

Abstract
The N348I mutation at the connection subdomain of HIV-1 reverse transcriptase (RT) confers clinically significant resistance to both nucleoside and non-nucleoside RT inhibitors (NNRTIs) by mechanisms that are not well understood. We used transient kinetics to characterize the enzymatic properties of N348I RT and determine the biochemical mechanism of resistance to the NNRTI nevirapine (NVP). We demonstrate that changes distant from the NNRTI binding pocket decrease inhibitor binding (increase Kd-NVP) by primarily decreasing the association rate of the inhibitor (kon-NVP). We characterized RTs mutated in either p66 (p66N348I/p51WT), p51 (p66WT/p51N348I), or both subunits (p66N348I/p51N348I). Mutation in either subunit caused NVP resistance during RNA-dependent and DNA-dependent DNA polymerization. Mutation in p66 alone (p66N348I/p51WT) caused NVP resistance without significantly affecting RNase H activity, whereas mutation in p51 caused NVP resistance and impaired RNase H, demonstrating that NVP resistance may occur independently from defects in RNase H function. Mutation in either subunit improved affinity for nucleic acid and enhanced processivity of DNA synthesis. Surprisingly, mutation in either subunit decreased catalytic rates (kpol) of p66N348I/p51N348I, p66N348I/p51WT, and p66WT/p51N348I without significantly affecting affinity for deoxynucleotide substrate (Kd-dNTP). Hence, in addition to providing structural integrity for the heterodimer, p51 is critical for fine tuning catalytic turnover, RNase H processing, and drug resistance. In conclusion, connection subdomain mutation N348I decreases catalytic efficiency and causes in vitro resistance to NVP by decreasing inhibitor binding.
Keywords: Antiviral Agents, Drug Resistance, Enzyme Inhibitors, Enzyme Kinetics, Enzyme Mechanisms, Enzyme Mutation, Human Immunodeficiency Virus, Pre-steady State Kinetics, Reverse Transcription, Non-nucleoside Reverse Transcriptase Inhibitor
Introduction
Human immunodeficiency virus type 1 (HIV-1) reverse transcriptase (RT) converts the viral single-stranded, positive sense RNA genome to a double-stranded DNA, which is integrated into the host genome. To achieve this task, RT possesses multiple enzymatic activities, including both DNA-dependent and RNA-dependent DNA polymerase activities and RNase H activity. RT is a heterodimer composed of 66-kDa (p66) and 51-kDa (p51) subunits. p66 is 560 amino acids long and comprises the spatially distinct polymerase and RNase H domains. The polymerase domain of p66 includes the fingers, palm, and thumb subdomains that resemble a clasping right hand connected to the RNase H domain through the connection subdomain. p51 contains the first 440 amino acids of p66 and is derived by HIV-1 protease-mediated cleavage of an RNase H domain from the p66/p66 homodimer. Because p51 of the heterodimer has no enzymatic function, it has been proposed that its role is simply to provide structural support to p66.
HIV-1 RT has been a prominent target of anti-AIDS therapies. There are two classes of approved drugs that target RT: the nucleoside RT inhibitors (NRTIs)3 and the non-nucleoside RT inhibitors (NNRTIs). NRTIs mimic deoxynucleotide triphosphate (dNTP) substrates required for DNA synthesis. Once integrated into the nascent viral DNA, NRTIs act as chain terminators because they lack a 3′-OH group required for formation of a phosphodiester bond with the incoming nucleotide (1, 2). On the other hand, NNRTIs are non-competitive RT inhibitors with respect to either dNTP or nucleic acid substrates and have been proposed to interfere with the chemical step of DNA synthesis by altering the precise geometry of the polymerase active site and by restricting the mobility of the p66 thumb (3–9). NNRTIs bind at the NNRTI binding pocket (NNIBP), a hydrophobic pocket at the base of the p66 thumb and close (∼10 Å) to the polymerase active site, formed primarily by residues from p66 and Glu-138 from p51. NNRTI binding results in conformational changes that shift the “primer grip” and overextend the p66 thumb (10). Both classes of drugs have been successfully used as key components of highly active antiretroviral therapies in combination with protease, integrase, and entry inhibitors. However, as is the case with all anti-AIDS drugs, prolonged use inevitably leads to the emergence of drug-resistant HIV strains.
RT uses two main strategies to develop NRTI resistance. The first strategy involves a selective interference with the incorporation of NRTIs into viral DNA (11–15). The second mechanism involves enhanced excision and removal of the chain terminator from the 3′-end of the blocked primer using ATP as a nucleophile (16–18). Unblocking the primer terminus frees the reactive hydroxyl group at the 3′-end of the primer and allows for continued DNA synthesis. Typically, NRTI resistance mutations are found at the NRTI- or ATP-binding sites (19–21).
NNRTI resistance is caused by mutations at, or close to, the NNIBP (5) that have been shown to reduce inhibitor binding through loss or change of contacts (22–25), through steric interactions (25, 26), or by interfering with the entry of the NNRTI because they change interactions at the entrance of the NNIBP (27, 28).
Until recently, almost all clinically relevant NRTI and NNRTI resistance mutations were found at the p66 fingers and palm subdomains. Lately, an increasing number of mutations at the connection subdomain and RNase H domain have been shown to enhance resistance to azidothymidine (AZT) (29, 30). We and others have recently shown that N348I is the first clinically significant single amino acid mutation found to confer resistance to multiple members of both NRTI and NNRTI classes of inhibitors (31–33). N348I often emerges as a result of AZT- and/or didanosine-containing therapy and is found in combination with AZT excision-enhancing mutations (32, 34). N348I is not a polymorphism; it appears in ∼10% of treatment-experienced individuals compared with less than 1% in treatment-naïve patients (31). N348I has also been detected at a high frequency in patients receiving AZT/lamivudine (2′,3′-dideoxy-3′-thiacytidine) treatment, and it has been recently suggested that N348I compensates for the antagonism between M184V and excision-enhancing mutations (35, 36). In cell culture experiments, HIV harboring N348I RT (HIV-1N348I) is resistant to NRTIs, including AZT and didanosine, and when in combination with excision-enhancing mutations enhances resistance to tenofovir (31, 32, 37). HIV-1N348I is also resistant to NNRTIs, such as nevirapine (NVP), and to a lesser extent to delavirdine (31–33). When in combination with the NNRTI resistance mutation Y181C, N348I enhances resistance to the second generation NNRTI etravirine (37).
Several studies have focused on the mechanism by which CSMs confer resistance to NRTIs. Nikolenko et al. (30, 38) proposed that a number of CSMs increase AZT resistance by reducing template RNA degradation, thereby preserving the RNA template and providing additional time for RT to excise AZT monophosphate. Yap et al. (31) showed that N348I reduces the rate of RNA template degradation by RT in either a WT background or in the presence of excision-enhancing AZT resistance mutations. Ehteshami et al. (39) proposed that CSMs N348I and A360V enhance resistance to AZT through both RNase H-dependent and -independent mechanisms. Specifically, addition of the CSMs N348I and A360V to the AZT-resistant M41L/D67N/T215Y RT enhanced AZT resistance by causing preservation of AZT-terminated RNA/DNA that can be unblocked by RT with CSMs (39).
