

Abstract
LC8 dynein light chain (DYNLL) is a highly conserved eukaryotic hub protein with dozens of binding partners and various functions beyond being a subunit of dynein and myosin Va motor proteins. Here, we compared the kinetic and thermodynamic parameters of binding of both mammalian isoforms, DYNLL1 and DYNLL2, to two putative consensus binding motifs (KXTQTX and XG(I/V)QVD) and report only subtle differences. Peptides containing either of the above motifs bind to DYNLL2 with micromolar affinity, whereas a myosin Va peptide (lacking the conserved Gln) and the noncanonical Pak1 peptide bind with Kd values of 9 and 40 μm, respectively. Binding of the KXTQTX motif is enthalpy-driven, although that of all other peptides is both enthalpy- and entropy-driven. Moreover, the KXTQTX motif shows strikingly slower off-rate constant than the other motifs. As most DYNLL partners are homodimeric, we also assessed the binding of bivalent ligands to DYNLL2. Compared with monovalent ligands, a significant avidity effect was found as follows: Kd values of 37 and 3.5 nm for a dimeric myosin Va fragment and a Leu zipper dimerized KXTQTX motif, respectively. Ligand binding kinetics of DYNLL can best be described by a conformational selection model consisting of a slow isomerization and a rapid binding step. We also studied the binding of the phosphomimetic S88E mutant of DYNLL2 to the dimeric myosin Va fragment, and we found a significantly lower apparent Kd value (3 μm). We conclude that the thermodynamic and kinetic fine-tuning of binding of various ligands to DYNLL could have physiological relevance in its interaction network.
Keywords: Calorimetry, Fluorescence, Kinetics, Ligand-binding Protein, Surface Plasmon Resonance (SPR), Avidity, Dynein Light Chain, Linear Peptide Motif
Introduction
LC8 dynein light chain (DYNLL)3 is a highly conserved small eukaryotic protein. It was originally discovered as a light chain of the dynein (1) and later of myosin Va (myoVa) (2) motor protein complexes. However, DYNLL has many interaction partners unrelated to motor proteins. Therefore, it has been suggested that DYNLL is a hub protein that plays important roles in the interactome of eukaryotic cells in various cellular events, including apoptosis, molecular, organelle, and nuclear transport, viral infection, cancer development, and transcription regulation (3, 4). More intensively studied DYNLL-binding proteins include neuronal nitric-oxide synthase (nNOS) (5), myoVa (2), Bcl-2-modifying factor (Bmf) (6), Bcl-2 interacting mediator (Bim) (7), dynein intermediate chain (DIC) (8), the Drosophila swallow mRNA localizing protein (9), and p21-activated protein kinase 1 (Pak1) (10, 11). Several solution and crystal structures of apo-DYNLL and complexes with binding peptides have been determined (12–20). DYNLL has a homodimeric structure, and the bound partner peptides lie in two identical grooves formed at the dimerization interface (12–20). Formerly, it was widely assumed that DYNLL could function as a cargo adapter on dynein and myoVa motors (6, 21, 22). However, this hypothesis is difficult to reconcile with the symmetric homodimeric structure of DYNLL and most of its partners, including myoVa and DIC. Instead, it has been suggested, based on the effect of DYNLL on its partner proteins, that one of the major roles of DYNLL dimers could be the ability to promote dimerization and stabilization of their interaction partners (2, 3, 23).
DYNLL has two mammalian isoforms (DYNLL1 and DYNLL2; previously known as DLC1 and DLC2 or LC8a and LC8b) (21, 22) that differ from each other only in six residues. All of these residues are located outside of the ligand binding grooves. Despite their similarity, DYNLL1 and DYNLL2 seem to discriminate binding partners in the cell (6, 19), although some in vitro studies do not support this finding (21, 24). The binding grooves are able to interact with short linear sequences, all of which are part of intrinsically disordered regions of the partner proteins. These binding motifs were divided into the following three classes based on sequence similarities: KXTQTX (e.g. Bmf), XG(I/V)QVD (e.g. nNOS), and noncanonical (e.g. myoVa and Pak1) (2, 14, 25–27). However, the functional relevance of this classification has never been investigated. A broad range of affinities of various binding peptides and protein fragments (Kd values between 100 nm and 100 μm), determined by different approaches, were reported (2, 26, 28–31). However, it is important to note that many of the partners were shown to exist as homodimers, and therefore DYNLL most likely forms dimer-dimer complexes with its partners (2, 23). Thus, the observed dissociation constants of monomeric peptides may not be used directly to describe the interaction with dimeric partners, which in fact are bivalent protein ligands (15). Accordingly, we have previously reported an affinity enhancement of dimeric myoVa fragments binding to DYNLL2, compared with a monomeric peptide (2), and the same was noted for a dimeric DIC fragment (26). However, the quantitative relationship between the affinity and the monomer-dimer state of the binding partner was not investigated in previous studies.
Regulation of the interactions of DYNLL as a hub protein is not well understood. Binding of the partners to DYNLL could be regulated by phosphorylation of Thr or Ser residues within the DYNLL-binding motif (32). Phosphorylation of Ser-88 of DYNLL could be another way of regulation by shifting the monomer-dimer equilibrium strongly to the monomer state, thus eliminating the binding grooves (29, 33). It is not clear which kinase is involved in this regulation; Pak1 was originally shown to phosphorylate DYNLL (10, 34); however, a recent study did not support its direct regulatory role (26).
Here, we report the kinetic and thermodynamic parameters of binding of DYNLL isoforms to various partners with monovalent and bivalent motifs. We found that the affinity is dramatically increased by the bivalent nature of the binding partners. We also found that the binding reaction of both monovalent and bivalent ligands can be best explained by a conformational selection model. Furthermore, we show that ligands can bind to the S88E phosphomimetic form of DYNLL by pulling the monomer-dimer equilibrium back to the dimer state.
EXPERIMENTAL PROCEDURES
Expression, Purification, and Synthesis of Proteins and Peptides
DYNLL2 (amino acid residues 1–89; Uniprot, Q96FJ2) and the monomeric fragment of myoVa (residues 1275–1297; Q9Y4I1-2) were cloned and expressed as described previously (2). The S88E DYNLL2 mutant was generated by PCR using a mutant oligonucleotide and the wild type DYNLL2 construct as a template. The dimeric myoVa fragment was constructed by amplifying the myoVa fragment (residues 1209–1320) by PCR and subsequently fusing coding sequences of a leucine zipper from a yeast transcription factor (GCN4; P03069) to the 5′ and 3′ ends using the megaprimer method (35). The leucine zippers increase the stability of the dimers. The PCR product was cloned into pET15b using NdeI and BglII/BamHI sites. Constructs of DYNLL1 (P63167), Bmf (Q96LC9), and Pak1 (Q13153) were kind gifts of Drs. I. Rodríguez-Crespo, M. G. Hinds, and R. Kumar, respectively. DYNLL1 (residues 1–89), Bmf (residues 1–159), and Pak1 (residues 203–268) were amplified by PCR and subcloned into pET15b (Novagen) vector using NdeI and EcoRI sites. The dimeric Bmf fragment was created by fusing a leucine zipper to the 3′ end of the sequence corresponding to Bmf residues 64–73. Expression vectors and subcloned protein fragments used in this work are listed in supplemental Table 1.
