

Abstract
Schizophrenia probably has a developmental origin. This review refers to three of our published series of studies related to this hypothesis: loss of dendritic spines on cerebral neocortical pyramidal neurons, decreased numerical density of glutamatergic neurons, and microgliosis. First, brains of schizophrenic patients and non-schizophrenic controls were obtained post mortem and blocks of multiple cortical areas impregnated with a Rapid Golgi method. Spines were counted on the dendrites of pyramidal neurons of which the soma was in layer III (which takes part in corticocortical connectivity) and which met strict criteria for impregnation quality. Data were obtained blind: diagnoses were only revealed by a third party after measurements were completed. The mean spine count in all cortical areas studied in the control series was 243 mm−1 of dendrite and in the schizophrenics 108. Measurements in frontal and temporal association cortex showed the greatest reduction in spine number in schizophrenia (299 in control frontal cortex and 101 in schizophrenics, and 276 mm−1 in control temporal cortex and 125 in schizophrenics). There was no correlation of spine loss with age at death. Our results support the concept of a neurodevelopmental defect in the neuropil affecting glutamatergic neurons in schizophrenia and may help to explain loss of cortical volume without loss of neurons. In a second part of our study we used an antibody to the kainate receptor subunit GluR 5/6/7 and showed a decrease in numerical density of presumed glutamatergic neurons in schizophrenic orbitofrontal cortex. Finally, as glia play a major role in the developing nervous system, we investigated whether schizophrenia was associated with glial changes in frontal and temporal cortex. Astroglia and microglia were identified in schizophrenic and control brains, using antibodies to glial fibrillary acidic protein (GFAP) and class II human leucocyte antigen (HLA-DR), respectively. Significant increases were found in microglial numerical density in schizophrenics compared with controls: 28% in frontal area 9 (115 cells mm−2 compared with 89), and a 57% increase in temporal area 22 (139 cells mm−2 compared with 88). For both areas, astroglia showed no significant differences between schizophrenics and controls. No significant differences were found in cortical thickness or total neuronal numerical density between the two groups. This specific increase in numerical density of microglia in temporal and frontal cortex of chronic schizophrenics, not related to aging, could be related to possible changes in cortical neuropil architecture as revealed by loss of dendritic spines.
Keywords: cortex, dendritic spines, glutamatergic neurons, microglia, neurodevelopment, post mortem, schizophrenia
Schizophrenia as a neurodevelopmental disease
Schizophrenia is probably a neurodevelopmental disorder with evidence of brain changes before clinical onset (Harrison, 1995, 1997; Ross & Pearlson, 1996; Raedler et al. 1998; Weinberger & Marenco, 2003). Behavioural and intellectual abnormalities in children who later developed schizophrenia support this neurodevelopmental hypothesis (Done et al. 1994).
Enlargement of the cerebral ventricles was for a long time the most consistent anatomical finding in schizophrenia using neuroimaging or at post mortem (Johnstone et al. 1976; Weinberger et al. 1979; Andreasen et al. 1982; Reveley et al. 1982; DeLisi et al. 1986; Pfefferbaum et al. 1988; Suddath et al. 1989), and may be fully developed before the onset of illness (Frangou & Murray, 1996). Studies showed a decrease in volume of cerebral cortex but no change in that of subcortical white matter (Suddath et al. 1989; Zipursky et al. 1992; Harvey et al. 1993), suggesting that there is no loss of axons or probably of somata of cortical neurons (Selemon et al. 1995; Selemon & Goldman-Rakic, 1999), but there is a loss in neuronal processes, such as axon terminals or dendritic branches or spines, in the cortical neuropil. More recent imaging studies have extended the original observations, showing that patients with schizophrenia have decreased grey matter in frontal, temporal and parietal cortices compared with controls (Honea et al. 2008). Furthermore, van Haren et al. (2008) observed that changes in brain volume throughout life were different between schizophrenics and controls. This was particularly evident in the first 20 years or so of illness (before the age of 45), when grey matter loss and lateral ventricle increase were most marked in patients relative to controls. Patients also showed increase in third ventricle volume over time. Poor outcome patients showed more brain tissue loss than good outcome patients. So, overall there seems to be loss of cerebral tissue, at least in the early stages of schizophrenia. But this may be of neuropil rather than of neuronal cell bodies.
To investigate possible changes in neuropil, we carried out studies of dendritic spines (Garey et al. 1993, 1995, 1998), in the light of our evidence of the large changes they undergo during normal development (Michel & Garey, 1984; Fig. 1) at a time when major changes are taking place in human cortical neuronal architecture (Mrzljak et al. 1992; Cao et al. 1996; Yan et al. 1996, 1997) and synaptogenesis (Huttenlocher, 1979, 1984; Huttenlocher & Dabholkar, 1997; Huttenlocher et al. 1982; Rakic et al. 1994). Similar and very extensive work, including a tentative molecular mechanism for spine changes, has been reported by Glantz & Lewis (1995, 2000),Kolluri et al. (2005),Hill et al. (2006) and Sweet et al. (2009). A recent review of dendritic spines summarizes current views on their dynamics and importance (Bhatt et al. 2009).
Fig. 1.
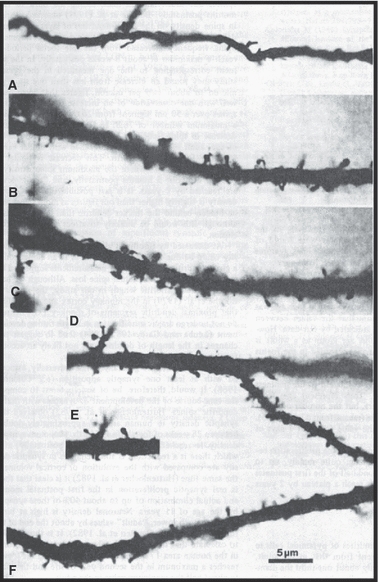
Micrographs of segments of apical dendrites of layer III cortical pyramidal neurons. (A) Fetus at 33 weeks' gestation. Narrow dendrite with few immature, hair-like spines. (B,C) Thicker dendrite at 3 months postnatal with more bulbous spines. (D,E) Spine numerical density is highest at 5 months. (F) 21 months: spine density has fallen to perinatal levels. From Michel & Garey (1984).
A loss of dendritic spines, particularly from cortical pyramidal neurons, could be consistent with the glutamate hypothesis of schizophrenia (Kim et al. 1980; Hirsch et al. 1997), as the N-methyl-d-aspartate (NMDA) subtype of glutamate receptor is present on their dendrites and probably dendritic spines (Petralia et al. 1994), and NMDA receptor blockade by phencyclidine (Vincent et al. 1979; Zukin & Zukin, 1979) results in schizophrenia-like symptoms (Luby et al. 1959). Positive and negative symptoms in schizophrenia may be associated with NMDA receptor hypofunction (Bennett, 2009).