The mechanism by which CSMs confer resistance to NNRTIs is largely unknown. Moreover, the effect of N348I on the biochemical aspects of DNA synthesis is also not well understood. Here we resolved the mechanism of NVP resistance using pre-steady state kinetics analysis to quantify NVP binding and dissociation from wild-type (WT) and N348I enzymes. We also determined how Ile-348 in p66 (Ile-348p66) or p51 (Ile-348p51) affects other enzymatic properties of RT relevant to drug resistance, including the efficiency of DNA polymerization, RNase H activity, processivity of DNA synthesis, and affinity for various substrates.
EXPERIMENTAL PROCEDURES
Enzymes and Nucleic Acids
We prepared subunit-specific RT mutants by expressing the two subunits from a single plasmid, allowing in situ folding and yielding proteins of superior quality compared with those obtained from reconstitution of separately expressed RT subunits. In our experience, this method of RT expression is a critical factor for optimizing transient kinetics and crystallographic data. The p66 and p51 sequences of RT (BH-10) were cloned in the pETDuet-1 vector (Novagen) using restriction sites PpumI and SacI for p51 and SacII and AvrII for p66. To produce mutant RT, sequences coding for p66 and p51 were independently subcloned and mutated using the QuikChange mutagenesis kit (Stratagene) before being sequentially cloned into a pETDuet-1 vector. Sequences coding for a hexahistidine tag and the 3C protease recognition sequence were added at the N terminus of p51. RT enzymes were expressed in BL21 Escherichia coli (Invitrogen) and purified by nickel affinity chromatography and mono Q anion exchange chromatography as described previously (40). Oligonucleotides were purchased from Integrated DNA Technologies (Coralville, IA). All reactions, with the exception of NVP susceptibility and RNase H assays, used Td31/Cy3-Pd18 as a template/primer. The Td31/Cy3-Pd18 template sequence was 5′-CCATAGCTAGCATTGGTGCTCGAACAGTGAC-3′(Td31), and the 5′ Cy3-labeled DNA primer sequence was 5′-Cy3-GTCACTGTTCGAGCACCA-3′ (Cy3-Pd18). For the NVP susceptibility assays, a 100-base-long template (T100; described in Ref. 40) annealed to the Pd18 primer was used. For the RNase H assays, a 22-base pair duplex (Cy3-Tr35/Pd22) consisting of a 5′ Cy3-labeled RNA template (Cy3-Tr35) of the sequence 5′-Cy3-GGAAAUCUCUAGCAGUGGCGCCCGAACAGGGACCU-3′ annealed to a DNA primer (Pd22) of the sequence 5′-AGGTCCCTGTTCGGGCGCCACT-3′ was used (39). Primers were annealed to templates at concentrations corresponding to a 1:3 molar ratio. Deoxynucleotide triphosphates and dideoxynucleotide triphosphates were purchased from Fermentas (Glen Burnie, MD). The concentrations of nucleotides and nucleic acids were calculated spectrophotometrically based on absorption at 260 nm.
Active Site Titration and Determination of Kd-DNA
We determined the active site concentrations of RT preparations using pre-steady state burst experiments. We preincubated a fixed concentration of RT (40 nm; determined by absorbance measurements) in RT buffer (50 mm NaCl and 50 mm Tris-HCl, pH 7.8) with increasing concentrations of Td31/Cy3-Pd18 followed by rapidly mixing with a solution of MgCl2 (5 mm) and dATP (50 μm) in RT buffer (41) using an RQF-3 rapid quench-flow instrument (KinTek Corp., Austin, TX) (all values are final concentrations). Reaction mixtures were then incubated at 37 °C for times spanning 0.005–5 s before quenching with EDTA (50 mm). Reaction products were resolved under denaturing conditions in polyacrylamide-urea DNA gels and quantitated by phosphorimaging and densitometry using Multi Gauge V3.0 (FujiFilm). First the amounts of extended primer (P) were plotted against time with GraphPad Prism 4, and the data were fit to the following biphasic equation,
where A is the amplitude of the burst phase that represents the enzyme-DNA complex at the start of the reaction, kobs is the observed burst rate constant for dNTP incorporation, kss corresponds to the steady state rate constant, and t is the reaction time. Next, the active site concentration and template/primer dissociation constant (Kd-DNA) were determined by plotting the amplitude (A) against template/primer concentration. The data were fit by non-linear regression to a quadratic equation,
 |
where [RT] is the concentration of actively binding polymerase molecules. Subsequent transient state biochemical experiments were performed using corrected active site concentrations.
Pre-steady State Kinetics of dNTP Incorporation
The optimal polymerase rates were obtained by pre-steady state kinetics analysis using single nucleotide incorporation assays. A solution containing RT (30 nm), Td31/Cy3-Pd18 (30 nm), and EDTA (0.5 mm) in RT buffer was mixed with a solution of MgCl2 (5 mm) and dATP (0.5–75 μm) for reaction times ranging between 0.005 and 5 s before quenching with EDTA (50 mm). Reaction products were resolved and quantitated as described above. Burst phase incorporation rates and substrate affinities at individual dATP concentrations were obtained from fitting the data to the burst equation (Equation 1). To determine optimal rates of dNTP incorporation (kpol) and dNTP binding to the enzyme-DNA complex (Kd-dATP), observed burst rates (kobs) were fit to the following hyperbolic equation.
Single Turnover Processivity Assays
Processivity assays were conducted under single turnover conditions as described by Patel et al. (42). Solutions containing Td31/Cy3-Pd18 (20 nm), WT (40 nm) or mutant enzyme (60 nm p66N348I/p51N348I), and EDTA (0.5 mm) in RT buffer were mixed with solutions containing dNTPs (50 μm) and MgCl2 (5 mm) for varying times (0.05–5 s) before quenching with EDTA (50 mm). Reaction products were resolved and quantitated as described above. To obtain the rate of Pd18 extension, results were plotted using a one-phase exponential decay equation,
where kapp is the apparent rate of product (P) formation, A0 is the amplitude, t is time, and C is a constant. The rates of nucleotide incorporation at individual positions along the template were obtained by fitting data to a double exponential equation,
where A is the concentration of 19-mer or higher length products, k1 is the rate of product generation, k2 is the rate of subsequent product elongation, and C is a constant.
RNase H Assays
RNase H assays were performed by incubating RNA/DNA duplex Cy3-Tr35/Pd22 (100 nm) with RT (200 nm) in RT buffer at 37 °C with MgCl2 (6 mm). Reactions were quenched after incubation (1–8 min) with equal volumes of 99% formamide containing trace amounts of bromphenol blue. Reaction products were resolved in denaturing polyacrylamide-urea DNA gels and quantitated as described above. The primary RNase H cleavage product is mainly 18 nucleotides from the 3′-end of the DNA primer (−18 nucleotides), and the secondary cleavage product is mainly 12 nucleotides from the 3′-end of the primer (−12 nucleotides) as reported previously (39, 43).
