± 0.13 |
| Y142H | 0.25 ± 0.13 | 0.26 ± 0.14 |
| Y142N | 0.10 ± 0.01 | 0.12 ± 0.02 |
| L144F | 0.40 ± 0.08 | 0.33 ± 0.03 |
| L144I | 0.23 ± 0.01 | 022 ± 0.03 |
| I145F | 0.18 ± 0.07 | 0.25 ± 0.09 |
| I145L | 0.54 ± 0.01 | 0.48 ± 0.08 |
Influence of the Bafilomycin Mutations on Inhibitor Binding
The influence of the mutations in the Vma3p of S. cerevisiae on the binding of archazolid or bafilomycin was tested by activity assays on isolated yeast vacuoles at inhibitor concentrations ranging from 0.1 nm to 3 μm. The IC50 value for wild type strain BMA64-1B was 6.6 nm for archazolid A and 4.7 nm for bafilomycin A1 (Table 3).
TABLE 3.
IC50 values for the inhibition of the wild type and mutant strains by bafilomycin A1 and archazolid A
Vacuolar membrane preparations and ATPase activity assays were carried out as described under “Experimental Procedures.” The activities were corrected for the residual activity at the point of maximal inhibition by bafilomycin A1 and archazolid A, respectively. IC50 values with the indicated standard deviations result from plotting the average values obtained from two or three independent vacuolar preparations.
| Mutation in Vma3p | IC50 |
Factor |
||
|---|---|---|---|---|
| Bafilomycin A1 | Archazolid A | Bafilomycin A1 | Archazolid A | |
| nm | ||||
| Control | ||||
| BMA64-1B | 4.7 ± 0.4 | 6.6 ± 0.5 | ||
| Helix 1 | ||||
| T32I | 38.4 ± 2.4 | 3.7 ± 0.9 | 8.0 | 0.6 |
| I39F | 5.8 ± 0.4 | 4.1 ± 0.4 | 1.2 | 0.6 |
| Helix 2 | ||||
| I54F | 1.7 ± 0.1 | 3.4 ± 0.4 | 0.4 | 0.5 |
| G61S | 316 ± 73 | 4.7 ± 0.9 | 67 | 0.7 |
| Y66F | 2.9 ± 0.4 | 5.6 ± 0.3 | 0.6 | 1 |
| Helix 4 | ||||
| F135L | 35.7 ± 8.8 | 3.1 ± 0.4 | 7.6 | 0.5 |
| V138A | 8.1 ± 1.0 | 4.6 ± 0.9 | 1.7 | 0.7 |
| V138T | 43.8 ± 7.1 | 8.6 ± 1.2 | 9.3 | 1.3 |
| L141F | 11.3 ± 1.0 | 4.2 ± 1.0 | 2.4 | 0.6 |
| L141I | 1.6 ± 0.2 | 3.7 ± 0.5 | 0.3 | 0.6 |
| Y142H | 38.3 ± 6.4 | 4.1 ± 1.4 | 8.1 | 0.6 |
| Y142N | 56 ± 9.7 | 0.7 ± 0.1 | 12 | 0.1 |
| L144F | 2.5 ± 0.4 | 4.5 ± 0.6 | 0.5 | 0.7 |
| L144I | 1.0 ± 0.1 | 0.8 ± 0.1 | 0.2 | 0.1 |
| I145F | 1.8 ± 0.2 | 4.4 ± 1.2 | 0.4 | 0.7 |
| I145L | 11.9 ± 1.5 | 8.8 ± 1.7 | 2.5 | 1.4 |
Regarding bafilomycin, our results in principle confirmed the data published for N. crassa (14). The most striking effect resulted from the G61S mutation that exhibited an ∼70-fold higher IC50 value compared with the wild type S. cerevisiae strain (Table 3 and Fig. 2A), being consistent with a 90-fold increase reported for N. crassa (14). Mutations F135L, Y142H, and Y142N had an ∼10-fold higher IC50 value, which was also in the range of the published results (14). A notably lower increase of the IC50 value for the T32I mutation in S. cerevisiae as compared with N. crassa had already been observed (13). In contrast to the results obtained for N. crassa, mutation I39F had virtually no influence on the binding of bafilomycin in S. cerevisiae (Table 3 and Fig. 2A). Interestingly, mutation I54F approximately halved the IC50 value for bafilomycin A1 as compared with the wild type strain, suggesting a V-ATPase slightly more sensitive toward bafilomycin (Table 3). However, the minor influence of the I54F mutation, in contrast to the corresponding mutation in N. crassa, is in line with previous data for S. cerevisiae (13, 14).
