

Abstract
This review presents an overview of human cortical malformation based on the insights gained from examination of human fetal brains. Examination at early stages of fetal brain development allows the identification of the specific pathways which are disrupted in human cortical malformation. Detailed examination of human fetal brains in parallel with studies of genetics and animal models is leading to new concepts of cortical malformations. Here we review a range of human cortical malformations based on a simple classification according to the developmental process thought to be disrupted: neuroblast proliferation, undermigration, overmigration, cortical maturation and destructive lesions. A single case example of a dated intrauterine injury illustrates the spectrum of malformations which may result at a single period in development. The recommended methods of examination of human fetal brain are described together with some of their pitfalls. Detailed neuropathological observations indicate the need for caution in the classification of malformations; radiological findings and pathology of the mature brain do not reflect the specific disruptive pathways of cortical malformations. While many insults may lead to the same pattern of malformation, a single insult can lead to multiple patterns of malformation. Our detailed studies of the human fetal brain suggest that the interface between the meninges and the radial glial end feet may be an intriguing new focus of interest in understanding cortical development.
Keywords: cortical malformations, human cerebral cortex, lissencephaly, neuronal migration disorders, pia, polymicrogyria
Introduction
This review presents an overview of human cortical malformation based on the insights gained from examination of human fetal brains. Examination at early stages of fetal brain development allows the identification of the specific pathways which are disrupted in human cortical malformation. Detailed examination of fetal brains in parallel with studies of genetics and mouse models is leading to new concepts of cortical malformations (Fallet-Bianco et al. 2008).
For several decades the seminal work of Rakic and his co-workers (Sidman & Rakic, 1973), has taught us the fundamental importance of the radial scaffold and the interaction between migrating cells and the radial glia in cortical development. However, disruption of the pial basement membrane associated with the radial glial end feet has been recently demonstrated in a number of cortical malformations associated with overmigration. Further, proliferation of pial basement membrane is a prominent feature of many forms of polymicrogyria (PMG) and signals from the meninges appear to be responsible for aspects of the regulation of cortical growth and development (Radakovits et al. 2009; Siegenthaler et al. 2009). This suggests that the interface between the meninges and the radial glia at the pial basement membrane may be an intriguing new focus of interest in human cortical development.
Human cortical malformations
Modern clinical imaging methods are of such sophistication that they can precisely define many malformations and have led to major advances in their understanding and classification. However, even the most detailed imaging cannot describe the tissue architecture of malformations and, more importantly, cannot elucidate the specific developmental pathways which were disrupted to cause them. This information is essential to the understanding of aetiology and timing of developmental disruptions and the functions of the genes involved. Meaningful classification of cortical malformations depends on knowledge of the basic underlying mechanisms.
The work presented here is based on experience of examination of the human fetal brain for clinical diagnostic purposes. The use of human material has limitations which do not apply to animal models, but models can never precisely reflect the processes of human brain development and in particular the timing of these events, which shows huge variation between species of differing gestational periods. Study of the developing human brain is invaluable, but every brain represents a personal tragedy for a family. The authors and readers of this review owe a great debt of gratitude to all those families who have given consent for the use of their babies’ brains to further our understanding.
Technical aspects of the examination of human fetal and neonatal brains
Human fetal brains are often autolysed by the time of autopsy due to the inevitable period of time between death and delivery. Additional delay between delivery and autopsy contributes further to artefact and autolysis. The immature brain may be extremely soft, even after fixation for several weeks, but this should not deter sampling for microscopic examination, which almost invariably adds valuable information. Unfortunately, these delicate brains tend to fragment during processing; the cortical surface being particularly vulnerable to mechanical disruption by handling. Careful processing by experienced and dedicated laboratory technical staff can minimize artefactual damage.
Fixation
In recent years we have had high rates of consent for fetal brain examination, particularly if the brain is examined within a few days so that it can be quickly reunited with the body without disrupting the family's funeral arrangements. To comply with families’ wishes we have experimented with reduced fixation times and found that fixation for only a few days can produce excellent results, particularly if strong (undiluted) formalin is used. It is apparent that delay between death and autopsy is more damaging to tissues than short fixation times.
Staining methods
Haematoxylin and eosin (H & E) is the standard stain but the anatomy of the cortical surface can be particularly enhanced by the use of the reticulin stain (a silver impregnation method) which demonstrates the collagen fibres in the pial basement membrane and around small blood vessels in the brain parenchyma. Reticulin demonstrates the integrity or otherwise of these membranes and highlights pial anomalies which may be too subtle to detect with H&E. It demonstrates the proliferation of collagen on the brain surface, common in polymicrogyria, and marks the buried pial surface in cobblestone lissencephaly and the capillary proliferation in response to injury. Immunocytochemical markers specific for endothelium, such as CD31 and CD34, work well in fetal tissue. Perl's stain for blood breakdown products indicates old haemorrhage and areas of tissue destruction which may not be readily seen with H & E. The macrophage marker CD68 also highlights areas of brain damage.
Many of the cell markers used in experimental anatomy depend on frozen tissue or perfusion fixation and are not applicable to human diagnostic material; however, improvements in antigen retrieval techniques are extending the application of experimental techniques to human tissue.
The normal developing cortex
The fetal cortex consists of a densely packed band of cells in which only the superficial marginal zone (layer 1) is readily distinguished. The immature neurons and glia do not show cytologically recognizable differentiation until after the end of gestation at full term.
Papillae of Retzius
In fetuses of 16–24 weeks gestation it is not uncommon to see irregularity of the cells of layer 1 and 2 of the cortex with projections of layer 2 cells into the relatively acellular layer 1 or molecular layer (Fig. 1). These are called ‘Papillae of Retzius’ and are commonly seen in the absence of any other abnormality. While widely regarded as an artefact specific to this gestational period, great care must be taken not to overlook genuine cortical malformation. It is necessary to check that the overlying brain surface is normal, the pia is intact and that blood vessels are normal. The deep cortical border should be well demarcated from the underlying white matter and without underlying neuronal ectopia. If these observations are normal, one may be reassured that the disturbance confined to layers 1 and 2 is most likely due to artefact.