Constructs were transformed in BL21-(DE3) Rosetta cells (Novagen). After induction, cells were grown at 37 °C for 3 h in 2YT medium. Glutathione S-transferase (GST) fusion and His-tagged proteins were purified on glutathione-Sepharose (Amersham Biosciences) and Ni2+-affinity columns (Bio-Rad), respectively. His- and GST-tagged proteins were digested by thrombin. DYNLL1, DYNLL2, DYNLL2 S88E mutant, and the Bmf protein were further purified on a HiTrapQ ion-exchange column (Amersham Biosciences).
Bmf (TSQEDKATQTL) and nNOS peptides (EMKDTGIQVDRDL) were synthesized in-house by solid-phase techniques using an ABI 431A peptide synthesizer and standard N-(9-fluorenyl)methoxycarbonyl chemistry. N-terminal fluorescein-labeled Bmf peptide was synthesized similarly, except that before the cleavage step 5(6)-carboxyfluorescein was coupled to the N-terminal amino group. Peptides were further purified by reverse-phase HPLC on a Jupiter 300 C18 column (Phenomenex). Concentrations were measured by absorbance at 280 nm using calculated extinction coefficients or by amino acid analysis. The identity of each peptide and protein was confirmed by mass spectrometry.
Isothermal Titration Calorimetry (ITC)
All titrations were carried out at 299 K in PBS supplied by 3 mm 2-mercaptoethanol (pH 7.4) using a Microcal VP-ITC apparatus. DYNLL1 or DYNLL2 (in 30 μm initial concentration) was titrated up to 5-fold excess of ligands. Depending on the Kd and ligand concentration, up to 40 injections were performed with 900-s time intervals between injections. The measured heat changes were corrected for dilution effects using data from similar control experiments. The Origin for ITC 5.0 (OriginLab) software package was used for data processing. Several binding models were tested, but the simplest one site binding (A + B = AB) model provided satisfactory results. The errors of the thermodynamic parameters reported are based on deviations from the theoretical best fit.
Surface Plasmon Resonance
Surface plasmon resonance (SPR) experiments were performed on a Biacore 3000 instrument equipped with research grade CM5 sensor chips. Dimeric myoVa and full-length Bmf (residues 1–159)-coated surfaces were prepared at 25 °C by the amine-coupling method. Flow cells were activated for 6 min with a solution containing 50 mm N-hydroxysuccinimide and 200 mm N-ethyl-N′-dimethylaminopropylcarbodiimide. Dimeric myoVa was diluted to 50 μg/ml in 10 mm sodium acetate (pH 5.0) and injected over the surface for 7 min at 10 μl/min flow rate. Excess reactive sites were subsequently blocked by injection of 35 μl of 1 m ethanolamine (pH 8.5). To monitor association, various concentrations of DYNLL2 or DYNLL2 S88E in running buffer (10 mm Hepes (pH 7.4), 0.15 m NaCl, 3 mm EDTA, and 0.005% Surfactant P20) were applied to either the ethanolamine blocked control or dimeric myoVa-coated surface at a flow rate of 10 μl/min for 15 min and the changes in the response units were recorded. Dissociation of the complexes was followed for up to 15 min after injection of DYNLL2 or DYNLL2 S88E free running buffer. The responses from the surface covered with ethanolamine were subtracted from the responses obtained with the dimeric myoVa-coated surfaces. Kinetic (kon and koff) and equilibrium parameters (Kd) were derived from the sensograms using BIAevaluation 3.1 software with fitting the data to a simple 1:1 Langmuir interaction model.
Fluorescence Anisotropy
Fluorescence anisotropy experiments were carried out at 23 °C in PBS supplied by 3 mm 2-mercaptoethanol using an FLS920 spectrofluorometer (Edinburgh Instruments). A CW 450 W xenon arc lamp was used as a 494-nm excitation source. Anisotropy was recorded at 540 nm. Fluorescein-labeled Bmf peptide was titrated with DYNLL2 and DYNLL2 S88E (the concentration of Bmf was held at 818 nm). Anisotropy values were plotted against DYNLL2 and DYNLL2 S88E concentrations, and the experimental data were fitted using
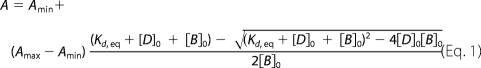 |
where Kd, eq is the dissociation constant; [D]0 is the total concentration of DYNLL; [B]0 is the total concentration of fluorescein-labeled Bmf; A is the measured anisotropy of the reaction mixture; and Amin and Amax are the anisotropy values of the solution when the total amount of fluorescein-labeled Bmf is free or complexed, respectively.
Stopped-flow Fluorescence Spectroscopy
Tryptophan fluorescence-based transient kinetic measurements were carried out using a KinTek SF-2004 stopped-flow apparatus. A xenon light source was used to excite the DYNLL isoforms at 297 nm (4 nm bandwidth). On the emission side, a 340-nm interference filter was used. Samples were in PBS buffer supplied by 3 mm 2-mercaptoethanol; the temperature was held at 23 °C. The premix concentration of DYNLL and of the partners were 3 μm and between 1 and 1000 μm, respectively. The mixing ratio was 1:1. At least three traces were averaged and corrected for photobleaching. Double exponentials were fitted (I = A1e−kobs1t + A2e−kobs2t, where I is the fluorescence intensity; A1 and A2 are the amplitudes of the fast and slow phases, respectively; kobs1 and kobs2 are the observed rate constants of the fast and slow phases, respectively) to the experimental data. Detailed data analysis and model fitting were performed by the KinTek Global Kinetic Explorer 2.2.563 software (36, 37). Data were imported as concentration series with average and σ values of each data point. Two possible minimal models were constructed and used for analysis (Schemes 1 and 2). The “induced fit” (IF) model assumes a first binding step followed by an isomerization step occurring in the ligand-bound form. The “conformational selection” (CS) model assumes that the apoprotein can adopt two states. One of the states is able to bind to the ligand, but the other one is not. The two states are in a conformational equilibrium. Binding of the ligand to the protein pulls the conformational equilibrium by consuming the apo-form in the binding-competent state. Both steps can be associated with a change in fluorescence in both models. Scaling factors were applied to each trace during the fitting procedure. Multiple attempts with different initial parameter values were carried out to find the best fits. Errors on each parameter were estimated using FitSpace explorer (37) by calculating the lower and upper bounds (reported in supplemental Table 2). The calculated boundaries reflect fits within χ2 multiplied by 1.1 (as suggested in Ref. 37).