FIGURE 2.

Influence of bafilomycin mutations in Vma3p of S. cerevisiae on inhibitor binding. V-ATPase activities were measured on isolated yeast vacuoles of wild type strain BMA64-1B (WT) and strains with mutations in Vma3p as indicated. Values are the average of three independent vacuolar preparations. Error bars are S.D. Absolute archazolid A- and bafilomycin A1-sensitive activities are shown in Table 2, IC50 values are indicated in Table 3.
Regarding the binding of archazolid, none of the mutations with a negative influence on the binding of bafilomycin mutations showed a decrease of sensitivity. In contrast, most of the bafilomycin mutations appeared to increase sensitivity of the V-ATPase toward archazolid slightly and mutation Y142N even led to a strong increase of sensitivity, with an IC50 value ∼10-fold lower as compared with the wild type strain (Table 3 and Fig. 2B). The finding that there was hardly any similarity regarding the effects of mutations on bafilomycin and archazolid binding was quite surprising, because the results of previous competition experiments had implied that plecomacrolides and archazolid at least partially share a common binding site (19).
Labeling of the V-ATPase with a Radioactive Analog of Archazolid
To demonstrate the binding of archazolid to subunit c, we performed cross-linking studies with a radioactive photoactivatable analog of the inhibitor. For this purpose a derivative with two diazirinyl groups (BD-archazolid), with or without 14C, was synthesized based on prior structure-function analysis (Fig. 3A) (31, 32). The non-radioactive derivative was tested for inhibitory efficiency on the purified V-ATPase from M. sexta (Fig. 3B). The IC50 value of BD-archazolid was 40-fold higher, compared with that for archazolid, but with 0.6 μm still in the low micromolar range. Therefore we used the [14C]-labeled isotopologue of this derivative in our cross-linking experiments. Due to the increased IC50 value of BD-archazolid a high concentration was needed for labeling, inevitably leading to a high overall background in the autoradiograms (Fig. 4). Nevertheless, a prominent labeling of a band at the molecular mass of subunit c could be observed (Fig. 4, A and B, black arrows). This labeling was drastically reduced after preincubation of the V-ATPase with 50 μm concanamycin A (Fig. 4, A and B). Because concanamycin A is known to bind to subunit c of the V-ATPase (12), this result clearly points to the specific labeling of subunit c by [14C]BD-archazolid. In addition, the identity of subunit c was confirmed by labeling with the photoaffinity labeling derivative of concanamycin, [14C]concanolid A (12, 23) (Fig. 4C, black arrow).4 Labeling of additional subunits by [14C]concanolid A is currently being investigated. Evaluation of the radioactivity of the excised bands of subunit c from the SDS-PAGE with a scintillation counter revealed a difference between the UV-treated and non-treated samples of 25 ± 0.25 cpm for [14C]concanolid A, comprising one [14C]-isotope, and 37 ± 0.37 cpm for [14C]BD-archazolid, which comprises two [14C]-isotopes (mean ± S.D.).
FIGURE 3.

A derivative of archazolid A created for photoaffinity labeling studies. Based on structure-function analysis (31, 32) a derivative for cross-linking studies, BD-archazolid, was created. A, structure of BD-archazolid. B, inhibition of the V-ATPase by BD-archazolid. Purification of the M. sexta V1VO holoenzyme and following activity assays were carried out as described under “Experimental Procedures.” Values represent the average of three independent preparations, with error bars corresponding to the S.D. V-ATPase activity sensitive to archazolid A was 1.8 ± 0.2 μmol/min/mg. The IC50 values were 0.016 ± 0.002 μm for the non-modified inhibitor and 0.6 ± 0.03 μm for BD-archazolid.
