

Abstract
The regioselective palladium-catalyzed cross-coupling reactions of 2,4,7-trichloroquinazoline with various aryl- and heteroarylboronic acids are reported. An efficient, sequential strategy was developed that provides access to novel, functionalized heterocycles.
Keywords: palladium catalysis, cross-coupling, heterocycles, regioselectivity, quinazoline
Palladium-catalyzed cross-coupling reactions represent a powerful method for the formation of highly substituted heterocycles.1 The regioselective activation of polyhalogenated heteroaromatics in such reactions has been extensively studied2 and can provide a versatile means for the synthesis of libraries containing functionalized substituents in specific positions of the heterocyclic scaffold. The quinazoline moiety is of particular importance, as it is a component of a variety of biologically active compounds, including potent tyrosine kinase inhibitors,3 antibacterial,4 and anticancer agents.5 For example, trisubstituted quinazoline derivatives such as A and B have been prepared as part of a series of liver X receptor (LXR) modulators (Figure 1).6
Figure 1.

Quinazoline derivatives designed as LXR modulators
Quinazolines are also components of several approved drugs, including erlotinib,7 used to treat several types of tumors, iressa,8 an epidermal growth factor receptor inhibitor approved for the treatment of nonsmall cell lung cancer, and prazosin,9 an α-adrenergic receptor blocker. While the regioselectivity for palladium-catalyzed alkylation,10 alkynylation,11 and arylation12 of 2,4-dichloroquinazolines has been previously explored, only the alkylation10 and alkynylation11 of 6-bromo-2,4-dichloroquinazoline have been reported. For cross-coupling reactions employing 2,4-dichloroquinazoline, exclusive selectivity for the most electrophilic C-4 position13 has been observed. Attempts to achieve monosubstitution with the more highly halogenated 6-bromo-2,4-dichloroquinazoline resulted in coupling at both the C-4 and C-6 positions in a ratio of 3:1, respectively. To our knowledge, a regioselective method for the sequential polyarylation of trihalogenated quinazolines has not been disclosed. Herein, we describe a versatile new protocol for the regioselective palladium-catalyzed cross-coupling reactions of 2,4,7-trichloroquinazoline14 with aryl- and heteroarylboronic acids.
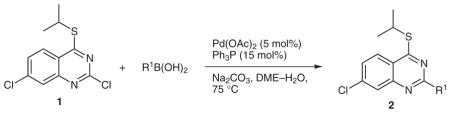
Initially, we set out to achieve the regioselective cross-coupling of 2,4,7-trichloroquinazoline by consecutive, Suzuki cross-couplings (Scheme 1, route A). We envisioned the first coupling to take place at the most electrophilic C-4 position, followed by substitutions at the C-2 and C-7 positions, respectively. However, coupling at C-4 proved to be low-yielding due to competitive hydrolysis at that site under the reaction conditions. In order to circumvent this problem, a new route was designed in which the C-4 position was first temporarily deactivated by a thioether, followed by a regioselective cross-coupling at C-2 (Scheme 1, route B). The C-4 position would then be functionalized via a palladium-catalyzed, copper(I)-mediated cross-coupling to provide compounds of type 3, which could undergo a Suzuki reaction to provide trifunctionalized quinazoline target compounds 4.
Scheme 1.

Selective cross-coupling strategies to minimize synthetic steps and maximize diversity in quinazolines 4
Thioether 1 was readily accessed by treatment of 2,4,7-trichloroquinazoline with 1.05 equivalents of isopropyl mercaptan and NaH. Substitution occurred exclusively at the electrophilic C-4 position. Subsequent regioselective C-2 arylation of 1 proceeded in excellent yields with most arylboronic acids in the presence of 5 mol% Pd(OAc)2 and 15 mol% Ph3P at 75 °C, furnishing diaryl products 2 (Table 1, entries 1–8).15 In the case of oxygen- and nitrogen-containing heterocyclic nucleophiles, slightly lower yields were obtained, presumably due to the decreased reactivity of these reagents,16 as shown by the need for prolonged reaction times (Table 1, entries 8–10). Excess boronic acid (1.5 equiv) was necessary for complete consumption of starting materials, and attempts to use stoichiometric amounts resulted in incomplete conversions. In addition, efforts to decrease the reaction time by increasing reaction temperatures resulted in poor regioselectivity and low overall yields.