SCHEME 1.

Induced fit model.
SCHEME 2.

Conformational selection model.
RESULTS
Comparison of Binding Properties of DYNLL1 and DYNLL2 Isoforms
Bmf and nNOS are known to bind in vivo to DYNLL2 (19) and DYNLL1 (5), respectively. They contain distinct types of DYNLL-binding motifs (Bmf, KATQTD and nNOS, TGIQVD), suggesting that the DYNLL isoforms could somehow discriminate between these sequences. To explore whether this isoform specificity can be detected in vitro, the thermodynamic and kinetic parameters of the association reactions were compared for the two pairs of complexes by ITC and by stopped-flow fluorescence spectroscopy. Although DYNLL1 showed slightly weaker and slower binding to the peptides than DYNLL2 (e.g. DYNLL1-Bmf, Kd, eq ≈1 μm, konC = 10.4 mm−1 s−1; DYNLL2-Bmf, Kd, eq ≈0.7 μm, konC = 21.7 mm−1 s−1), both peptides were able to bind to both isoforms with rather similar dissociation constants (Table 1 and Fig. 1; see below for detailed explanation of the kinetic analysis). Therefore, if isoform-specific target selection exists in vivo, one should assume the existence of additional “specificity-determining” factors. Because of the in vitro similarity of the two isoforms, only DYNLL2 was used hereafter in all measurements.
TABLE 1.
Thermodynamic and kinetic parameters of DYNLL binding to various partners
| Partner | Isoform | Kd, eqa | ΔHa | −TΔSa | konCb | koffCb |
|---|---|---|---|---|---|---|
| nm | kJ mol−1 | kJ mol−1 | m−1s−1 | s−1 | ||
| Bmf (DKATQTL) | DYNLL1 | 1050 ± 40 | −46.1 ± 0.2 | 11.9 | 10400 | –c |
| Bmf (DKATQTL) | DYNLL2 | 735 ± 22 | −51.3 ± 0.1 | 16.1 | 21700 | –c |
| Bmf (dimeric) | DYNLL2 | 3.46 ± 0.46 | −81.1 ± 0.1 | 32.7 | 13000 | –c |
| nNOS (DTGIQVD) | DYNLL1 | 7000 ± 230 | −18.7 ± 0.1 | −10.8 | 46800 | 4.22 × 10−1 |
| nNOS (DTGIQVD) | DYNLL2 | 5410 ± 150 | −22.3 ± 0.1 | −7.91 | 58400 | 4.37 × 10−1 |
| Myosin Va (DKNTMTD) | DYNLL2 | 8850 ± 670 | −23.9 ± 0.4 | −4.93 | 6590 | 1.03 × 10−1 |
| Myosin Va (dimeric) | DYNLL2 | 37 ± 5 | −28.5 ± 0.1 | −13.9 | 4020 | –c |
| Pak1 (DVATSPI) | DYNLL2 | 42700 ± 5300 | −21.5 ± 4.1 | −3.48 | 16400 | 6.41 × 10−1 |
a Kd, eq, ΔH, and −TΔS values were calculated from ITC measurements (Fig. 1). Errors are the standard error of fitting.
b konC and koffC were calculated from stopped-flow experiments (Fig. 4 and supplemental Fig. 2).
c The koffC values for dimeric binding and high affinity binding of monomeric Bmf could not be determined with sufficient accuracy. The standard error of konC and koffC values and more detailed kinetic data can be found in supplemental Table 2.
FIGURE 1.

Thermodynamic data showing monomeric or dimeric Bmf fragment binding to DYNLL isoforms. Heat changes were recorded in ITC experiments upon titration of DYNLL1 (A) and DYNLL2 (B) with a monomeric Bmf fragment. In both cases, the dissociation constants (0.74 and 1.05 μm, respectively), the entropy change, and the enthalpy change were very similar (see Table 1). C, dimeric Bmf fragment (Kd = 3.5 nm) showed ∼200-fold stronger binding to DYNLL2 than the monomeric Bmf.
Comparison of Various DYNLL-binding Motifs
The high diversity observed among the DYNLL-binding motifs suggests that in the cell, where several interacting partners could be present at the same time and therefore compete for a common groove on DYNLL, the different dissociation constants together with the local concentrations of the partners would determine the distribution of DYNLL among the interacting proteins. Thermodynamic parameters and kinetic constants of complex formation were determined by ITC and stopped-flow measurements, respectively (Table 1 and Figs. 1 and 4). The consensus class 1 type Bmf peptide (KATQTL) interacted with the highest affinity (Kd, eq ≈ 0.7 μm) with DYNLL2. The nNOS peptide (TGIQVD) representing class 2 consensus motif bound somewhat weaker (Kd, eq ≈5 μm), whereas the noncanonical myoVa peptide (KNTMTD) has almost the same affinity as the nNOS peptide (Kd, eq ≈9 μm). The most divergent sequence from the consensus motifs in Pak1 (VATSPI) has the lowest affinity (Kd, eq ≈40 μm). Interestingly, the association of DYNLL2 to nNOS, myoVa, and Pak1 is favorable both entropically and enthalpically, whereas the interaction with Bmf is enthalpy-driven and entropically disfavored. Accordingly, the free energy of binding of Bmf to DYNLL2 has a higher contribution from enthalpy change (−50 kJ/mol) than those of other partners. The association rate constants of the peptides are in the range of 6–58 mm−1s−1. The off-rates of the complexes are between 0.1 and 0.6 s−1 for nNOS, myoVa, and Pak1. Interestingly, the off-rate constant of Bmf is about 100 times lower (∼0.002 s−1). Our conclusion is that the two putative classes of motifs can bind to DYNLL2 with similar strength but with different interaction patterns and mechanism.
FIGURE 4.

Tryptophan fluorescence-based transient kinetic measurements. A, fluorescence change of Trp-54 in DYNLL isoforms was monitored following rapid mixing of DYNLL (3 μm monomer concentration (Conc)) with different peptides at a series of peptide concentrations (post-mixing concentrations indicated). Trp-54 was selectively excited at 297 nm, and the emission was monitored at 340 nm. Biphasic binding transients were observed. The reactions of nNOS with DYNLL1 are shown. Curves represent global best-fits based on the conformational selection model, fitted by Kintek Explorer Pro software. B, as an alternative to global fit analysis, double exponential approximations to the recorded transients were also applied. In this analysis, the determined kobs1 (fast phase observed rate constant) values showed linear dependence on peptide concentration under pseudo first-order conditions, as exemplified by the Bmf-LeuZ data. (Linear fitting was performed by weighting each data point with the associated standard error.) C, kobs2 (slow phase observed rate constant) values were in a narrow range (0.01–0.06) in every experiment and showed no obvious dependence on the peptide concentration. D, under pseudo first-order conditions, the fractional amplitudes of the fast (A1) and slow (A2) phases were around 70 and 30%, respectively. (The vertical black lines in B–D represent a practical lower limit of pseudo first-order conditions) (DYNLL2/BmF-LeuZ concentration ratio of 1:5).