Labeling of the V-ATPase with the Fluorescent DCCD Derivative NCD-4
The fluorescent carbodiimide NCD-4 (33) specifically binds, like DCCD, to the essential glutamate within helix 4 of subunit c, leading to inhibition of the V-ATPase (5). We wanted to find out whether there is any interference between archazolid and NCD-4, which would indicate a binding of the macrolactone close to the essential glutamate.
The incubation of either the V-ATPase holoenzyme or the VO complex with NCD-4 revealed a prominent band of appropriate size in the SDS-PAGE (Fig. 5, lane 1). In the holoenzyme this label was completely impeded by preincubation with archazolid A (Fig. 3B), whereas bafilomycin A1 (Fig. 6B) had no pronounced effect (Fig. 5, lanes 3 and 4). Preincubation of the VO complex with the inhibitors led to similar results, except that archazolid A did not completely prevent labeling of subunit c (Fig. 5, lane 4). This is probably due to a better accessibility of the c-ring in the free VO complex or the higher abundance of subunit c in the VO sample, resulting in labeling of some c subunits with NCD-4. The different effects of bafilomycin A1 and archazolid A were unexpected because previous data had shown that bafilomycin A1, B1, and archazolid A as well as concanamycin prevent labeling of subunit c with a radioactive derivative of concanamycin A (125I-concanolide A) (12, 19).
FIGURE 5.
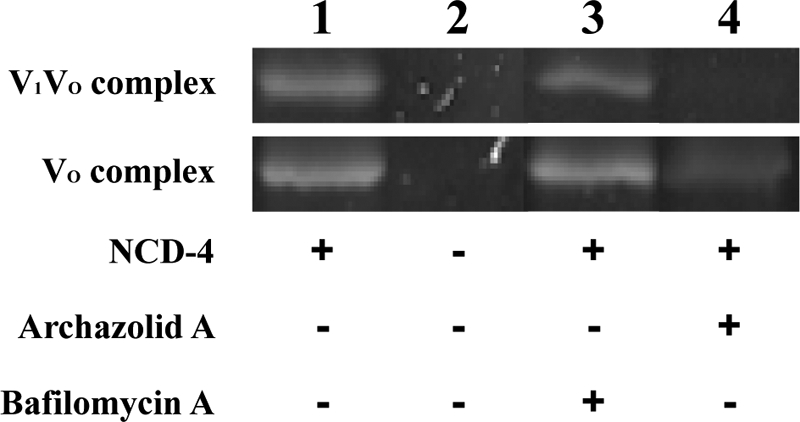
NCD-4 labeling of the V-ATPase subunit c in the presence of archazolid and bafilomycin. Purification of M. sexta V-ATPase and NCD-4 labeling was carried out as described under “Experimental Procedures.” Either V1VO holoenzyme or the VO complex was preincubated for 10 min at 25 °C with 50 μm bafilomycin A1 (lane 3) or archazolid A (lane 4), respectively. Then NCD-4 was added to a final concentration of 0.1 mm and the sample was incubated for 3 h at 25 °C. Control reactions were performed using dimethyl sulfoxide for preincubation, followed by the addition of NCD-4 or pure ethanol (lanes 1 and 2). Of each sample ∼20 μg of protein was subjected to SDS-PAGE and NCD-4 labeling of subunit c was documented under UV light (Versadoc, Bio-Rad). The IC50 values are 3.0 ± 0.2 μm for NCD-4 (see Fig. 6B), 16 ± 0.2 nm for archazolid A (see Fig. 3B), and 9.0 ± 0.8 nm for bafilomycin A (see Fig. 6B).
FIGURE 6.

