

Abstract
A new complex salt of composition [Co(phen)3]3(V4O12)2Cl·27H2O (phen = 1,10-phenanthroline and [V4O12]4- = tetrameric dodecaoxotetravanadate ion) was synthesized by reacting appropriate salts in aqueous medium. The complex salt has been characterized by elemental analyses, thermogravimetric analysis (TGA), cyclic voltammetry (CV), FT-IR and UV/Vis spectroscopies, solubility product and conductance measurements. Single crystal X-ray structure determination revealed ionic structure consisting of three complex cations, [Co(phen)3]3+, two [V4O12]4- anions, one chloride and twenty seven lattice waters. Detailed structural and spectroscopic analyses of [Co(phen)3]3(V4O12)2Cl·27H2O show that the large anion is stabilized by the large cationic metal complex as there is preferred shape compatibility that leads to a large number of lattice stabilizing non-covalent interactions.
Keywords: Cobalt(III); Second sphere coordination chemistry; 1,10-Phenanthroline; Polyoxovanadate; X-ray crystallography
1. Introduction
Anions are essential to life with roles in biological processes, industry and agriculture [1-9]. Some anionic species exit only in solution but cannot be isolated as solid salts due to solvent stabilization or lattice inhibition. This lattice inhibition can be removed by selection of suitable counter ions. According to D.H. McDaniel, the counter ion affects the formation of complex in a number of ways like lattice dominating, lattice energy limiting, insulating effects, polarization effect, shape and size [10]. Solid metal complexes are stabilized by large counter ions, preferably ions of the same but opposite charge. The importance of shape and size of cation in precipitating the desired complexes is well illustrated by halocuprate and iodoplatinate(II) salts [10, 11]. Counter ions which can have non-covalent interactions with anions in addition to the electrostatic interaction are useful in synthesis of new and unusual anionic species like [Hg2(SCN)7]3- and [HgBr5]3- [12, 13]. In these complexes, in addition to the large counter anion, non-covalent interaction like N-H…X play crucial roles in lattice stabilization.
Anionic species containing polyoxotransition metals have been studied extensively due to their varied geometries and oxidation states. Vanadium, in the form of organic–inorganic metal hybrid vanadium oxides or complexes with polyoxovanadate have shown ability to adopt a variety of coordination geometries and oxidation states [14] with potential utilization in catalysis and materials. These vanadates are good electron acceptors, electron relay stations [15] and some are used in photolytic water splitting [16, 17]. The formation of polyoxovanadates depends upon coordination preferences of the transition metal used and geometrical constraints of the polydentate ligands. Anhydrous alkali metal metavanadates are composed of infinite chains of corner sharing VO4 tetrahedra in MVO3 (M = NH4, K, Rb, Cs); KVO3·H2O and Ca(VO3)2 [18] contain chains of edge-linked VO5 units while forming M2V2O6 with metals like Mg, Zn, Pb, Cd and Cu [19-23]. For transition metals with large organic ligands, oxovanadates V4O124- have been reported, [Co4O4(dpaH)4(CH3COO)2]2·V4O12·5H2O [24] and [MII(phen)3]2V4O12·phen·22H2O (MII = Co, Ni) [25]. The majority of complexes containing polyoxovanadates reported have been prepared under isothermal conditions except a few like Ni(C8H22N4)](VO3)2 and [MII(phen)3]2V4O12·phen·22H2O (MII = Co, Ni). In the present study we have utilized cationic tris(1,10-phenanthroline)cobalt(III) complex for isolation of peroxovanadate (V4O12)4- as there is no report in the literature of stabilization of [V4O12]4- by trivalent or complexed metal ion. It is also envisaged that oxygens bonded to vanadium(V) will form hydrogen bonds (through second sphere interactions) with C-H groups originating from 1,10-phenanthroline coordinated to cobalt(III) to generate an intricate network that would stabilize the crystal lattice. This study reports synthesis, spectroscopic and X-ray structural study of [Co(phen)3]3(V4O12)2Cl·27H2O, the first report of a stabilization of rare [V4O12]4- by [Co(phen)3]3+.
2. Experimental
2.1. Materials
Analytical grade reagents were used without purification. [Co(phen)3]Cl3 was prepared by reacting [Co(NH3)5Cl]Cl2 and 1,10-phenanthroline monohydrate according to the method described in the literature [26].
2.2. Instruments and measurements
C, H and N were estimated microanalytically by automatic PERKIN ELMER 2400 CHN elemental analyzer. Cobalt was estimated by volumetric method [27]. UV/Visible spectra were recorded in water using a HITACHI 330 spectrophotometer. Infrared spectrum was recorded using a PERKIN ELMER spectrum RX FT-IR system using a KBr pellet. Conductance measurements were performed on a Pico Conductivity Meter (Model CNO4091201, Lab India) in aqueous medium at 25 °C using doubly-distilled water.