Nevirapine Susceptibility Assays
Drug susceptibility steady state assays were carried out by measuring the extension of Td100/Pd18 in the presence of increasing concentrations of NVP. Reactions of 50 μl containing RT (2 nm), MgCl2 (5 mm), Td100/Pd18 (10 nm), and dNTPs (0.8 μm of each with 0.5 μCi/nmol α-32P-labeled dTTP) in RT buffer were initiated by the addition of MgCl2 (6 mm), incubated at room temperature for 15 min, and terminated by the addition of EDTA (50 mm). Reaction products were then passed through a charged nylon filter using a vacuum manifold apparatus (Whatman, GE Healthcare). Extended primers, radiolabeled by the incorporation of [32P]dTMP, bound to the membrane, whereas the unincorporated [α-32P]dTTP was filtered through. Radioactive filters were exposed to phosphor screens followed by phosphorimaging and analysis using the Multi Gauge V3.0 software (FujiFilm). Dose-response curves of triplicate samples were plotted using GraphPad Prism 4 to determine IC50 values for NVP.
Gel-based NVP susceptibility assays were performed using Td31/Cy3-Pd18 or Tr31/Cy3-Pd18 (RNA template of corresponding sequence). RT (20 nm) was preincubated with increasing concentrations of NVP in RT buffer for 5 min at 37 °C before the addition of template/primer (60 nm), dNTPs (1 μm), and MgCl2 (5 mm). Reactions were allowed to proceed for 5 min at 37 °C before being quenched with EDTA (50 mm). Reaction products were resolved on denaturing polyacrylamide-urea DNA gels, and fluorescent bands corresponding to full-extension products were quantified as described above. IC50 values were derived from dose-response curves using GraphPad Prism 4.
Nevirapine Binding Assays
Solutions of RT (30 nm active sites), Td31/Cy3-Pd18 (30 nm), and EDTA (5 mm) in RT buffer were incubated with various concentrations of NVP for 10 min at room temperature before initiating reactions with MgCl2 (0.5 mm) and saturating dATP (100 μm). Reactions were allowed to proceed for times between 0.005 and 5 s before quenching with EDTA (50 mm). Reaction products were fit to the burst equation (Equation 1) to calculate burst amplitudes (A) for each data set. Burst amplitudes were then fit to the following hyperbolic equation (44) to determine the dissociation constant for NVP (Kd-NVP),
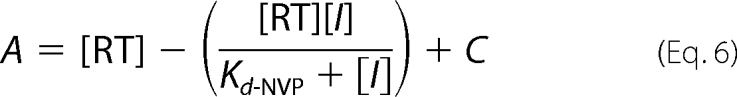 |
where [RT] is the concentration of actively binding polymerase molecules, [I] is the concentration of inhibitor, and C is a constant.
The apparent rate of NVP binding to RT (kapp-NVP) was obtained by premixing a solution of RT (30 nm) and Td31/Cy3-Pd18 (30 nm) in RT buffer with saturating NVP (40 μm) for increasing amounts of time (0.2–5 s). Reactions were then initiated by addition of MgCl2 (5 mm) and saturating dATP (50 μm), allowed to proceed for 0.2 s, and quenched with EDTA (250 mm). Reaction products were resolved and quantitated as described above and plotted with GraphPad Prism 4 using an equation for one-phase exponential decay,
where P is the reaction product, A0 is the amount of product in the absence of NVP, t is the preincubation time with NVP before addition of MgCl2/dATP, and C is a constant.
The kapp-NVP values obtained were next inserted into the following equation to derive the NVP association constant (kon-NVP) (8).
The kon-NVP and Kd-NVP values were then used to calculate the NVP dissociation rate (koff-NVP) through the following equation.
Molecular Modeling
A molecular model of p66N348I/p51N348I was generated using starting protein coordinates from the crystal structure of HIV-1 RT in complex with NVP (Protein Data Bank code 1VRT) (3). The coordinates were initially processed by the Protein Preparation tool (Schrödinger Molecular Modeling Suite, New York, NY), which deletes unwanted water molecules, sets charges and atom type of metal ions, corrects misoriented Gln and Asn residues, and optimizes hydrogen atom orientations. Amino acid side chains were substituted as needed using the Maestro software (Schrödinger Molecular Modeling Suite). To ensure that any observed changes in the structure of the mutant were not due to the protocols used, the WT structure was also treated in parallel using the same protocols. Molecular dynamics simulations of the WT and p66N348I/p51N348I models were performed to obtain optimally stable structures using the software Impact, interfaced with Maestro, at constant temperature, and the OPLS_2005 force field. The molecular dynamics simulations were performed for 1000 steps with 0.001-ps intervals. The temperature relaxation time was 0.01 ps. The Verlet integration algorithm was used in the simulations. Visualization of the molecules, comparisons, and figure preparation were carried out using PyMOL.
RESULTS
Effect of N348I on Enzyme-Template/Primer Complex Formation
We determined the effect of N348I on DNA/DNA binding affinity using a pre-steady state kinetics assay where we titrated the amount of nucleic acid that can be bound to, and extended by, RT (Fig. 1). These experiments showed the Kd-DNA for the double mutant (p66N348I/p51N348I) to be 9.9 nm, slightly lower than that for WT (12.5 nm; Table 1). Subunit-specific experiments were performed to further dissect the effect of the mutation in either RT subunit. These experiments determined the Kd-DNA values for p66N348I/p51WT and p66WT/p51N348I to be 4.8 and 5.4 nm, respectively. Hence, the presence of either Ile-348p66 or Ile-348p51 resulted in an increased affinity for double-stranded DNA. These results were consistent with gel shift assays, which also showed that the mutant enzymes bind DNA/DNA and RNA/DNA oligonucleotides slightly more efficiently than does WT RT (data not shown).
FIGURE 1.

Calculation of dissociation constants of WT and N348I mutant RTs for nucleic acid (Kd-DNA). Enzymes (40 nm; as determined by absorbance measurements at 280 nm) were preincubated with increasing concentrations of Td31/Cy3-Pd18 duplex DNA. The RT-DNA complex was rapidly mixed with MgCl2 and dATP and incubated for times spanning from 0.005 to 5 s before quenching with EDTA (50 mm final concentration). The burst amplitudes were calculated for each concentration of DNA by fitting the corresponding time courses to the burst equation (see “Experimental Procedures”). The amplitudes were plotted as a function of the DNA concentrations and fit to a quadratic equation. The fit of the data yielded the following Kd values for each enzyme: WT (■), Kd-DNA = 12.5 nm; p66N348I/p51N348I (□), Kd-DNA = 9.9 nm; p66N348I/p51WT (●), Kd-DNA = 4.8 nm; and p66WT/p51N348I (○), Kd-DNA = 5.4 nm.
TABLE 1.