Suppression of archazolid binding monitored with NCD-4 labeling of V-ATPase subunit c. A, purification of the M. sexta V1VO holoenzyme and NCD-4 labeling was carried out as described under “Experimental Procedures.” V-ATPase was preincubated for 1 h at 25 °C with an excess (0.5 mm) of bafilomycin A1 (Baf A), concanamycin A (Con A), concanolide A (Ccl A), apicularen A (Api A), or the solvent dimethyl sulfoxide (DMSO). The samples were then incubated for 10 min at 25 °C in the presence of 50 μm archazolid A (lanes 1, 3, 5, 7, and 9) or dimethyl sulfoxide (lanes 2, 4, 6, and 8). After that, all samples were additionally incubated for 1 h at 25 °C with 0.1 mm NCD-4. Of each sample ∼20 μg of protein was subjected to SDS-PAGE and NCD-4 labeling of subunit c was documented under UV light (Versadoc, Bio-Rad). B, inhibition curves of the inhibitors used in A. Purification of the M. sexta V1VO holoenzyme and following activity assays were carried out as described under “Experimental Procedures.” The NCD-4 sensitive V-ATPase activity was 2.2 ± 0.4 μmol/min/mg, the IC50 value was 3.0 ± 0.2 μm. Other IC50 values were 15 ± 1.5 nm for apicularen A (taken from Huss et al. 19), 9.0 ± 0.8 nm for bafilomycin A, 7.4 ± 0.5 nm for concanamycin A (taken from Huss et al. (12)), and 3.1 ± 0.6 nm for concanolid A.4 Values represent the average of at least two independent preparations, with error bars correspond to S.D.
To find out the reason for the apparent discrepancy, we preincubated the V-ATPase of M. sexta with different inhibitors, followed in the first step by incubation with archazolid A and in a second step by incubation with NCD-4 (Fig. 7A). After preincubation with an excess of the plecomacrolides, bafilomycin A1, concanamycin A, and concanolide A, respectively, the addition of archazolid A no longer prevented binding of NCD-4 to subunit c (Fig. 6A, lanes 3, 5, and 7; for IC50 values, see Fig. 6B). In contrast, preincubation with apicularen A, a member of the benzolactone enamides family of V-ATPase inhibitors, which is predicted to have a binding site totally different from archazolid and the plecomacrolides (12, 19, 32), did not impede the binding of archazolid A as indicated by a clear prevention of NCD-4 labeling (Fig. 6A, lane 9). Taken together, it is thus very likely that binding sites for archazolid and the plecomacrolides overlap significantly, but to a smaller extent than expected before (19).
FIGURE 7.

Influence of new mutations in Vma3p on the binding of archazolid and bafilomycin. V-ATPase activities were measured on isolated yeast vacuoles of wild type strain BMA64-1B (WT) and strains with mutations in Vma3p as indicated. Values are the average of two independent vacuolar preparations. The error bars correspond to S.D. Absolute archazolid A- and bafilomycin A1-sensitive activities are shown in Table 2, IC50 values in Table 3.
Influence of Further Selected Mutations on Inhibitor Binding
As a consequence of the results described above we selected amino acids in the equatorial region of Vma3p (Tyr-66, Val-138, Leu-141, Leu-144, Ile-145), which we presumed to interact with archazolid for further mutagenesis studies.
Regarding bafilomycin most of the mutations we established rendered the V-ATPase only slightly more resistant (V138A, L141F, and I145L) or slightly more sensitive (Y66F, L141I, L144F, L144I, and I145F) to the inhibitor (Table 3). Thus it is not likely that these amino acids interact directly with bafilomycin. In contrast, V138T exhibited a more striking effect with an ∼10-fold increase of the IC50 value for bafilomycin (Table 3 and Fig. 7A). On the other hand, L144I notably reduced the IC50 value ∼5-fold indicating a V-ATPase more sensitive toward the inhibitor (Table 3 and Fig. 7A).
Concerning archazolid, the additional mutations in general also only led to slight changes of the IC50 value and in most cases made the V-ATPase more sensitive toward the inhibitor (Table 3). Remarkably, the L144I mutation revealed the strongest effect with an IC50 value 10-fold lower then in the wild type strain, clearly pointing to a V-ATPase more sensitive toward archazolid (Table 3 and Fig. 7B).
DISCUSSION
Along with the identification of archazolid A as a novel V-ATPase inhibitor the first information about its potential binding site was obtained. In a competition assay archazolid A as well as bafilomycin A1, B1, and concanamycin A were able to prevent labeling of the VO subunit c with the radioactive concanamycin derivative 125I-concanolide A (12, 19). In conclusion, archazolid was believed to have a binding site and inhibition mechanism similar to the plecomacrolide antibiotics (12, 19). From mutagenesis studies, mainly in N. crassa, a model of the plecomacrolide binding site was deduced in which inhibitor binding occurs at the interface of two adjacent c subunits of the VO complex. Amino acids participating in binding of the inhibitor are primarily located in helices 1 and 2 as well as in the highly conserved region of helix 4 close to the essential glutamate (11, 13, 14).