Table 1.
Selective Suzuki Reaction of 1
 | |||
|---|---|---|---|
| Entry | R1 | Time (h) | Yield (%) |
| 1 | 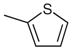 |
24 | 99 |
| 2 |  |
33 | 92 |
| 3 | 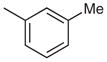 |
36 | 90 |
| 4 | 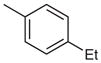 |
33 | 95 |
| 5 | 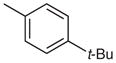 |
25 | 93 |
| 6 | 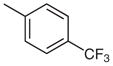 |
13 | 89 |
| 7 | 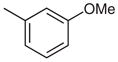 |
17 | 89 |
| 8 | 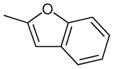 |
33 | 79 |
| 9 | 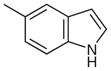 |
48 | 66 |
| 10 | 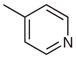 |
48 | 53 |
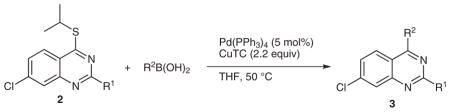
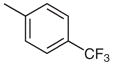
Stage 2 functionalization at the C-4 position was performed using the palladium-catalyzed, copper(I)-mediated desulfitative coupling conditions reported by Liebeskind and coworkers.17 These reactions were carried out in the presence of excess copper(I) thiophene-2-carboxylate (CuTC) and boronic acid. All desulfitative arylations were achieved in excellent yields and complete selectivity for the C-4 position (Table 2).
Table 2.
Palladium-Catalyzed, Copper(I)-Mediated Coupling of 2
 | ||||
|---|---|---|---|---|
| Entry | R1 | R2 | Time (h) | Yield (%) |
| 1 | 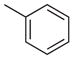 |
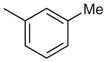 |
26 | 89 |
| 2 | 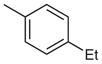 |
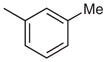 |
22 | 83 |
| 3 | 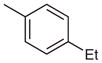 |
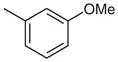 |
19 | 89 |
| 4 | 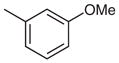 |
 |
19 | 87 |
| 5 | 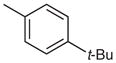 |
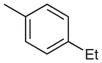 |
19 | 86 |
| 6 | 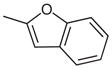 |
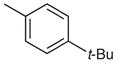 |
27 | 91 |
| 7 | 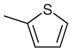 |
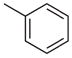 |
13 | 97 |
| 8 | 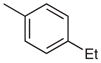 |
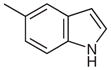 |
48 | 66 |
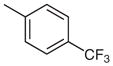
The third and final cross-coupling reaction was initially carried out using the conditions reported in Table 1. In all cases, incomplete consumption of starting material resulted, even upon heating to reflux and increasing the equivalents of boronic acid. Finally, in order to ensure the complete consumption of starting material, 10 mol% Pd(OAc)2, 30 mol% Ph3P, and 4.0 equivalents of boronic acid were used. These conditions resulted in a successful cross-coupling at position C-7 in good yields for both aryl- and heteroarylboronic acids (Table 3).
Table 3.