Pre-steady state RT kinetics
| Kd-DNA | kpol | Kd-dATP | kpol/Kd-dATP | |
|---|---|---|---|---|
| nm | s−1 | μm | μm−1s−1 | |
| WT | 12.5 | 24.4 ± 0.9 | 1.3 ± 0.4 | 18.8 |
| p66N348I/p51N348I | 9.9 | 5.7 ± 0.3 | 1.0 ± 0.3 | 5.7 |
| p66N348I/p51WT | 4.8 | 8.6 ± 0.4 | 0.9 ± 0.2 | 9.6 |
| p66WT/p51N348I | 5.4 | 8.4 ± 0.3 | 0.8 ± 0.4 | 10.5 |
Pre-steady State Kinetics of Single Nucleotide Incorporation
Kinetics of pre-steady state dATP incorporation were determined as described previously (8). Both WT and p66N348I/p51N348I exhibited a biphasic pattern of nucleotide incorporation (initial burst phase followed by steady state phase; Fig. 2). The kinetics constants (kpol, Kd-dATP, and catalytic efficiency or kpol/Kd-dATP) estimated from pre-steady state kinetic analyses are shown in Table 1. The p66N348I/p51N348I enzyme displayed a ∼4-fold decrease in kpol relative to WT without a significant change in Kd-dATP (Table 1). Hence, WT RT had a catalytic efficiency (kpol/Kd-dATP) over 3-fold greater than the N348I double mutant enzyme. Interestingly, the presence of Ile-348p66 or Ile-348p51 did not affect affinity for dATP but did decrease the turnover number (kpol) (Figs. 2 and 3 and Table 1). Moreover, the decreases in catalytic efficiencies for Ile-348p66 and Ile-348p51 appear to be additive (Table 1).
FIGURE 2.

Pre-steady state kinetics of dATP incorporation by WT (A) and p66N348I/p51N348I (B) RTs are shown. 30 nm RT (active sites) were preincubated with 30 nm Td31/Cy3-Pd18. Reactions were initiated by adding 10 mm MgCl2 plus varying concentrations of dATP (0.5 μm, ■; 1 μm, ▾; 2.5 μm, ●; 10 μm, ♢; 20 μm, □; and 50 μm, ○) before being quenched with EDTA. All stated concentrations are the final concentrations after mixing. C, pre-steady state burst dependence of dATP incorporation by WT (■) or p66N348I/p51N348I (□) RTs. The first turnover rates were obtained from the burst phases and were then fit to a quadratic equation as a function of dATP concentration. The WT enzyme displayed a Kd-dATP of 1.3 ± 0.4 μm and a maximum incorporation rate of 24.4 ± 0.9 s−1. The double mutant RT (p66N348I/p51N348I) showed a similar Kd-dATP of 1.0 ± 0.3 μm and a reduced maximum incorporation rate of 5.7 ± 0.3 s−1. Error bars represent the standard deviation between results of at least three experiments.
FIGURE 3.

Pre-steady state kinetics of dATP incorporation by subunit-specific mutant enzymes p66N348I/p51WT (A) and p66WT/p51N348I (B) are shown. 30 nm RT (concentration of active sites) and 30 nm Td31/Cy3-Pd18 were preincubated in RT buffer. Reactions were initiated by adding 10 mm MgCl2 plus increasing concentrations of dATP (0.5 μm, ■; 1 μm, ▾; 2.5 μm, ●; 10 μm, ♦; 20 μm, □; and 50 μm, ○) and quenched with EDTA. C, pre-steady state burst dependence of dATP incorporation by the site-directed mutant enzymes. p66N348I/p51WT (●) showed a Kd-dATP of 0.9 ± 0.2 μm and a maximum incorporation rate of 8.6 ± 0.4 s−1. p66WT/p51N348I (○) showed a Kd-dATP of 0.8 ± 0.4 μm and a maximum incorporation rate of 8.4 ± 0.3 s−1. Error bars represent the standard deviation between results of at least three experiments.
Single Turnover Processivity Assays
We applied a single turnover processivity assay developed by Patel et al. (42) to quantify relative processivities of DNA polymerization (Fig. 4). In this assay, we monitored rates of consecutive nucleotide incorporations under single turnover conditions where enzyme dissociation from the nucleic acid prohibits elongation of the primer strand. The rate of single nucleotide incorporation (k1) and the rate of processive DNA synthesis (k2) were calculated at several template positions for each enzyme. The ratio of the rate of processive DNA synthesis to the rate of nucleotide incorporation (k2/k1) is referred to here as the processivity index. Our experiments clearly demonstrated a reproducible ∼2-fold enhancement in processivity for p66N348I/p51N348I relative to the WT enzyme (Table 2). Moreover, both Ile-348p66 and Ile-348p51 slightly enhanced processivity. These results were consistent with trap-based experiments performed in our laboratory (data not shown) and by others (39).
FIGURE 4.

Single turnover processivity assays. 20 nm Td31/Cy3-Pd18 was combined with 40 nm RT in RT buffer before rapidly mixing with saturating NVP (20 μm) for varying incubation times (0.05–1 s). Next, MgCl2 (10 mm) and saturating dATP (50 μm) were rapidly added, and reactions were allowed to proceed for 200 ms before quenching with EDTA. Extension of the 18-mer primer into 19-mer (A), 20-mer (B), and 21-mer products (not shown) by WT (■), p66N348I/p51N348I (□), p66N348I/p51WT (●), or p66WT/p51N348I (○) was quantified to determine rates of product appearance and subsequent processive extension of those products.
TABLE 2.
Single turnover processivity (-fold change over WT)
| 19-mer | 20-mer | 21-mer | Average | |
|---|---|---|---|---|
| WT | 1 | 1 | 1 | 1 |
| p66N348I/p51N348I | 3.3 | 1.5 | 1.1 | 2.0 |
| p66N348I/p51WT | 1.8 | 0.89 | 1.1 | 1.3 |
| p66WT/p51N348I | 2.9 | 0.76 | 1.2 | 1.6 |
RNase H Assays
To determine whether Ile-348p66 or Ile-348p51 alters the RNase H activity of RT, we monitored time course RNase H cleavages of 5′-Cy3-RNA/DNA. Cleavage patterns produced by WT and p66N348I/p51N348I are similar to those reported previously (39). The primary RNA cleavage typically occurs 17–18 nucleotides upstream from the polymerase active site positioned at the 3′ primer end. The enzyme then repositions to make a secondary cleavage ∼12 nucleotides upstream from the 3′ primer end. Over time, the secondary cleavage products are further processed into increasingly smaller fragments. Our experiments demonstrated that both the primary and the secondary cleavage activities of p66N348I/p51N348I were slower than those noted for WT (Fig. 5). At extended incubation times, we observed the accumulation of secondary cleavage products in the case of p66N348I/p51N348I, consistent with a more significant impairment of postsecondary cleavages compared with the effects on primary cleavage (Fig. 5). Interestingly, the secondary cleavage products in the case of p66N348I/p51WT appear comparable with WT. However, p66WT/p51N348I generated cleavage products similar to those of the double mutant p66N348I/p51N348I RT.
FIGURE 5.

