

Abstract
Intraneuronal accumulation of ubiquitin conjugates is a pathological feature of neurodegenerative disorders such as Alzheimer’s disease (AD). Previous reports propose that accumulation of ubiquitinated species in AD is a result of inhibition of proteasomal activity by amyloid-β (Aβ) peptides, which leads to blocking of ubiquitin-dependent protein degradation by the proteasome. Here, we provide additional insight into proteasomal dysfunction by Aβ peptides by revealing that aggregated forms of Aβ(1−42) peptides (especially small oligomers) are, in fact, competitive substrates for the chymotrypsin-like activity of the human 20S (h20S) proteasome. In addition to examining the kinetics of the h20S proteasome activity in the presence or absence of Aβ peptides, we use gel electrophoresis, LC-MS, and TOF-MS/MS analyses to examine the degradation of Aβ(1−42) by the h20S proteasome. The observed peptide fragments resulting from proteolytic cleavage of Aβ were consistent with predicted cleavage sites from proteasome degradation. These results support that the interaction of Aβ peptides with the proteasome may play a mechanistic role in proteasomal dysfunction in AD pathology. These results may also reveal a previously unknown natural pathway for clearance of Aβ in normal or diseased cells.
Keywords: Amyloid, degradation, proteasome, kinetics, Alzheimer’s disease
Many neurodegenerative disorders are characterized by the intraneuronal accumulation of misfolded amyloidogenic proteins (1). In Alzheimer’s disease (AD), for instance, amyloid-β (Aβ) peptides are the major component in senile plaques (2,3) and are believed to play a central role in the development of the disease (4). These peptides are derived from the proteolytic cleavage of amyloid precursor protein (APP) by a series of secretase enzymes (5,6); accumulation of Aβ is believed to be a result of an imbalance between the production and clearance of this peptide in the brain. Although significant effort has revealed the mechanistic details for the origin of Aβ in AD (7), natural pathways for degradation and removal of this peptide remain unclear. Previous reports provide some evidence that the proteolytic enzymes neprilysin, insulin-degrading enzyme, and presequence peptidase could be involved in Aβ clearance in the body (8−11). Identification of other natural enzymes that can degrade Aβ peptides may provide a more complete picture for this important disease-related biological process and may also lead to new treatment strategies that target the removal of Aβ in AD (12). Here, we demonstrate that aggregated forms of Aβ(1−42) peptides are competitive substrates for the human 20S (h20S) proteasome. We show that the proteasomal degradation machinery is capable of cleaving Aβ(1−42) peptides in a dose-dependent manner, without significantly affecting the overall catalytic function of the proteasome. These results challenge previous reports that Aβ peptides cause proteasomal dysfunction through inhibition of its enzymatic activity (13−15).
The proteasome is a large protein complex comprised of a barrel-shaped 20S functional proteolytic subunit and two 19S regulatory domains (16). The 20S domain is characterized as having three different types of proteolytic activity: a chymotrypsin-like, a trypsin-like, and a peptidyl-glutamyl-peptide activity that collectively function to degrade misfolded and oxidized proteins (17). Previous work proposed that Aβ(1−40) peptides localize inside the 20S subunit along the active proteolytic site and inhibit the chymotrypsin-like activity of the proteasome (14,15). Aβ(1−42) peptides were subsequently reported to impair all three types of proteolytic activity of the 20S proteasome in a dose-dependent manner (13).
In order to provide a more detailed mechanistic analysis for the reported (13−15) decrease in activity of the 20S proteasome by Aβ(1−42), we formulated three different preparations of Aβ to determine which assembly state of the peptide (monomers, oligomers, or fibrils) had the most significant effect on the chymotrypsin-like activity of purified h20S proteasome. Figure 1 shows that preparations of Aβ peptides comprising ∼18% oligomers (i.e., trimers and tetramers) and no fibrils inhibited proteasome activity more effectively than Aβ preparations containing essentially all Aβ monomers or a significant fraction (∼76%) of Aβ fibrils (see Figure S1 in the Supporting Information for the characterization of these Aβ preparations). The proteasome activity (as determined by its ability to cleave a fluorogenic peptide substrate, LLVY-AMC) was attenuated by 35% when exposed to a 10 μM solution of the oligomeric Aβ preparation compared with the reaction in the absence of Aβ, while exposure to 10 μM Aβ in monomeric or fibrillar form reduced only 23% and 15% of the proteasome activity, respectively (Figure 1). Additionally, Figure S2 in the Supporting Information shows that proteasome activity was suppressed by concentrations of oligomeric Aβ peptides as low as 1 μM. These results are in agreement with previous reports on the effect of Aβ oligomers on proteasome activity (13).
