

Abstract
We review the elasticity of flexible and stiff polymer networks with permanent cross-links and synthesize these results into a unifying polymer chain network model. This framework is then used to address how the network elasticity becomes modified when the network cross-linking is thermoreversible in nature, changes in the stability of the network with deformation, and the effect of a variable rate of network deformation on the non-linear elastic response. Comparisons are made between this class of simplified network models with elasticity measurements performed on flexible chain and stiff fiber networks, both with permanent and associative cross-links. Although these network models are highly idealized, they are apparently able to capture many aspects of the elastic properties of diverse real networks.
I. Introduction
Synthetic and natural networks of polymers and self-assembling molecules are ubiquitous in manufacturing and biology, and the study of network elasticity has a long and distinguished scientific history.1 The theory of flexible polymer networks has received particular attention, but even in this case the quantitative role of interchain interactions in the dry rubber state (the so-called “entanglement” effect) has been slow to develop and the topic remains one of scientific and technological interest.1 Networks of stiff fibers have seen a large upsurge of interest recently because many networks of biological origin (and thus many biological materials) are comprised of such networks, which have an elasticity quite distinct from their flexible network counterparts. Specifically, the elasticity of flexible polymer networks is often characterized at moderate deformations by strain softening and positive normal stresses while stiff fiber networks often exhibit strain stiffening and negative normal stresses under deformation.2,3 The elasticity of these different classes of networks could thus not be more different from each other. Moreover, many real networks are comprised of network junctions or cross-links that involve a reversible association-dissociation process so that the junctions are not fixed for all time, although their time-averaged number may be an invariant. Deforming these associating networks leads to diverse complicating effects such as the breakdown of the network and subsequent slow recovery following cessation of an applied stress, an elastic response on the rate of network deformation, etc. Irreversible deformation effects such as fracture and plastic flow can also arise when network materials are subjected to large deformations.
There are diverse applications in manufacturing and biology relating to the control of the elastic properties of networks and it is clearly important to have effective and computationally tractable analytic models to help characterize these networks and to organize methods to control their properties through engineering of molecular structure and the control of thermodynamic conditions of gel formation. In our discussion below, we first review some models of this kind that have been found successful for describing networks of flexible polymer chains with fixed network junctions. A new and potentially useful model of the elasticity of stiff fiber networks is introduced in the course of this development based on molecular modeling and analytical arguments. This model reduces in its simplified form neglecting entanglement interactions to the phenomenological Fung model4–11 that is widely utilized in modeling biological materials when entanglement interactions are neglected in our model.
After a review and synthesis of prior results, we generalize our modeling to describe networks whose junctions form through thermally reversible association. This development requires an explanation of how these transient junctions influence the elasticity of the resulting network and we utilize results from rigidity percolation theory to develop a simplified model of the elasticity of such thermoreversible systems. We also consider changes in the thermodynamic stability of self-assembled networks with deformation and the thermodynamics of the network self-assembly based on highly simplified models. Comparison of special cases of our model to experimental observations on permanent and thermally reversible systems under diverse conditions show rather good agreement in many cases, indicating that these models offer a practical approach to describing the elasticity of both rubbery cross-linked materials and complex fiber gels formed by self-assembly.
In the following section, we summarize some models that have proven effective in describing flexible polymer networks, including effects associated with the interaction between network chains (‘chain entanglement’). We then address how the elasticity becomes modified when the cross-linking is associative and where the network forms by self-assembly. Finally, in Section III, we compare our idealized flexible and stiff chain models of network elasticity to experimental observations to illustrate the modeling results and the different types of elasticity that networks exhibit.
II. Elasticity of polymer networks with permanent junctions
A. Networks of flexible cross-linked chains (‘rubber’)
Models of the elasticity of rubbery materials composed of flexible chains have concentrated on minimal aspects of rubbery materials. In particular, the classical rubber elasticity models of Wall and Flory,12 James and Guth,13 Edwards,14 and many others have focused on the consequences of network connectivity. Classical theories treat rubbery materials as idealized networks of random walk chains whose junctions deform approximately affinely in response to a macroscopic deformation, thereby changing the entropy of the system. More recent work has emphasized inter-chain interactions or ‘entanglement’ interactions defined in terms of the topological constraint of chain un-crossability and correlations arising from molecular packing.
A minimal statistical mechanical model of rubber elasticity must incorporate three main features of the network chains: 1) A connected network of flexible chains,12–14 2) ‘Entanglement’ constraints,15–18 3) Finite volume of chains.19 The localization model (LM) of rubber elasticity is a minimal model that directly addresses these effects and we briefly summarize the essential ideas of this model,20–22 which also forms the foundation for our generalizations below for other more complex networks.