Effect of mutations on RNase H Activity. 100 nm Cy3-Tr35/Pd22 DNA/RNA was incubated with 200 nm active RT for varying times (1–8 min) at 37 °C in RT buffer. Reactions were quenched with equal volumes of 99% formamide containing trace amounts of bromphenol blue. The control (no added MgCl2) shows the full-length, uncleaved RNA template. The cleavage positions relative to the 3′ terminus of the DNA primer are indicated to the left of the gel images.
Nevirapine Susceptibility
Drug susceptibility assays have shown that viruses harboring the N348I RT mutation are resistant to NVP (31, 32). We found the NVP resistance of N348I to be higher at the virus level in cell-based assays than with assays using purified RT enzymes (Fig. 6), similar to the results reported by Yap et al. (31) and Hachiya et al. (32). Our in vitro filter binding and gel-based NVP susceptibility assays using DNA/DNA or RNA/DNA substrates showed that p66N348I/p51N348I is ∼2–3-fold resistant to NVP relative to WT (Fig. 6 and Table 3). Interestingly, both Ile-348p66 and Ile-348p51 conferred NVP resistance in gel-based and filter binding assays using either DNA/DNA or RNA/DNA substrates (∼2-fold resistance for p66N348I/p51WT and ∼3-fold resistance for p66WT/p51N348I) (Table 3).
FIGURE 6.

A, filter binding assay for NVP susceptibility. DNA/DNA duplex (10 nm) was incubated with HIV-1 RT (2 nm) in RT buffer in the presence of varying amounts of inhibitor. All dNTPs were present at a final concentration of 0.8 μm each with 0.5 μCi of [α-32P]dTTP. Reactions were initiated by the addition of 6 mm MgCl2 and quenched with EDTA. Products were passed through a charged nylon filter using a vacuum manifold apparatus. B, WT (■) RT displayed an IC50 of 187 ± 3 nm, and p66N348I/p51N348I (□) displayed an IC50 of 356 ± 3 nm. In the case of the subunit-specific mutants, the p66N348I/p51WT (●) enzyme displayed an IC50 of 401 ± 4 nm, and the p66WT/p51N348I (○) RT showed an IC50 of 448 ± 2 nm. Error bars represent the standard deviation between results of at least three experiments.
TABLE 3.
NVP susceptibility (-fold resistance)
| Filter binding assay, DNA/DNA | Gel-based assay |
||
|---|---|---|---|
| DNA/DNA | RNA/DNA | ||
| WT | 1 | 1 | 1 |
| p66N348I/p51N348I | 1.9 | 2.7 | 2 |
| p66N348I/p51WT | 2.1 | 1.9 | 2 |
| p66WT/p51N348I | 2.4 | 3.1 | 2.7 |
Determination of Kd-NVP
The suppression of polymerization burst amplitude by NVP represents the amount of inhibitor bound to the RT-DNA complex (8). We therefore determined the dissociation constant of NVP (Kd-NVP) from various RTs by measuring the effect of the inhibitor on the pre-steady state burst phase of polymerization as described by Johnson and co-workers (8). Burst amplitude suppression values were plotted using a hyperbolic equation to obtain Kd-NVP (Fig. 7). The Kd-NVP for WT RT was 0.08 μm, whereas Kd-NVP for p66N348I/p51N348I was 2-fold greater (0.16 μm) (Fig. 7 and Table 4), indicating a decreased affinity of p66N348I/p51N348I for NVP. Additional experiments with the subunit-specific mutant enzymes showed that the binding affinity of NVP for p66N348I/p51WT was reduced 1.5-fold relative to WT. However, the Kd-NVP of p66WT/p51N348I was similar to that of WT (Table 4).
FIGURE 7.

Determination of NVP dissociation equilibria (Kd-NVP). Single nucleotide dATP incorporation assays were performed under pre-steady state conditions. RT (30 nm active sites) was incubated with 30 nm Td31/Cy3-Pd18 and varying concentrations of NVP. Reactions were initiated by adding MgCl2 (10 mm) plus saturating dATP before being quenched with EDTA. The burst amplitudes for RT WT and p66N348I/p51N348I were plotted against NVP concentration and fit to a hyperbolic equation (see “Experimental Procedures”). The Kd-NVP for WT (■) was determined to be 0.08 ± 0.01 μm, and the Kd-NVP for p66N348I/p51N348I (□) was 0.16 ± 0.04 μm. The subunit-specific mutant enzyme p66N348I/p51WT (●) had a Kd-NVP of 0.12 ± 0.02 μm, and the Kd-NVP of p66WT/p51N348I (○) was 0.07 ± 0.01 μm.
TABLE 4.
Kinetics of NVP binding
-Fold change relative to WT is in parentheses.
| Kd-NVP | kapp-NVP | kon-NVP | koff-NVP | |
|---|---|---|---|---|
| μm | s−1 | 104m−1s−1 | s−1 | |
| WT | 0.08 ± 0.01 (1) | 2.3 ± 0.6 (1) | 5.7 (1) | 0.005 (1) |
| p66N348I/p51N348I | 0.16 ± 0.04 (2) | 1.3 ± 0.4 (0.6) | 3.4 (0.6) | 0.005 (1) |
| p66N348I/p51WT | 0.12 ± 0.02 (1.5) | 2.0 ± 0.6 (0.9) | 5.0 (0.9) | 0.006 (1.2) |
| p66WT/p51N348I | 0.07 ± 0.01 (1) | 0.6 ± 0.1 (0.25) | 1.5 (0.3) | 0.001 (0.2) |
Calculation of kon-NVP and koff-NVP
The increased Kd-NVP for p66N348I/p51N348I and p66N348I/p51WT demonstrated that the mechanism of NVP resistance involves differential binding of the inhibitor. However, Kd-NVP is an equilibrium constant (Kd-NVP = koff-NVP/kon-NVP) that does not distinguish whether the differences are due to a decreased rate of association (kon-NVP) or to an enhanced rate of dissociation (koff-NVP). To resolve the specific effects on NVP binding, we determined the kon-NVP and koff-NVP for WT and mutant enzymes (Fig. 8 and Table 4). To calculate kon-NVP values, we first estimated the apparent rates of NVP binding (kapp-NVP). The WT kapp-NVP was almost ∼2-fold larger than that of p66N348I/p51N348I and comparable with that of p66N348I/p51WT. However, the kapp-NVP for p66WT/p51N348I was 4-fold slower than that of the WT (Fig. 8 and Table 4). We then calculated kon-NVP using our experimentally determined Kd-NVP and kapp-NVP values (Table 4). The kon-NVP value for WT was almost twice as high as that for p66N348I/p51N348I (5.7 × 104 and 3.4 × 104 m−1 s−1, respectively). Interestingly, p66WT/p51N348I displayed the largest decrease in kon-NVP without a significant change in Kd-NVP.
FIGURE 8.