Figure 1.

Relative chymotrypsin-like activity of the h20S proteasome with or without the presence of 10 μM fibrillar, oligomeric, or monomeric Aβ(1−42). Statistically significant differences were found for all three assembly states of Aβ(1−42) compared with activity in the absence of Aβ peptide. Statistical significance denoted as *p < 0.01, **p < 0.001. Each data point represents the average from three independent runs.
Kinetic analysis of the hydrolysis of the proteasome substrate LLVY-AMC as a function of the concentration of oligomeric Aβ(1−42) peptides showed that the Aβ peptides inhibited the chymotrypsin-like activity of the h20S proteasome in a dose-dependent manner (Figure 2). We incubated various concentrations of LLVY-AMC substrate (0−200 μM) in the presence of different concentrations of oligomeric Aβ(1−42) peptides (0−5 μM) for 20 min in buffer prior to addition of purified h20S proteasome. We subsequently incubated this proteasome solution for 30 min prior to calculating the velocity of product formation. Table 1 presents the values for Vmax and observed KM (KM,obs, the concentration of substrate that produces half-maximal velocity in the presence of a competitor) for the LLVY-AMC substrate as a function of the concentration of oligomeric Aβ peptides; these values were estimated using standard Michaelis−Menten analysis (18). Interestingly, we found that the KM,obs of substrate LLVY-AMC increased from 5.9 ± 0.8 to 38 ± 9.1 μM upon the addition of increasing concentrations of Aβ(1−42), while Vmax remained essentially constant at all concentrations of Aβ (from 0.031 ± 0.001 to 0.037 ± 0.003 μM·min−1). The apparent inhibition constant, Ki, for Aβ(1−42) and the proteasome was 0.83 ± 0.40 μM (estimated by averaging the Ki values from all three sets of Aβ concentrations), which represents the concentration of Aβ required to decrease the maximal rate of the reaction to half of the uninhibited value. The constant values for Vmax with concomitant increasing KM,obs values as a function of increasing Aβ concentrations suggests that Aβ(1−42) might be a competitive inhibitor for the proteasome (18); that is, Aβ(1−42) interacts competitively with the same site on the h20S proteasome as the LLVY-AMC substrate. Alternatively, the kinetic behavior shown in Figure 2 could also arise if the Aβ(1−42) peptides acted as a substrate for the proteasome.
Figure 2.

Kinetic analysis of the chymotrypsin-like activity of the h20S proteasome in the presence of 0, 0.5, 1, or 5 μM oligomeric Aβ(1−42) peptides.
Table 1. Values for Kinetic Parameters of the Chymotrypsin-Like Activity of the Proteasome in the Presence of Different Concentrations of Oligomeric Aβ(1−42) Peptides.
| Aβ conc (μM) | KM,obs (μM) | Ki (μM) | Vmax (× 10−2 μM/min) |
|---|---|---|---|
| 0 | 5.9 ± 0.8a | 3.1 ± 0.1 | |
| 0.5 | 8.4 ± 1.3 | 1.2 | 3.1 ± 0.1 |
| 1 | 20 ± 4.2 | 0.4 | 3.5 ± 0.2 |
| 5 | 38 ± 9.1 | 0.9 | 3.7 ± 0.3 |
KM,obs equals the apparent KM of the LLVY-AMC substrate when no Aβ is present.