Pyramidal cells in layer III are glutamatergic (Fonnum, 1984) and are involved in projections between various cortical areas (Lund et al. 1975), and might therefore be expected to be important in higher cognitive function that is disturbed in schizophrenia.
If subtle neuronal abnormalities occur in schizophrenia, one might expect changes in glial cells, which have important functions in the CNS. Most studies have concluded that there is no increase in astroglia, thus suggesting a developmental as opposed to a degenerative process (Falkai et al. 1988; Stevens et al. 1988; Harrison, 1995). We therefore studied glia in another cohort of schizophrenic and control brains, concentrating on microglia (Radewicz et al. 2000), using immunocytochemistry for identifying specific glial cells.
Specimens
Our studies formed part of the Charing Cross Prospective Schizophrenia Project in which authorization was obtained in life for neuropsychological evaluation of long-stay schizophrenic patients and subsequent neuropathological study of their brains post mortem. Clinical histories and neuropsychological assessments of the patients, diagnosed using DSM IIIR criteria, were available. Control material was obtained from routine autopsies or donated bodies, after relevant authorization; for none of the control brains was there a history of psychiatric disease. The brains were coded so that the nature of each specimen was unknown to the investigator. The examination was therefore conducted ‘blind’ to eliminate assessment bias.
Dendritic spines
Methods
For the study of spines (Garey et al. 1998) the brains of eight schizophrenic patients (one female, seven males; average age 70 years, range 44–83) and 11 non-schizophrenic controls (two females, nine males: average age 70 years, range 50–78) were fixed in 4% buffered paraformaldehyde, within a post mortem interval (PMI: the time from death to fixation of the specimens) of 4–96 h. Blocks of neocortical tissue were removed from all cerebral lobes, and prepared for microscopy using the Rapid Golgi technique. and sectioned at 100 μm. The relative thickness of the sections helped maintain the integrity of the impregnated neurons, and allowed more complete analysis of their dendritic arbors.
Qualitative observations
Many densely impregnated neurons and neuroglia were present in the Golgi sections (Fig. 2). Typical pyramidal neurons were found with somata in all layers except layer I, but most commonly in layers II, III and V. The neurons of which the soma was in layer III tended to form a more homogeneous population than those in other layers, and there was less fragmentation of their dendrites, particularly the apical dendrite that was, of necessity, shorter than that of layer V pyramids. A single apical dendrite could always be identified extending from the superior pole of the soma, usually as far as layer I where it ramified, although often it branched earlier. Several basal dendrites typically formed a skirt around the inferior pole of the soma. All dendrites bore numerous spines.
Fig. 2.
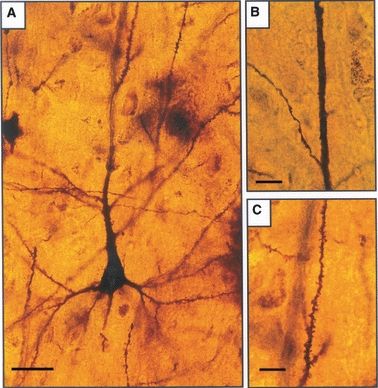
Layer III pyramidal neurons, Golgi method. (A) From a control brain, showing spiny dendrites. (B) Spineless segment of apical dendrite in a schizophrenic brain. (C) Higher power of spiny apical dendrite in a control brain. Scale bars: A: 25 μm; B: 15 μm; C: 10 μm.
Various degrees of loss of dendritic spines were detectable on the pyramidal neurons in most specimens of schizophrenic cortex (Fig. 2). In these cases the apical and basal dendrites and their branches usually had some segments that were almost completely smooth, whereas others had a few long, thin or short, stubby spines, usually occurring singly or in clusters of up to four.
Spine density measurements
In a given section, 10 adjacent pyramidal cells that complied with the following sampling criteria were analysed. All had their cell body in layer III, they had complete or near complete dendritic arbors, and were unobscured by surrounding cells. Each was drawn at 400× magnification using a microscope fitted with a drawing tube, and the total length of the dendrites for that cell measured. At a magnification of 1000× the number of spines on all the dendrites was counted under oil immersion. As some of the spines were obscured by their own dendrite, only those spines protruding laterally from the dendritic shafts into surrounding clear neuropil were counted. The spine density of a pyramidal neuron was calculated as the total number of spines divided by the total length of its dendrites, and was expressed as the number of spines per millimeter of dendrite. The mean spine density of the 10 neurons was then calculated.
Over all areas of cortex studied, a mean numerical density of 108 spines mm−1 was calculated on layer III pyramidal neurons in schizophrenic cortex, compared to 243 spines mm−1 for non-schizophrenic controls, a highly significant difference (Student's t-test, P < 0.0001). Analysis of blocks of frontal association cortex (areas 9, 10, 11 and 45) showed 101 spines mm−1 in schizophrenics and 299 in controls. Temporal association cortex from areas 20, 21, 22, 38 and 42 showed 125 spines mm−1 in schizophrenics and 276 in controls (Fig. 3). There was, therefore, consistency in frontal and temporal cortices, where functional changes have been demonstrated in schizophrenia. In primary motor or sensory areas (1, 2, 3, 4, 17) and in parietal and cingular cortex (areas 7 and 21) the effect was less marked (schizophrenics: 103 spines mm−1; controls: 160). There was no obvious difference between left and right hemispheres.
Fig. 3.
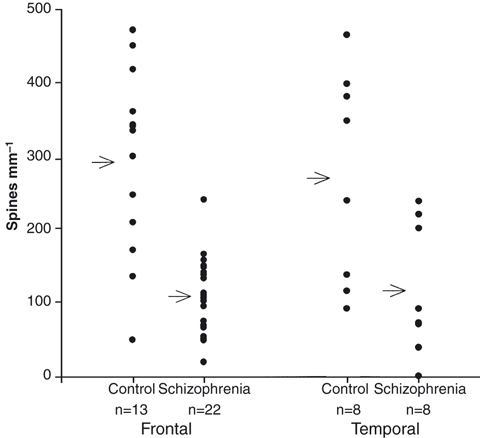
Scatter plots of spine numerical densities on layer III pyramidal apical dendrites from control and schizophrenic frontal and temporal cortex. Arrows indicate the mean for the blocks studied in each group.
It was important to match age and spine count in the schizophrenic group to determine whether loss of spines in schizophrenic cortex could be due to older age (Jacobs et al. 1997). In the frontal cortex, all schizophrenic patients, with one exception, had a lower spine count than a matched control of the same or greater age. In temporal cortex the same was true except over age 70, when all counts were low. Regression analysis of spine density vs. age showed a marked reduction in spine number with age in controls, but in schizophrenics the regression curve was essentially flat throughout the age range. We therefore concluded that there was no correlation of spine loss with age in schizophrenics.