Suzuki Reaction of 3
 | |||||
|---|---|---|---|---|---|
| Entry | R1 | R2 | R3 | Time (h) | Yield (%) |
| 1 | 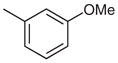 |
 |
 |
34 | 81 |
| 2 | 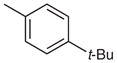 |
 |
 |
22 | 86 |
| 3 | 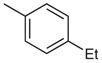 |
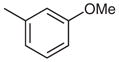 |
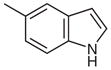 |
34 | 66 |
In conclusion, we have demonstrated that temporary deactivation of the C-4 position by substitution of the chlorine atom with isopropyl mercaptan allows for the subsequent regioselective palladium-catalyzed cross-coupling at the C-2 position in 2,7-dichloroquinazoline 1. Furthermore, palladium-catalyzed, copper(I)-mediated desulfitative coupling at the C-4 position, followed by the final palladium-catalyzed cross-coupling at the C-7 position, provides convenient access to the desired tricarbosubstituted quinazolines. This simple and efficient sequential coupling route to highly substituted quinazolines enables the orchestration of regioselective palladium-catalyzed cross-coupling reactions for the preparation of focused libraries of kinase inhibitors.18
2,7-Dichloro-4-(isopropylthio)quinazoline (1)
To a solution of 2,4,7-trichloroquinazoline14 (0.300 g, 1.28 mmol) in freshly distilled and degassed THF (13.0 mL) cooled to 0 °C was added a premixed solution of i-PrSH (0.12 mL, 1.28 mmol) and NaH (0.032 g, 1.35 mmol) in THF (2.0 mL) dropwise. The mixture was stirred for 16 h, warmed to r.t., poured into ice cold H2O, extracted with EtOAc (5 × 25 mL), and washed with H2O. The combined organic extracts were dried (MgSO4) and concentrated to give a light yellow residue. The residue was purified by chromatography on SiO2 (1:50, EtOAc–hexanes) to provide 1 (0.291 g, 83%) as a light yellow crystalline solid; mp 89.1–90.1 °C (EtOAc). IR (ATR): 3075, 2965, 2881, 1551, 1459, 1321, 1224, 1133, 852 cm−1. 1H NMR (300 MHz, DMSO-d6): δ = 8.07 (d, 1 H, J = 9.0 Hz), 7.98 (d, 1 H, J = 2.1 Hz), 7.72 (dd, 1 H, J = 9.0, 2.1 Hz), 4.18 (sept, 1 H, J = 6.9 Hz), 1.46 (d, 6 H, J = 6.9 Hz). 13C NMR (75 MHz, DMSO-d6): δ = 174.8, 156.0, 149.7, 140.1, 128.9, 126.6, 125.9, 120.2, 36.0, 22.2 (2 C). MS (EI): m/z (%) = 272 (33) [M]+, 230 (100), 195 (48), 161 (37). HRMS (EI): m/z calcd for C11H10Cl2N2S: 271.9942; found: 271.9946.
General Procedure for Compounds of Type 2
To a reaction vial was added 1 (1.0 equiv), Pd(OAc)2 (0.05 equiv), Ph3P (0.15 equiv), Na2CO3 (3.1 equiv), and R1B(OH)2 (1.5 equiv). The reaction mixture was flushed with N2. Freshly distilled and degassed DME and H2O (DME–H2O, 10:1) were added via syringe to generate a 0.1 M solution of 1, and the reaction mixture was stirred at 75 °C under a N2 atmosphere for the required time. H2O was added, and the mixture was extracted with CH2Cl2. The combined organic layers were washed with brine, dried (MgSO4), and concentrated under reduced pressure. The crude residue was purified by chromatography on SiO2 (EtOAc–hexanes or THF–toluene) to give the desired products of type 2.