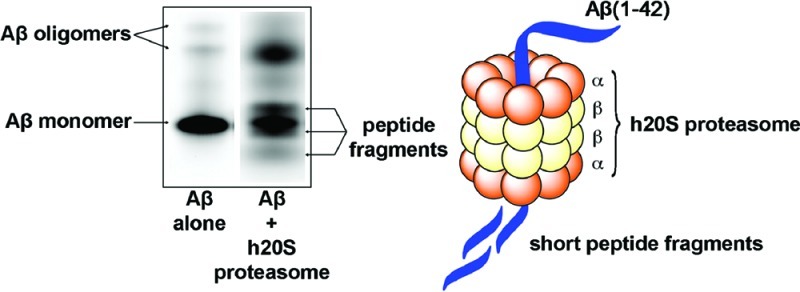
To examine whether Aβ(1−42) peptides could be degraded by the proteasome, we analyzed by 16% SDS−PAGE gel the reaction of the h20S proteasome with oligomeric Aβ(1−42) carrying a fluorophore on the N-terminus (Figure 3). We found three new fluorescent gel bands (presumably representing N-terminal fragments of Aβ) after the fluorescently labeled Aβ(1−42) was incubated with the h20S proteasome for 24 h at 37 °C; these bands were not present when the Aβ peptides were incubated in the absence of the h20S proteasome. Additionally, incubation of the fluorescently labeled Aβ(1−42) with the h20S proteasome in the presence of commercial proteasome inhibitors lactacystin, epoxomicin, bortezomib, or MG-132 resulted in significant reduction in the cleavage of the Aβ peptides compared with incubation of the peptide and h20S proteasome alone (Figure 3). Gel analysis of four commercial Aβ-derived peptides (also carrying fluorophores on their N-termini) suggested that the electrophoretic mobility of Aβ fragments did not necessarily correlate with the molecular weights of the peptides (see Figure S3 in the Supporting Information). We, therefore, could not accurately determine the size of the N-terminal fragments of fluorescently labeled Aβ(1−42) from the gel shown in Figure 3.
Figure 3.

Fluorescence gel image of aggregated Hilyte fluor 488-labeled Aβ(1−42) peptides incubated with or without the presence of h20S proteasome and in the presence of h20S proteasome and four different proteasome inhibitors. The arrows indicate Aβ monomers, small oligomers of Aβ (i.e., trimers and tetramers), or the new fluorescent bands that presumably represent the N-terminal fragments of Aβ formed from reaction with the h20S proteasome.
In order to determine the identity of some of the peptide fragments from degradation of Aβ(1−42) by the proteasome, we incubated label-free oligomeric Aβ(1−42) peptides with the h20S proteasome and analyzed the reaction by RP-HPLC (Figure 4A). Digestion of Aβ(1−42) by the h20S proteasome resulted in the formation of several prominent UV-active peaks by HPLC. In contrast, the HPLC trace of Aβ(1−42) peptide that was incubated in the absence of h20S proteasome only showed one prominent peak. The major UV-active peaks in both samples were analyzed by mass spectrometry (MS), leading to the assignment of many fragments of Aβ in the sample that contained the h20S proteasome (Figure 4B) (19). In order to further support that the h20S proteasome can degrade Aβ(1−42) peptides, we again incubated label-free oligomeric Aβ(1−42) peptides with the h20S proteasome and analyzed the reaction mixture by TOF-MS/MS. This analytical method made it possible to conclusively identify the observable peptide fragments from degradation of Aβ by the proteasome, since it provided information on the peptide sequence of each species. This method again revealed many small peptide fragments resulting from reaction of oligomeric Aβ(1−42) peptides with the h20S proteasome (Figure 4C). The identification of several N-terminal peptide fragments indicated that N-terminal residues 6−20 of Aβ(1−42) are most susceptible to cleavage by the proteasome. Many peptide fragments identified by TOF-MS/MS were consistent with the peptide fragments identified by LC-MS, such as Aβ(1−7), Aβ(1−9), Aβ(7−14), Aβ(7−15), Aβ(8−15), and Aβ(1−15). We attribute the absence of C-terminal fragments in these analyses to the known poor detection signal of hydrophobic fragments of Aβ by MS (20). The poor detection of C-terminal fragments may be a result of their general propensity to aggregate into fibrils (21), which may not be conducive to MS analysis.
Figure 4.