Glutamatergic neurons
Methods
We used blocks fixed in 4% paraformaldehyde from the orbitofrontal cortex (area 11 of Brodmann, 1909/2006) from nine schizophrenics (five males, four females) and eight controls (three males, five females) (Garey et al. 2006). The blocks were embedded in wax, and sectioned at 10 μm, before mounting on glass sides. Sections per block were immunostained with a mouse monoclonal antibody recognizing the kainate receptor subunit GluR5/6/7 (Chemicon) and described as labelling specific neurons in primate neocortex (Huntley et al. 1993). Slides were microwaved in citrate buffer (pH 6) for 5 min to enhance the presentation of the antigen (Newman & Gentleman, 1997) and incubated overnight in the kainate receptor antibody diluted 1 : 200. A standard ABC (Vectastain) routine was then used, and the staining visualized using a metal-enhanced diaminobenzidine substrate. Kainate receptor-positive cells in a column from the pia to the white matter were counted at ×200 independently by two observers. Differences between the schizophrenic and control brains were tested for significance by using the non-parametric Mann–Whitney test.
Kainate receptor-positive neurons
Cells labelled for kainate receptor were visible in all layers. Most were pyramidal neurons (Fig. 4). Nissl-stained sections revealed no significant difference between left and right cortices or between schizophrenics and controls. There were no significant differences in the number of immunopositive neurons between left and right sides of control or schizophrenic brains. However, there were significantly fewer kainate receptor-positive neurons in the schizophrenics than the controls. Using both left and right cortex, the lower numerical density of stained neurons in the schizophrenic group was highly significant (488 compared with 618; 21% reduction; P = 0.005).
Fig. 4.

Micrograph of schizophrenic orbitofrontal cortex labelled for kainate receptor.
Glia
Methods
For the glia study (Radewicz et al. 2000) eight schizophrenics and 10 controls were used. The mean age of the schizophrenics was 80 (range 65–89) and of the controls 72 (range 55–90). Neuropathological examination was carried out in each case. Parkinson and Huntington diseases were excluded on clinical history and by examination of the substantia nigra and caudate nucleus. Possible Alzheimer disease was assessed by clinical history, macroscopic examination of the brain, and a modified CERAD procedure (Mirra et al. 1991) using immunocytochemical screening of sections from cingulate, hippocampal, frontal, occipital and parietal cortex for plaques and tangles with antibodies against β-amyloid protein (1E8) and Tau protein (AT8). Three brains with histological evidence of Alzheimer disease were excluded from the quantitative study.
Cortical blocks were removed from frontal area 9 and temporal area 22 (Brodmann, 1909/2006) of both hemispheres and fixed in 4% paraformaldehyde or 10% formalin. Sections were cut at 20 μm, mounted and incubated with the primary antibody solution. A polyclonal rabbit anti-bovine antibody against glial fibrillary acidic protein (GFAP) (Dako; 1 : 800) was used to visualize astroglia, and a monoclonal anti-HLA-DR antibody (LN-3, ICN; 1 : 5) for microglia. A standard ABC (Vectastain) routine was then followed. The reaction product was visualized with a solution of diaminobenzidine, cobalt chloride and hydrogen peroxide (Fig. 5). Separate sections were stained with cresyl fast violet to determine cortical cytoarchitecture and thickness.
Fig. 5.
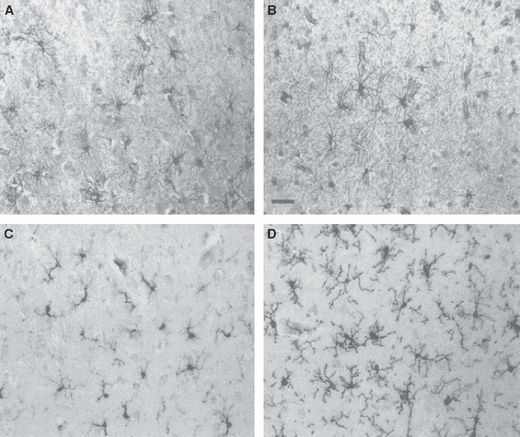
GFAP staining for astroglia (A,B) and LN3 staining for microglia (C,D) in layer III of the temporal cortex of control (A,C) and schizophrenic (B,D) subjects. Scale bar = 40 μm. There is no obvious astrogliosis in schizophrenia, but there is microgliosis. From Radewicz et al. (2000).
Microglia
The laminar distribution of microglia in controls was similar in areas 9 and 22, with a similar numerical density in cortical layers I–V and a significantly greater density in layer VI and into white matter. In both areas, significant increases in microgliosis were observed in the schizophrenic group (Figs 5 and 6). In area 9 there was a 28% increase in microglia in schizophrenics (115 cells mm−2) vs. controls (89). In area 22, the increase was even greater, at 57% (139 cells mm−2 in schizophrenics vs. 88 in controls). A comparison of microglial density in individual layers showed that in both areas it was evident for all layers in the schizophrenic group. In area 22, increases were highly significant (67% for layer I, 68% for layer II, 70% for layer III, 65% for layer IV, 53% for layer V and 35% for layer VI). We found no differences in microglial density or laminar distribution between hemispheres for either diagnostic group.
Fig. 6.
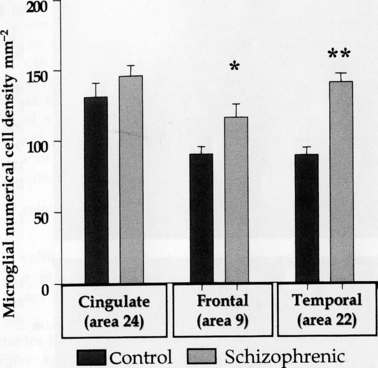
Significantly increased microglial numerical density in frontal and temporal, but not in cingulate, cortex in schizophrenia. Student's t-test for independent variables: *P < 0.05; **P < 0.01. From Radewicz et al. (2000).
So, the greatest microgliosis in schizophrenics was in temporal cortex where the density was similar to that in Alzheimer disease. However, in our cases with diagnosed Alzheimer disease, not used for quantification, the microglia had an ‘activated’ morphology with an enlarged cell body and thickened, elongated processes. ‘Resting’ microglia, with small oval cell bodies and multiple long, thin processes, were evident throughout the cortex of controls. Microglia in schizophrenic cortex resembled those in the controls. Thus, although the number of microglia was greatly increased in the schizophrenic group, their morphology was not drastically altered.