In our present study we wanted to investigate to which extent the archazolid binding site is related to the binding site described for the plecomacrolides. The labeling of subunit c with radioactive derivatives of archazolid as presented in Fig. 4 confirmed binding of the inhibitor to this subunit as it had been deduced from previous competition assays (19).
To find out the amino acids of subunit c participating in binding of archazolid, we successfully established the known bafilomycin mutations for N. crassa and also several selected mutations in subunit c of S. cerevisiae via site-directed mutagenesis. All the yeast strains mutated in subunit c assembled a functional V-ATPase with at least 30% of wild type ATPase activity (Fig. 1 and Tables 1 and 2). This result is somewhat contradictory to the previously published result that most of the bafilomycin mutations disabled V-ATPase function in S. cerevisiae (13). The discrepancy might be explained by our usage of a non-tagged version of subunit c expressed by its native promoter that probably leads to an optimal expression of the mutant proteins. In addition, the yeast strain used in our study might be more tolerant to low V-ATPase activity and can thus cope with more severe mutations in subunit c.
Concerning the binding of bafilomycin in S. cerevisiae, we expected a similar effect as published for N. crassa because of the high sequence identity of c subunits between these two species. In general, we could confirm this expectation as all mutations known so far except I39F also showed an influence on bafilomycin binding in S. cerevisiae (Table 3). In addition, we found that valine 138 also participates in binding of bafilomycin as its mutation to threonine increases the IC50 value for the inhibitor about 10-fold (Table 3 and Fig. 7A). Fig. 8 shows that valine 138 (highlighted in olive) is the “missing link” between tyrosine 142 (highlighted in blue) and the other amino acids forming the bafilomycin binding site (all in cyan). The 5-fold decrease of the IC50 value exhibited by the L144I mutation points to a V-ATPase more sensitive to bafilomycin. This effect is quite surprising, because leucine 144 does not reside within the proposed bafilomycin binding site (14) (Fig. 8). But as the side chain of amino acid 144 is located in close proximity to tyrosine 66 in helix 2, the L144I mutation might have an indirect effect on the arrangement of the bafilomycin amino acids in helix 2 (Fig. 8, B and C).
FIGURE 8.

Model of the inhibitor binding sites in Vma3p of the V-ATPase from S. cerevisiae. The Vma3 amino acid sequence was fitted onto the E. hirae K-ring structure (4). Presented here are a top (C) and a side view (B) of the region of interest between two adjacent Vma3 subunits and the full, putatively decameric Vma3-ring structure (A). The alternating Vma3 subunits are colored in light or dark gray. Threonine 32, isoleucine 39, isoleucine 54, glycine 61, phenylalanine 135 (all cyan), and valine 138 (olive) participate in binding of bafilomycin, tyrosine 142 (blue) is involved in binding of both inhibitors, whereas glutamate 137 (red) is only part of the archazolid binding site. Further mutations in the putative binding site of archazolid revealed that amino acid leucine 144 (orange) is also involved in inhibitor binding, whereas tyrosine 66, leucine 141, and isoleucine 145 (all beige) are not.
In view of the assumption that archazolid and the plecomacrolides share the same binding site within the V-ATPase subunit c (19), we supposed that the bafilomycin mutations should also alter the affinity of the V-ATPase for archazolid. Surprisingly, most of the mutations had little influence on archazolid binding, making the yeast V-ATPase slightly more sensitive to this inhibitor (Table 3). In contrast, the Y142N and L144I notably changed the IC50 value resulting in a V-ATPase, which was ∼10-fold more sensitive to archazolid (Table 3 and Figs. 2B and 7B).
The increase in sensitivity achieved by the two mutations may be explained by an alteration of the archazolid binding site that leads to a higher affinity for the inhibitor. As the Y142H mutation has a smaller influence then the mutation Y142N, it is likely that the binding of archazolid is facilitated in the absence of a ring structure at position 142. Concerning bafilomycin, in contrast, the ring structure seems to be advantageous for binding, as its absence increases the IC50 value of the inhibitor (Table 3 and Fig. 2A). Regarding leucine 144, the exchange to isoleucine obviously alters the archazolid binding site more than the corresponding mutation to phenylalanine (Table 3). As both, tyrosine 142 and leucine 144, are located in the middle of the membrane with the leucine slightly oriented to the luminal side (Fig. 8), one may assume that archazolid interacts with the equatorial region of the c-ring (Fig. 9A).