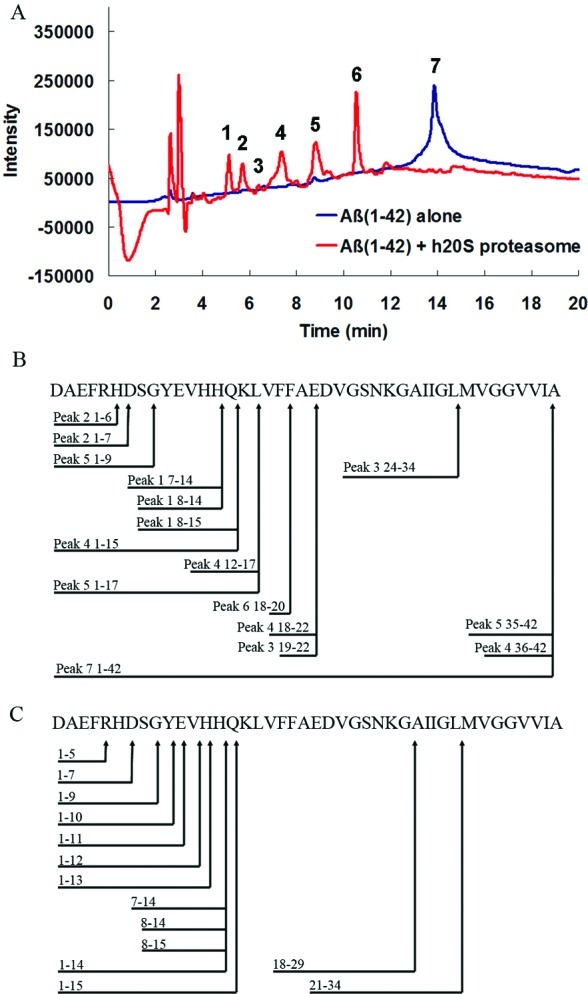
RP-HPLC and mass spectrometry (MS) analyses of peptide fragments formed from reaction of Aβ(1−42) peptides with the h20S proteasome: (A) RP-HPLC traces of Aβ(1−42) with (red) or without (blue) incubation with h20S proteasome; (B) summary of the amino acid sequence of the observed fragments of Aβ(1−42) and the proposed h20S proteasome cleavage sites as estimated by LC-MS analysis of each HPLC peak (for a list of masses, see Table S1 in the Supporting Information); (C) summary of the amino acid sequence of the observed fragments of Aβ(1−42) and the proposed h20S proteasome cleavage sites as estimated by TOF-MS/MS (for a list of masses, see Table S2 in the Supporting Information).
These results from gel electrophoresis and from MS analyses strongly support that Aβ(1−42) can act as a substrate for the h20S proteasome. To examine whether the observed peptide fragments were consistent with the known proteolytic activity of the proteasome, we compared the fragmentation pattern of Aβ(1−42) shown in Figure 4B,C to two predictive models for cleavage of Aβ(1−42) peptides by the proteasome (22): PAProC (23,24) and MAPPP (25,26). This comparison revealed that the observed cleavage sites on Aβ were, to a large extent, consistent with these proteasome cleavage prediction models (see Figure S4 in the Supporting Information). These results, therefore, suggest that the observed degradation of Aβ arises from the normal proteolytic function of the proteasome.
In summary, this work provides new insight into the impairment of the cellular proteasomal degradation machinery that is typically associated with AD and other amyloidogenic neurodegenerative disorders (27,28) . In contrast to previous reports suggesting that Aβ peptides act as inhibitors of proteasome activity (13−15), the results presented here suggest that the impairment of proteasomal function by Aβ may arise from the competition of natural proteasomal substrates with increasing concentrations of toxic oligomeric Aβ peptides within the cells of patients with AD. In addition to supporting the hypothesis that Aβ peptides play a central role in AD pathology (4) (perhaps through their effect on proteasome function (29,30)), these results reveal that the proteasome may also potentially play a role in the natural degradation and clearance of Aβ peptides in cells. Finally, these findings may contribute to efforts aimed at clearing Aβ peptides as a therapeutic strategy for the treatment of AD (12).