Thermogravimetric analysis (TGA) was conducted with a Mettler Toledo TGA/SDTA 851 instrument calibrated using high purity indium and high purity zinc standards. The experiment was conducted under nitrogen flow between 308 and 873 K and under air flow between 873 and 1173 K. The flow was maintained at 70 ml min-1. The specimen, about 10 mg, contained in a 70 μl alumina pan was carried from 308 to 1173 K at variable heating rate: the heating rate was 1 K min-1 when the weight loss was higher than 2 μg s-1, and 20 K min-1 when the weight loss was less than 1 μg s-1.
Electrochemical measurements were conducted on a potentiostat (CH Instuments 440a) with a glass encased 3-mm diameter glassy carbon electrode (Cypress Systems), Pt wire, and Ag/AgCl (3 M KCl) as the working, auxiliary, and reference electrodes, respectively. The working electrode was polished on a polishing pad with 0.05 μm alumina powder, rinsed and sonicated prior to use. All solutions were bubbled with N2 for 5 min to remove O2 prior to scanning.
2.3. Synthesis of [Co(phen)3]3(V4O12)2Cl·27H2O
Tris(1,10-phenanthroline)cobalt(III) chloride (0.5 g, 0.7 mmol) was dissolved in water (10 mL) and in another beaker (0.248 g, 2.1 mmol) ammonium metavanadate was dissolved in water (30 mL). Upon mixing the two solutions, no color change or precipitation occurred. When it was allowed to evaporate slowly at room temperature, yellow single crystals appearing after a few hours were allowed to grow. The crystals were separated and dried in air. The yield was 0.487 g (66.25%). The composition was established by elemental analyses. [Co(phen)3]3(V4O12)2Cl·27H2O Found (%): C, 41.57; H, 4.02; N, 8.05; Co, 5.67; Cl, 1.09; Calculated (%): C, 41.67; H, 4.05; N, 8.10; Co, 5.70; Cl, 1.14.
2.4. X-ray structure determination
Single crystal X-ray diffraction data for the complex salt were collected on a Nonius Kappa CCD diffractometer using graphite-monochromated MoKα radiation (λ = 0.71069 Ǻ). All intensities were corrected for Lorentz, polarization and adsorption [28]. The structure was solved by direct methods with the SIR97 program [29] and refined by block matrix least squares (four blocks) using the SHELXL-97 program [30]. Non-hydrogen atoms were refined anisotropically. Hydrogens of phenanthroline were given calculated positions, while those belonging to water were not included in the refinement. All other calculations were performed using WinGX [31] and PARST [32]. Final R-values together with selected refinement details are given in table 1.
Table 1.
Crystal data and refinement parameters of [Co(phen)3]3(V4O12)2Cl·27H2O
| Chemical formula | 3(CoC36H24N6)·2(V4O12)·Cl·27(H2O) |
| Mr | 3112.03 |
| Cell setting, space group | Triclinic, P-1 |
| Temperature (K) | 295 |
| a, b, c (Å) | 15.4063(2), 18.8746(3), 25.1206(4) |
| α, β, γ (°) | 97.7540(8), 103.5560(7), 101.4310(7) |
| V (Å3) | 6833.14(18) |
| Z | 2 |
| Dx (Mg m−3) | 1.746 |
| Radiation type | Mo Kα |
| μ (mm−1) | 0.99 |
| Crystal form, color | Prismatic, orange |
| Crystal size (mm) | 0.29 × 0.14 × 0.09 |
| No. of measured, independent and observed reflections | 42624, 28163, 10204 |
| Criterion for observed reflections | I > 2σ(I) |
| θmax (°) | 27.0 |
| Refinement on | F2 |
| R[F2 > 2σ(F2)], wR(F2), S | 0.101, 0.361, 0.98 |
| No. of reflections / No. of parameters | 28163 / 1776 |
| Weighting scheme | Calculated w = 1/[σ2(Fo2) + (0.1928P)2], where P = (Fo2 + 2Fc2)/3 |
| Δρmax, Δρmin (e Å−3) | 0.99, -0.69 |
3. Results and discussion
3.1. Synthesis
The complex salt, [Co(phen)3]3(V4O12)2Cl·27H2O was obtained by reaction of tris(1,10-phenanthroline)cobalt(III) chloride and ammonium metavanadate in water in 1:3 molar ratio (scheme 1); metavanadate tetramerizes to form [V4O12]4- in the presence of [Co(phen)3]3+.
Scheme 1.
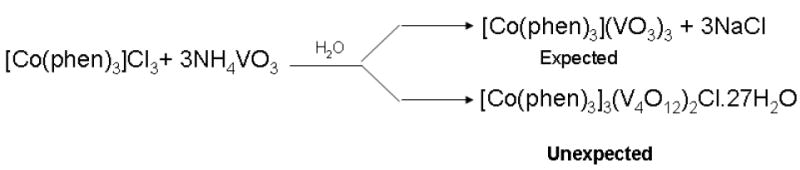
Schematic representation of chemical reaction.