Fig. 1.
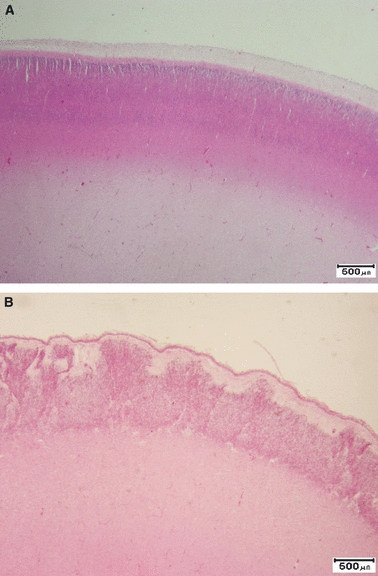
(A) Normal cortex at 25 weeks gestation. There is a densely packed uniform band of cells. Although the cortical laminae are not yet clearly seen, there is a clear distinction from the underlying white matter. The superficial cortex has a well demarcated marginal layer. (B) Papillae of Retzius. There are groups of cells of layer 2 extending into the marginal zone. The deep cortical border is well demarcated from the white matter and the pial surface is intact. H&E scale bar: 500 μm.
Classification of cortical malformations
The disorders described will be set out according to a very simple classification based on the developmental process thought to have been primarily disrupted:
neuroblast proliferation, growth and differentiation;
-
neuronal migration;
undermigration;
overmigration;
hind brain migration defects;
cortical maturation and folding;
destructive lesions.
Neuroblast proliferation, growth and differentiation
Microcephaly
Microcephaly is defined as a small head. There is an important clinical distinction between microcephaly due to damage or atrophy of the brain and microcephaly vera, in which the brain is small due to a genetic cause while the rest of the body is of normal size. In microcephaly vera the brain structure is either normal or has mild simplification of the gyral pattern. Mutations have been identified in several genes, all of which are implicated in cell division and cell cycle regulation; it has been suggested that molecular evolution of these genes may have been a driving force for the increase in size of the brain during human evolution (Mochida, 2009).
Cortical dysplasia
This term is applied to focal areas of cortical malformation in which neuronal orientation and lamination are disrupted and dysmorphic neurons are seen; these may be giant cells or balloon cells, both the result of abnormal regulation of cell growth (Crino, 2005). Several classification schemes have been devised. Type 1 (sometimes called architectural dysplasia), is characterized by abnormal distribution of neurons. Type II (cytoarchitectural dysplasia) is subdivided into type IIa, in which dysmorphic neurons are identified, and type IIb, where dysmorphic neurons and balloon cells are seen (Fig. 2).
Fig. 2.
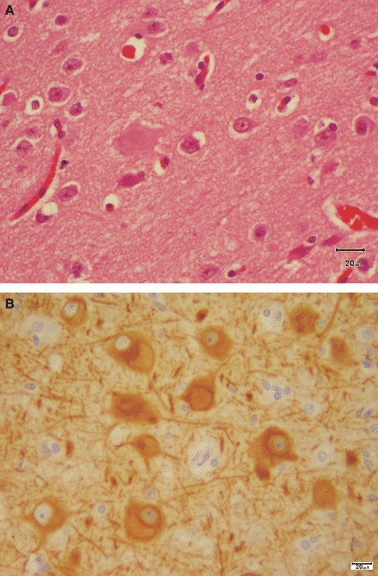
Focal cortical dysplasia. (A) This section is stained with H&E and shows giant and balloon cells. (B) MAP-2 stain shows the pale cytoplasmic inclusions of balloon cells corresponding to the pale, glassy cytoplasm in H&E stains. Scale bar: 20 μm.
Tuberous sclerosis complex
Tuberous sclerosis complex (TSC) is associated with both malformations and benign neoplasms (Boer et al. 2008), indicating defects in neuronal migration as well as early cell growth and proliferation. The disease results from mutations of the genes TSC1 or TSC2.
The giant cells of TSC and balloon cells of cortical dysplasias both express proteins indicating a mixed glial and astrocytic lineage; they also express phospho-S6, a protein involved in ribosomal protein translation and regulation of cell growth (Crino, 2005). Few fetal examples have been described.
Figure 3 shows a fetus of 27 weeks with a collection of abnormal cells at the junction of cortex and white matter. The abnormal cells extend, often along vessel walls, to the subpial surface. Use of a number of cell markers shows a mixed population of cells, small oval deeply staining cells intermingled with giant cells, which themselves show heterogeneity of protein expression.
Fig. 3.
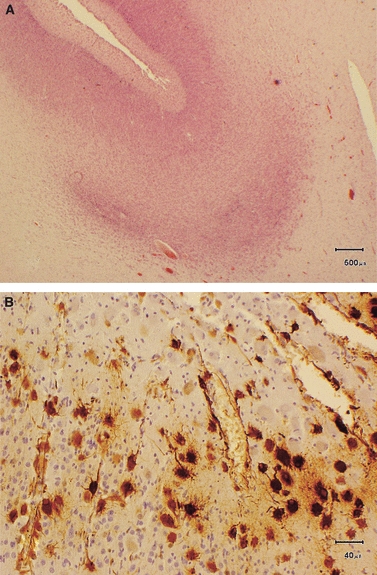
Fetus of 27 weeks gestation with tuberous sclerosis (TS). (A) Low-power section stained with H&E shows an abnormal mass of cells at the junction of deep cortex with the white matter. Scale bar: 500 μm. (B) A higher-power section stained with vimentin shows that only a proportion of the abnormal giant cells express this protein. The vimentin-positive cells form foot processes around blood vessels. Scale bar: 40 μm.