7-Chloro-4-(isopropylthio)-2-(thiophen-2-yl)quinazoline(Table 1, Entry 1)
Mp 122.7–124.7 °C (DMSO). IR (ATR): 2973, 2917, 2855, 1524, 1437, 1327, 1236, 988, 837, 773, 714 cm−1. 1H NMR (300 MHz, DMSO-d6): δ = 8.06 (dd, 1 H, J = 3.6, 1.2 Hz), 8.00 (d, 1 H, J = 8.7 Hz), 7.95 (d, 1 H, J = 2.1 Hz), 7.84 (dd, 1 H, J = 5.1, 1.5 Hz), 7.61 (dd, 1 H, J = 9.0, 2.1 Hz), 7.26 (dd, 1 H, J = 4.8, 3.6 Hz), 4.30 (sept, 1 H, J = 6.9 Hz), 1.53 (d, 6 H, J = 6.9 Hz). 13C NMR (75 MHz, DMSO-d6): δ = 171.2, 156.0, 149.0, 142.8, 139.2, 131.7, 129.8, 128.8, 127.6, 127.0, 125.8, 120.0, 35.7, 22.4 (2 C). ESI-MS: m/z (%) = 321 ([M + 1]+ 100), 277 (65). ESI-HRMS: m/z calcd for C15H14ClN2S2 [M + 1]: 321.0287; found: 321.0271.
General Procedure for Compounds of Type 3
To a reaction vial was added a compound of type 2 (1.0 equiv), CuTC (2.2 equiv), and R2B(OH)2 (2.2 equiv). The reaction mixture was flushed with N2 and freshly distilled and degassed THF was added via syringe to generate a 0.06 M solution of 2. The reaction mixture was stirred vigorously at 50 °C under a N2 atmosphere for the required time. A sat. aq solution of NaHCO3 was added, and the solution was extracted with CH2Cl2. The combined organic layers were washed with NaHCO3, dried (MgSO4), and concentrated under reduced pressure. The residue was then purified by chromatography on SiO2 (EtOAc–hexanes or THF–toluene) to provide the corresponding products of type 3.
7-Chloro-2-phenyl-4-m-tolylquinazoline (Table 2, Entry 1)
IR (ATR): 3058, 3030, 2914, 2851, 1556, 1532, 1336, 913, 766, 695, 682 cm−1. 1H NMR (300 MHz, DMSO-d6): δ = 8.63–8.56 (m, 2 H), 8.22 (d, 1 H, J = 2.1 Hz), 8.11 (d, 1 H, J = 9.0 Hz), 7.73 (dd, 1 H, 9.0, 2.1 Hz), 7.70 (s, 1 H), 7.67 (d, 1 H, J = 7.6 Hz), 7.62–7.57 (m, 3 H), 7.54 (d, 1 H, J = 7.5 Hz), 7.49 (d, 1 H, J = 7.4 Hz), 2.47 (s, 3 H). 13C NMR (75 MHz, DMSO-d6): δ = 168.3, 160.0, 151.9, 139.1, 138.1, 137.1, 136.6, 131.2, 131.0, 130.4, 129.1, 128.8 (2 C), 128.6, 128.4, 128.3 (2 C), 127.3, 127.2, 119.8, 21.1. MS (EI): m/z (%) = 330 (17) [M]+, 329 (43) [M – 1]+, 238 (43), 91 (100). HRMS (EI): m/z calcd for C21H15ClN2: 330.0924; found: 330.0930.
General Procedure for Compounds of Type 4
To a reaction vial was added a compound of type 3 (1.0 equiv), Pd(OAc)2 (0.10 equiv), Ph3P (0.30 equiv), Na2CO3 (6.2 equiv), and R3B(OH)2 (4.0 equiv). The reaction mixture was flushed with N2. Freshly distilled and degassed DME and H2O (DME–H2O, 10:1) were added via syringe to generate a 0.1 M solution of 3, and the reaction mixture was sealed and heated at reflux for the required time. H2O was added, and the mixture was extracted with CH2Cl2. The combined organic layers were washed with brine, dried (MgSO4), and concentrated under reduced pressure. The crude residue was purified by chromatography on SiO2 (EtOAc–hexanes or THF–toluene) to give the desired products of type 4.