The newly formed complex salt has been characterized by elemental analyses, TGA, cyclic voltammetry studies, spectroscopies (UV/Visible and FT-IR) and conductance measurements. The presence of one chloride ion was confirmed by its gravimetric analysis using AgNO3 solution. The complex salt is sparingly soluble in water and other common solvents. The crystal structure has been unambiguously established by single crystal X-ray crystallography.
3.2. Thermogravimetric analysis
Thermogravimetric analysis of the complex salt showed five weight loss steps. The first and second steps in the TGA curve correspond to weight loss of 12.8% and 2.8% at 363 K and 411 K, respectively (Supplementary Material), which can be ascribed to loss of free water (calculated weight loss was 15.62% and observed weight loss was 15.60% in the first two steps). The third and fourth weight loss of 5.3 and 4.3%, respectively, take place between 425 and 873 K, corresponding to pyrolysis of organic matter. The fifth weight loss (41.3%) corresponds to oxidation of combustible material at 873 K, when the atmosphere changed from nitrogen to air. In the pan, at the end of analysis, 33.5% of ash remains.
3.3. Cyclic voltammetry studies
The electrochemical properties of [Co(phen)3]3+ have been studied [33], with CV reported to have an Epc and Epa of 0.114 and 0.179 V vs. Ag/AgCl, respectively. CV of [Co(phen)3]3(V4O12)2Cl·27H2O showed a similar CV response at a glassy carbon electrode in 0.1 M KCl aqueous solution with a scan rate of 100 mV/s. A CV of the chloride salt showed similar behavior. [Co(phen)3]3+ undergoes a one-electron reduction to the 2+ form with a peak potential (Epc) of 0.143 V vs. Ag/AgCl as shown in Supplementary Material. On the reverse scan, Co(phen)32+ is reoxidized at Epa of 0.215 V. The peak potential separation (ΔE) of 72 mV indicates that this is the reversible one electron couple mentioned earlier. No electro-activity was seen for V5+ species in the potential range studied (0.6 to -0.9 V), even though the standard reduction potential (E0) of VO2+ to V3+ species is 0.115 V vs. Ag/AgCl. The ratio of Ipa/Ipc was close to 1 (0.988), further indicating that only the reversible couple contributes to Ipc as the irreversible reduction of V4+ species would be expected to contribute to Ipc.
3.4. Measurement of solubility product
The solubility product (Ksp) of the complex salt determined in water at 25 °C was 8.08 × 10-22 as compared to that of [Co(phen)3]Cl3 (2.7 × 10-3), showing affinity of the complex cation is greater for metavanadate than chloride. This increase in affinity may be due to the increased interactions between cations and anions.
3.5. Molar conductance measurements
Molar conductance of complex salt was measured in water in the concentration range 0-100 × 10-4 M at 25 °C. The limiting molar conductance at infinite dilution (Λo) was obtained by plotting the square root of concentration versus molar conductance, when extrapolated to zero. The Λo obtained is 882 Scm2mol-1. The high value of molar conductance revealed that the complex salt consists of a large number of ions and unusual composition of complex salt.
3.6. Spectroscopic characterization
Vibrational spectrum of newly synthesized complex has been recorded in the region 400-4000 cm-1. The peak assignments have been made in consultation with literature values [34-36]. IR spectral bands at 1634-1582, 3340 and 488 cm-1 were assigned to ν(C=C)/ν(C=N), ν(O-H) of H2O and ν(Co-N). These peaks are characteristic for 1,10-phenanthroline attached to cobalt(III) and water of crystallization. The IR spectrum of individual polyoxovanadate is characteristic due to the relatively rigid structure of V4O124- ion. IR bands at 942 and 905 cm-1 were assigned to symmetrical and asymmetric vibrations of V-Ot (terminal oxygens) while bands at 815 and 640 cm-1 were assigned to asymmetrical and symmetric vibrations of the bridging V-Ob-V. Similar IR bands have been reported for [Co(phen)3]2V4O12·phen·22H2O and [Ni(phen)3]2V4O12·phen·22H2O [25]. For [Co4O4(dpaH)4(CH3COO)2]2V4O12·5H2O [24] bands due to V-O, V-O-V were reported at 950-770 cm-1.
For Co(III) low spin complexes, it is possible to observe d-d transitions in the visible region because transitions corresponding to t2g6 → t2g5 eg1 with promoted electron maintaining its spin are possible. Therefore, two electronic transitions 1A1g → 1T1g and 1A1g → 1T2g are reported for familiar orange–yellow color for coordination compounds containing Co(III) which were observed around 340 and 470 nm. 1,10-Phenanthroline is also coordinated to Co(III) and transitions around 224, 264, 290 nm arise from phen [34, 37, 38]. The electronic spectrum of the title complex salt in water show absorption maxima (λmax) at 219, 273, 280, 349 and 456 nm, close to reported values.