In the control group, a slight increase in microglial density was evident with increasing age, but no correlation between microglia and age was found in schizophrenics. Because there was a significant difference in PMI between schizophrenics and controls, microglial numerical density was compared with PMI, but no significant correlation between the two was found for either the control or schizophrenic groups. Total cell density in area 22 was quantified to assess whether neuronal changes might account for increase in microglial density in schizophrenics: only area 22 was assessed, as the most significant alterations in microglia were in this area. There were no significant differences between schizophrenics and controls in total cell density in either hemisphere. Microglia constituted 7% of the total cell population for controls and 12% for schizophrenics. Thus it appears that the microglial increase in schizophrenic temporal cortex was a singular event and not associated with changes in other cortical cell densities.
Astroglia
Although not quantified, GFAP revealed astroglial cell bodies and processes of similar morphology and with no obvious differences in numerical density or in individual layers in control and schizophrenic cases in frontal and temporal cortex (Fig. 5). In cases with Alzheimer pathology, astroglia appeared larger with more processes throughout the cortex.
Discussion
We described exuberant spine production in human perinatal cortex (Michel & Garey, 1984; Fig. 1). Apical dendrites of layer III pyramidal neurons in primary visual cortex undergo considerable changes in the late neonatal and early postnatal period. At 33 weeks of gestation we found narrow dendritic shafts with a few hair-like spines. Three months postnatally there were thicker shafts with numerous bulbous spines. At 5 months, spines reached their highest density, and only at 21 months postnatally did they return to perinatal levels.
In schizophrenia we found a consistent and significant reduction in the number of dendritic spines on pyramidal neurons of which the soma was in layer III, which take part in corticocortical connectivity. Spines are known to be highly dynamic and able to grow, change shape and retract according to local neural activity (Amaral & Pozzo-Miller, 2009). Measurements in temporal and frontal association cortex showed the greatest reduction in spine number (Fig. 3). Essentially similar results were reported by Glantz & Lewis (2000) for prefrontal area 46. Kolluri et al. (2005) have since demonstrated that reduction in spine density on pyramidal neurons in prefrontal cortex in schizophrenia is specific to layer III with little or no change in other layers. Notably, the total number of layer III pyramidal neurons does not appear to be reduced in schizophrenia, although packing density is increased, so the implication is that lower densities of dendritic spines reflect lower numbers per neuron, and thus reduced neuropil (Dorph-Petersen et al. 2009), support for the ‘reduced neuropil hypothesis’ of Selemon & Goldman-Rakic (1999).
As a reduction in the number of spines has been reported in the cerebral cortex of elderly subjects (Scheibel et al. 1975), we took the effect of age into consideration by comparing the schizophrenics as a group with age-matched controls, and by comparing matched pairs of schizophrenics and controls. We found no evidence that low spine counts in schizophrenia were related to age, compared with a normal population.
Schizophrenics are more likely to develop Alzheimer disease than the general population (Prohovnik et al. 1993), and a decreased spine density has also been reported in Alzheimer disease (Mehraein et al. 1975; Catalá et al. 1988; el Hachimi & Foncin, 1990). Our calculation of two of these results (Mehraein et al. 1975; Catalá et al. 1988) showed means of 335–652 spines mm−1 of apical dendrite in controls and 228–364 in patients with Alzheimer disease. Thus, mean spine densities in Alzheimer disease were much higher than that of our schizophrenics and suggest that their decreased spine density was not due to concomitant Alzheimer disease.
Neuroleptics may have toxic effects in animals (Sommer & Quandt, 1970), and this might lead to loss of dendritic spines (Benes et al. 1985) in some areas but not in others (Uranova et al. 1991). Several of the patients described here were not on neuroleptic treatment but still had low spine counts. The effect of altered social environment must also be considered. The schizophrenics had spent long periods in institutions, compared with controls from relatively rich social environments. The effects of social deprivation in humans are unknown, although rats housed in isolation showed no difference in cortical spine densities compared to rats kept in a social environment (Connor & Diamond, 1982). Other factors which could affect spine density in schizophrenics include the effects of alcohol (Ferrer et al. 1986), but this can be documented and does not concern the in-patient population described here.
Postmortem delay could result in tissue damage and loss of dendritic spines, but we found no relationship between PMI and the number of spines. Also, the Golgi technique may only partially impregnate some neurons. This was overcome as far as possible by following strict criteria for selecting and analysing cells and using similar technical procedures in all cases. To avoid the difficulties of counting spines in front of and behind the dendritic shafts, we only counted those in the clear zones of neuropil flanking the shafts. It follows that such counts are underestimates of the true population. However, as a common procedure was used in all brains, the results should be consistent in relation to each other and truly reflect differences between schizophrenic and non-schizophrenic brains. Other difficulties experienced in the counting of spines included detection of two separate but closely spaced spines, spines lying close to the parent dendrite and the detection of weakly stained spines. These difficulties were minimized by counting at high magnification and following strict criteria across all samples. Variability between individual cells was minimized by taking the mean value of 10 cells in each block.
There was no obvious difference between left and right hemispheres in terms of spine loss. This is consistent with some imaging studies of schizophrenics, which showed no difference in reduction in cortical volume between left and right sides (Suddath et al. 1989; Zipursky et al. 1992), but is in contrast to the results of other studies, which showed that the left hemisphere was more affected by structural and functional abnormalities (Crow et al. 1989). The variability between individual brains could reflect individual differences in brain structure and the heterogeneity of schizophrenia (Liddle et al. 1992). A further stage of the analysis of the present results should relate the spine counts to precise clinical diagnosis, including the prospective neuropsychological data available on these patients.
The nature of the changes which could lead to a schizophrenia-specific spine loss is unknown. Cortical spine densities increase, and then decrease to the normal adult level during the first months of life (Michel & Garey, 1984; Fig. 1), and the reduced spine density in schizophrenia could be the result of some event at a critical point during development. Loss of spines on pyramidal neurons could imply a loss of NMDA receptors which are present on their dendrites or spines (Petralia et al. 1994). As pyramidal neurons are intercortical association cells (Lund et al. 1975), it is also possible that communication between neocortical areas may be disrupted as a result of loss of spines. Spines may be lost as a result of reduced synaptic transmission, suggested by a decrease in the synaptic protein synapsin I in schizophrenic cortex (Browning et al. 1993). Also, afferent axon terminals may be lost, and we have shown by electron microscopy ‘naked’ postsynaptic densities on the dendrites in schizophrenic cortex (Ong & Garey, 1993). There may be reduced glutamate receptors in schizophrenic cortex: electron microscopy has shown abnormal excitatory synapses with vesicles that clump away from unusually short postsynaptic densities where glutamate receptor would be situated (Ong & Garey, 1993). We also find reduced glutamate receptor immunoreactivity (Von Bussmann et al. 1996; Garey et al. 2006), and expression of glutamate receptor gene is reduced in hippocampus (Harrison et al. 1991). Excessive pruning of prefrontal synapses, perhaps involving excitatory glutamatergic inputs to pyramidal neurons, has been suggested as a possible aetiological factor in schizophrenia (Keshavan et al. 1994). Fewer dopaminergic fibres innervate pyramidal cells in the cingulate cortex of schizophrenics, while more innervate GABAergic non-pyramidal cells (Benes et al. 1995). As spines of pyramidal cells receive dopaminergic synapses as well as glutaminergic ones (Smiley et al. 1992), it is possible that spine loss accounts for at least some of the lost dopaminergic innervation.