FIGURE 9.

Model of the inhibitor binding sites in Vma3p of the V-ATPase from S. cerevisiae. To illustrate that the size of archazolid and bafilomycin matches the size of their putative binding sites, the three-dimensional structures of the inhibitors were positioned at the Vma3p-ring model. A, the alternating Vma3p subunits are colored in light or dark gray. Threonine 32, isoleucine 39, isoleucine 54, glycine 61, and phenylalanine 135 (all cyan), tyrosine 66, leucine 141, and leucine 145 (all beige), glutamate 137 (red), valine 138 (olive), tyrosine 142 (blue), and leucine 144 (orange). Three-dimensional space fill structure of archazolid (green) and bafilomycin (yellow) is shown. B, right panel, top view on the planar macrolactone ring of archazolid (green) and bafilomycin (yellow); left panel, view of the molecules rotated to the right 90°.
The findings from the mutagenesis studies, which mark a striking difference in the binding sites of archazolid and bafilomycin, were additionally supported by the results obtained from the labeling experiments with NCD-4. Labeling of subunit c was prevented by preincubation with archazolid but not with bafilomycin (Fig. 5) or concanamycin (Fig. 6A). Altogether, these results clearly show that the binding site of archazolid is more distinct from the plecomacrolide binding site than expected before (19). The archazolid binding site obviously comprises the essential glutamate in helix 4, whereas the plecomacrolides do not interfere with this region of the V-ATPase.
As can be seen in the model of a ring of c subunits of S. cerevisiae, the amino acids that contribute to bafilomycin binding (highlighted in cyan) are located on the opposite side of the essential glutamate 137 (highlighted in red) at the interface between two adjacent c subunits (Fig. 8). The only exception is tyrosine 142 (highlighted in blue), which is oriented toward the membrane facing side of helix 4. In addition, leucine 144 (highlighted in orange) is also oriented in this direction.
Based on this model we tend to propose that binding of archazolid occurs within one single c subunit and not at the interface of two c subunits. Fig. 8A also indicates that the plecomacrolide binding site mainly resides in the cytosolic half of the membrane bilayer as it had been shown in the three-dimensional model for the c-ring from N. crassa before (14). In contrast, tyrosine 142, leucine 144, and the essential glutamate 137, are located in the middle of the membrane. Combining these factors, it is very likely that the binding site of archazolid is located within the triangle of tyrosine 142, leucine 144, and the essential glutamate at the equatorial region of a single c subunit (Fig. 9A).
By modeling the proposed inhibitor binding sites for bafilomycin and archazolid at the crystal structure of the K-ring of the V-ATPase from E. hirae with bound DCCD (PDB code 2DB4) (Fig. 10), it becomes obvious that the bulky molecule archazolid (green) covers both, the binding site of DCCD (red) and all the amino acids involved in archazolid binding at the membrane facing side. In contrast, bafilomycin (yellow) is restricted to the interface of two adjacent subunits and may not interfere with DCCD.
FIGURE 10.

Inhibitor binding sites with respect to the binding site of DCCD at the K-ring of E. hirae (PDB code 2DB4). Shown here are a top (A) and a side view (B) of the region of interest in the K-ring with the three-dimensional space fill structures of archazolid (green), bafilomycin (yellow) and DCCD at glutamate 139 (red). The alternating K subunits are colored in light or dark gray. Colored amino acid residues correspond to the sites mutated in S. cerevisiae. Valine 34, alanine 41, leucine 56, glycine 63, and methionine 137 (all cyan), tyrosine 68, isoleucine 143, and valine 147 (all beige), threonine 140 (olive), leucine 144 (blue), and phenylalanine 146 (orange).