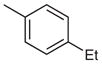
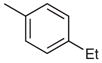
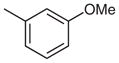
2-(4-tert-Butylphenyl)-4-(4-ethylphenyl)-7-(3-methoxyphenyl)-quinazoline (Table 3, Entry 2)
Mp 89.2–91.0 °C (EtOAc). IR (ATR): 3063, 2959, 2866, 1552, 1528, 1338, 852, 773 cm−1. 1H NMR (300 MHz, DMSO-d6): δ = 8.55 (d, 2 H, J = 8.6 Hz), 8.37 (d, 1 H, J = 1.7 Hz), 8.18 (d, 1 H, J = 8.7 Hz), 8.03 (dd, 1 H, J = 8.8, 1.8 Hz), 7.86 (d, 2 H, J = 8.1 Hz), 7.61 (d, 2 H, J = 8.6 Hz), 7.52 (d, 2 H, J = 8.2 Hz), 7.51–7.49 (m, 1 H), 7.47 (dd, 2 H, J = 7.1, 1.7 Hz), 7.11–7.03 (m, 1 H), 3.89 (s, 3 H), 2.78 (q, 2 H, J = 7.5 Hz), 1.35 (s, 9 H), 1.30 (t, 3 H, J = 7.6 Hz). 13C NMR (75 MHz, DMSO-d6): δ = 167.6, 159.9, 159.6, 153.6, 151.8, 146.3, 145.4, 140.0, 134.9, 134.5, 130.4, 130.1 (2 C), 128.2 (2 C), 128.1 (2 C), 127.6, 126.9, 125.6, 125.5 (2 C), 120.2, 119.7, 114.7, 112.7, 55.3, 34.7, 31.1 (3 C), 28.1, 15.5. MS (EI): m/z (%) = 472 (100) [M]+, 457 (79). HRMS (EI): m/z calcd for C33H32N2O: 472.2515; found: 472.2510.
Supplementary Material
Acknowledgments
We gratefully acknowledge financial support provided by the NIH (P41GM081275).
Footnotes
This paper is dedicated to Prof. Gerry Pattenden on the occasion of his 70th birthday.
Supporting Information for this article is available online at http://www.thieme-connect.com/ejournals/toc/synlett.
References and Notes
- 1.Kalinin VN. Synthesis. 1992:413. [Google Scholar]
- 2.Schröter S, Stock C, Bach T. Tetrahedron. 2005;61:2245. [Google Scholar]
- 3.(a) Fry DW, Kraker AJ, McMichael A, Ambroso LA, Nelson JM, Leopold WR, Connors RW, Bridges AJ. Science. 1994;265:1093. doi: 10.1126/science.8066447. [DOI] [PubMed] [Google Scholar]; (b) Uckun FM, Sudbeck EA, Mao C, Ghosh S, Liu XP, Vassilev AO, Navara CS, Narla RK. Curr Cancer Drug Targets. 2001;1:59. doi: 10.2174/1568009013334287. [DOI] [PubMed] [Google Scholar]; (c) Strawn LM, Shawver LK. Exp Opin Invest Drugs. 1998;7:553. doi: 10.1517/13543784.7.4.553. [DOI] [PubMed] [Google Scholar]
- 4.Bedi PMS, Kumar V, Mahajan MP. Bioorg Med Chem Lett. 2004;14:5211. doi: 10.1016/j.bmcl.2004.07.065. [DOI] [PubMed] [Google Scholar]
- 5.Foster BA, Coffey HA, Morin MJ, Rastinejad F. Science. 1999;286:2507. doi: 10.1126/science.286.5449.2507. [DOI] [PubMed] [Google Scholar]
- 6.Bernotas RC, Ullrich JW, Travins JM, Wrobel JE, Unwalla RJ. 2009020683. WO. 2009;A2
- 7.Gundla R, Kazemi R, Sanam R, Muttineni R, Sarma JARP, Dayam R, Neamati N. J Med Chem. 2008;51:3367. doi: 10.1021/jm7013875. [DOI] [PubMed] [Google Scholar]