We also determined the binding properties of an almost full-length Bmf polypeptide (residues 1–159), missing only the C-terminal highly hydrophobic putative membrane anchor (38) to DYNLL2 by SPR and found only little differences (Kd, eq ≈4 μm) compared with the Bmf peptide (Kd, eq ≈0.7 μm). This result confirms that the short linear binding motif is the only determinant of the binding affinity, and no secondary DYNLL-binding site is present, at least on Bmf.
Dimeric State of the Interacting Partners Considerably Increases Affinity to DYNLL
Most binding partners of DYNLL (myoVa, DIC, swallow, and nNOS) exist as homodimers. Moreover, DYNLL promotes dimerization as was observed with swallow, DIC, and myoVa (2, 15, 23, 39). Hence, it is important to characterize dimer-to-dimer interactions of DYNLL complexes to get more insight into their biological role. To collect relevant data, we compared the kinetic and thermodynamic properties of monomeric (monovalent ligand) and dimeric (bivalent ligand) forms of two partners (Table 1). First, we investigated the in vitro binding properties of DYNLL2 and a stable dimeric fragment of myoVa. We have previously shown that a similar myoVa fragment with only one Leu zipper at the N-terminal end has a dimerization Kd of ∼30 nm (3). A monomeric peptide corresponding to the minimal DYNLL-binding motif of myoVa was used as a control. ITC measurements showed that the stoichiometry of the complexes was 1:1 (dimer to dimer) and 2:1 (two monomers to a dimer) for the dimeric and monomeric myoVa construct, respectively. The dimeric fragment has an ∼250-fold stronger affinity to DYNLL2 (Kd, eq ≈40 nm) relative to the monomeric one (Kd, eq ≈9 μm). This effect is most likely the consequence of avidity, but other causes, such as the contribution of a secondary DYNLL-binding site on the myoVa fragment, cannot be completely ruled out. To analyze directly the contribution of avidity to the affinity increase, an artificial dimeric DYNLL-binding sequence, a Leu zipper dimerized Bmf motif (connected by a six-residue linker) was constructed and compared with the monomeric Bmf peptide (Table 1 and Fig. 1). The observed increase in affinity was similar (∼200-fold, Kd, eq = 3 nm) to that of myoVa. These results confirm that the enhanced affinity of dimeric partner binding to DYNLL can be achieved by avidity effect.
Affinity enhancement by avidity could be attributed to the decrease in the observed off-rate constants with almost unchanged on-rate constants (40–43). To investigate this assumption, we first determined the rate constants by stopped-flow fluorescence spectroscopy (Table 1 and supplemental Table 2). Binding of the dimeric myoVa was also measured by SPR because its high affinity to DYNLL prevented determination of the koff values with sufficient accuracy by the stopped-flow method (Table 2 and Fig. 2). The results indicate that the off-rate constant of dimeric myoVa is ∼450-fold lower than that of monomeric myoVa, although less than 2-fold decrease was detected in the on-rate constant (Table 1).
TABLE 2.
Comparison of the binding of wild type and monomeric mutant DYNLL2 to monomeric and dimeric partners
FIGURE 2.

SPR measurements. SPR assay was used to investigate the interaction between the dimeric myoVa and DYNLL2 (A) or DYNLL2 S88E (B). The dimeric myoVa was immobilized on CM5 sensor chip. 10, 5, 2, 1, 0.5, 0.1μm and 20, 17.5, 15, 12.5, 10, 7.5, and 5 μm concentrations of DYNLL2 and DYNLL2 S88E, respectively, were used to monitor the association and dissociation. Data were analyzed with simple 1:1 Langmuir interaction model. Similar observed off-rate constants were obtained with the two DYNLL2 constructs (DYNLL2 S88E, 3.41 × 10−4 ± 4.5 × 10−5 s−1; WT, 2.24 × 10−4 ± 1.0 × 10−5 s−1), although the observed on-rate constant of the DYNLL2 S88E (147 ± 64 m−1s−1) was much smaller than the observed on-rate constant of the WT DYNLL2 (4840 ± 1290 m−1s−1).
Binding Properties of the S88E DYNLL2 Mutant
It has been previously shown that phosphorylation of DYNLL at Ser-88 could inhibit partner binding (10) by promoting dissociation of DYNLL dimers to monomers (29, 33). The monomer-dimer equilibrium of the phosphomimetic DYNLL2 S88E mutant is strongly shifted toward the monomer state (29, 33). Binding affinities of the wild type and the mutant DYNLL2 were determined using a fluorescein-labeled peptide corresponding to the binding motif of Bmf by a fluorescence anisotropy titration assay (Table 2 and Fig. 3). The observed Kd value of the S88E mutant was 30-fold weaker (110 μm) than that of wild type DYNLL2 (∼3.5 μm). Such a weak interaction probably does not have any physiological significance. The dimeric nature of the binding partners causes avidity, and this could affect the observed affinity to the S88E mutant as well. This hypothesis was tested by SPR and an immobilized dimeric myoVa fragment as a model (Table 2 and Fig. 2).
FIGURE 3.

Fluorescence anisotropy measurements of Bmf peptide binding to DYNLL2 and DYNLL2 S88E. Fluorescein-labeled Bmf peptide was titrated with increasing concentrations of DYNLL2 (■) and DYNLL2 S88E mutant (▴). The inset contains the same DYNLL2 titration data (■) in the lower concentration range. Fluorescein was excited at 494 nm, and the anisotropy was monitored at 540 nm. The concentration of fluorescein-labeled Bmf peptide was held at 818 nm. In the case of DYNLL2 the observed dissociation constant was 3.58 ± 0.4 μm, although DYNLL2 S88E showed much weaker binding (Kd = 110 ± 20 μm).
The calculated dissociation constants for wild type and S88E mutant DYNLL2 were 50 nm and 2.7 μm, respectively. It is noteworthy that the ∼50-fold weaker Kd value in case of the mutant mainly resulted from a change in the observed on-rate constant, whereas the off-rate constant was almost unchanged. The monomeric form of the S88E mutant is unable to bind its partners because the binding groove formation is coupled to dimerization (29, 33). Because only the dimeric DYNLL is functional in binding, complex formation depletes the small amount of dimers in equilibrium with monomers in the mutant. Therefore, the monomer-dimer equilibrium is pulled toward dimers. In the case of the S88E mutant, the low observed kon value probably results from a very low degree of dimer formation at the applied concentration.