Disruptions of neuronal migration
Undermigration
Failed or incomplete neuronal migration may be generalized, as in type 1 lissencephaly and double cortex syndrome, or may be focal and represented by clusters of ectopic neurons forming heterotopic nodules in the subcortical or periventricular zones.
Failed migration results from a defect in one of the many processes involving cell recognition and interaction essential for migration, or from inherent cytoskeletal defects within the migrating cells. To migrate, a cell requires the cytoskeletal structure which permits formation and withdrawal of cell processes and relocation of the nucleus. The fundamental importance of the microtubular system has been demonstrated by the association of mutations in tubulin genes with malformations involving failed or impaired migration. Identification of mutations in tubulin genes has led to recognition of a much wider spectrum of malformation patterns than was previously anticipated (Jaglin & Chelly, 2009). Tubulins are cytoskeletal proteins involved in a wide range of functions, including roles in the cell cycle, morphology and polarity of cells, mobility, intracellular trafficking and axonal growth. The tubulin-related disorders exhibit a wide range of brain malformations including microcephaly, pachygyria, heterotopia and agenesis of the corpus callosum (Fallet-Bianco et al. 2008; Jaglin et al. 2009).
Type 1 lissencephaly
Type 1 lissencephaly is characterized by a smooth brain surface with coarse, if any, gyration. The cortex is thick with poor lamination and an indistinct deep border with the underlying white matter (Fig. 4). Periventricular nodular heterotopias may be seen beneath the ependymal lining of the lateral ventricles. The olivary nuclei are commonly malformed, reflecting abnormal migration across the tissues of the brainstem. Brainstem surface migration is usually not affected and the brainstem leptomeninges and cerebellar cortex are normal.
Fig. 4.
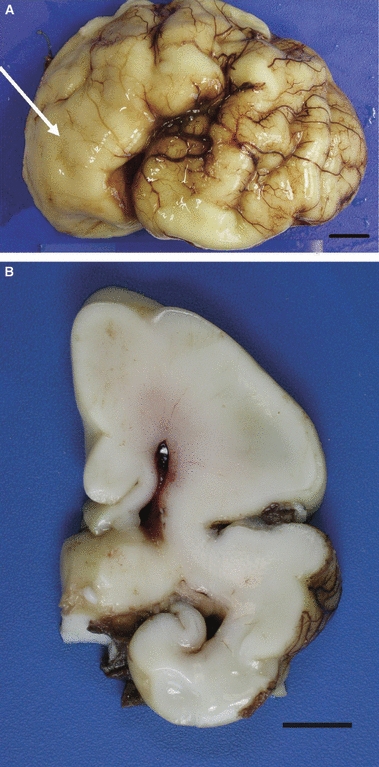
Type 1 lissencephaly in a 34-week fetus. (A) Lateral view of the brain shows poor gyration which is more marked anteriorly (arrow). (B) A coronal slice shows a thick, but simple cortex and small ventricles. Scale bar: 1 cm.
Overmigration syndromes
The overmigration syndromes all share disruptions of the basement membrane asociated with migration of neurons from the cortex into the overlying leptomeninges. The developmental mechanisms in individual syndromes are likely to be diverse. The significance of primary defects in basement membrane proteins has long been recognized in the dystroglycanopathies and cobblestone cortex (Muntoni et al. 2004), but several other distinct genetic syndromes have been described, including GPR56, MARCKS and TUBB2B mutations. Overmigration is also a feature of destructive lesions and polymicrogyria (PMG) – see below.
The demonstration, in TUBB2B mutation, of focal breaches in the basement membrane and neuronal overmigration leading to a polymicrogyra-like pattern led to the suggestion that radial glial cells play a role in development of the pial basement membrane, as indicated in GPR56 mutation (Li et al. 2008).
However, it may be a bit more complicated than that. First, in several mutations associated with cortical dysplasia the pial basement membrane appears to develop normally; breaches develop later (Blackshear et al. 1997; Hu et al. 2007; Li et al. 2008), suggesting that defects arise as a result of instability of the basement membrane during remodelling and brain growth. Secondly, it appears that the basement membrane per se may influence cortical development (Zarbalis et al. 2007; Radakovits et al. 2009; Siegenthaler et al. 2009). Foxc1 is a protein expressed in all three meningeal layers; homozygous mouse mutants show normal early formation of the pial basement membrane, which later develops defects associated with detached radial glial end feet leading to disorganization of cortical neurons in the marginal zone (Zarbalis et al. 2007). However, in this model there does not appear to be the extensive overmigration characteristic of cobblestone lissencephaly, instead the dysplasia is intracortical.
In dystroglycanopathies and GPR56 mutation, migration of neurons through the hindbrain basement membranes is also defective (Muntoni et al. 2004; Koirala et al. 2009).
Generalized overmigration (type II lissencephaly or cobblestone cortex)
Animal and human studies show three distinct patterns of generalized overmigration; loss of integrity of the pial basement membrane appears to be critical to all. In the mouse model of cobblestone cortex associated with dystroglycanopathy a continuous basement membrane forms but later develops breaches through which glial end feet protrude. Subsequently, neurons are found on the brain surface outside the basement membrane (Hu et al. 2007). It is notable that the original cortical plate in these mice does not appear to form normally, in that a marginal zone is not seen; a situation comparable to the early human cobblestone malformation (Fig. 5A–C). In contrast, in the mouse GPR56 model the cortex develops normally until E13.5, after which defects are observed in the basement membrane followed by neuronal overmigration into the subarachnoid space (Li et al. 2008). Interestingly, in both forms of overmigration the hippocampus is spared (Fig. 5D) (Li et al. 2008).
Fig. 5.