Despite the fact that most of the bafilomycin mutations have only negligible influence on archazolid, the prevention of archazolid binding by preincubation with different plecomacrolide antibiotics as shown in Fig. 6A points to the interference of both inhibitor classes. As amino acid tyrosine 142 contributes to the binding of both inhibitor classes, it is most probable that the binding sites overlap in this region of the V-ATPase (Fig. 9A). Taken together, this more precise model of the archazolid binding site helps to explain the apparent discrepancy between the previous finding that archazolid prevents labeling of subunit c with [125I]-concanolide A (19) and the results described in the present study.
Taking into account that the effect of the mutations in subunit c of N. crassa on the related molecules bafilomycin and concanamycin is already different (14), the disparity between the effects of archazolid and bafilomycin is not surprising. Archazolid is a bulkier molecule than bafilomycin or concanamycin and thus may not fit into the interface between two c subunits (Fig. 9B). In this regard, one could presume that archazolid inhibits V-ATPase function by blocking the rotation of the c-ring relative to subunit a, as it was proposed for bafilomycin A1 before (13). As the binding site of archazolid comprises the conserved glutamate residue that is essential for proton translocation, it appears, however, also possible that this inhibitor directly prevents the transport of protons by shielding the glutamate residue. To elucidate the binding site and inhibition mechanism of archazolid in more detail, it will now be interesting to investigate further mutations in the VO complex.
Supplementary Material
Acknowledgments
We thank Nicole Meyer, Johanna Bahr, and Katja Finsterbusch for help in the mutagenesis studies, as well as Gundula Key and Martin Dransmann for excellent technical assistance.
This work was supported in part by grants of the VolkswagenStiftung (Funding Initiative “Interplay between Molecular Conformations and Biological Function” (to D. M., H. W., and M. H.), Deutsche Forschungsgemeinschaft Grant SFB 431 (H. W. and M. H.) and BMBF MetabolitGenoMik Grant 0313105 (to S. G.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental data.
C. Nardmann, T. Bender, S. Grond, P. von Zezschwitz, H. Wieczorek, and M. Huss, manuscript in preparation.
- V-ATPase
- vacuolar ATPase
- DCCD
- N,N′-dicyclohexylcarbodiimide
- BD-archazolid
- bis-[4-(3-trifluoromethyldiazirin-3-yl)benzoyl]-archazolid A
- [14C]concanolid A
- 23-O-(3-trifluoromethyldiazirin-3-yl)-[1-14C]benzoyl-21-desoxyconcanolide A
- MES
- 4-morpholineethanesulfonic acid.
REFERENCES
- 1.Beyenbach K. W., Wieczorek H. (2006) J. Exp. Biol. 209, 577–589 [DOI] [PubMed] [Google Scholar]
- 2.Forgac M. (2007) Nat. Rev. Mol. Cell Biol. 8, 917–929 [DOI] [PubMed] [Google Scholar]
- 3.Muench S. P., Huss M., Song C. F., Phillips C., Wieczorek H., Trinick J., Harrison M. A. (2009) J. Mol. Biol. 386, 989–999 [DOI] [PubMed] [Google Scholar]
- 4.Murata T., Yamato I., Kakinuma Y., Leslie A. G., Walker J. E. (2005) Science 308, 654–659 [DOI] [PubMed] [Google Scholar]
- 5.Finbow M. E., Eliopoulos E. E., Jackson P. J., Keen J. N., Meagher L., Thompson P., Jones P., Findlay J. B. (1992) Protein Eng. 5, 7–15 [DOI] [PubMed] [Google Scholar]
- 6.Harrison M., Powell B., Finbow M. E., Findlay J. B. (2000) Biochemistry 39, 7531–7537 [DOI] [PubMed] [Google Scholar]