Two-step Binding Kinetics
A single buried tryptophan residue (Trp-54) is found in each DYNLL monomer that responds to ligand binding. On rapid mixing of DYNLL2 with ligands using a stopped-flow apparatus, biphasic binding transients were observed (Fig. 4A and supplemental Fig. 1). In double exponential fits to traces recorded under pseudo first-order conditions, the observed rate constants of the fast phase (kobs1) showed linear dependence on peptide ligand concentration (Fig. 4B), whereas the observed rate constants of the slow phase (kobs2) were in all cases in the same range (0.01–0.05 s−1) with no obvious concentration dependence (Fig. 4C). This suggests that the observed fluorescence changes arose from a combination of a second-order association and a first-order step, a conformational change of either ligand-free or ligand-bound DYNLL2. Under pseudo first-order conditions, ∼70% of the total amplitude change came from the first phase and ∼30% from the second phase (Fig. 4D).
Besides the exponential approximations, experimental data were also fitted globally using the two possible minimal models, IF and CS (see under “Experimental Procedures”). Detailed parameters obtained by either models can be found in supplemental Table 2. Generally, the kinetic parameters of binding could be determined with acceptable quality. The parameters of the isomerization steps were less reliable. Regarding the high affinity binding of dimers or Bmf, the koff value of binding was uncertain (see the error values in supplemental Table 2). The rate constants of the binding steps determined by either the IF and CS models had similar values, which also indicates that these values are well constrained by the data, and the uncertainties of the isomerization step may have little effect on the determined parameters of the binding step. The kinetic parameters reported in this work (Table 1) are those obtained using the CS model, which provided slightly better fitting than the IF model. Kd, eq values were robustly determined by our ITC experiments (Table 1 and Fig. 1). The overall dissociation constants were also calculated from the IF model as shown in Equation 3,
and also from the CS model shown in Equation 4,
where Kb = kon/koff and Ki = k+i/k− i (supplemental Table 2). The obtained Kd, eq values were in line with those determined by ITC measurements except the high affinity partners where the off-rate constants were not reliable by the stopped-flow method.
DISCUSSION
DYNLL Isoforms Have Similar Binding Properties to Their Various Targets
To understand the robust interactome of the cell, it is important to characterize and quantify individual protein-protein interactions. In this study, we have described the binding properties of the LC8 dynein light chain hub protein to various binding partners. The ubiquitous eukaryotic DYNLL has two mammalian paralogs, DYNLL1 and DYNLLL2, with 93% sequence similarity. In a previous study, Bim and Bmf, two pro-apoptotic proteins sharing similar KXTQTX type DYNLL-binding motifs, were found in vivo to be associated exclusively with DYNLL1 and DYNLL2, respectively (6, 7); however in an in vitro GST pulldown assay, no discrimination was observed (19). On the other hand, nNOS having a XG(I/V)QTD motif is a known binding partner of DYNLL1 only (5). Here, we quantitatively compared the binding properties of the two DYNLL isoforms to the two distinct types of binding motifs (XG(I/V)QVD in nNOS and KXTQTX in Bmf) in vitro. Only small differences in affinities were observed, which cannot explain the in vivo observed partner specificities. It is likely that other factors or proteins mediate the specificity and distinct cellular localization of DYNLL isoforms. The six-residue differences between the two paralogs are located on the external surface of DYNLL homodimers, and they might be part of a potential recognition site for the yet unidentified specificity determining protein(s). Day et al. (19) found that His-41 and Tyr-41 on the surface of DYNLL1 and DYNLL2, respectively, determine the specific localization of the isoforms to their respective cytoskeletal structures. Future studies are needed to clarify the structural basis of in vivo isoform specificity of DYNLL binding partners.
Fine-tuning of Affinities Is Achieved through Diversity of Binding Motifs
The linear DYNLL-binding motifs were originally divided into two classes, XG(I/V)QVD and KXTQTX (44). The most conserved position is the Gln, which is generally in the vicinity of Thr or Ile/Val residues (19, 45). It is important to note that mixed sequences (IQT in p53BP1 and VQT in Nup159) can be found in several DYNLL-binding motifs. Moreover, there are at least two motifs that lack the central Gln, myoVa, and Pak1; the myoVa sequence (KNTMTD) is otherwise more similar to the KXTQTX consensus, whereas the Pak1 sequence (VATSPI) is clearly a noncanonical one. We compared the dissociation constants of representative sequences from the two putative canonical classes and the two noncanonical sequences to DYNLL2, and we found relatively small differences (Table 2). This narrow range of affinities could hardly determine the specific binding of DYNLL2 when more partners are present simultaneously. The hierarchy of binding affinities proposed in a previous study (26) is corroborated by our data except that myoVa binds five times stronger than Pak1. We conclude that there is a continuum of binding affinities of the various partners irrespective of the putative type of motifs. However, the binding mechanisms of the various classes seem to be different. Binding of the KXTQTX motif was found to be enthalpically favored and entropically disfavored, whereas the motifs from the other class or having noncanonical sequence (myoVa and Pak1) are both enthalpy- and entropy-driven (28). Moreover, the KXTQTX motif has a lower off-rate constant than the other class. It is not yet clear whether there is any biological relevance of the different binding mechanisms.
Barbar and co-workers (17) proposed that positive cooperativity between the two ligand-binding sites of DYNLL could be a mechanism to promote simultaneous binding of ligands containing the same motif. This could be one factor for determining optimal binding strength and specificity. However, our results suggest that, in agreement with the previous qualitative data of Williams et al. (15, 26), the dimer state of the partners and hence avidity is more important in this respect (see below). Differences in binding motifs may thus be a “fine-tuning mechanism” in the interaction network of DYNLL.
Importance of Avidity, Several Hundredfold Stronger Binding to Dimer Partners of DYNLL
To understand the interaction between dimeric DYNLL and dimeric partners, several factors should be taken into account, including coupled equilibria, such as the monomer-dimer equilibrium of DYNLL. The dimer stability of DYNLL was found to be submicromolar (26). Because DYNLL is predominantly dimeric under physiological conditions, and targets are able to bind only to dimeric DYNLL, the monomer-dimer equilibrium affects ligand binding only if it is strongly shifted by a regulatory signal (e.g. reversible phosphorylation). Another important factor could be the above-mentioned cooperativity of the two DYNLL-binding sites; however, our ITC and stopped-flow data can be well explained without the incorporation of cooperativity into the models. Therefore, we analyzed all data in this work by the simplifying assumption that ligand binding to the two grooves at the opposite sides of a DYNLL dimer is independent. The third factor to be taken into account is the stoichiometry of the complexes. We observed a 1:1 (dimer-to-dimer) stoichiometry in case of a complex of dimeric myoVa fragment and DYNLL2 by ITC measurement. The same stoichiometry was found by others in the case of isolated chick brain full-length myoVa (46) and in Tctex-DIC-DYNLL crystal structure (15).