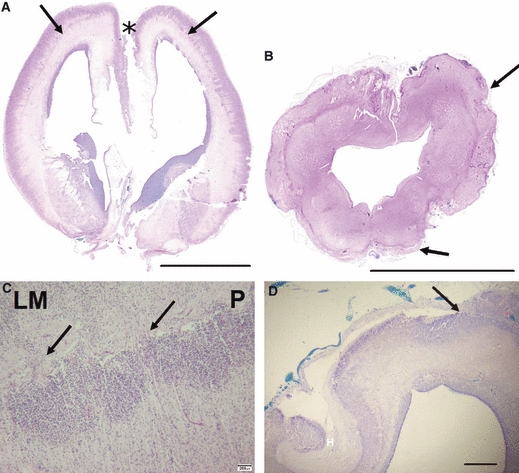
Cobblestone lissencephaly, 23 weeks gestation fetus. (A) Coronal section through the brain, stained with H&E, shows a line running throughout the cerebral wall (arrows) where the original pia was to be found (see higher power picture C.) Note there is also adhesion between the medial frontal lobes (asterisk). Scale bar: 1 cm. (B) A section through the hindbrain (scale bar: 1 cm) shows thickening of the leptomeninges around the brainstem and cerebellum (arrows) which contain abnormal clusters of migrating neurones. (C) The original pia is marked (P). Cells of the cortical plate form dense groups abutting directly on the pial basement membrane without a molecular layer. Migrating cells may be seen streaming through defects in the basement membrane (arrows) into the leptomeninges (LM). Scale bar: 200 μm. (D) A section of the hippocampus (H) showing the junction of normal and abnormal cortex. Here, just lateral to the hippocampus, the leptomeninges become buried in the dysplastic cobblestone cortex (Luxol fast blue stain). Scale bar: 1 mm.
A similar sequence of events is seen in the MARCKS-deficient mouse (Blackshear et al. 1997) where cortex and basement membrane are normal at E9.5 but by E13, basement membrane defects are apparent and cells are seen in the subarachnoid space. In this model it appears that defects in the cytoskeleton, cell adhesion and polarity lead to deformed aberrant radial glial end feet. It is suggested that lack of apicobasal polarity leads to impaired interaction with the pial basement membrane and overmigration of neurons into the leptomeninges (Weimer et al. 2009).
A third pattern of cortical malformation is associated with defective pial expression of Foxc1 protein, a protein normally expressed in all layers of the meninges (Zarbalis et al. 2007). In this model, initial cortical development is also normal, but later, at E18.5, basement membrane defects appear and there is disorganization of glial end feet. This malformation differs from the cobblestone cortex in that there appears to be no extracortical migration; the cortical dysplasia is limited to disorganization of neurons within the marginal layer.
Disruption of hindbrain migration pathways
Brainstem and cerebellar development depends on two migration pathways. Certain of the cells destined for the cerebellar cortex and basal brainstem nuclei, such as the inferior olivary nuclei, are generated in the rhombic lip at the lateral angle of the fourth ventricle and migrate to their final destination through the surface basement membranes.
The cells destined for the cerebellar cortical granule layers spread over the entire cerebellum from 10 weeks of gestation (Sidman & Rakic, 1973). Cells of the brainstem nuclei also originate in the rhombic lip. These cells migrate on the surface and then penetrate the brainstem to form its nuclei (Altman & Bayer, 1978a,b;). Another population of cells migrates from the rhombic lip within the parenchyma of the brainstem and developing cerebellum, independent of interaction with the leptomeninges. These latter pathways may be disrupted in both type I and type II lissencephaly, but the surface leptomeningeal pathways are only disrupted in the cobblestone type and not in classical lissencephaly.
Disruption of the surface migratory pathways indicates not only dependence on the integrity of the basement membrane or extracellular matrix but also highly selective adhesion to its constituent proteins. The cobblestone complex appears to be due to defective glycosylation of alpha-dystroglycan, while migrating cerebellar granule cells of GPR56 mutant mice, which have malformations of the rostral cerebellum, show selective deficiency in adhesion to laminin-1 and fibronectin but normal adhesion to collagen IV (Koirala et al. 2009).
Cortical maturation and folding
After the cortical plate has formed, it undergoes further maturation, including the acquisition of a glial population and development of intercellular connections and dendritic growth. The closely packed cells of the immature cortex become separated by increasing amounts of neuropil. Morphological differentiation and lamination become better defined. The most obvious maturational change is gyration, which occurs between 21 and 40 weeks in the human brain. If the cortex does not form normally there may be few simplified gyri (pachygyria) or virtually complete absence, as in the lissencephalies. There are also syndromes in which gyri appear to be too small and too numerous (polymicrogyria).
Polymicrogyria
Based on imaging, polymicrogyria (PMG) is characterized by an irregular, bumpy surface of the cortex, with apparent thickening and a stippled appearance of the grey–white matter junction (Barkovich, 2005). Different patterns of PMG have been described, including a number of bilateral PMG syndromes (Leventer et al. 2010). PMG is a highly heterogeneous malformation, resulting from both genetic and destructive events, including infection, hypoxia-ischaemia, and trauma (Jansen & Andermann, 2005). In many cases even combined analysis of imaging, pathology and genetic studies is unable to identify the underlying aetiology.
The delineation of different PMG patterns on imaging has facilitated the orientation of genetic studies, at least to a certain degree. However, it has been shown that a single bilateral PMG syndrome may have multiple genetic aetiologies (Villard et al. 2002; Santos et al. 2008) and that genetic causes may also give rise to asymmetrical PMG patterns (Jaglin et al. 2009).
The features of polymicrogyria may be difficult to recognize on the brain surface as gyri often appear coarse; the cut surface usually shows a cortex which appears abnormally thick, formed by ‘piling upon each other of many small gyri with fused surfaces’ (Friede, 1989). The neuropathological description of PMG generally remains limited to the description of a two- or four-layered cortex, which is generally thought to correlate respectively to early vs. late insults to the developing brain. The concurrence of both two- and four-layered cortex in the same patient is not at all uncommon and can be seen both in cases with defined genetic lesions and in those with destructive lesions. Moreover, in early fetal cases it can be very difficult or impossible to define the number of layers in the PMG cortex. Making the distinction between these forms may be of little value.