3.7. Description of the structure
3.7.1. Coordination geometry and bonding
The asymmetric unit is large, being constituted by three [Co(phen)3]3+, two dodecaoxotetravanadates, one chloride and 27 co-crystallized waters. The high water content is confirmed by TGA analysis, as reported above. The chloride is disordered over two equivalent positions, as well as a number of co-crystallized solvent molecules. A selection of relevant bond distances and angles is reported in table 2. The geometry around the three cobalts is slightly distorted octahedral with N-Co-N angles involving nitrogen of the same 1,10-phenanthroline being < 90°. The Co-N distances are consistent with those found in recently reviewed Co(III)-1,10-phenanthroline complexes [17-21]. The oxidation states of Co have been checked in the frame of the bond valence sum method [39] using the Co-N parameters reported in the byparm2009.cif table [40]. The calculated values are 3.5, 3.6 and 3.4 for Co1, Co2 and Co3, respectively. V-O distances are similar to the mean bond lengths obtained from a CSD search on compounds containing the oxovanadate anion, 1.779(6) and 1.632(6) Å for exocyclic and endocyclic bonds, respectively (values calculated on 16 hits). The eight-membered vanadate rings are almost planar, as shown by the total puckering amplitude index 0.16(9), according to Cremer & Pople [41]) and by values of the endocyclic torsion angles, ranging from 0.7 to 38°. ORTEPIII [42] views of one cobalt complex and one oxovanadate are shown in figure 1.
Table 2.
Selected bond lengths and angles (Å and °) for [Co(phen)3]3(V4O12)2Cl·27H2O.
| Co1-N1 | 1.931(8) | Co2-N10 | 1.950(9) |
| Co1-N2 | 1.936(8) | Co2-N11 | 1.938(9) |
| Co1-N3 | 1.952(8) | Co2-N12 | 1.922(8) |
| Co1-N4 | 1.960(9) | Co3-N13 | 1.951(9) |
| Co1-N5 | 1.959(9) | Co3-N14 | 1.955(9) |
| Co1-N6 | 1.935(8) | Co3-N15 | 1.937(9) |
| Co2-N7 | 1.932(9) | Co3-N16 | 1.961(9) |
| Co2-N8 | 1.954(9) | Co3-N17 | 1.955(9) |
| Co2-N9 | 1.930(8) | Co3-N18 | 1.971(9) |
| V1-O1 | 1.642(10) | V4-O12 | 1.604(7) |
| V1-O2 | 1.769(8) | V5-O13 | 1.623(8) |
| V1-O3 | 1.767(10) | V5-O14 | 1.628(10) |
| V1-O4 | 1.600(9) | V5-O15 | 1.774(8) |
| V2-O2 | 1.774(9) | V5-O16 | 1.786(11) |
| V2-O5 | 1.630(10) | V6-O16 | 1.762(11) |
| V2-O6 | 1.773(10) | V6-O17 | 1.643(9) |
| V2-O7 | 1.616(7) | V6-O18 | 1.624(8) |
| V4-O3 | 1.765(11) | V10-O19 | 1.599(12) |
| V3-O6 | 1.773(11) | V10-O20 | 1.773(10) |
| V3-O8 | 1.651(10) | V10-O21 | 1.731(1) |
| V3-O9 | 1.758(8) | V10-O24 | 1.619(12) |
| V3-O10 | 1.630(9) | V11-O20 | 1.758(10) |
| V4-O9 | 1.786(8) | V11-O22 | 1.628(12) |
| V4-O11 | 1.622(10) | V11-O23 | 1.595(11) |
| N1-Co1-N2 | 84.6(3) | N8-Co2-N12 | 87.9(4) |
| N1-Co1-N3 | 94.9(3) | N9-Co2-N10 | 84.2(4) |
| N1-Co1-N5 | 94.2(4) | N9-Co2-N11 | 91.5(4) |
| N1-Co1-N6 | 87.8(3) | N10-Co2-N11 | 88.9(4) |
| N2-Co1-N3 | 89.7(3) | N10-Co2-N12 | 93.7(4) |
| N2-Co1-N4 | 93.1(3) | N11-Co2-N12 | 85.1(4) |
| N2-Co1-N6 | 93.6(4) | N13-Co3-N14 | 84.1(3) |
| N3-Co1-N4 | 84.8(4) | N13-Co3-N16 | 92.5(4) |
| N3-Co1-N5 | 92.1(4) | N13-Co3-N17 | 94.4(3) |
| N4-Co1-N5 | 88.0(4) | N13-Co3-N18 | 89.7(3) |
| N4-Co1-N6 | 92.7(4) | N14-Co3-N15 | 92.6(4) |
| N5-Co1-N6 | 84.6(4) | N14-Co3-N16 | 88.9(4) |
| N7-Co2-N8 | 85.1(4) | N14-Co3-N18 | 93.6(4) |
| N7-Co2-N9 | 89.2(4) | N15-Co3-N16 | 83.9(4) |
| N7-Co2-N10 | 93.5(4) | N15-Co3-N17 | 89.1(4) |
| N7-Co2-N12 | 94.5(4) | N15-Co3-N18 | 94.0(4) |
| N8-Co2-N9 | 94.2(4) | N16-Co3-N17 | 94.1(4) |
| N8-Co2-N11 | 92.8(4) | N17-Co3-N18 | 83.4(4) |
Figure 1.