The affinity enhancement phenomenon caused by dimerization is called avidity. It has been widely studied in antibodies but less studied in other protein-protein interactions (47). It was observed in the complex of DYNLL with a dimeric DIC fragment, but only qualitatively analyzed (15). In this study, we compared the affinities of two pairs of monomeric and dimeric DYNLL-binding motifs (of myoVa and Bmf) and found >200-fold affinity enhancement in both cases. In case of a myoVa dimer, kinetic analysis by stopped-flow and SPR measurements showed that the increased affinity is due to a decreased rate of dissociation of the complex in line with the avidity hypothesis (47). The avidity effect could explain the previously described wide range of binding affinities (100 nm to 100 μm) determined for either monomeric or dimeric ligands (2, 26, 28–31, 33).
Dimer to Monomer Transition of DYNLL Could Be an Effective Way of Regulation
DYNLL2 monomers are structurally incapable of binding to their partners, even though complex formation was observed in this work at very high concentrations of a monomeric partner (Kd >110 μm). Dimeric myoVa showed an ∼50 times higher apparent affinity indicating the presence of avidity effect with the S88E mutant. In this case, the observed Kd values is in the same range as that of the wild type DYNLL with monomeric ligand. As ligands bind only to DYNLL dimers (a strong assumption), Le Chatelier's principle shifts the monomer-dimer equilibrium toward dimer formation. Detailed thermodynamic analysis of the S88E mutant interactions has not been attempted. The dissociation constant of the S88E mutant dimer was determined by Barbar and co-workers (48) using NMR titration and reported to be >1 mm. These results are consistent with the hypothesis that control of the monomer-dimer equilibrium of DYNLL could have a regulatory role by strongly affecting target selection. Under physiological conditions monomeric DYNLL2 could bind, although with diminished apparent affinity, to dimeric partners that locally have high concentration; however, they would not interact with monomeric partners.
In an in vitro study, weak or no interaction was found between the phosphomimetic S88E mutant and Bim or Pak1 (29). Others were able to detect relatively strong interactions in case of the DIC and swallow (33). Recently, a phosphomimetic S90E mutant of Chlamydomonas flagella LC8, which is analogous to the human DYNLL S88E mutant, was found to be largely monomeric in vitro but dimeric in vivo. This mutation did not cause significant change in flagellar generation and motility (49). The crystal structure of a homologous phosphomimetic DYNLL mutant bound to a fragment of swallow has also been determined (48). The mutant was dimeric in this complex, and its structure was largely identical to that of wild type DYNLL-swallow complex (48).
It is still not clear which kinase phosphorylates the vertebrate DYNLL isoforms in vivo. Previous studies showed that Pak1 not only binds to but also phosphorylates DYNLL1 (10, 34). However, a recent study questioned the regulatory role of Pak1 (26). We confirmed in this work that Pak1 was unable to phosphorylate DYNLL1 and DYNLL2 in an in vitro assay; instead, it most likely phosphorylated a thrombin recognition site in the recombinant His-tagged proteins (supplemental Fig. 2). It is now concluded that Pak1 is not involved directly in regulation of the DYNLL interaction network, and Ser-88 must be phosphorylated in the cell by a yet unidentified kinase. Nevertheless, it is clear the phosphorylation of Ser-88 will affect DYNLL binding to its targets and could provide an effective way of regulation.
Binding of Ligands to DYNLL Occurs in Two Steps
In this work, we report the first kinetic analysis of the binding reactions of mono- and bivalent ligands to DYNLL. Two-phase binding was observed in all cases, irrespective of the valence of the ligands. Two possible minimal models, the IF or the CS can be used to describe this behavior of the system.
Structures of both apo-DYNLL (17) and several DYNLL-peptide complexes have been determined by x-ray crystallography (13, 15, 16). Comparison of these structures revealed a ligand-dependent widening of the binding grooves by ∼1 Å for KXTQTX (swallow) ligands and by ∼2 Å for XG(I/V)QVD (nNOS) ligands (17). Therefore, the conformational change of DYNLL must be an important requirement for ligand binding. However, these structures provide no information on the order of the ligand binding and the conformational change. Backbone dynamics of DYNLL and DYNLL-peptide complexes were investigated by 15N NMR relaxation experiments, both in the apo- and ligand-bound forms (50, 51). The amino acid residues of the target-binding grooves of apo-DYNLL displayed conformational exchanges on the millisecond-to-microsecond time scale. The existence of slow time scale exchanges was also indicated by backbone amide hydrogen-deuterium exchange studies (50, 51). However, these experiments are probing conformational exchange at a minutes-to-hours time scale. The alternative (dynamic) conformations were suggested to be responsible for the ability of DYNLL to bind to diverse targets. Similar experiments with DYNLL-peptide complexes indicated that although the overall backbone dynamics of DYNLL remains largely unchanged, the conformational breathing of the partner binding groove is significantly reduced in the complex. However, some parts of DYNLL showed flexibility even after complex formation (50). More recent results showed that the degree of structural ordering of DYNLL upon partner binding was ligand-dependent and correlates with the entropy change of the binding reaction (28). There was only a minimal change found in backbone dynamics relative to apo-DYNLL if ligand binding was entropically favored. Entropically disfavored binding was consistent with higher ordering of DYNLL in the complex (28). These results also confirm that there is a difference in the binding mechanism of the different classes of motifs. The presence of increased conformational motions of DYNLL in the apo-form compared with that in the DYNLL-ligand complex revealed by the NMR experiments is consistent with our CS model. Moreover, in stopped-flow experiments, based on the comparison of χ2/degrees of freedom values, slightly better fits were obtained in almost every case using the CS model, and the three-dimensional confidence contour plots also indicated a better fit (supplemental Fig. 3). Therefore, we assume that DYNLL-ligand binding occurs via the CS mechanism. A similar binding mechanism has been demonstrated for ubiquitin and its target proteins (52). Finally, it must be noted that, based on our data, the existence of more complex reaction schemes (like the combination of IF and CS) cannot be ruled out.
Physiological Relevance of the Thermodynamic and Kinetic Fine-tuning of the DYNLL Interaction Network
What is the functional significance of avidity? In general, the binding affinity and specificity are significantly enhanced. Based on our quantitative analysis of complex formation of representative members of the DYNLL binding partners as monovalent and bivalent ligands, it is likely that at physiological protein concentrations (mostly submicromolar to micromolar in range) (53) the DYNLL-binding sites would be saturated only by dimeric interacting proteins. Indeed, most of the known binding partners of DYNLL are either dimeric or promoted to form dimers upon DYNLL binding by physically holding the two interacting polypeptide chains close to each other (54). Higher apparent affinity of DYNLL binding to dimeric protein fragments was first noted by studying DYNLL2 and myoVa interactions (2) and later attributed to avidity (15). This hypothesis questioned the original proposal of heterodimeric cargo adapter role of DYNLL on dynein and myoVa motor complexes (6, 7, 21, 55–58).