There have been suggestions that certain cortical cells, in particular GABAergic neurons, may migrate abnormally in schizophrenia (Akbarian et al. 1993). However, we were unable to detect any change in cell numbers, density or distribution for GABAergic neurons in schizophrenia (Leclercq et al. 1996; Leclerq et al. 1998).
We also investigated possible astroglial or microglial changes associated with schizophrenia. The study of gliosis in schizophrenia has centred largely on histological studies classing all glial types together, or has focused on astrogliosis. We categorized sub-types using immunocytochemical techniques. We found an increase in HLA-DR immunoreactive microglia in frontal and temporal cortex of chronic schizophrenics (Radewicz et al. 2000), with more marked increases in the temporal. Microglia are sensitive markers for the condition of neurons in their close proximity (Streit, 1996). Their morphology can indicate subtle alterations, injury or aberrant functioning of neurons. Tissue damage produces microglial activation characterized by a graded series of responses. First, microglia proliferate at the site of injury. Secondly, they alter their morphology to become activated cells. Ultimately, the cells may become amoeboid and phagocytic.
In schizophrenics, proliferation of microglia in frontal and temporal cortices could be attributed to several factors. One, which is consistent with a developmental hypothesis of schizophrenia, is a response to perinatal injury, following which increased numbers of microglia might remain in the adult grey matter. They could serve a continued neuroprotective function for neighbouring neurons. They might be associated with the maintenance of altered synaptic connectivity, such as the decreased number of dendritic spines on pyramidal cells that we describe here, as microglia have been shown to remodel the CNS (Gehrmann et al. 1993). One might postulate that the disease may be due to an early or even congenital error or insult that could mobilize microglia to help protect non-affected neurons: ‘damage limitation’. An elevated microglial number might then persist into adult life. Microglia may regulate malfunctioning synaptic mechanisms in the cortex in many ways. Important questions are whether remodelling of dendritic spines is a continuous process in schizophrenia or merely the reflection of an earlier event, and whether the observed increase in microglia is present at the onset of the disease or progresses during the course of the illness. Recent results would tend to confirm the lack of astrogliosis in schizophrenia, and that there is microgliosis (Wierzba-Bobrowicz et al. 2005), at least in some cases such as suicides (Bernstein et al. 2009). However, none of our subjects died from suicide.
Our results support the concept of a neurodevelopmental deficit in schizophrenia, particularly in glutamatergic neurons, involving subtle alterations to neuropil microcircuits rather than neuronal number or position. It remains to be determined whether sub-syndromes of the disease affect different areas of cortex or are associated with different degrees of change.
Acknowledgments
Our original work was supported by grants from the Stanley Foundation and Glaxo Wellcome. It was done in close collaboration with T. R. E. Barnes, M. Bauer, A. Davis, S. M. Gentleman, S. R. Hirsch, C. Hornstein, M. Kanani, P.D. Leclercq, A. Mortimer, W. Y. Ong, T. S. Patel, K. Radewicz R. Reynolds and K.A. von Bussmann. I thank C. J. Bruton and M. C. Royston for valuable help at various stages, K. MacRae for statistical advice, and S. M. Ansell, T. B. Bull, C. M. Lee, C. T. Lee and L. B. Moran for expert technical assistance.
References
- Akbarian S, Bunney WE, Potkin SG, et al. Altered distribution of nicotinamide-adenine dinucleotide phosphate-diaphorase cells in frontal lobe of schizophrenics implies disturbances of cortical development. Arch Gen Psychiatry. 1993;50:169–177. doi: 10.1001/archpsyc.1993.01820150007001. [DOI] [PubMed] [Google Scholar]
- Amaral MD, Pozzo-Miller L. The dynamics of excitatory synapse formation on dendritic spines. Cellscience. 2009;5:19–25. [PMC free article] [PubMed] [Google Scholar]
- Andreasen NC, Smith MR, Jacoby CG, et al. Ventricular enlargement in schizophrenia: definition and prevalence. Am J Psychiatry. 1982;139:292–296. doi: 10.1176/ajp.139.3.292. [DOI] [PubMed] [Google Scholar]
- Benes FM, Paskevich PA, Davidson J, et al. Synaptic rearrangements in medial prefrontal cortex of haloperidol-treated rats. Brain Res. 1985;348:15–20. doi: 10.1016/0006-8993(85)90353-1. [DOI] [PubMed] [Google Scholar]
- Benes FM, Todtenkopf MS, Taylor JB. A shift in tyrosine hydroxylase-immunoreactive varicosities (TH-IRv) from pyramidal (PN) to nonpyramidal (NP) neurons occurs in layer II of the anterior cingulate cortex of schizophrenics. Abstr Soc Neurosci. 1995;21:259. [Google Scholar]
- Bennett M. Positive and negative symptoms in schizophrenia: the NMDA receptor hypofunction hypothesis, neuregulin/ErbB4 and synapse regression. Aust N Z J Psychiatry. 2009;43:711–721. doi: 10.1080/00048670903001943. [DOI] [PubMed] [Google Scholar]
- Bernstein HG, Steiner J, Bogerts B. Glial cells in schizophrenia: pathophysiological significance and possible consequences for therapy. Expert Rev Neurother. 2009;9:1059–1071. doi: 10.1586/ern.09.59. [DOI] [PubMed] [Google Scholar]
- Bhatt DH, Zhang S, Gan WB. Dendritic spine dynamics. Annu Rev Physiol. 2009;71:261–282. doi: 10.1146/annurev.physiol.010908.163140. [DOI] [PubMed] [Google Scholar]