NVP binding kinetics: apparent binding rates of nevirapine (kapp-NVP). RT (30 nm active sites) was first incubated in RT buffer with 30 nm Td31/Cy3-Pd18. This solution was rapidly mixed with an equal volume of saturating NVP (40 μm) for the indicated times before then being mixed with saturating dATP (50 μm) and MgCl2 (10 mm). The reactions were quenched with 250 mm EDTA, pH 8.0. Reaction products were fit to a single exponential decay equation to obtain observed binding rates. The kapp for RT WT (■) was 2.3 ± 0.6 s−1, the kapp for p66N348I/p51N348I (□) was 1.3 ± 0.4 s−1, the kapp for p66N348I/p51WT (●) was 2.0 ± 0.6 s−1, and the kapp for p66WT/p51N348I (○) was 0.6 ± 0.1 s−1.
Molecular Modeling
The molecular dynamics simulations of WT and p66N348I/p51N348I models resulted in structures that were highly similar to the starting coordinates (root mean square deviation of Cα, ∼1 Å) (Fig. 9). The N348I substitution interrupted a hydrogen bond network involving the amide side chains of residues Asn-348, Gln-330, and Gln-340 (Fig. 9A, top inset). This change did not appear to significantly affect the structure of the connection subdomain. However, we observed a noticeable change at the floor of NNIBP in the molecular model of the NVP complex with p66N348I/p51N348I (Fig. 9A, bottom inset, arrow) that may affect access to this cavity.
FIGURE 9.

A, superposition of molecular models of WT RT (multicolored schematic; see below) and p66N348I/p51N348I RT (magenta schematic). The molecular models were constructed using molecular dynamics simulations as described in the text. Overall, the structures are very similar (∼1-Å root mean square deviation for Cα), but the mutations cause noticeable differences at the floor of the NNRTI binding pocket (indicated by an arrow in inset C). In the connection subdomain of p66 (yellow), the N348I mutation affects the hydrogen bond network between residues 348, Gln-340, and Gln-330. These changes appear to affect the geometry of the NNRTI binding pocket, resulting in a decreased association rate (kon) of NVP (cyan spheres) with the N348I RT. B and C, alternate views of the NNRTI binding pockets of WT and N348I RTs. When NVP (cyan sticks) is bound to RT, the access to the NNRTI binding pocket is completely obstructed by the surrounding residues in the WT enzyme (B) but not in N348I RT (C). The WT RT color scheme shown in A is as follows: fingers in blue, palm in red, thumb in green, connection in yellow, RNase H in orange, and p51 in brown. Figures were made using PyMOL.
DISCUSSION
Previous studies have shown that changes at regions distant to the RT active sites can affect enzymatic functions (45–53). Moreover, it has been shown that NNRTI resistance can be conferred by mutation on residues of the p51 subunit alone (54). The present study with subunit-specific N348I mutants provides new mechanistic insights into how remote mutations in either subunit affect enzyme activity and drug resistance. It also establishes that, in addition to providing structural integrity (55, 56), p51 is critical in fine tuning RT enzymatic activities.
Based on simple primer extension assays, it was previously reported that WT and N348I RTs have comparable polymerase activities (31). However, our in-depth transient kinetics analysis clearly demonstrated a significantly (>3-fold) decreased catalytic efficiency for p66N348I/p51N348I. This reduction in catalytic efficiency is consistent with previous reports by us and others that HIV-1N348I virus has a reduced replicative capacity (32, 33). Hence, N348I imparts a fitness cost to the virus, placing it in the special category of “desirable” mutations that would weaken the virus once acquired.
Interestingly, Ile-348p66 and Ile-348p51 each decreased catalytic efficiency, providing unexpected evidence that changes in p51 can affect the catalytic step of DNA synthesis without affecting dNTP binding (Table 1). We also found that Ile-348p66 and Ile-348p51 each enhanced processivity (Fig. 4 and Table 2). Consistent with this finding, both Ile-348p66 and Ile-348p51 slightly enhanced DNA binding affinity (Fig. 1 and Table 1). Mutation in either subunit may improve RT-nucleic acid interactions by changing the positioning and/or flexibility of enzyme regions that are in contact with DNA. Such changes may not significantly alter the Kd-DNA but could affect proper alignment of DNA with dNTP substrates or stability of the catalytic complex. The enhanced processivity of N348I RT may also contribute to NRTI resistance as it is possible that this property could provide more opportunities for unblocking chain-terminated primers or NNRTI dissociation (38, 39). The molecular details of these effects are not yet fully understood and should be addressed by structural studies of N348I RT.
Regarding the effect of N348I on RNase H function, we confirmed a previously reported defect of p66N348I/p51N348I in processing RNA/DNA (31, 39). Moreover, using subunit-specific mutants, we were able to demonstrate that the RNase H defect is due primarily to Ile-348p51 (Fig. 5), further expanding our understanding of how p51 contributes to multiple enzymatic functions.
Our reported increase in the NVP IC50 of p66N348I/p51N348I is consistent with values published elsewhere (2–3-fold increase) (31, 57). These in vitro changes are smaller than those observed in cell-based assays for viruses harboring the same mutation (typically 7–8-fold and up to 27-fold) (31–34). Such quantitative differences between enzymatic and cell-based estimates of drug resistance are not uncommon. For example, the excision-enhancing mutations cause up to 40-fold AZT resistance in cell-based assays compared with typically ∼2-fold changes observed in enzymatic assays depending on the sequence and length of the templates (longer templates provide more opportunities for inhibition) (17, 18, 58–60). During reverse transcription of the entire HIV genome in vivo, sequence-specific effects on inhibitor susceptibility may also contribute to stronger resistance. Importantly, the magnitude of N348I resistance to NVP in vitro mirrors the observed changes in Kd-NVP, indicating that the decreased binding of the inhibitor accounts for the observed resistance in vitro.
Our work clearly shows that the mechanism of N348I resistance to NVP involves a straightforward decrease in binding affinity for this inhibitor. Less clear is the role of RNase H in NVP resistance. Nikolenko et al. (38) proposed that the decrease in RNase H activity of CSMs preserves the RNA template and provides more time for NNRTIs to dissociate from the RT, resulting in the resumption of DNA synthesis and enhanced NNRTI resistance. Our data indicate that NVP resistance cannot always be linked to a defect in RNase H: we found N348I to be equally susceptible to NVP during extension of RNA/DNA and DNA/DNA template/primers (DNA/DNA is not a substrate for RNase H cleavage). Also, p66N348I/p51WT was resistant to NVP even though it did not have a significant RNase H defect. Hence, NVP resistance of CSMs is expected to be affected by RNase H-independent mechanisms and possibly by RNase-dependent mechanisms. It is also possible that resistance is affected by sequence-specific effects on inhibitor susceptibility (57).
Three broad classes of NNRTI resistance mechanisms have been proposed (for a review, see Ref. 56). (a) Resistance mutations, such as L100I and G190A, prevent NNRTI binding through direct steric interactions (55, 61). (b) Others, such as Y181C and Y188L, cause a loss of or change in the direct interactions of the inhibitor at the NNIBP. (c) K103N and K101E interfere with inhibitor entry into the pocket (27, 28). All three mechanisms involve residues at the NNRTI binding pocket. Because Ile-348p66 and Ile-348p51 are distant from the NNIBP, we can rule out any direct interactions with NVP. Hence, N348I differs from other NNRTI resistance mutations because it affects resistance to NVP through indirect structural effects that primarily decrease the association rate (kon-NVP) of the inhibitor (Table 4).