Friede (1989) described three characteristic microscopic features: abnormal arrangement of cells and an intracortical fibre plexus, excessive folding of all or only the upper layers, and fusion of gyral surfaces, with large sinusoid intracortical vessels marking the seams between adjacent fused gyri. Among many patterns of human PMG, we have seen fusion of adjacent gyri in association with bilateral periventricular nodular heterotopia (Fig. 6).
Fig. 6.
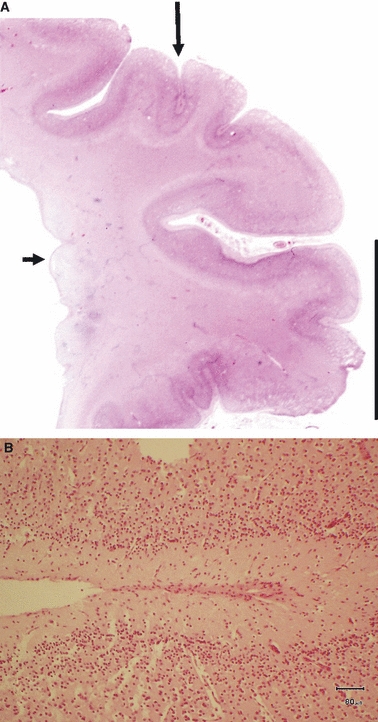
(A) Bilateral periventricular nodular heterotopia in a term neonate. One subependymal nodule is indicated with a short arrow. This section of the cerebral hemisphere shows focal polymicrogyria in the cortex overlying the periventricular nodules (long arrow). H&E scale bar: 1 cm. (B) A higher power image shows focal adhesion of adjacent gyri which appear to be ‘zipping up’ from the depth of a sulcus. Scale bar: 80 μm.
Our experience with examination of fetal examples, however, has shown that true fusion of the molecular layers does not seem to be a prerequisite for the diagnosis of PMG, as it is not uncommon to see a folded and undulating neuronal band beneath a straight molecular layer without any evidence of true fusion (Fig. 7). It therefore seems more appropriate to limit the neuropathological definition of PMG to the presence of one or more undulating or festooning cortical neuronal layers. The observation of this pattern at 18 weeks, well before the onset of normal gyration, is further evidence that this is not always an abnormality of folding or fusion of adjacent gyri but an earlier inherent anomaly of cortical formation (Fig. 8). This brings into question the signals for folding and when they are operative.
Fig. 7.
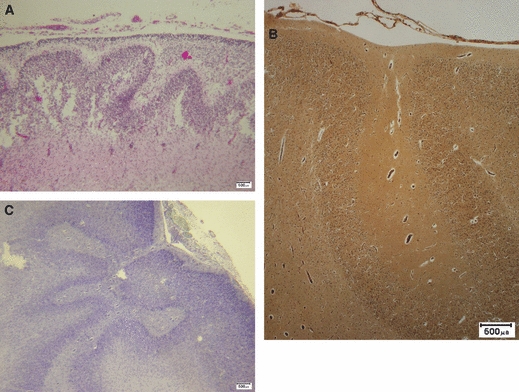
Three examples of polymicrogyria characterized by undulating cortical laminae beneath a smooth cortical surface. (A) Fetus (H&E); (B) adult reticulin stain, (C) Adult Luxol fast blue and cresyl violet stain. Scale bars: 500 μm.
Fig. 8.
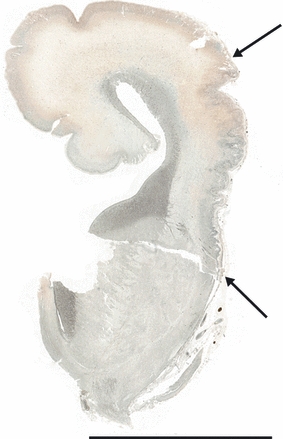
Coronal section of the brain of an 18 weeks gestation fetus. Focal cortical irregularity and festooning of the neuronal band is seen in the area which will become the perisylvian cortex (arrows). Reticulin stain. Scale bar: 1 cm.
The leptomeninges overlying the PMG lesion can be normal but often appear thickened, with vascular proliferation and leptomeningeal heterotopia (Fig. 9). This can be encountered both in genetically proven PMG and in PMG resulting from destructive causes. The abnormal leptomeninges may extend out on both sides of the PMG lesion. It remains to be clarified whether this leptomeningeal thickening participates in the generation of the PMG cortex or represents a secondary change, or whether both mechanisms co-exist. Leptomeningeal thickening with neuroglial heteropia was also recently described in TUBB2B mutations (Jaglin et al. 2009) and GPR56 mutations. The malformation associated with the GPR56 mutation is described as bilateral frontoparietal polymicrogyria, but closer examination of the early stages of development of this malformation in the mouse model has shown defects in the basement membrane and neuronal overmigration (see above). These observations blur the definitions between the classically described malformations and indicate the need for caution with classification systems.
Fig. 9.
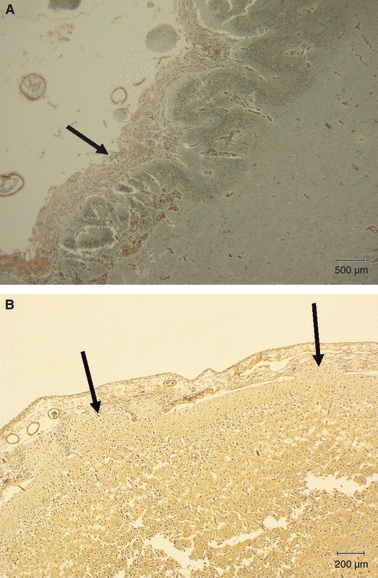
Polymicrogyria. (A) Polymicrogyria associated with cytomegalovirus infection. Note the considerable thickening of the pial basement membrane over the abnormally festooned neuronal layer of the cortex. It is the festooning of the neuronal band which leads to classification as polymicrogyria. Reticulin stain. Scale bar: 500 μm. (B) 36 weeks gestation fetus. There are focal glioneuronal heterotopia (arrows) in the greatly thickened leptomeninges overlying an area of polymicrogyria. Reticulin stain. Scale bar: 200 μm.