- Brodmann K. In: Vergleichende Lokalisationslehre der Grosshirnrinde in ihren Prinzipien dargestellt auf Grund des Zellenbaues. 3rd edn. Garey LJ, translator. Leipzig: Barth 1909; 1909/2006. Brodmann's Localisation in the Cerebral Cortex, New York: Springer 2006. [Google Scholar]
- Browning MD, Dudek EM, Rapier JL, et al. Significant reductions in synapsin but not synaptophysin specific activity in the brains of some schizophrenics. Biol Psychiatry. 1993;34:529–535. doi: 10.1016/0006-3223(93)90195-j. [DOI] [PubMed] [Google Scholar]
- Cao QL, Yan XX, Luo XG, et al. Prenatal development of parvalbumin immunoreactivity in the human striate cortex. Cereb Cortex. 1996;6:620–630. doi: 10.1093/cercor/6.4.620. [DOI] [PubMed] [Google Scholar]
- Catalá I, Ferrer I, Galofré E, et al. Decreased numbers of dendritic spines on cortical pyramidal neurons in dementia. A quantitative Golgi study on biopsy samples. Hum Neurobiol. 1988;6:255–259. [PubMed] [Google Scholar]
- Connor JR, Diamond MC. A comparison of dendritic spine number and type on pyramidal neurons of the visual cortex of old adult rats from social or isolated environments. J Comp Neurol. 1982;2:10. doi: 10.1002/cne.902100111. [DOI] [PubMed] [Google Scholar]
- Crow TJ, Ball J, Bloom SR, et al. Schizophrenia as an anomaly of development of cerebral asymmetry. A postmortem study and a proposal concerning the genetic basis of the disease. Arch Gen Psychiatry. 1989;46:1145–1150. doi: 10.1001/archpsyc.1989.01810120087013. [DOI] [PubMed] [Google Scholar]
- DeLisi LE, Goldin LR, Hamovit JR, et al. A family study of the association of increased ventricular size with schizophrenia. Arch Gen Psychiatry. 1986;43:148–153. doi: 10.1001/archpsyc.1986.01800020058007. [DOI] [PubMed] [Google Scholar]
- Done DJ, Crow TJ, Johnstone EC, et al. Childhood antecedents of schizophrenia and affective illness: social adjustments at age 7 and 11. Br Med J. 1994;309:699–703. doi: 10.1136/bmj.309.6956.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorph-Petersen KA, Delevich KM, Marcsisin MJ, et al. Pyramidal neuron number in layer 3 of primary auditory cortex of subjects with schizophrenia. Brain Res. 2009;1285:42–57. doi: 10.1016/j.brainres.2009.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falkai P, Bogerts B, Rozumek M. Limbic pathology in schizophrenia: the entorhinal region – a morphometric study. Biol Psychiatry. 1988;24:515–521. doi: 10.1016/0006-3223(88)90162-x. [DOI] [PubMed] [Google Scholar]
- Ferrer I, Fábregues I, Rairiz J, et al. Decreased numbers of dendritic spines on cortical pyramidal neurons in human chronic alcoholism. Neurosci Lett. 1986;69:115–119. doi: 10.1016/0304-3940(86)90425-8. [DOI] [PubMed] [Google Scholar]
- Fonnum F. Glutamate: a neurotransmitter in mammalian brain. J Neurochem. 1984;42:1–11. doi: 10.1111/j.1471-4159.1984.tb09689.x. [DOI] [PubMed] [Google Scholar]
- Frangou S, Murray RM. Imaging as a tool in exploring the neurodevelopment and genetics of schizophrenia. Brit Med Bull. 1996;52:587–596. doi: 10.1093/oxfordjournals.bmb.a011569. [DOI] [PubMed] [Google Scholar]
- Garey LJ, Ong WY, Barnes TRE, et al. Dendritic spine loss on pyramidal cells of temporal cortex in schizophrenia. Neuropathol Appl Neurobiol. 1993;19:186–187. [Google Scholar]
- Garey LJ, Ong WY, Patel TS, et al. Reduction in dendritic spine number on cortical pyramidal neurons in schizophrenia. Abstr Soc Neurosci. 1995;21:237. [Google Scholar]
- Garey LJ, Ong WY, Patel TS, et al. Reduced dendritic spine density on cerebral cortical pyramidal neurons in schizophrenia. J Neurol Neurosurg Psychiatry. 1998;65:446–453. doi: 10.1136/jnnp.65.4.446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garey LJ, Von Bussmann KA, Hirsch SR. Decreased numerical density of kainate receptor-positive neurons in the orbitofrontal cortex of chronic schizophrenics. Exp Brain Res. 2006;173:234–242. doi: 10.1007/s00221-006-0396-8. [DOI] [PubMed] [Google Scholar]
- Gehrmann J, Matsumoto Y, Kreutzberg GW. Microglia: intrinsic immunoeffector cell of the human brain. Brain Res Rev. 1993;20:269–287. doi: 10.1016/0165-0173(94)00015-h. [DOI] [PubMed] [Google Scholar]
- Glantz LA, Lewis DA. Assessment of spine density on layer III pyramidal cells in the prefrontal cortex of schizophrenic subjects. Abstr Soc Neurosci. 1995;21:239. [Google Scholar]
- Glantz LA, Lewis DA. Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch Gen Psychiatry. 2000;57:65–73. doi: 10.1001/archpsyc.57.1.65. [DOI] [PubMed] [Google Scholar]
- el Hachimi KH, Foncin JF. Loss of dendritic spines in Alzheimer's disease. C R Acad Sci III. 1990;311:387–402. [PubMed] [Google Scholar]
- van Haren NEM, Pol HEH, Schnack HG, et al. Progressive brain volume loss in schizophrenia over the course of the illness: Evidence of maturational abnormalities in early adulthood. Biol Psychiatry. 2008;63:106–113. doi: 10.1016/j.biopsych.2007.01.004. [DOI] [PubMed] [Google Scholar]
- Harrison PJ. On the neuropathology of schizophrenia and its dementia: neurodevelopmental, neurodegenerative, or both? Neurodegen. 1995;4:1–12. doi: 10.1006/neur.1995.0001. [DOI] [PubMed] [Google Scholar]
- Harrison PJ. Schizophrenia: a disorder of neuro-development. Curr Opin Neurobiol. 1997;7:285–289. doi: 10.1016/s0959-4388(97)80018-9. [DOI] [PubMed] [Google Scholar]
- Harrison PJ, McLaughlin D, Kerwin RW. Decreased hippocampal expression of a glutamate receptor gene in schizophrenia. Lancet. 1991;337:450–452. doi: 10.1016/0140-6736(91)93392-m. [DOI] [PubMed] [Google Scholar]
- Harvey I, Ron MA, Du Boulay G, et al. Reduction of cortical volume in schizophrenia on magnetic resonance imaging. Psychol Med. 1993;23:591–604. doi: 10.1017/s003329170002537x. [DOI] [PubMed] [Google Scholar]