Methods
Materials
Lyophilized Aβ(1−42) was purchased from GL Biochem Ltd. N-terminal labeled Hilyte fluo Aβ(1−17), Aβ(25−35), Aβ(1−42), and Aβ(1−40) were purchased from AnaSpec. Hexafluoroiospropanol (HFIP) was purchased from Sigma Aldrich. The purified human 20S proteasome was purchased from Boston Biochem. Fluorogenic peptide N-succinyl-Leu-Leu-Val-Tyr-AMC (LLVY-AMC) was purchased from Enzo Life Science. Monoclonal anti-Aβ IgG (clone 6E10, Convance Cat. no. SIG-39320, derived from residues 1−17 of Aβ peptide as antigens) and rabbit polyclonal anti-mouse IgG (anti-mouse IgG H+L conjugated with alkaline phosphatase) were obtained from Abcam (Cambridge, MA). Water (18.2 μΩ/cm) was filtered through a NANOPure Diamond (Barnstead) water purification system before preparation of all aqueous solutions.
Preparation of Aβ(1−42)
Monomeric Aβ was prepared as previously described with some modifications (31). Briefly, Aβ(1−42) was initially solubilized in 100% 1,1,1,3,3,3-hexafluoro-2-propanal (HFIP) to 1 mM concentration at RT for 21 h on a shaker. The solution was sonicated and vortexed before it was diluted in cold nanopure water (2:1 H2O/HFIP). Aliquoted fractions were lyophilized for 2 days to remove HFIP and water, followed by storage in parafilm-sealed eppendorf tubes at −80 °C with desiccant until used. Monomeric Aβ was obtained by suspension of the peptides in nanopure water to a concentration of 100 μM immediately prior to use. For preparation of Aβ oligomers, the peptides were suspended in nanopure water to a concentration of 100 μM and incubated for 3 days at 4 °C. Fibrillary Aβ was obtained by dissolving Aβ in nanopure water to a concentration of 100 μM and incubating for 3 days at 37 °C. Western blot analysis using a mouse monoclonal antibody raised against residues 1−17 of Aβ (clone 6E10) revealed incubation conditions that resulted in preparations of Aβ that comprised different distributions of assembly states (see Figure S1 in the Supporting Information).
Proteasome Activity Assay
The proteasome chymotrypsin-like activity assay was carried out using fluorogenic peptide substrate Suc-Leu-Leu-Val-Tyr-AMC (LLVY-AMC). A 100 μL reaction mixture containing 100 ng of purified human 20S proteasome and 0 or 10 μM Aβ(1−42) (in monomeric, oligomeric, or fibrillar form) were preincubated in assay buffer (20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 0.5 mM ethylenediaminetetraacetic acid (EDTA), 0.035% SDS, pH 7.8) at 37 °C for 20 min before addition of 10 μM LLVY-AMC substrate. The reaction was incubated at 37 °C for an additional 30 min and the fluorescence of hydrolyzed 7-amino-4-methyl-coumarin (AMC) was detected with a 360/465 nm filter set in a spectrofluorimeter (Spectra max Gemini EM, Molecular Device).
Kinetic Analysis of Proteasome Activity with or without Aβ(1−42)
Kinetic analysis of proteasome chymotrypsin-like activity was performed using a 100 μL reaction mixture containing 40 ng of purified human 20S proteasome, LLVY-AMC substrate (0−200 μM), and Aβ(1−42) (0, 0.5, 1, and 5 μM) in assay buffer (20 mM HEPES, 0.5 mM EDTA, 0.035% SDS, pH 7.8). The solutions were incubated at 37 °C, and the formation of AMC was measured at 1 min intervals for 2 h with a 360/465 nm filter set in a spectrofluorimeter. The rate of substrate hydrolysis was calculated, and kinetic parameters KM,obs, Vmax, and Ki were determined by fitting the data to the Michaelis-Menten equation (18) using Origin 7 SR1 software (OriginLab Corp., Northampton, MA).