Some clues on the possible structural effects of this mutation are provided by the comparison of molecular models for WT and p66N348I/p51N348I enzyme complexes with NVP that revealed intriguing changes at the NNIBP. Although NVP appears fully enclosed in the WT NNIBP (Fig. 9B), it seems that changes at the floor of NNIBP in the p66N348I/p51N348I-NVP complex may be responsible for the observed decrease in NVP binding (Fig. 9, A, bottom left inset, and C). The details of these structural changes will be addressed in future crystallographic studies.
In conclusion, we have determined that N348I, a drug resistance mutation at the connection subdomain of RT, causes NVP resistance primarily by reducing the association rate of the inhibitor and thus decreasing its binding at the NNIBP. Moreover, this mutation significantly affects several enzymatic properties of RT, including catalytic turnover and efficiency, DNA binding and processivity of DNA synthesis, RNase H activity, and NNRTI resistance.
This work was supported, in whole or in part, by National Institutes of Health Grants AI076119 and AI074389 (to S. G. S.) and AI079801. This work was also supported by a grant from the Korea Food and Drug Administration and the Ministry of Knowledge and Economy, Bilateral International Collaborative Research and Development Program, Republic of Korea.
- NRTI
- nucleoside RT inhibitor
- NNRTI
- non-nucleoside RT inhibitor
- NVP
- nevirapine
- NNIBP
- NNRTI binding pocket
- AZT
- azidothymidine
- CSM
- connection subdomain mutation.
REFERENCES
- 1.De Clercq E. (2004) J. Clin. Virol. 30, 115–133 [DOI] [PubMed] [Google Scholar]
- 2.Parniak M. A., Sluis-Cremer N. (2000) Adv. Pharmacol. 49, 67–109 [DOI] [PubMed] [Google Scholar]
- 3.Ren J., Esnouf R., Garman E., Somers D., Ross C., Kirby I., Keeling J., Darby G., Jones Y., Stuart D., Stammers D. (1995) Nat. Struct. Biol. 2, 293–302 [DOI] [PubMed] [Google Scholar]
- 4.Ding J., Das K., Hsiou Y., Sarafianos S. G., Clark A. D., Jr., Jacobo-Molina A., Tantillo C., Hughes S. H., Arnold E. (1998) J. Mol. Biol. 284, 1095–1111 [DOI] [PubMed] [Google Scholar]
- 5.Kohlstaedt L. A., Wang J., Friedman J. M., Rice P. A., Steitz T. A. (1992) Science 256, 1783–1790 [DOI] [PubMed] [Google Scholar]
- 6.Rodgers D. W., Gamblin S. J., Harris B. A., Ray S., Culp J. S., Hellmig B., Woolf D. J., Debouck C., Harrison S. C. (1995) Proc. Natl. Acad. Sci. U.S.A. 92, 1222–1226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hsiou Y., Ding J., Das K., Clark A. D., Jr., Hughes S. H., Arnold E. (1996) Structure 4, 853–860 [DOI] [PubMed] [Google Scholar]
- 8.Spence R. A., Kati W. M., Anderson K. S., Johnson K. A. (1995) Science 267, 988–993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rittinger K., Divita G., Goody R. S. (1995) Proc. Natl. Acad. Sci. U.S.A. 92, 8046–8049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tantillo C., Ding J., Jacobo-Molina A., Nanni R. G., Boyer P. L., Hughes S. H., Pauwels R., Andries K., Janssen P. A., Arnold E. (1994) J. Mol. Biol. 243, 369–387 [DOI] [PubMed] [Google Scholar]
- 11.Sluis-Cremer N., Arion D., Parniak M. A. (2000) Cell. Mol. Life Sci. 57, 1408–1422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martín-Hernández A. M., Domingo E., Menéndez-Arias L. (1996) EMBO J. 15, 4434–4442 [PMC free article] [PubMed] [Google Scholar]
- 13.Gao G., Orlova M., Georgiadis M. M., Hendrickson W. A., Goff S. P. (1997) Proc. Natl. Acad. Sci. U.S.A. 94, 407–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gao H. Q., Boyer P. L., Sarafianos S. G., Arnold E., Hughes S. H. (2000) J. Mol. Biol. 300, 403–418 [DOI] [PubMed] [Google Scholar]
- 15.Sarafianos S. G., Das K., Clark A. D., Jr., Ding J., Boyer P. L., Hughes S. H., Arnold E. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 10027–10032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meyer P. R., Matsuura S. E., So A. G., Scott W. A. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 13471–13476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meyer P. R., Matsuura S. E., Mian A. M., So A. G., Scott W. A. (1999) Mol. Cell 4, 35–43 [DOI] [PubMed] [Google Scholar]
- 18.Meyer P. R., Matsuura S. E., Tolun A. A., Pfeifer I., So A. G., Mellors J. W., Scott W. A. (2002) Antimicrob. Agents Chemother. 46, 1540–1545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang H., Chopra R., Verdine G. L., Harrison S. C. (1998) Science 282, 1669–1675 [DOI] [PubMed] [Google Scholar]
- 20.Tuske S., Sarafianos S. G., Clark A. D., Jr., Ding J., Naeger L. K., White K. L., Miller M. D., Gibbs C. S., Boyer P. L., Clark P., Wang G., Gaffney B. L., Jones R. A., Jerina D. M., Hughes S. H., Arnold E. (2004) Nat. Struct. Mol. Biol. 11, 469–474 [DOI] [PubMed] [Google Scholar]
- 21.Das K., Bandwar R. P., White K. L., Feng J. Y., Sarafianos S. G., Tuske S., Tu X., Clark A. D., Jr., Boyer P. L., Hou X., Gaffney B. L., Jones R. A., Miller M. D., Hughes S. H., Arnold E. (2009) J. Biol. Chem. 284, 35092–35100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Das K., Ding J., Hsiou Y., Clark A. D., Jr., Moereels H., Koymans L., Andries K., Pauwels R., Janssen P. A., Boyer P. L., Clark P., Smith R. H., Jr., Kroeger Smith M. B., Michejda C. J., Hughes S. H., Arnold E. (1996) J. Mol. Biol. 264, 1085–1100 [DOI] [PubMed] [Google Scholar]
- 23.Ren J., Esnouf R., Hopkins A., Ross C., Jones Y., Stammers D., Stuart D. (1995) Structure 3, 915–926 [DOI] [PubMed] [Google Scholar]
- 24.Ren J., Nichols C., Bird L., Chamberlain P., Weaver K., Short S., Stuart D. I., Stammers D. K. (2001) J. Mol. Biol. 312, 795–805 [DOI] [PubMed] [Google Scholar]
- 25.Ren J., Nichols C. E., Chamberlain P. P., Weaver K. L., Short S. A., Stammers D. K. (2004) J. Mol. Biol. 336, 569–578 [DOI] [PubMed] [Google Scholar]
- 26.Sarafianos S. G., Das K., Hughes S. H., Arnold E. (2004) Curr. Opin. Struct. Biol. 14, 716–730 [DOI] [PubMed] [Google Scholar]