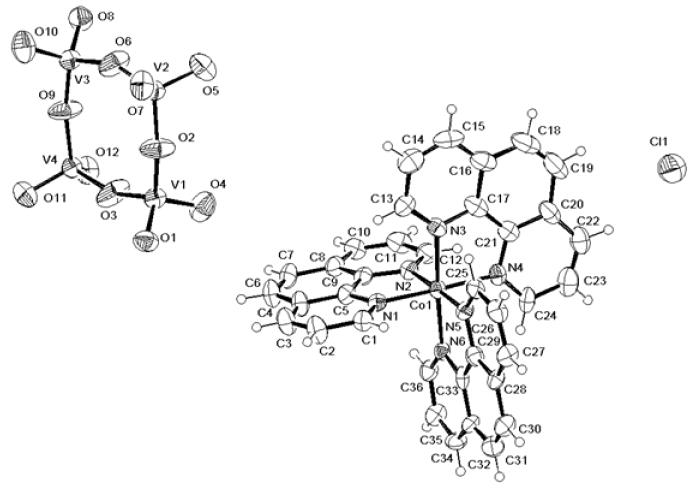
ORTEPIII view of one cobalt complex cation, one polyoxovanadate anion and one chloride. Ellipsoids are drawn at 40% probability. For the sake of clarity, the other ions and all lattice water molecules constituting the asymmetric unit are omitted.
3.7.2. Packing
The crystal packing, due to the high number of water molecules involved, is quite complicated. Table 3 reports short O⋯O contacts inside the crystal lattice, ranging from 2.55(3) to 2.98(3) Å, which are typical for hydrogen bonding of medium strength. In addition, phen C–H groups were involved in C–H⋯O/Cl type interactions [43-46] (as many as thirty two significant hydrogen bonds have been observed which are given in table 4) defining the second-sphere coordination around the three cobalt complex cations. Three complex cations with C-H…O contacts are shown in figure 2. These C-H…O interactions (H…A distance ranging from 2.33-2.69 Å and angles A…H-D ranging from 132-160°) are of comparable strength to commonly employed N-H…O interaction (H…A distance ranging from 2.08-2.76 Å angles A…H-D ranging from 130-179°) [47] in anion binding studies. These C-H…O interactions play a crucial role in binding [V4O12]4- as nineteen C-H…O interactions are between oxygens originating from [V4O12]4- and C-H of 1,10-phenanthroline. No significant π-π interactions have been found, since the centroids of the stacked phenanthroline moieties are located more than 3.8 Å apart. The packing diagram of the complex salt is shown in figure 3.
Table 3.
Short O⋯O/Cl contacts (Å) for [Co(phen)3]3(V4O12)2Cl·27H2O.
| O14…O19W | 2.768(17) | O8W…O22W | 2.824(23) |
| O17…O5W | 2.900(13) | O9W…O16W | 2.755(16) |
| O17…O10W | 2.775(15) | O9W…O17W | 2.798(20) |
| O18…O27W | 2.679(21) | O9W…O21W | 2.844(21) |
| O24…O31W | 2.818(22) | O10W…O16W | 2.940(18) |
| O1W…O15W | 2.892(16) | O16W…O27W | 2.730(28) |
| O2W…O4W | 2.777(15) | O18W…O20W | 2.799(16) |
| O2W…O6W | 2.868(16) | O18W…O32W | 2.870(32) |
| O2W…O14W | 2.787(20) | O19W…O20W | 2.822(22) |
| O3W…O5W | 2.698(13) | O26W…O29W | 2.821(33) |
| O5W…O19W | 2.780(19) | O26W…O31W | 2.785(35) |
| O6W…O18W | 2.816(17) | O27W…O34W | 2.912(37) |
| O8W…O21W | 2.897(18) | ||
| O4…O24Wi | 2.737(18) | O33W…O22Wiv | 2.554(27) |
| O23W…O23i | 2.728(26) | O3W…O22v | 2.693(15) |
| O1W…O11ii | 2.735(12) | O28W…O30Wvi | 2.778(40) |
| O8W…O8ii | 2.776(14) | O23W…O11vi | 2.810(23) |
| O15W…O10ii | 2.690(16) | O25W…O32Wvi | 2.732(34) |
| O16W…O8ii | 2.747(17) | O14W…O5vii | 2.748(17) |
| O22W…O5ii | 2.742(20) | O28W…O23viii | 2.693(20) |
| O1W…O24iii | 2.738(16) | O32W…O25Wix | 2.732(34) |
| O6W…Cl1iii | 2.781(13) | O1…O4Wx | 2.782(15) |
| O15W…O26Wiii | 2.801(23) | O4W…O24Wxi | 2.815(23) |
| O17W…O14iii | 2.790(14) | O29W…O18Wxi | 2.989(25) |
| O25W…O22Wiv | 2.984(26) | O30W…O19xi | 2.841(28) |
Symmetry codes: 1-x,-y,-z;
x,y+1,z+1;
1-x,-y,1-z;
x+1,y,z;
x,y,1+z;
x,y+1,z;
x+1,y,z+1;
x+1,y+1,z+1;
x,y-1,z;
1-x,-y-1,1-z;
2-x,-y,1-z
Table 4.