Digestion Assay of Fluorophore-Labeled Aβ(1−42) Using Gel Electrophoresis Analysis
A 1 mM solution of Hilyte fluor 488 labeled Aβ(1−42) was prepared in 1.0% NH4OH and further diluted in nanopure water to a final concentration of 100 μM. The fluorescently labeled Aβ was allowed to incubate at 4 °C for 3 days before use. A 4 μM solution of fluorescently labeled oligomeric Aβ peptides were incubated with 0 or 5 μg of h20S proteasome in 10 μL of assay buffer (20 mM HEPES, 0.5 mM EDTA, 0.035% SDS, pH 7.8) for 24 h at 37 °C. For reactions in the presence of proteasome inhibitors, 100 μM solutions of lactacystin, epoxomicin, bortezomib, or MG-132 in assay buffer were preincubated with 5 μg of h20S proteasome for 10 min at 37 °C, followed by addition of a solution of fluorescently labeled oligomeric Aβ peptides (to give a final concentration of 4 μM peptide) and continued incubation for 24 h at 37 °C. Reactions were stopped by adding 10 μL of 2× SDS sample loading buffer, followed by boiling for 10 min. Aliquots (10 μL) of these reaction mixtures were analyzed on 16% SDS−polyacrylamide gels with 8 M urea. The fluorescent gel bands were detected using a Typhoon 9400 variable mode imager (GE Healthcare Bio-Sciences Corp., Piscataway, NJ).
LC/MS and TOF-MS/MS Analyses of the Degradation of Aβ(1−42) Peptides by the Proteasome
A solution containing 400 μM oligomeric Aβ was incubated with 1.4 μg of h20S proteasome in 100 μL of assay buffer (20 mM HEPES, 0.5 mM EDTA, 0.035% SDS, pH 7.8) for 24 h at 37 °C. The proteasome protein was removed by centrifugation at 12,000 × g for 5 min in a centrifugal dialysis unit (Millipore Microcon YM-50). A Thermo LCQdeca mass spectrometer coupled with a HP1100 LC system was used for LC-MS analysis. The UV detection wavelength was set at 215 nm and positive ion mode electrospray ionization (ESI) was employed. A 5-μm Brownlee Spheri-5 Phenyl (250 mm × 4.6 mm) column (Applied Biosystems Inc., Foster City, CA) was used for separation with a flow rate of 1.0 mL/min. LC mobile A consisted of 5% acetonitrile in water with 0.1% TFA, and LC mobile phase B consisted of 90% acetonitrile with 0.1% TFA. The LC gradient started from 15% mobile phase B and was increased to 95% mobile phase B in 25 min, then lowered to 15% mobile phase B in 2 min and held at 15% mobile phase B for 3 min. Approximately 10% of the LC flow (∼0.10 mL/min) was introduced to the ESI source and the remaining was diverted to the waste. Xcalibur 1.2 software was used for data acquisition and processing.
For peptide sequencing, a digested sample of 8 μL of Aβ(1−42) treated with h20S was analyzed using an integrated system consisting of an autosampler Tempo nano LC system (AB Sciex) and a quadrupole time-of-flight (QqTOF) mass spectrometer (QSTAR Elite, AB Sciex), equipped with a nanoelectrospray ionization source. The sample was injected into the mass spectrometer through a C18 column at an ion spray voltage of 2300 eV. The amino acid composition produced by the QSTAR was searched using Mascot Daemon version 2.2.0 (Matrix Science Ltd.) against human_genome_NCBI database with a threshold of p < 0.5.
Acknowledgments
This work was partially supported by the Alzheimer’s Association (Grant NIRG-08-91651) and the Alzheimer’s Disease Research Center (Grant NIH 3P50 AG005131). We also acknowledge the NSF for a CAREER Award to J.Y. (Grant CHE-0847530) and for support of the mass spectrometry facility at UCSD (Grant CHE-0116662). We would like to thank Dr. Yongxuan Su for help with MS analyses.
Supporting Information Available
Characterization of different assembly states of Aβ(1−42), results from the inhibition of the chymotrypsin-like activity of the h20S proteasome with different concentrations of oligomeric Aβ(1−42), gel electrophoresis analysis of fluorescently labeled Aβ or Aβ-derived peptides, and comparison of predicted cleavage sites of Aβ(1−42) from LC/MS and TOF-MS/MS analyses. This material is available free of charge via the Internet at http://pubs.acs.org.