The grey–white matter junction in PMG can be either sharp or blurred, with heterotopic neurons or nodules populating the transition zone between grey and white matter.
PMG can occur as an isolated finding or can be associated with other CNS anomalies including periventricular nodular heterotopia, excessive scattered neurons in the underlying white matter, and brainstem or cerebellar abnormalities. PMG can also be part of a multiple congenital anomaly syndrome with additional abnormalities outside the CNS.
As highlighted above, multiple questions with respect to the development of PMG, including the role of the leptomeninges, the mechanisms of premature folding, and the mechanisms by which different genetic or acquired causes result in a similar cortical abnormality, remain unanswered. It is hoped that the integration of progress in the fields of imaging, genetics and animal models with human neuropathology will assist in finding at least part of the answers. To date, it has not been possible to determine the primary insult, genetic or destructive, associated with each of the observed subtypes of PMG.
Destructive lesions
While destructive lesions may appear to be of no interest to the neurobiologist, they are of great importance in human diagnostic pathology. A well documented injury may teach us about the effects of a single insult at a specific point in brain development. It is clear that a single insult can cause multiple disruptions, leading to a variety of malformation patterns and, conversely, that multiple forms of insult (genetic, infectious and ischaemic) may lead to the same malformation.
A single timed intrauterine injury
The following case illustrates the effect of an ischaemic insult at 24 weeks of gestation. The mother has very generously given consent for the case to be described for the purposes of education.
The mother was involved in a road traffic accident when she was 24 weeks pregnant. Post-injury fetal ultrasound brain scan and a repeat at 28 weeks were normal. However, by 30 weeks the fetal MRI scan showed reduction in supratentorial brain volume, prominent extraaxial spaces and bilateral ‘porencephalic cysts’.
The pregnancy was terminated 12 weeks after the injury at almost 36 weeks gestation. The brain was small, two-thirds of the expected weight, and there was bilateral infarction of middle cerebral artery territory (Fig. 10).
Fig. 10.
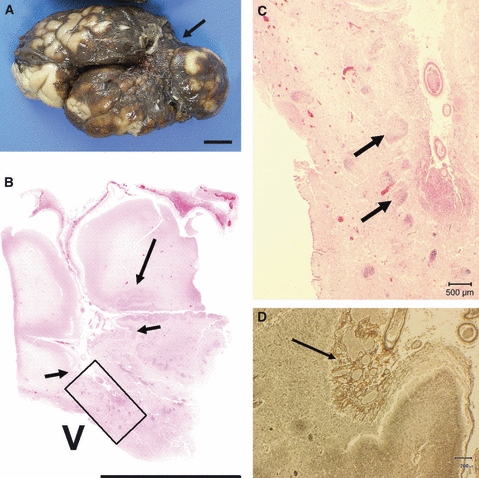
Fetus 36 weeks; 12 weeks after maternal trauma. (A) Lateral view of the whole fixed brain showing a large area of collapse which is old infarction in middle cerebral artery territory (arrow). Scale bar: 1 cm. (B) H&E stained section of the middle cerebral artery territory shows a large area of infarction (between small arrows) and polymicrogyria at the junction of damaged brain with normal cortex (long arrow). Scale bar = 1 cm. V = ventricle. (C) This is the area marked with a square in (B). Beneath the infarcted cortex there are numerous groups of migrating cells within the white matter which have failed to reach the cortex (arrows). Scale bar: 500 μm. (D) High-power image of cortex in the infarcted middle cerebral artery territory. Note that there is florid thickening and increased vascularity of the leptomeninges over the damaged cortex (arrow). Reticulin stain. Scale bar: 200 μm.
Coronal slices showed bilateral middle cerebral artery infarction. There was PMG in the frontal and occipital watershed zones. Histology of the cerebral hemispheres showed, apart from necrosis, failure of neuronal migration with subcortical heterotopia, polymicrogyria, overmigration into the leptomeninges, transmantle dysplasia and schizencephaly. There was middle cerebral artery infarction as well as watershed zone damage. The damage was the result of fetal cerebral ischaemia, rather than direct trauma to the baby or the brain.
This case illustrates that a single timed insult can lead to a variety of malformations, the nature of which depends on the timing and the developmental processes which are disrupted. Further, it shows that the arterial territories and watershed zones are established by 24 weeks. It has generally been assumed that the fetal brain is not susceptible to watershed damage until term (Huang & Castillo, 2008).
Schizencephaly
Schizencephaly is one of the more common cortical malformations with a prevalence of 1.5 per 100 000. Defined as a split in the brain wall extending through the full thickness from the ventricle to the leptomeningeal surface, it was first proposed as the result of primary failure of development of a small focus of the germinal matrix (Yakovlev & Wadsworth, 1946). EMX2 mutation has been proposed as a cause, but this has not been established (Tietjen et al. 2007). Consensus opinion is that schizencephaly is the result of a destructive event (Norman, 1981); and Friede, (1989) does not differentiate schizencephaly from porencephaly, describing both as circumscribed hemispheric necrosis occurring in utero. Barkovich (Barkovich & Kjos, 1992) suggested the cause was ischaemia before 25 weeks of gestation. Most cases result from a vascular disruption; attempted abortion, amniocentesis, chorionic villus sampling and motor vehicle accidents have been described in association with the condition. Twenty-three of 43 cases of ‘isolated schizencephaly’ were bilateral (Curry et al. 2005).