Hydrogen bonding parameters for C–H…O/Cl contacts (Å and °).
| D–H …A | D–H | D…A | H…A | D–H…A |
|---|---|---|---|---|
| C11-H7…O21 | 0.930 | 3.55(1) | 2.67 | 158 |
| C22-H14…Cl1 | 0.930 | 3.54(2) | 2.66 | 156 |
| C43-H29…O7 | 0.930 | 3.23(2) | 2.55 | 130 |
| C73-H49…O15 | 0.930 | 3.15(1) | 2.45 | 133 |
| C85-H57…O33W | 0.930 | 3.24(3) | 2.51 | 136 |
| C102-H68…O18 | 0.930 | 3.38(2) | 2.69 | 132 |
| C51-H35…O3i | 0.930 | 3.24(1) | 2.39 | 151 |
| C54-H36…O12i | 0.930 | 3.51(1) | 2.63 | 158 |
| C1-H1…O2i | 0.930 | 3.03(1) | 2.33 | 132 |
| C30-H20…O10i | 0.930 | 3.36(1) | 2.60 | 139 |
| C7-H5…O12ii | 0.930 | 3.20(1) | 2.45 | 138 |
| C12-H8…O22iii | 0.930 | 3.28(1) | 2.51 | 141 |
| C13-H9…O24Wiii | 0.930 | 3.36(2) | 2.67 | 131 |
| C63-H43…O5Wiv | 0.930 | 3.50(2) | 2.67 | 149 |
| C49-H33…O2Wiv | 0.930 | 3.26(2) | 2.47 | 143 |
| C25-H17…Cl1v | 0.930 | 3.30(1) | 2.54 | 138 |
| C31-H21…O13vi | 0.930 | 3.41(1) | 2.57 | 150 |
| C36-H24…Cl2vi | 0.930 | 3.28(1) | 2.49 | 144 |
| C34-H22…O16vi | 0.930 | 3.25(1) | 2.48 | 139 |
| C97-H65…O5Wvi | 0.930 | 3.26(1) | 2.51 | 136 |
| C62-H42…O22vii | 0.930 | 3.20(2) | 2.38 | 147 |
| C37-H25…O9vii | 0.930 | 3.19(1) | 2.47 | 134 |
| C55-H37…O4vii | 0.930 | 3.38(2) | 2.66 | 135 |
| C57-H38…O4vii | 0.930 | 3.40(2) | 2.68 | 135 |
| C59-H40…O8Wviii | 0.930 | 3.40(1) | 2.65 | 139 |
| C79-H53…O13ix | 0.930 | 3.25(2) | 2.52 | 136 |
| C91-H61…O28Wx | 0.930 | 3.42(3) | 2.58 | 149 |
| C84-H56…O25Wx | 0.930 | 3.39(2) | 2.61 | 142 |
| C87-H59…O8Wxi | 0.930 | 3.28(2) | 2.39 | 158 |
| C106-H70…O6xii | 0.930 | 3.30(1) | 2.41 | 160 |
| C103-H69…O7xii | 0.930 | 3.46(1) | 2.58 | 158 |
| C108-H72…O9Wxiii | 0.930 | 3.29(2) | 2.54 | 138 |
Symmetry codes : -x,-y-1,-z;
1-x,-y-1,-z;
1-x,-y,-z;
x-1,y,z-1;
-x,-y,-z;
1-x,-y,1-z;
x-1,y,z;
x-1,y-1,z-1;
2-x,-y,1-z;
2-x,1-y,1-z;
1-x,1-y,1-z;
x+1,y+1,z+1;
x+1,y,z
Figure 2.
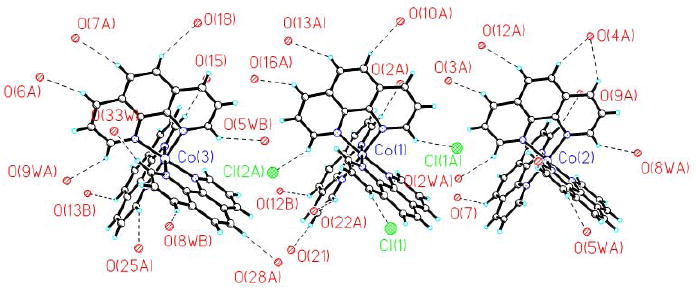
C-H…O interactions between complex cations and hydrogen bond acceptor groups from anions and lattice waters.
Figure 3.