- 27.Hsiou Y., Ding J., Das K., Clark A. D., Jr., Boyer P. L., Lewi P., Janssen P. A., Kleim J. P., Rösner M., Hughes S. H., Arnold E. (2001) J. Mol. Biol. 309, 437–445 [DOI] [PubMed] [Google Scholar]
- 28.Ren J., Nichols C. E., Chamberlain P. P., Weaver K. L., Short S. A., Chan J. H., Kleim J. P., Stammers D. K. (2007) J. Med. Chem. 50, 2301–2309 [DOI] [PubMed] [Google Scholar]
- 29.Delviks-Frankenberry K. A., Nikolenko G. N., Boyer P. L., Hughes S. H., Coffin J. M., Jere A., Pathak V. K. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 10943–10948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nikolenko G. N., Delviks-Frankenberry K. A., Palmer S., Maldarelli F., Fivash M. J., Jr., Coffin J. M., Pathak V. K. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 317–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yap S. H., Sheen C. W., Fahey J., Zanin M., Tyssen D., Lima V. D., Wynhoven B., Kuiper M., Sluis-Cremer N., Harrigan P. R., Tachedjian G. (2007) PLoS Med. 4, e335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hachiya A., Kodama E. N., Sarafianos S. G., Schuckmann M. M., Sakagami Y., Matsuoka M., Takiguchi M., Gatanaga H., Oka S. (2008) J. Virol. 82, 3261–3270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gupta S., Fransen S., Paxinos E. E., Stawiski E., Huang W., Petropoulos C. J. (2010) Antimicrob. Agents Chemother. 54, 1973–1980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hachiya A., Shimane K., Sarafianos S. G., Kodama E. N., Sakagami Y., Negishi F., Koizumi H., Gatanaga H., Matsuoka M., Takiguchi M., Oka S. (2009) Antiviral Res. 82, 115–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.von Wyl V., Ehteshami M., Symons J., Bürgisser P., Nijhuis M., Demeter L. M., Yerly S., Böni J., Klimkait T., Schuurman R., Ledergerber B., Götte M., Günthard H. F. (2010) J. Infect. Dis. 201, 1054–1062 [DOI] [PubMed] [Google Scholar]
- 36.Radzio J., Yap S. H., Tachedjian G., Sluis-Cremer N. (2010) AIDS 24, 659–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sluis-Cremer N., Moore K., Radzio J., Sonza S., Tachedjian G. (2010) AIDS 24, 317–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nikolenko G. N., Delviks-Frankenberry K. A., Pathak V. K. (2010) J. Virol. 84, 5238–5249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ehteshami M., Beilhartz G. L., Scarth B. J., Tchesnokov E. P., McCormick S., Wynhoven B., Harrigan P. R., Götte M. (2008) J. Biol. Chem. 283, 22222–22232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Michailidis E., Marchand B., Kodama E. N., Singh K., Matsuoka M., Kirby K. A., Ryan E. M., Sawani A. M., Nagy E., Ashida N., Mitsuya H., Parniak M. A., Sarafianos S. G. (2009) J. Biol. Chem. 284, 35681–35691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Spence R. A., Anderson K. S., Johnson K. A. (1996) Biochemistry 35, 1054–1063 [DOI] [PubMed] [Google Scholar]
- 42.Patel S. S., Wong I., Johnson K. A. (1991) Biochemistry 30, 511–525 [DOI] [PubMed] [Google Scholar]
- 43.Götte M., Rausch J. W., Marchand B., Sarafianos S., Le Grice S. F. (2010) Biochim. Biophys. Acta 1804, 1202–1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang L. Z., Kenyon G. L., Johnson K. A. (2004) J. Biol. Chem. 279, 38424–38432 [DOI] [PubMed] [Google Scholar]
- 45.Ghosh M., Jacques P. S., Rodgers D. W., Ottman M., Darlix J. L., Le Grice S. F. (1996) Biochemistry 35, 8553–8562 [DOI] [PubMed] [Google Scholar]
- 46.Sarafianos S. G., Das K., Tantillo C., Clark A. D., Jr., Ding J., Whitcomb J. M., Boyer P. L., Hughes S. H., Arnold E. (2001) EMBO J. 20, 1449–1461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Julias J. G., McWilliams M. J., Sarafianos S. G., Arnold E., Hughes S. H. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 9515–9520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wisniewski M., Palaniappan C., Fu Z., Le Grice S. F., Fay P., Bambara R. A. (1999) J. Biol. Chem. 274, 28175–28184 [DOI] [PubMed] [Google Scholar]
- 49.Krebs R., Immendörfer U., Thrall S. H., Wöhrl B. M., Goody R. S. (1997) Biochemistry 36, 10292–10300 [DOI] [PubMed] [Google Scholar]
- 50.Powell M. D., Ghosh M., Jacques P. S., Howard K. J., Le Grice S. F., Levin J. G. (1997) J. Biol. Chem. 272, 13262–13269 [DOI] [PubMed] [Google Scholar]
- 51.Palaniappan C., Wisniewski M., Jacques P. S., Le Grice S. F., Fay P. J., Bambara R. A. (1997) J. Biol. Chem. 272, 11157–11164 [DOI] [PubMed] [Google Scholar]
- 52.Ghosh M., Williams J., Powell M. D., Levin J. G., Le Grice S. F. (1997) Biochemistry 36, 5758–5768 [DOI] [PubMed] [Google Scholar]
- 53.Jacques P. S., Wöhrl B. M., Ottmann M., Darlix J. L., Le Grice S. F. (1994) J. Biol. Chem. 269, 26472–26478 [PubMed] [Google Scholar]
- 54.Boyer P. L., Ding J., Arnold E., Hughes S. H. (1994) Antimicrob. Agents Chemother. 38, 1909–1914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sarafianos S. G., Marchand B., Das K., Himmel D. M., Parniak M. A., Hughes S. H., Arnold E. (2009) J. Mol. Biol. 385, 693–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Singh K., Marchand B., Kirby K. A., Michailidis E., Sarafianos S. G. (2010) Viruses 2, 606–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Biondi M. J., Beilhartz G. L., McCormick S., Götte M. (2010) J. Biol. Chem. 285, 26966–26975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Quan Y., Liang C., Inouye P., Wainberg M. A. (1998) Nucleic Acids Res. 26, 5692–5698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Meyer P. R., Matsuura S. E., Zonarich D., Chopra R. R., Pendarvis E., Bazmi H. Z., Mellors J. W., Scott W. A. (2003) J. Virol. 77, 6127–6137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Girouard M., Diallo K., Marchand B., McCormick S., Götte M. (2003) J. Biol. Chem. 278, 34403–34410 [DOI] [PubMed] [Google Scholar]
- 61.Menéndez M. A., Pérez de Oteyza C. (2001) Enferm. Infecc. Microbiol. Clin. 19, 287–290 [DOI] [PubMed] [Google Scholar]