Destructive lesions of the brain wall may be seen in watershed zones (Fig. 11), although in the fetus, middle cerebral artery territory is more commonly involved. Lesions occurring before 20 weeks are associated with impaired neuronal migration which results in the cleft being lined by dysplastic heterotopic neurons.
Fig. 11.
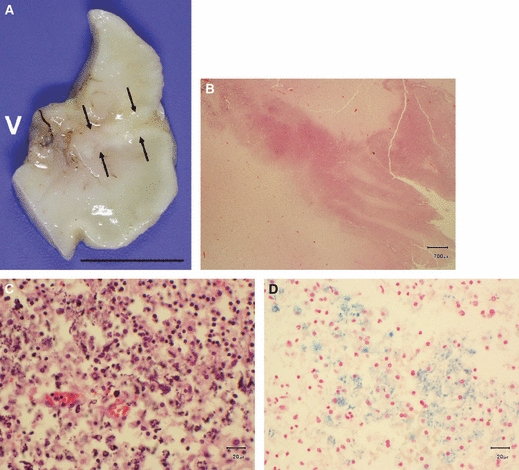
Fetus of 26 weeks gestation with infarctions in watershed territories. (A) The naked eye appearance of a fixed brain slice shows an area where a band of white tissue, resembling the cortex, forms a fold dipping almost the full depth of the brain wall towards the ventricular lining (V). Scale bar: 1 cm. (B) Higher power image shows an abnormally folded band of neurons around the edges of cleft (H&E). Scale bar: 700 μm. (C) There is focal calcification in cells within the damaged brain wall suggesting old brain destruction as a cause for the cleft (H&E). Scale bar: 20 μm. (D) Perl's stain showing altered blood pigment in an area of old damage. Scale bar: 20 μm.
Larger cystic infarcts may have dysplastic cortex at the border with normal brain in the ischaemic penumbra (Squier et al. 2003) if the infarct occurs prior to cortical maturation. Collapse of a large, fluid-filled infarct cavity may lead to the appearance of a cleft-like disruption of the brain wall and fulfil the criteria for schizencephaly (Fig. 12).
Fig. 12.
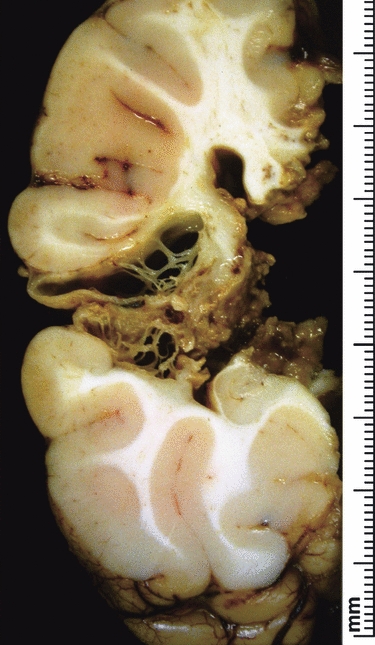
Fixed slice of a hemisphere surgically removed for treatment of epilepsy. The brain slice shows a collapsed cyst in the territory of the middle cerebral artery. The resulting appearance is of a full thickness cleft through the brain wall, which is lined by irregular membranes representing the remnants of the original brain wall.
Conclusion
Observation of very early stages of cortical malformations indicates the need for caution in their classification; the appearances seen in the mature brain by pathology or radiology do not necessarily reflect the specific pathways which have been disrupted in the development of malformations. Many insults may lead to the same pattern of malformation and a single insult can lead to multiple patterns of malformation.
Acknowledgments
We are indebted to the mothers who gave consent for us to study their babies’ brains. The sections were prepared and stained by the staff of the Neuropathology Laboratory at the John Radcliffe Hospital, without whose dedication and experience these studies could not have been undertaken. We are grateful to Drs Steve Gould and Colene Bowker and all the paediatric pathologists who refer brains for diagnosis.
References
- Altman J, Bayer SA. Prenatal development of the cerebellar system in the rat. I. Cytogenesis and histogenesis of the deep nuclei and the cortex of the cerebellum. J Comp Neurol. 1978a;179:23–48. doi: 10.1002/cne.901790104. [DOI] [PubMed] [Google Scholar]
- Altman J, Bayer SA. Prenatal development of the cerebellar system in the rat. II. Cytogenesis and histogenesis of the inferior olive, pontine gray, and the precerebellar reticular nuclei. J Comp Neurol. 1978b;179:49–75. doi: 10.1002/cne.901790105. [DOI] [PubMed] [Google Scholar]
- Barkovich J. Malformations of cerebral cortical develop-ment. In: Barkovich AJ, editor. Pediatric Neuroimaging. 4th edn. Lippincott, Williams and Wilkins; 2005. [Google Scholar]
- Barkovich AJ, Kjos BO. Schizencephaly: correlation of clinical findings with MR characteristics. Am J Neuroradiol. 1992;13:85–94. [PMC free article] [PubMed] [Google Scholar]
- Blackshear PJ, Silver J, Nairn AC, et al. Widespread neuronal ectopia associated with secondary defects in cerebrocortical chondroitin sulfate proteoglycans and basal lamina in MARCKS-deficient mice. Exp Neurol. 1997;145:46–61. doi: 10.1006/exnr.1997.6475. [DOI] [PubMed] [Google Scholar]
- Boer K, Troost D, Jansen F, et al. Clinicopathological and immunohistochemical findings in an autopsy case of tuberous sclerosis complex. Neuropathology. 2008;28:577–590. doi: 10.1111/j.1440-1789.2008.00920.x. [DOI] [PubMed] [Google Scholar]
- Crino PB. Molecular pathogenesis of focal cortical dysplasia and hemimegalencephaly. J Child Neurol. 2005;20:330–336. doi: 10.1177/08830738050200041101. [DOI] [PubMed] [Google Scholar]
- Curry CJ, Lammer EJ, Nelson V, et al. Schizencephaly: heterogeneous etiologies in a population of 4 million California births. Am J Med Genet A. 2005;137:181–189. doi: 10.1002/ajmg.a.30862. [DOI] [PubMed] [Google Scholar]
- Fallet-Bianco C, Loeuillet L, Poirier K, et al. Neuro-pathological phenotype of a distinct form of lissencephaly associated with mutations in TUBA1A. Brain. 2008;131(Pt 9):2304–2320. doi: 10.1093/brain/awn155. [DOI] [PubMed] [Google Scholar]