X.Z. and J.Y. conceived and designed the experiments, X.Z. performed the experiments, X.Z. and J.Y. co-wrote the manuscript and Supporting Information.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Taylor J. P.; Hardy J.; Fischbeck K. H. (2002) Toxic proteins in neurodegenerative disease. Science 296, 1991–1995. [DOI] [PubMed] [Google Scholar]
- Selkoe D. J. (2001) Alzheimer’s disease: Genes, proteins, and therapy. Physiol. Rev. 81, 741–766. [DOI] [PubMed] [Google Scholar]
- Kang J.; Lemaire H.-G.; Unterbeck A.; Salbaum J. M.; Masters C. L.; Grzeschik K.-H.; Multhaup G.; Beyreuther K.; Muller-Hill B. (1987) The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature 325, 733–736. [DOI] [PubMed] [Google Scholar]
- Haass C.; Selkoe D. J. (2007) Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid beta-peptide. Nat. Rev. Mol. Cell. Biol. 8, 101–112. [DOI] [PubMed] [Google Scholar]
- Haass C.; Selkoe D. J. (1993) Cellular processing of beta-amyloid precursor protein and the genesis of amyloid beta-peptide. Cell 75, 1039–1042. [DOI] [PubMed] [Google Scholar]
- Nunan J.; Small D. H. (2000) Regulation of APP cleavage by alpha-, beta- and gamma-secretases. FEBS Lett. 483, 6–10. [DOI] [PubMed] [Google Scholar]
- Selkoe D. J. (1994) Normal and abnormal biology of the beta-amyloid precursor protein. Annu. Rev. Neurosci. 17, 489–517. [DOI] [PubMed] [Google Scholar]
- Farris W.; Mansourian S.; Leissring M. A.; Eckman E. A.; Bertram L.; Eckman C. B.; Tanzi R. E.; Selkoe D. J. (2004) Partial loss-of-function mutations in insulin-degrading enzyme that induce diabetes also impair degradation of amyloid beta-protein. Am. J. Pathol. 164, 1425–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckman E. A.; Reed D. K.; Eckman C. B. (2001) Degradation of the Alzheimer’s amyloid beta peptide by endothelin-converting enzyme. J. Biol. Chem. 276, 24540–24548. [DOI] [PubMed] [Google Scholar]
- Shirotani K.; Tsubuki S.; Iwata N.; Takaki Y.; Harigaya W.; Maruyama K.; Kiryu-Seo S.; Kiyama H.; Iwata H.; Tomita T.; Iwatsubo T.; Saido T. C. (2001) Neprilysin degrades both amyloid beta peptides 1−40 and 1−42 most rapidly and efficiently among thiorphan- and phosphoramidon-sensitive endopeptidases. J. Biol. Chem. 276, 21895–21901. [DOI] [PubMed] [Google Scholar]
- Hama E.; Shirotani K.; Masumoto H.; Sekine-Aizawa Y.; Saido T. C. (2001) Clearance of extracellular and cell-associated amyloid beta peptide through viral expression of neprilysin in primary neurons. J. Biochem. 130, 721–726. [DOI] [PubMed] [Google Scholar]
- Malito E.; Hulse R.; Tang W. J. (2008) Amyloid beta-degrading cryptidases: insulin degrading enzyme, presequence peptidase, and neprilysin. Cell. Mol. Life Sci. 65, 2574–2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng B. P.; Green K. N.; Chan J. L.; Blurton-Jones M.; LaFerla F. M. (2008) A-beta inhibits the proteasome and enhances amyloid and tau accumulation. Neurobiol. Aging 29, 1607–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregori L.; Hainfeld J. F.; Simon M. N.; Goldgaber D. (1997) Binding of amyloid beta protein to the 20 S proteasome. J. Biol. Chem. 272, 58–62. [DOI] [PubMed] [Google Scholar]
- Gregori L.; Fuchs C.; Figueiredo-Pereira M. E.; Van Nostrand W. E.; Goldgaber D. (1995) Amyloid beta-protein inhibits ubiquitin-dependent protein degradation in vitro. J. Biol. Chem. 270, 19702–19708. [DOI] [PubMed] [Google Scholar]
- Hilt W.; Wolf D. H. (1996) Proteasomes: Destruction as a programme. Trends Biochem. Sci. 31, 96–102. [PubMed] [Google Scholar]
- Heinemeyer W.; Fischer M.; Krimmer T.; Stachon U.; Wolf D. H. (1997) The active sites of the eukaryotic 20 S proteasome and their involvement in subunit precursor processing. J. Biol. Chem. 272, 25200–25209. [DOI] [PubMed] [Google Scholar]
- Segel I. H. (1975) Enzyme kinetics: behavior and analysis of rapid equilibrium and steady-state enzyme systems, pp100−159, Wiley-Interscience, New York. [Google Scholar]
- Since the Aβ peptides likely continue to aggregate over the course of these studies and affect the UV signal of the parent Aβ peptide on the HPLC instrument, quantification of the time-dependent degradation of Aβ(1−42) by the h20S proteasome was not possible using this method of analysis.