As a first approximation, the localization model (LM) takes the free energy ΔFnetwork of the network per unit volume to be proportional to the number of chains per unit volume νd where ΔFchain is the chain free-energy of deformation,
| (1) |
This result, which derives explicitly from classical elasticity network model calculations, assumes that each chain sees an equivalent molecular environment arising from its interaction with surrounding chains and this approximation thus amounts to a mean field approximation23 when applied more generally. For a cross-linked network, the number of chains per unit volume is taken to be proportional to the number of cross-links per unit volume, the cross-link density.
The second basic approximation is to assume that the network chains are Gaussian chains in our estimation of the chain connectivity contribution to ΔFnetwork. This approximation has its limitations, especially for large network extensions and for cases where the network chains are stiff or semi-flexible. (The treatment of finite extensibility effects and chain semi-flexibility are relatively well-understood and we return to incorporating these effects below.) For the present, we are concerned with understanding the basic nature of non-classical ‘entanglement’ contributions to network elasticity arising from inter-chain interactions that exist at high polymer concentrations.
Before initiating our modeling of the entanglement contribution to ΔFnetwork, we make some general physical observations that constrain our theoretical development:
Dry rubbery materials are normally nearly incompressible because of strong repulsive inter-segment interactions, despite the random coil nature of the polymers.
Confinement of network chains to a volume on the order of the hard core volume of the chains alters the average chain entropy relative to an unconfined chain.
The question is then how one calculates this entropy change under deformation conditions.
The Feynman-Kac functional limit theorem (FKLT) implies that confining a chain by any means leads to a universal change in the chain free energy of the flexible polymer chains, Fconfin ∼ <R2>/ξ2, where <R2> is the mean squared dimensions of the unconfined chain and ξ is the localization length describing the scale over which the chain is localized.24 <R2> is proportional to the chain length, N, so that Fconfin is extensive in the chain length. This limiting scaling relation is also known as ‘ground state dominance’25 based on a quantum analogy with the Brownian chain model. More generally, we have the more general scaling relation, Fconfin ∼ <R>df/ξdf for generalized random walks (having independent steps, but whose variance in step length is not finite) where <R> is the mean chain size and df is the fractal dimension of the chain, i.e., <R> ∼ N1/df. Evidently, we again obtain a confinement contribution Fconfin that is extensive in N. In the present instance, this localization effect derives from the hard core repulsive interactions between a given chain and its surrounding chains so that Fconfin is entropic in nature. In other words, hard core excluded volume interactions confine the chains to ‘tube-like’ regions localized around some average chain conformation and this chain confinement gives rise to a change of the entropy per link of the polymer chain. The FKFLT provides the fundamental mathematical underpinning of the tube model of polymer melts and rubber elasticity.
So far we have based our model on two fundamental limit theorems of broad mathematical and physical significance- the central limit theorem describing the statistical properties of random walk chains and the FKLT describing how the free energy of these chains changes with confinement (or the mathematical equivalent of this effect). This provides a sound foundation for a general theory of flexible polymer networks with strong localizing inter-chain interactions in the melt state. The problem of calculating how Fconfin becomes modified by macroscopic deformation of the rubbery material is more difficult. It seems reasonable to assume that under quasi-equilibrium conditions of deformation the FKLT relation still applies and the crux of the LM then reduces to estimating how ξ varies with deformation. There is certainly no reason to believe that ξ should vary affinely with the extent of macroscopic deformation λi along the laboratory-fixed axes, as might reasonably be argued for the coordinates defining the chain junctions in the network.
Gaylord and Douglas20,21 approached this basic problem by assuming, as Edwards23 had done before, that the network chains are contained within a random tube with local harmonic confining potential that is composed of segments that are oriented along three directions (x, y, z) in the lab-fixed frame. The harmonic tube model for the inter-chain interaction potential is chosen simply for mathematical convenience. The FKLT ensures that essentially any reasonable confining potential will lead to the same limiting results. We note that Heinrich et al. have also developed a popular tube model of rubber elasticity,26 based on the same chain localization concept, but these authors do not invoke the packing arguments to specify the molecular parameters in their model and the change in elasticity with network deformation and swelling. McLeish and coworkers27–29 have considered the tube model to describe the unusual scattering behavior in stretched polymer networks and have reviewed recent applications of tube model to modeling various aspects of the phenomenology of entangled polymer chain melts.
A random tube model can now be constructed by viewing the random tube as consisting of straight tube sections lying along the macroscopic deformation axes. The distribution function describing the distribution of the chain monomers within a random tube segment then factorizes into a product of Gaussian functions defined in terms of coordinates along the tube axis and a coordinate normal to the tube axis. Because of the separability property of random chains, the random tube segments can be imagined to be aligned along the three macroscopic deformation axes with equal probability, a construction first introduced by James and Guth in their approximate treatment of finite- extensibility effects on network elasticity.