- 7.Nishi T., Forgac M. (2002) Nat. Rev. Mol. Cell Biol. 3, 94–103 [DOI] [PubMed] [Google Scholar]
- 8.Páli T., Whyteside G., Dixon N., Kee T. P., Ball S., Harrison M. A., Findlay J. B., Finbow M. E., Marsh D. (2004) Biochemistry 43, 12297–12305 [DOI] [PubMed] [Google Scholar]
- 9.Huss M., Wieczorek H. (2009) J. Exp. Biol. 212, 341–346 [DOI] [PubMed] [Google Scholar]
- 10.Bowman E. J., Bowman B. J. (2005) J. Bioenerg. Biomembr. 37, 431–435 [DOI] [PubMed] [Google Scholar]
- 11.Bowman B. J., Bowman E. J. (2002) J. Biol. Chem. 277, 3965–3972 [DOI] [PubMed] [Google Scholar]
- 12.Huss M., Ingenhorst G., König S., Gassel M., Dröse S., Zeeck A., Altendorf K., Wieczorek H. (2002) J. Biol. Chem. 277, 40544–40548 [DOI] [PubMed] [Google Scholar]
- 13.Bowman E. J., Graham L. A., Stevens T. H., Bowman B. J. (2004) J. Biol. Chem. 279, 33131–33138 [DOI] [PubMed] [Google Scholar]
- 14.Bowman B. J., McCall M. E., Baertsch R., Bowman E. J. (2006) J. Biol. Chem. 281, 31885–31893 [DOI] [PubMed] [Google Scholar]
- 15.Wang Y., Inoue T., Forgac M. (2005) J. Biol. Chem. 280, 40481–40488 [DOI] [PubMed] [Google Scholar]
- 16.Sasse F., Steinmetz H., Höfle G., Reichenbach H. (2003) J. Antibiot. 56, 520–525 [DOI] [PubMed] [Google Scholar]
- 17.Hassfeld J., Farès C., Steinmetz H., Carlomagno T., Menche D. (2006) Org. Lett. 8, 4751–4754 [DOI] [PubMed] [Google Scholar]
- 18.Menche D., Hassfeld J., Li J., Rudolph S. (2007) J. Am. Chem. Soc. 129, 6100–6101 [DOI] [PubMed] [Google Scholar]
- 19.Huss M., Sasse F., Kunze B., Jansen R., Steinmetz H., Ingenhorst G., Zeeck A., Wieczorek H. (2005) BMC Biochem. 6, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roberts C. J., Raymond C. K., Yamashiro C. T., Stevens T. H. (1991) Methods Enzymol. 194, 644–661 [DOI] [PubMed] [Google Scholar]
- 21.Uchida E., Ohsumi Y., Anraku Y. (1985) J. Biol. Chem. 260, 1090–1095 [PubMed] [Google Scholar]
- 22.Kane P. M. (1995) J. Biol. Chem. 270, 17025–17032 [PubMed] [Google Scholar]
- 23.Bender T., Huss M., Wieczorek H., Grond S., von Zezschwitz P. (2007) Eur. J. Org. Chem. 3870–3878 [Google Scholar]
- 24.Dröse S., Boddien C., Gassel M., Ingenhorst G., Zeeck A., Altendorf K. (2001) Biochemistry 40, 2816–2825 [DOI] [PubMed] [Google Scholar]
- 25.Dröse S., Bindseil K. U., Bowman E. J., Siebers A., Zeeck A., Altendorf K. (1993) Biochemistry 32, 3902–3906 [DOI] [PubMed] [Google Scholar]
- 26.Jansen R., Kunze B., Reichenbach H., Höfle G. (2000) Eur. J. Org. Chem. 913–919 [Google Scholar]
- 27.Wieczorek H., Cioffi M., Klein U., Harvey W. R., Schweikl H., Wolfersberger M. G. (1990) Methods Enzymol. 192, 608–616 [DOI] [PubMed] [Google Scholar]
- 28.Kelley L. A., Sternberg M. J. (2009) Nat. Protoc. 4, 363–371 [DOI] [PubMed] [Google Scholar]
- 29.Nelson H., Nelson N. (1990) Proc. Natl. Acad. Sci. U.S.A. 87, 3503–3507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Noumi T., Beltrán C., Nelson H., Nelson N. (1991) Proc. Natl. Acad. Sci. U.S.A. 88, 1938–1942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Menche D., Hassfeld J., Sasse F., Huss M., Wieczorek H. (2007) Bioorg. Med. Chem. Lett. 17, 1732–1735 [DOI] [PubMed] [Google Scholar]
- 32.Menche D., Hassfeld J., Steinmetz H., Huss M., Wieczorek H., Sasse F. (2007) Eur. J. Org. Chem. 1196–1202 [Google Scholar]
- 33.Chadwick C. C., Thomas E. W. (1983) Biochim. Biophys. Acta 730, 201–206 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.