Among the DYNLL binding partners, only Bim and Bmf were reported so far to be monomeric (59). However, one can speculate that their hydrophobic C-terminal part might function as a membrane-anchoring site (38), and therefore, they can behave as a “quasi-bivalent” ligand for DYNLL to utilize avidity to increase binding affinity on the surface of membranes. Interestingly, the KXTQTX motif of Bmf showed strikingly slower dissociation than other motifs (koffC ≈10−3, in contrast to koffC ≈10−1). The apparent koffC value of a DYNLL-dimeric partner interaction can be low due to the avidity effect, so the resulting complexes can have a longer lifetime in the cell, where this kind of stability might be an important requirement for their function. It is possible that the sequence of Bmf has been evolutionarily optimized for lower koffC values, because Bmf is not dimeric. The kinetic difference between the KXTQTX and other DYNLL-binding motif classes seems to be an important feature, but it is also possible that the slow off-rate is a specific property of pro-apoptotic proteins within this class.
In conclusion, we propose that the two distinguishable kinetic and thermodynamic mechanisms of complex formation (KXTQTX motif and others motif classes) represent a fine-tuning mechanism in the interaction network of DYNLL. The low off-rate of the bivalent binding of dimers and hence longer lifetime of the complexes could be ideal for DYNLL to function as a dimerization and/or sequestration hub protein that is also involved in formation of supramolecular structures.
Supplementary Material
Acknowledgments
We give special thanks to Ferenc Tölgyesi for helping in ITC measurements, to Katalin Kékesi for MS analysis, and to András Patthy for peptide synthesis.
This work was supported by Hungarian Scientific Research Fund OTKA K61784, NI68466, and NK81950 (L. N.), K71915, NNF78783 (M. K.), and National Development Agency Grant TAMOP-4.2.1.B-09/1/KMR. M. K. is a Bolyai Fellow of the Hungarian Academy of Sciences.

The on-line version of this article (available at http://www.jbc.org) contains supplemental “Experimental Procedures,” Tables 1 and 2, and Figs. 1–3.
- DYNLL
- LC8 dynein light chain
- CS
- conformational selection
- DIC
- dynein intermediate chain
- IF
- induced fit
- ITC
- isothermal titration calorimetry
- myoVa
- myosin Va
- SPR
- surface plasmon resonance
- nNOS
- nitric-oxide synthase
- Bmf
- Bcl-2-modifying factor
- Bim
- Bcl-2 interacting mediator.
REFERENCES
- 1.King S. M., Patel-King R. S. (1995) J. Biol. Chem. 270, 11445–11452 [DOI] [PubMed] [Google Scholar]
- 2.Hódi Z., Németh A. L., Radnai L., Hetényi C., Schlett K., Bodor A., Perczel A., Nyitray L. (2006) Biochemistry 45, 12582–12595 [DOI] [PubMed] [Google Scholar]
- 3.Barbar E. (2008) Biochemistry 47, 503–508 [DOI] [PubMed] [Google Scholar]
- 4.Hodi Z., Rapali P., Radnai L., Molnar T., Szenes A., Kardos J., Buday L., Stafford W., Nyitray L. (2007) FEBS J. 274(s1), 106–106 [Google Scholar]
- 5.Jaffrey S. R., Snyder S. H. (1996) Science 274, 774–777 [DOI] [PubMed] [Google Scholar]
- 6.Puthalakath H., Villunger A., O'Reilly L. A., Beaumont J. G., Coultas L., Cheney R. E., Huang D. C., Strasser A. (2001) Science 293, 1829–1832 [DOI] [PubMed] [Google Scholar]
- 7.Puthalakath H., Huang D. C., O'Reilly L. A., King S. M., Strasser A. (1999) Mol. Cell 3, 287–296 [DOI] [PubMed] [Google Scholar]
- 8.Makokha M., Hare M., Li M., Hays T., Barbar E. (2002) Biochemistry 41, 4302–4311 [DOI] [PubMed] [Google Scholar]
- 9.Schnorrer F., Bohmann K., Nüsslein-Volhard C. (2000) Nat. Cell Biol. 2, 185–190 [DOI] [PubMed] [Google Scholar]
- 10.Vadlamudi R. K., Bagheri-Yarmand R., Yang Z., Balasenthil S., Nguyen D., Sahin A. A., den Hollander P., Kumar R. (2004) Cancer Cell 5, 575–585 [DOI] [PubMed] [Google Scholar]
- 11.Lu J., Sun Q., Chen X., Wang H., Hu Y., Gu J. (2005) Biochem. Biophys. Res. Commun. 331, 153–158 [DOI] [PubMed] [Google Scholar]
- 12.Tochio H., Ohki S., Zhang Q., Li M., Zhang M. (1998) Nat. Struct. Biol. 5, 965–969 [DOI] [PubMed] [Google Scholar]
- 13.Liang J., Jaffrey S. R., Guo W., Snyder S. H., Clardy J. (1999) Nat. Struct. Biol. 6, 735–740 [DOI] [PubMed] [Google Scholar]
- 14.Fan J., Zhang Q., Tochio H., Li M., Zhang M. (2001) J. Mol. Biol. 306, 97–108 [DOI] [PubMed] [Google Scholar]
- 15.Williams J. C., Roulhac P. L., Roy A. G., Vallee R. B., Fitzgerald M. C., Hendrickson W. A. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 10028–10033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Benison G., Karplus P. A., Barbar E. (2007) J. Mol. Biol. 371, 457–468 [DOI] [PubMed] [Google Scholar]
- 17.Benison G., Karplus P. A., Barbar E. (2008) J. Mol. Biol. 384, 954–966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang W., Lo K. W., Kan H. M., Fan J. S., Zhang M. (2003) J. Biol. Chem. 278, 41491–41499 [DOI] [PubMed] [Google Scholar]
- 19.Day C. L., Puthalakath H., Skea G., Strasser A., Barsukov I., Lian L. Y., Huang D. C., Hinds M. G. (2004) Biochem. J. 377, 597–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Makokha M., Huang Y. J., Montelione G., Edison A. S., Barbar E. (2004) Protein Sci. 13, 727–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Naisbitt S., Valtschanoff J., Allison D. W., Sala C., Kim E., Craig A. M., Weinberg R. J., Sheng M. (2000) J. Neurosci. 20, 4524–4534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pfister K. K., Fisher E. M., Gibbons I. R., Hays T. S., Holzbaur E. L., McIntosh J. R., Porter M. E., Schroer T. A., Vaughan K. T., Witman G. B., King S. M., Vallee R. B. (2005) J. Cell Biol. 171, 411–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang L., Hare M., Hays T. S., Barbar E. (2004) Biochemistry 43, 4611–4620 [DOI] [PubMed] [Google Scholar]