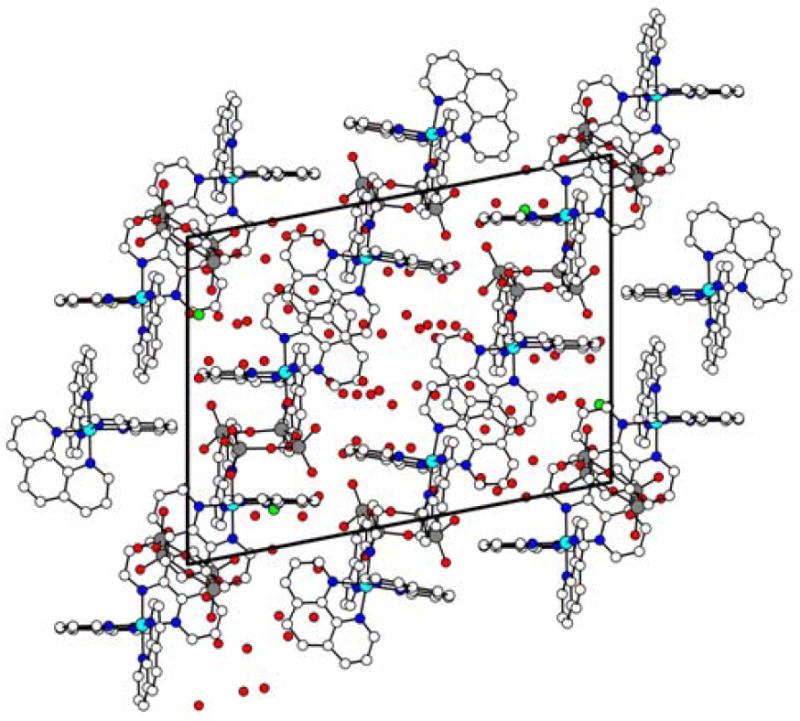
Packing diagram of the complex salt (viewed along a).
All related structures reported are characterized by high content of co-crystallized solvent with the number of waters ranging from 5 to 22. In particular, the structure of the Co(II) salt contains, besides 22 water molecules, a free co-crystallized phenanthroline. A possible explanation is that two cumbersome objects (the cation and the anion), very different in shape, when put together in the crystal lattice leaves large empty spaces that have to be occupied by the solvent of crystallization to assure efficient packing and thereby increasing stability of crystal lattice. Thus, O⋯O/Cl short contacts and C-H⋯O/Cl hydrogen bonds are driving forces for stabilization of the crystal lattice and formation of the complex salt.
4. Conclusions
A new complex salt [Co(phen)3]3(V4O12)2Cl·27H2O has been synthesized and characterized by physico-chemical, spectroscopic and X-ray structural studies. Single crystal X-ray structure determination revealed an ionic structure consisting of three complex cations, two V4O124- anions, one chloride and twenty seven lattice waters. The crystal lattice is stabilized by hydrogen bonding, C-H⋯O/Cl non-covalent interactions in addition to the electrostatic forces. Detailed structural and spectroscopic analyses of [Co(phen)3]3(V4O12)2Cl·27H2O show the large anion [V4O12]4- was stabilized by the large [Co(phen)3]3+.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge the financial support of CSIR vide grant no- 01(2020) 2005/ EMR-II, the US National Science Foundation (CHE-0516928), and the US National Institute of Health (1R01DK078652-01A2).
Footnotes
Supplementary data Crystallographic data have been deposited with the Cambridge Crystallographic Data Center as supplementary publication CCDC 733583. Copies of the data can be obtained free of charge on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK. Fax: +44 1223 336033; deposit@ccdc.cam.ac.uk.
References
- 1.Vieira L, Lavan A, Dragger F, Cabantchik ZI. J Biol Chem. 1994;269:16254. [PubMed] [Google Scholar]
- 2.Beer PD, Gale PA. Angew Chem Int Ed. 2001;40:486. [PubMed] [Google Scholar]
- 3.Bossi E, Giovannardi S, Binda F, Forlani G, Peres A. J Physiol. 2002;541:343. doi: 10.1113/jphysiol.2001.013457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stibor I, Zlatušková P. Top Curr Chem. 2005;255:31. [Google Scholar]
- 5.Kubik S, Goddard R, Otto S, Pohl S, Reyheller C, Stüwe S. Biosens Bioelectron. 2005;20:2364. doi: 10.1016/j.bios.2004.01.035. [DOI] [PubMed] [Google Scholar]
- 6.Davis F, Collyer SD, Higson SP. Top Curr Chem. 2005;255:97. [Google Scholar]
- 7.Gale P. Coord Chem Rev. 2006;250:2917. [Google Scholar]
- 8.Goel A, Brennan N, Brady N, Kenny PTM. Biosens Bioelectron. 2007;22:2047. doi: 10.1016/j.bios.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 9.Briza T, Kejik Z, Cisaova I, Kralova J, Martasek P, Karl V. Chem Commun. 2008:1901. doi: 10.1039/b718492a. [DOI] [PubMed] [Google Scholar]
- 10.McDaniel DH. Annual Reports in Inorganic and General Synthesis. 1972:1. [Google Scholar]
- 11.Basolo F. Coord Chem Rev. 1968;3:213. [Google Scholar]
- 12.Bala R, Sharma RP, Sharma R, Kariuki BM. Inorg Chem Commun. 2006;9:852. [Google Scholar]
- 13.Sharma RP, Sharma R, Kumar A, Venugopalan P, Brando P, Felix V. Inorg Chem Commun. 2009;12:945. [Google Scholar]