- Friede RL. Dysplasias of the cerebral cortex. In: Friede RL, editor. Developmental Neuropathology. 2 edn. Berlin, Heidelberg: Springer; 1989. pp. 330–346. [Google Scholar]
- Hu H, Yang Y, Eade A, et al. Breaches of the pial basement membrane and disappearance of the glia limitans during development underlie the cortical lamination defect in the mouse model of muscle-eye-brain disease. J Comp Neurol. 2007;502:168–183. [PubMed] [Google Scholar]
- Huang BY, Castillo M. Hypoxic-ischemic brain injury: imaging findings from birth to adulthood. Radiographics. 2008;28:417–439. doi: 10.1148/rg.282075066. [DOI] [PubMed] [Google Scholar]
- Jaglin XH, Chelly J. Tubulin-related cortical dysgeneses: microtubule dysfunction underlying neuronal migration defects. Trends Genet. 2009;25:555–566. doi: 10.1016/j.tig.2009.10.003. [DOI] [PubMed] [Google Scholar]
- Jaglin XH, Poirier K, Saillour Y, et al. Mutations in the beta-tubulin gene TUBB2B result in asymmetrical polymicro-gyria. Nat Genet. 2009;41:746–752. doi: 10.1038/ng.380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen A, Andermann E. Genetics of the polymicrogyria syndromes 4. J Med Genet. 2005;42:369–378. doi: 10.1136/jmg.2004.023952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koirala S, Jin Z, Piao X, et al. GPR56-regulated granule cell adhesion is essential for rostral cerebellar development. J Neurosci. 2009;29:7439–7449. doi: 10.1523/JNEUROSCI.1182-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leventer RJ, Jansen A, Pilz DT, et al. Clinical and imaging heterogeneity of polymicrogyria: a study of 328 patients. Brain. 2010;133(Pt 5):1415–1427. doi: 10.1093/brain/awq078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Jin Z, Koirala S, et al. GPR56 regulates pial basement membrane integrity and cortical lamination. J Neurosci. 2008;28:5817–5826. doi: 10.1523/JNEUROSCI.0853-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochida GH. Genetics and biology of microcephaly and lissencephaly. Semin Pediatr Neurol. 2009;16:120–126. doi: 10.1016/j.spen.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muntoni F, Brockington M, Torelli S, et al. Defective glycosylation in congenital muscular dystrophies. Curr Opin Neurol. 2004;17:205–209. doi: 10.1097/00019052-200404000-00020. [DOI] [PubMed] [Google Scholar]
- Norman MG. On the morphogenesis of ulegyria. Acta Neuropathol (Berl) 1981;53:331–332. doi: 10.1007/BF00690375. [DOI] [PubMed] [Google Scholar]
- Radakovits R, Barros CS, Belvindrah R, et al. Regulation of radial glial survival by signals from the meninges. J Neurosci. 2009;29:7694–7705. doi: 10.1523/JNEUROSCI.5537-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos NF, Secolin R, Brandao-Almeida IL, et al. A new candidate locus for bilateral perisylvian polymicrogyria mapped on chromosome Xq27. Am J Med Genet A. 2008;146A:1151–1157. doi: 10.1002/ajmg.a.32270. [DOI] [PubMed] [Google Scholar]
- Sidman RL, Rakic P. Neuronal migration, with special reference to developing human brain: a review. Brain Res. 1973;62:1–35. doi: 10.1016/0006-8993(73)90617-3. [DOI] [PubMed] [Google Scholar]
- Siegenthaler JA, Ashique AM, Zarbalis K, et al. Retinoic acid from the meninges regulates cortical neuron generation. Cell. 2009;139:597–609. doi: 10.1016/j.cell.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squier W, Salisbury H, Sisodiya S. Stroke in the developing brain and intractable epilepsy: effect of timing on hippocampal sclerosis. Dev Med Child Neurol. 2003;45:580–585. doi: 10.1017/s0012162203001075. [DOI] [PubMed] [Google Scholar]
- Tietjen I, Bodell A, Apse K, et al. Comprehensive EMX2 genotyping of a large schizencephaly case series. Am J Med Genet A. 2007;143A:1313–1316. doi: 10.1002/ajmg.a.31767. [DOI] [PubMed] [Google Scholar]
- Villard L, Nguyen K, Cardoso C, et al. A locus for bilateral perisylvian polymicrogyria maps to Xq28. Am J Hum Genet. 2002;70:1003–1008. doi: 10.1086/339433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weimer JM, Yokota Y, Stanco A, et al. MARCKS modulates radial progenitor placement, proliferation and organization in the developing cerebral cortex. Development. 2009;136:2965–2975. doi: 10.1242/dev.036616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakovlev PI, Wadsworth RC. Schizencephalies: a study of the congenital clefts in the cerebral mantle. I. Clefts with fused lips. J Neuropathol Exp Neurol. 1946;5:116–130. doi: 10.1097/00005072-194604000-00003. [DOI] [PubMed] [Google Scholar]
- Zarbalis K, Siegenthaler JA, Choe Y, et al. Cortical dysplasia and skull defects in mice with a Foxc1 allele reveal the role of meningeal differentiation in regulating cortical development. Proc Natl Acad Sci U S A. 2007;104:14002–14007. doi: 10.1073/pnas.0702618104. [DOI] [PMC free article] [PubMed] [Google Scholar]