- Trimpin S.; Deinzer M. (2005) Solvent-free mass spectrometry fro hydrophobic peptide sequence analysis and protein conformation studies. Biotechniques 39, 799–805. [DOI] [PubMed] [Google Scholar]
- Pike C. J.; Walencewicz-Wasserman A. J.; Kosmoski J.; Cribbs D. H.; Glabe C. G.; Cotman C. W. (1995) Structure-activity analyses of β-amyloid peptides: contributions of the β25−35 region to aggregation and neurotoxicity. J. Neurochem. 64, 253. [DOI] [PubMed] [Google Scholar]
- Saxova P.; Buus S.; Brunak S.; Kesmir C. (2003) Predicting proteasomal cleavage sites: a comparison of available methods. Int. Immunol. 15, 781–787. [DOI] [PubMed] [Google Scholar]
- Kuttler C.; Nussbaum A. K.; Dick T. P.; Rammensee H.-G.; Schild H.; Hadeler K.-P. (2000) An algorithm for the prediction of proteasomal cleavages. J. Mol. Biol. 298, 417–429. [DOI] [PubMed] [Google Scholar]
- Nussbaum A. K.; Kuttler C.; Hadeler K.-P.; Rammensee H.-G.; Schild H. (2001) PAProC: A prediction algorithm for proteasomal cleavages available on the WWW. Immunogenetics 53, 87–94. [DOI] [PubMed] [Google Scholar]
- Holzhütter H.-G.; Frömmel C.; Kloetzel P.-M. (1999) A theoretical approach towards the identification of cleavage-determining amino acid motifs of the 20s proteasome. J. Mol. Biol. 286, 1251–1265. [DOI] [PubMed] [Google Scholar]
- Holzhütter H.-G.; Kloetzel P.-M. (2000) A kinetic model of vertebrate 20S proteasome accounting for the generation of major proteolytic fragments from oligomeric peptide substrates. Biophys. J. 1196–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller J. N.; Hanni K. B.; Markesbery W. R. (2000) Impaired proteasome function in Alzheimer’s disease. J. Neurochem. 75, 436–439. [DOI] [PubMed] [Google Scholar]
- Keck S.; Nitsch R.; Grune T.; Ullrich O. (2003) Proteasome inhibition by paired helical filament-tau in brains of patients with Alzheimer’s disease. J. Neurochem. 85, 115–122. [DOI] [PubMed] [Google Scholar]
- LaFerla F. M.; Green K. N.; Oddo S. (2007) Intracellular amyloid-beta in Alzheimer’s disease. Nat. Rev. Neurosci. 8, 499–509. [DOI] [PubMed] [Google Scholar]
- Almeida C. G.; Takahashi R. H.; Gouras G. K. (2006) beta-amyloid accumulation impairs multivesicular body sorting by inhibiting the ubiquitin-proteasome system. J. Neurosci. 26, 4277–4288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlgren K. N.; Manelli A. M.; Stine W. B.; Baker L. K.; Krafft G. A.; LaDu M. J. (2002) Oligomeric and fibrillar species of amyloid-beta peptides differentially affect neuronal viability. J. Biol. Chem. 277, 32046–32053. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.