To calculate the free energy change with deformation, the junction positions are taken to deform affinely, Ri = λi·Ri0, where Ri0 is the initial distance between a given network chain end along the ith macroscopic deformation coordinate direction. This argument leads to the classical affine network model of rubber elasticity. Of course, the affine deformation assumption is an approximation and other models of rubber elasticity take this as a starting point of their development.30,31 It is the present authors' opinion that non-affine contributions to the network elasticity do not really address the inter-chain interaction effects responsible for ‘entanglement’ contributions to the elasticity of dry rubbers.
To estimate ξ(λi), we argue that the hard core volume of the chain and localizing tube are comparable and invariant to deformation (see Fig. 1). The assumption of affine displacement is taken to mean that that length of the tube segments along the deformation axis deform affinely,
Fig. 1.

Molecular dynamics simulation of chain deformation under macroscopic extension (the macroscopic deformation direction is along the horizontal direction); left chain is before deformation and the right is after deformation. Red spheres indicate chain segments and the gray segments are those of surrounding chains in the proximity of the illustrated test chain. The number of chain segments in the test chain environment is nearly invariant, consistent with an invariance of the tube volume with deformation. This figure is for schematic purposes only.
| (2) |
where L0 is the length of the undeformed tube. The invariance of the tube volume with this deformation implies that the length of the tube times its cross-sectional area is the same before and after deformation, . This implies that , where the deformation scaling exponent β equals, β = − ½ and where ξx0 = ξy0 = ξz0 ≡ ξ0. This scaling relation is obviously quite different from an affine variation, ξx = λxξ0. Rubinstein and Panyukov32 have made arguments that β is positive (β = ½), as in the affine assumption model. Their calculation is based on a model of topological invariance of the network under macroscopic deformation in a model that does not consider chain packing effects.
These considerations lead to the LM20,21 expression for the free-energy density of a dry rubber,
| (3) |
where the classical network theory shear modulus Gc is proportional to the cross-link density (νx) and thermal energy (kBT),
| (4) |
In the affine network model of Wall and Flory the prefactor is C0 = 1 and in the classical non-affine ‘phantom model’ of James and Guth,13 Deam and Edwards,14 where the network junctions can fluctuate with the constraint of excluded volume interactions, C0 = ½. More generally, C0 depends on the details on network structure (dangling ends, network functionality, etc.)33 and is considered to be a measurable parameter characterizing a given network.34 In the absence of other information, we take the ‘phantom’ elasticity model ‘frontfactor’ value, C0 ≈ ½. The entanglement contribution Ge to the free energy density of the network,
| (5) |
includes a cross-term proportional to Gc [and thus νx; see eqn (4)] with a network parameter γ describing the influence of cross-linking on the inter-chain entanglement interaction. The cross-link independent contribution to Ge is identified with the plateau modulus of the polymer melt, .20,21 Much is known about the dependence of on molecular parameters35 (see below) and this phenomenology is consistent with the localization model.36 We note that eqn (3) is consistent with the Valanis-Landel separable form of the strain energy density of rubbery materials, a property of the strain energy density that has been established to be a good approximation for many rubbery materials.37 The Valanis-Landel property greatly simplifies calculation of the deformation properties of rubbers.38 Appendix A describes the stress strain relations deduced from the LM model that we use in our comparisons to experiment below. Appendix B discusses the predictions for describing the effect of network swelling on rubber network elasticity. This broad accord of the LM model with these network observations is encouraging.
An explicit derivation of the stress σ for uniaxial deformation of an incompressible rubber material (where λx = λ; λy = λ−1/2; λz = λ−1/2) is given in Appendix A for the LM model. In particular, σ has the form σLM = Gc (λ − λ−2) + Ge (1 − λ−3/2), which is eqn (A.2). In our comparisons to experiments below we often refer to the reduced stress I(λ) = σ/(λ − λ−2), which is the observed stress relative to that predicted by the classical rubber elasticity theory. In a ideal incompressible ‘neo-Hookean’ rubber, I(λ) is a constant that is equal the classical shear modulus, Gc.
B. Generalized rod polymer network elasticity model with permanent cross-links
No single model adequately describes the elasticity of all network materials. This situation is not surprising when one considers the different physical nature of the various network types. Of particular importance is the rigidity of the filaments that comprise the network; classical rubber elasticity models are simply not applicable to gels consisting of more rigid fibers. Relatively stiff fiber networks are observed in the context of many fiber networks (e.g., the microtubule network within the cellular cytoplasm and the fibrous collagen network in the extracellular matrix of connective tissues, carbon nanotube networks, etc.) found in biology and manufacturing. Our goal is to develop effective minimal models of network elasticity that can be used to describe the deformation behavior of both synthetic and natural flexible polymer networks or fiber network gels that are commonly found in biological materials. Below we focus on the extreme models of networks composed of ideally flexible and stiff polymer chains both with and without entanglement interactions. We also emphasize both the effects of junctions formed either as a consequence of permanent cross-links or associative cross-link interactions because permanent and associative networks are common.