- Hill JJ, Hashimoto T, Lewis DA. Molecular mechanisms contributing to dendritic spine alterations in the prefrontal cortex of subjects with schizophrenia. Mol Psychiatry. 2006;11:557–566. doi: 10.1038/sj.mp.4001792. [DOI] [PubMed] [Google Scholar]
- Hirsch SR, Das I, Garey LJ, et al. A pivotal role for glutamate in the pathogenesis of schizophrenia, and its cognitive dysfunction. Pharmacol Biochem Behav. 1997;56:797–802. doi: 10.1016/s0091-3057(96)00428-5. [DOI] [PubMed] [Google Scholar]
- Honea RA, Meyer-Lindenberg A, Hobbs KB, et al. Is gray matter volume an intermediate phenotype for schizophrenia? A voxel-based morphometry study of patients with schizophrenia and their healthy siblings. Biol Psychiatry. 2008;63:465–474. doi: 10.1016/j.biopsych.2007.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntley GW, Rogers SW, Moran T, et al. Selective distribution of kainate receptor subunit immunoreactivity in monkey neocortex revealed by a monoclonal antibody that recognizes glutamate receptor subunits GluR5/6/7. J Neurosci. 1993;13:2965–2981. doi: 10.1523/JNEUROSCI.13-07-02965.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttenlocher PR. Synaptic density in human frontal cortex: developmental changes and effects of aging. Brain Res. 1979;163:195–205. doi: 10.1016/0006-8993(79)90349-4. [DOI] [PubMed] [Google Scholar]
- Huttenlocher PR. Synapse elimination and plasticity in developing human cerebral cortex. Am J Ment Defic. 1984;88:488–496. [PubMed] [Google Scholar]
- Huttenlocher PR, Dabholkar AS. Regional differences in synaptogenesis in human cerebral cortex. J Comp Neurol. 1997;387:167–178. doi: 10.1002/(sici)1096-9861(19971020)387:2<167::aid-cne1>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Huttenlocher PR, de Courten C, Garey LJ, et al. Synaptogenesis in human visual cortex – evidence for synapse elimination during normal development. Neurosci Lett. 1982;33:247–252. doi: 10.1016/0304-3940(82)90379-2. [DOI] [PubMed] [Google Scholar]
- Jacobs B, Driscoll L, Schall M. Life-span dendritic and spine changes in areas 10 and 18 of human cortex: a quantitative Golgi study. J Comp Neurol. 1997;386:661–680. [PubMed] [Google Scholar]
- Johnstone EC, Crow TJ, Frith CD, et al. Cerebral ventricular size and cognitive impairment in chronic schizophrenia. Lancet. 1976;ii:924–926. doi: 10.1016/s0140-6736(76)90890-4. [DOI] [PubMed] [Google Scholar]
- Keshavan MS, Anderson S, Pettegrew JW. Is schizophrenia due to excessive synaptic pruning in the prefrontal cortex? The Feinberg hypothesis revisited. J Psychiatr Res. 1994;28:239–265. doi: 10.1016/0022-3956(94)90009-4. [DOI] [PubMed] [Google Scholar]
- Kim JS, Kornhuber HH, Schmid-Burgk W, et al. Low cerebrospinal fluid glutamate in schizophrenic patients and a new hypothesis on schizophrenia. Neurosci Lett. 1980;20:379–382. doi: 10.1016/0304-3940(80)90178-0. [DOI] [PubMed] [Google Scholar]
- Kolluri N, Sun Z, Sampson AR, et al. Lamina-specific reductions in dendritic spine density in the prefrontal cortex of subjects with schizophrenia. Am J Psychiatry. 2005;162:1200–1202. doi: 10.1176/appi.ajp.162.6.1200. [DOI] [PubMed] [Google Scholar]
- Leclercq P, Royston MC, Garey LJ, et al. A positional analysis of a subset of cortical neurons in the frontal cortex of schizophrenic brains. Neuropathol Appl Neurobiol. 1996;22:164. [Google Scholar]
- Leclerq PD, Garey LJ, Gentleman SM, et al. An automated position analysis of parvalbumin-immunoreactive cells in the cerebral cortex in control and schizophrenic post-mortem brain. Schizophr Res. 1998;29:91. [Google Scholar]
- Liddle PF, Friston KJ, Frith CD, et al. Patterns of cerebral blood flow in schizophrenia. Br J Psychiatry. 1992;160:179–186. doi: 10.1192/bjp.160.2.179. [DOI] [PubMed] [Google Scholar]
- Luby ED, Cohen BD, Rosenbaum F, et al. Study of a new schizophrenomimetic drug Sernyl. Arch Neurol Psychiatry. 1959;81:363–369. doi: 10.1001/archneurpsyc.1959.02340150095011. [DOI] [PubMed] [Google Scholar]
- Lund JS, Lund RD, Hendrickson AE, et al. The origin of efferent pathways from the primary visual cortex, area 17, of the macaque monkey as shown by retrograde transport of horseradish peroxidase. J Comp Neurol. 1975;164:287–303. doi: 10.1002/cne.901640303. [DOI] [PubMed] [Google Scholar]
- Mehraein P, Yamada M, Tarnowska-Dziduszco E. Quantitative study on dendrites and dendritic spines in Alzheimer's disease and senile dementia. Adv Neurol. 1975;12:453–458. [PubMed] [Google Scholar]
- Michel AE, Garey LJ. The development of dendritic spines in the human visual cortex. Hum Neurobiol. 1984;3:223–227. [PubMed] [Google Scholar]
- Mirra SS, Heyman A, McKeel D, et al. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology. 1991;41:479–486. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- Mrzljak L, Uylings HB, Kostovic I, et al. Prenatal development of neurons in the human prefrontal cortex. II. A quantitative Golgi study. J Comp Neurol. 1992;316:485–496. doi: 10.1002/cne.903160408. [DOI] [PubMed] [Google Scholar]
- Newman SJ, Gentleman SM. Microwave antigen retrieval in formaldehyde-fixed human brain tissue. Methods Mol Biol. 1997;72:145–152. doi: 10.1385/0-89603-394-5:145. [DOI] [PubMed] [Google Scholar]
- Ong WY, Garey LJ. Ultrastructural features of biopsied temporopolar cortex (area 38) in a case of schizophrenia. Schizophr Res. 1993;10:15–27. doi: 10.1016/0920-9964(93)90072-q. [DOI] [PubMed] [Google Scholar]