- 8.(a) Wakeling AE, Guy SP, Woodburn JR, Ashton SE, Curry BJ, Barker AJ, Gibson KH. Cancer Res. 2002;62:5749. [PubMed] [Google Scholar]; (b) Baselga J, Rischin D, Ranson M, Calvert H, Raymond E, Kieback DG, Kaye SB, Gianni L, Harris A, Bjork T, Averbuch SD, Feyereislova A, Swaisland H, Rojo F, Albanell J. J Clin Oncol. 2002;20:4292. doi: 10.1200/JCO.2002.03.100. [DOI] [PubMed] [Google Scholar]; (c) Twombly R. J Natl Cancer Inst. 2002;94:1596. doi: 10.1093/jnci/94.21.1596. [DOI] [PubMed] [Google Scholar]
- 9.da Silva JFM, Walters M, Al-Damluji S, Ganellin CR. Bioorg Med Chem. 2008;16:7254. doi: 10.1016/j.bmc.2008.06.037. [DOI] [PubMed] [Google Scholar]
- 10.(a) Mangalagiu I, Benneche T, Undheim K. Tetrahedron Lett. 1996;37:1309. [Google Scholar]; (b) Qingbo L, Mangalagiu I, Benneche T, Undheim K. Acta Chem Scand. 1997;51:302. [Google Scholar]
- 11.Mangalagiu I, Benneche T, Undheim K. Acta Chem Scand. 1996;50:914. [Google Scholar]
- 12.Charpiot B, Brun J, Donze I, Naef R, Stefani M, Mueller T. Bioorg Med Chem Lett. 1998;8:2891. doi: 10.1016/s0960-894x(98)00508-3. [DOI] [PubMed] [Google Scholar]
- 13.(a) Eicher T, Hauptmann S. The Chemistry of Heterocycles. Wiley-VCH; Weinheim: 2003. [Google Scholar]; (b) Garcia Y, Schoenebeck F, Legault CY, Merlic CA, Houk KN. J Am Chem Soc. 2009;131:6632. doi: 10.1021/ja9004927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Curd HS, Landquist JK, Rose FL. J Chem Soc. 1948:1759. [PubMed] [Google Scholar]
- 15.Exclusive coupling at the C-2 position was determined by 2D NMR experiments.
- 16.(a) Barder TE, Walker SD, Martinelli JR, Buchwald SL. J Am Chem Soc. 2005;127:4685. doi: 10.1021/ja042491j. [DOI] [PubMed] [Google Scholar]; (b) Billingsley K, Buchwald SL. J Am Chem Soc. 2007;129:3358. doi: 10.1021/ja068577p. [DOI] [PubMed] [Google Scholar]
- 17.Liebeskind LS, Srogl J. Org Lett. 2002;4:979. doi: 10.1021/ol0200091. [DOI] [PubMed] [Google Scholar]
- 18.(a) Decornez H, Gulyás-Forró A, Papp Á, Szabó M, Sármay G, Hajdú I, Cseh S, Dormán G, Kitchen DB. ChemMedChem. 2009;4:1273. doi: 10.1002/cmdc.200900164. [DOI] [PubMed] [Google Scholar]; (b) Peng J, Lin W, Jiang D, Yuan S, Chen Y. J Comb Chem. 2007;9:431. doi: 10.1021/cc0601534. [DOI] [PubMed] [Google Scholar]; (c) Wipf P, Minion DJ, Halter RJ, Berggren MI, Ho CB, Chiang GG, Kirkpatrick L, Abraham R, Powis G. Org Biomol Chem. 2004;2:1911. doi: 10.1039/b405431h. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.