- 24.Lo K. W., Kogoy J. M., Rasoul B. A., King S. M., Pfister K. K. (2007) J. Biol. Chem. 282, 36871–36878 [DOI] [PubMed] [Google Scholar]
- 25.Rodríguez-Crespo I., Yélamos B., Roncal F., Albar J. P., Ortiz de Montellano P. R., Gavilanes F. (2001) FEBS Lett. 503, 135–141 [DOI] [PubMed] [Google Scholar]
- 26.Lightcap C. M., Sun S., Lear J. D., Rodeck U., Polenova T., Williams J. C. (2008) J. Biol. Chem. 283, 27314–27324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lo K. W., Naisbitt S., Fan J. S., Sheng M., Zhang M. (2001) J. Biol. Chem. 276, 14059–14066 [DOI] [PubMed] [Google Scholar]
- 28.Hall J., Hall A., Pursifull N., Barbar E. (2008) Biochemistry 47, 11940–11952 [DOI] [PubMed] [Google Scholar]
- 29.Song C., Wen W., Rayala S. K., Chen M., Ma J., Zhang M., Kumar R. (2008) J. Biol. Chem. 283, 4004–4013 [DOI] [PubMed] [Google Scholar]
- 30.Wagner W., Fodor E., Ginsburg A., Hammer J. A., 3rd. (2006) Biochemistry 45, 11564–11577 [DOI] [PubMed] [Google Scholar]
- 31.Nyarko A., Hare M., Hays T. S., Barbar E. (2004) Biochemistry 43, 15595–15603 [DOI] [PubMed] [Google Scholar]
- 32.Lei K., Davis R. J. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 2432–2437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Song Y., Benison G., Nyarko A., Hays T. S., Barbar E. (2007) J. Biol. Chem. 282, 17272–17279 [DOI] [PubMed] [Google Scholar]
- 34.Yang Z., Vadlamudi R. K., Kumar R. (2005) J. Biol. Chem. 280, 654–659 [DOI] [PubMed] [Google Scholar]
- 35.Sarkar G., Sommer S. S. (1990) BioTechniques 8, 404–407 [PubMed] [Google Scholar]
- 36.Johnson K. A., Simpson Z. B., Blom T. (2009) Anal. Biochem. 387, 20–29 [DOI] [PubMed] [Google Scholar]
- 37.Johnson K. A., Simpson Z. B., Blom T. (2009) Anal. Biochem. 387, 30–41 [DOI] [PubMed] [Google Scholar]
- 38.O'Connor L., Strasser A., O'Reilly L. A., Hausmann G., Adams J. M., Cory S., Huang D. C. (1998) EMBO J. 17, 384–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rodríguez-Crespo I., Straub W., Gavilanes F., Ortiz de Montellano P. R. (1998) Arch. Biochem. Biophys. 359, 297–304 [DOI] [PubMed] [Google Scholar]
- 40.Kramer R. H., Karpen J. W. (1998) Nature 395, 710–713 [DOI] [PubMed] [Google Scholar]
- 41.Pabbisetty K. B., Yue X., Li C., Himanen J. P., Zhou R., Nikolov D. B., Hu L. (2007) Protein Sci. 16, 355–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhou H. X. (2003) J. Mol. Biol. 329, 1–8 [DOI] [PubMed] [Google Scholar]
- 43.Zhou H. X. (2006) Biophys. J. 91, 3170–3181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Navarro-Lérida I., Martínez Moreno M., Roncal F., Gavilanes F., Albar J. P., Rodríguez-Crespo I. (2004) Proteomics 4, 339–346 [DOI] [PubMed] [Google Scholar]
- 45.Lajoix A. D., Gross R., Aknin C., Dietz S., Granier C., Laune D. (2004) Mol. Divers 8, 281–290 [DOI] [PubMed] [Google Scholar]
- 46.Espindola F. S., Suter D. M., Partata L. B., Cao T., Wolenski J. S., Cheney R. E., King S. M., Mooseker M. S. (2000) Cell Motil. Cytoskeleton 47, 269–281 [DOI] [PubMed] [Google Scholar]
- 47.Mammen M., Choi S. K., Whitesides G. M. (1998) Angew. Chem. Int. Ed. Engl. 37, 2755–2794 [DOI] [PubMed] [Google Scholar]
- 48.Benison G., Chiodo M., Karplus P. A., Barbar E. (2009) Biochemistry 48, 11381–11389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang P., Yang C., Wirschell M., Davis S. (2009) J. Biol. Chem. 284, 31412–31421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fan J. S., Zhang Q., Tochio H., Zhang M. (2002) J. Biomol. NMR 23, 103–114 [DOI] [PubMed] [Google Scholar]
- 51.Benison G., Barbar E. (2009) Methods Enzymol. 455, 237–258 [DOI] [PubMed] [Google Scholar]
- 52.Wlodarski T., Zagrovic B. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 19346–19351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ghaemmaghami S., Huh W. K., Bower K., Howson R. W., Belle A., Dephoure N., O'Shea E. K., Weissman J. S. (2003) Nature 425, 737–741 [DOI] [PubMed] [Google Scholar]
- 54.Lightcap C. M., Kari G., Arias-Romero L. E., Chernoff J., Rodeck U., Williams J. C. (2009) PLoS One 4, e6025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fejtova A., Davydova D., Bischof F., Lazarevic V., Altrock W. D., Romorini S., Schöne C., Zuschratter W., Kreutz M. R., Garner C. C., Ziv N. E., Gundelfinger E. D. (2009) J. Cell Biol. 185, 341–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Alonso C., Miskin J., Hernáez B., Fernandez-Zapatero P., Soto L., Cantó C., Rodríguez-Crespo I., Dixon L., Escribano J. M. (2001) J. Virol. 75, 9819–9827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Su Y., Qiao W., Guo T., Tan J., Li Z., Chen Y., Li X., Li Y., Zhou J., Chen Q. (2010) Cell. Microbiol. 12, 1098–1107 [DOI] [PubMed] [Google Scholar]
- 58.Varma D., Dawn A., Ghosh-Roy A., Weil S. J., Ori-McKenney K. M., Zhao Y., Keen J., Vallee R. B., Williams J. C. (2010) Proc. Natl. Acad. Sci. U.S.A. 107, 3493–3498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hinds M. G., Smits C., Fredericks-Short R., Risk J. M., Bailey M., Huang D. C., Day C. L. (2007) Cell Death Differ. 14, 128–136 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.