- Petralia RS, Wang YX, Wenthold RJ. The NMDA receptor subunits NR2A and NR2B show histological and ultrastructural localization patterns similar to those of NR1. J Neurosci. 1994;14:6102–6120. doi: 10.1523/JNEUROSCI.14-10-06102.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfefferbaum A, Zipursky RB, Lim KO, et al. Computed tomographic evidence for generalized sulcal and ventricular enlargement in schizophrenia. Arch Gen Psychiatry. 1988;45:633–640. doi: 10.1001/archpsyc.1988.01800310037005. [DOI] [PubMed] [Google Scholar]
- Prohovnik I, Dwork AJ, Kaufman MA, et al. Alzheimer-type neuropathology in elderly schizophrenia patients. Schizophr Bull. 1993;19:805–816. doi: 10.1093/schbul/19.4.805. [DOI] [PubMed] [Google Scholar]
- Radewicz K, Garey LJ, Gentleman SM, et al. Increase in HLA-DR immunoreactive microglia in frontal and temporal cortex of chronic schizophrenics. J Neuropathol Exp Neurol. 2000;59:137–150. doi: 10.1093/jnen/59.2.137. [DOI] [PubMed] [Google Scholar]
- Raedler TJ, Knable MB, Weinberger DR. Schizophrenia as a developmental disorder of the cerebral cortex. Curr Opin Neurobiol. 1998;8:157–161. doi: 10.1016/s0959-4388(98)80019-6. [DOI] [PubMed] [Google Scholar]
- Rakic P, Bourgeois JP, Goldman-Rakic PS. Synaptic development of the cerebral cortex: implications for learning, memory, and mental illness. Prog Brain Res. 1994;102:227–243. doi: 10.1016/S0079-6123(08)60543-9. [DOI] [PubMed] [Google Scholar]
- Reveley AM, Reveley MA, Clifford CA, et al. Cerebral ventricular size in twins discordant for schizophrenia. Lancet. 1982;1:540–541. doi: 10.1016/s0140-6736(82)92047-5. [DOI] [PubMed] [Google Scholar]
- Ross CA, Pearlson GD. Schizophrenia, the heteromodal association cortex and development: potential for a neurogenetic approach. Trends Neurosci. 1996;19:171–176. doi: 10.1016/s0166-2236(96)10022-9. [DOI] [PubMed] [Google Scholar]
- Scheibel ME, Lindsay RD, Tomiyasu U, et al. Progressive dendritic changes in aging human cortex. Exp Neurol. 1975;47:392–403. doi: 10.1016/0014-4886(75)90072-2. [DOI] [PubMed] [Google Scholar]
- Selemon LD, Goldman-Rakic PS. The reduced neuropil hypothesis: a circuit based model of schizophrenia. Biol Psychiatry. 1999;45:17–25. doi: 10.1016/s0006-3223(98)00281-9. [DOI] [PubMed] [Google Scholar]
- Selemon LD, Rajkowska G, Goldman-Rakic PS. Abnormally high neuronal density in the schizophrenic cortex. A morphometric analysis of prefrontal area 9 and occipital area 17. Arch Gen Psychiatry. 1995;52:805–818. doi: 10.1001/archpsyc.1995.03950220015005. [DOI] [PubMed] [Google Scholar]
- Smiley JF, Williams SM, Szigeti K, et al. Light and electron microscopic characterization of dopamine-immunoreactive axons in human cerebral cortex. J Comp Neurol. 1992;321:325–335. doi: 10.1002/cne.903210302. [DOI] [PubMed] [Google Scholar]
- Sommer H, Quandt J. Langzeitbehandlung mit Chlorpromazin im Tierexperiment. Fortschr Neurol Psychiatr. 1970;38:466–491. [PubMed] [Google Scholar]
- Stevens JR, Casanova MF, Bigelow L. Gliosis and schizophrenia. Biol Psychiatry. 1988;24:727–729. doi: 10.1016/0006-3223(88)90154-0. [DOI] [PubMed] [Google Scholar]
- Streit WJ. The role of microglia in brain injury. Neurotoxicology. 1996;17:671–678. [PubMed] [Google Scholar]
- Suddath RL, Casanova MF, Goldberg TE, et al. Temporal lobe pathology in schizophrenia: a quantitative magnetic resonance imaging study. Am J Psychiatry. 1989;146:464–472. doi: 10.1176/ajp.146.4.464. [DOI] [PubMed] [Google Scholar]
- Sweet RA, Henteleff RA, Zhang W, et al. Reduced dendritic spine density in auditory cortex of subjects with schizophrenia. Neuropsychopharm. 2009;34:374–389. doi: 10.1038/npp.2008.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uranova NA, Orlovskaya DD, Apel K, et al. Morphometric study of synaptic patterns in the rat caudate nucleus and hippocampus under haloperidol treatment. Synapse. 1991;7:253–259. doi: 10.1002/syn.890070402. [DOI] [PubMed] [Google Scholar]
- Vincent JP, Kartalovski B, Geneste P, et al. Interaction of phencyclidine (‘angel dust’) with a specific receptor in rat brain membranes. Proc Natl Acad Sci U S A. 1979;76:4678–4682. doi: 10.1073/pnas.76.9.4678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Bussmann KA, Rodway A, Gentleman SM, et al. Decreased glutamate binding sites in the left orbitofrontal cortex of chronic schizophrenics. Schizophr Res. 1996;18:175. [Google Scholar]
- Weinberger DR, Marenco S. Schizophrenia as a neurodevelopmental disorder. In: Hirsch SR, Weinberger DR, editors. Schizophrenia. 2nd edn. Hoboken, NJ: Wiley-Blackwell; 2003. pp. 326–348. [Google Scholar]
- Weinberger DR, Torrey EF, Neophytides AN, et al. Lateral cerebral ventricular enlargement in chronic schizophrenia. Arch Gen Psychiatry. 1979;36:735–739. doi: 10.1001/archpsyc.1979.01780070013001. [DOI] [PubMed] [Google Scholar]
- Wierzba-Bobrowicz T, Lewandowska E, Lechowicz W, et al. Quantitative analysis of activated microglia, ramified and damage of processes in the frontal and temporal lobes of chronic schizophrenics. Folia Neuropathol. 2005;43:81–89. [PubMed] [Google Scholar]
- Yan XX, Garey LJ, Jen LS. Prenatal development of NADPH-diaphorase-reactive neurons in human frontal cortex. Cereb Cortex. 1996;6:737–745. doi: 10.1093/cercor/6.5.737. [DOI] [PubMed] [Google Scholar]
- Yan XX, Cao QL, Luo XG, et al. Prenatal development of calbindin D-28K in human visual cortex. Cereb Cortex. 1997;7:57–62. doi: 10.1093/cercor/7.1.57. [DOI] [PubMed] [Google Scholar]
- Zipursky RB, Lim KO, Sullivan EV, et al. Widespread cerebral gray matter volume deficits in schizophrenia. Arch Gen Psychiatry. 1992;49:195–205. doi: 10.1001/archpsyc.1992.01820030027004. [DOI] [PubMed] [Google Scholar]
- Zukin SR, Zukin RS. Specific [3H]phencyclidine binding in rat central nervous system. Proc Natl Acad Sci U S A. 1979;76:5372–5376. doi: 10.1073/pnas.76.10.5372. [DOI] [PMC free article] [PubMed] [Google Scholar]