- 14.Li GH, Shi Z, Xu YH, Feng SH. Inorg Chem. 2003;34:1170. doi: 10.1021/ic025808u. [DOI] [PubMed] [Google Scholar]
- 15.Coronado E, Gomez-Garcia CJ. Chem Rev. 1998;98:273. doi: 10.1021/cr970471c. [DOI] [PubMed] [Google Scholar]
- 16.Kudo A, Omori K, Kato H. J Am Chem Soc. 1999;121:11459. [Google Scholar]
- 17.Ye J, Zou Z, Arakawa H, Oshikiri M, Shimoda M, Matsushita A, Shishido T. J Photochem Photobiol. 2002;A148:79. [Google Scholar]
- 18.Evans HT., Jr Z Kristallogr. 1960;114:257. [Google Scholar]
- 19.Bouloux J-C, Galy J. Bull Soc Chim Fr. 1969:739. [Google Scholar]
- 20.Ng HN, Calvo C. Can J Chem. 1972;50:3619. [Google Scholar]
- 21.Calvo C, Manolescu D. Acta Cryst. 1973;B29:1743. [Google Scholar]
- 22.Bouloux J-C, Prez G, Galy J. Bull Soc Fr Miner Cris t. 1972;95:130. [Google Scholar]
- 23.Angenault J, Rimsky A, Acad CR. Sci Peris Ser C. 1968;267:227. [Google Scholar]
- 24.Zhang X, You W, Zhu Z, Dang L, Sun Z, Zheng X. Inorg Chem Commun. 2006;9:526. [Google Scholar]
- 25.Zurkova L, Kucsera R, Gyepes R, Sivak M. Monatsh Chem. 2003;134:1071. [Google Scholar]
- 26.Pfeiffer P, Werdelmann Br. Z Anorg Allg Chem. 1950;31:263. [Google Scholar]
- 27.Schlessinger GG. Inorg Synth. 1967;9:160. [Google Scholar]
- 28.Blessing RH. Acta Cryst. 1995;A51:339. doi: 10.1107/s0108767394005726. [DOI] [PubMed] [Google Scholar]
- 29.Altomare A, Burla MC, Camalli M, Cascarano GL, Giacovazzo C, Guagliardi A, Moliterni AGG, Polidori G, Spagna R. J Appl Crystallogr. 1999;32:115. [Google Scholar]
- 30.Sheldrick GM. SHELX97 Program for Crystal Structure Refinement. University of Göttingen; Germany: 1997. [Google Scholar]
- 31.Farrugia LJ. J Appl Crystallogr. 1999;32:837. [Google Scholar]
- 32.Nardelli M. J Appl Crystallogr. 1995;28:659. [Google Scholar]
- 33.Carter MT, Bard AJ. J Am Chem Soc. 1987;109:7528. [Google Scholar]
- 34.Atanassova MS, Dimitrov GD. Spectrochim Acta. 2003;A59:1655. doi: 10.1016/s1386-1425(02)00397-9. [DOI] [PubMed] [Google Scholar]
- 35.Nakamoto K. Infrared and Raman Spectra of Inorganic and Coordination Compounds. 5. John Wiley & Sons; New York: 1997. [Google Scholar]
- 36.Nyquist RA, Kagel RO. Infrared Spectra of Inorganic Compounds. Academic Press Inc; London: 1971. [Google Scholar]
- 37.Hendry P, Ludi A. Adv Inorg Chem. 1990;35:117. [Google Scholar]
- 38.Thomos C, Pringle K, Dsacon GB. J Chem Edu. 1989;68:516. [Google Scholar]
- 39.Altermatt D, Brown ID. Acta Cryst. 1985;B41:244. [Google Scholar]
- 40.Brown I David. Brockhouse Institute for Materials Research. McMaster University; Hamilton, Ontario Canada: Copyright. [Google Scholar]
- 41.Cremer D, Pople JA. J Am Chem Soc. 1975;97:1354. [Google Scholar]
- 42.Burnett MN, Johnson CK. ORTEP-III: Oak Ridge Thermal Ellipsoids Plot Program for Crystal Structure Illustrations. Oak Ridge National Laboratory Report ORNL-6895; TN: 1996. [Google Scholar]
- 43.Desiraju GR. Acc Chem Res. 2002;35:565. doi: 10.1021/ar010054t. [DOI] [PubMed] [Google Scholar]
- 44.Thallapally PK, Katz AK, Carell HL, Desiraju GR. CrystEngComm. 2003;5:87. [Google Scholar]
- 45.Desiraju GR. Acc Chem Res. 1991;24:290. [Google Scholar]
- 46.Czugler M, Bathori N. CrystEngComm. 2004;6:494. [Google Scholar]
- 47.Wu B, Liang J, Yang J, Jia C, Yang X-J, Zhang H, Tang N, Janiak C. Chem Commun. 2008:1762. doi: 10.1039/b719019k. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.