The literature describing the elasticity of semi-flexible polymer networks and finite chain extensibility effects on polymer networks is extensive and we do not attempt to review these developments here since these effects are described adequately in other recent publications. Moreover, these effects can be readily incorporated into our (mean field) network model through a consideration of how the single chain elasticity is affected by the semi-flexibility and finite extensibility constraints. However, it is unclear whether molecular thread models, such as ours, that involve either ideally flexible or needle-like chain filaments can describe essential aspects of the non-linear elasticity of real rubbery materials and fiber networks. We simply assume the validity of these molecular chain models, the applicability of a mean field theoretical description, and then focus on the types of elasticity that these models predict in comparison to well-accepted experimental studies of network elasticity in flexible polymer and stiff fiber networks. Next, we introduce a molecular model of stiff polymer networks that directly extends the classical flexible chain network just described. As with our development of the flexible chain network elasticity model, our development relies heavily on former studies of network elasticity and the effort emphasizes the integration of fragmentary results into a more coherent picture of network elasticity and the comparative nature of flexible and stiff polymer networks.
We base our development of a minimal molecular model of the elasticity of a network of cross-linked rigid rods on a previous calculation by Vilgis, Boue, and Edwards39 who formulated a rod network theory based on a rigorous extension of the Deam and Edwards14 theory of the elasticity of a network of flexible ideal chains. In particular, they introduced a model in which needle-like rigid rods are tethered by freely pivoting junctions (tetrafunctional) to form a three-dimensional rod network. Curiously, they found that the elasticity of this rod network exactly recovers the functional form of the classical flexible chain elasticity model in the limit of small network deformations [See eqn (3)]. The entropic elasticity in this case derives entirely from the deformation of the junction positions rather than from stretching network chains (which are inextensible in the rod network model). Qualitatively new behavior was revealed at higher network deformation, however. The free energy of the rod network was found to increase exponentially with the square of the deformation parameter λ [see Eqn. (22)]. This is a “softer” increase in the free energy than the power-law singular increase found in the free energy of network deformation due to finite extensibility in flexible and semi-flexible network models. We analytically extend the results of Vilgis et al.39 by demanding that the limiting small and large deformation behavior of rod network be recovered. In particular, we propose the relation,
| (6) |
which by construction exactly recovers the asymptotic large and small deformation rod network elasticity results of Vilgis et al.39
Unfortunately, the analytic complexity of solving the non-linear integral equations involved in the mean field solution of the end-tethered rod network elasticity problem did not allow for a clear molecular parameter interpretation of the network ‘non-linear stiffening parameter’ b in eqn (6). We note, however, that eqn (6) is a familiar empirical relation in the non-linear elasticity of many real materials. In particular, eqn (6) is equivalent to the Fung hyperelasticity model,4–11 which provides a phenomenological description of the non-linear elasticity of diverse biological materials. This is first time that this widely discussed relation (i.e., the Fung model) has been deduced from a molecular model. The resulting stress-strain relationship in this model, considered below in our comparison to measurements, is described in Appendix B.
We formally introduce a rod network model that includes the effect of chain confinement by adding a generalized localization contribution to the network elasticity, , where νp = ½ and νp = 1 for networks comprised of ideal flexible and rigid chains, respectively,20,21 where radius of gyration Rg of an uncross-linked polymer chain in solution scales with chain mass as, Rg ∝ Mνp. Combining this contribution of the network elasticity with eqn (6) provides a minimal molecular model for the elasticity of rod polymer networks with interchain entanglement interactions. In particular, the proposed strain energy for a rod network is then,
| (7) |
We can expect local chain packing effects to be substantially less important in stiff polymer networks, but having stiff polymer chains should on the other hand amplify interactions between chains associated with the constraints of chain uncrossability and volume exclusion. Our comparisons to experiment below provide an opportunity to study these entanglement effects in stiff fiber networks within the frame of our model.
III. Elasticity of self-assembled networks
Many networks are formed by a self-assembly process involving the thermally reversible association of network chains. This process may involve the self-assembly of the network fibers, which in turn, exhibit branching to create a network (perhaps with other molecules that regulate the fiber branching process). For example, common gelatin networks self-assemble through the formation of stiff triple helices of the gelatin molecules that are linked by flexible chain ‘links’ where the chain helices are not well organized.40 The associative interactions between the monomers within the polymer network can be disrupted by heating or applied stress. Since networks of polymers with associative interactions and self-assembled fiber networks of bundled molecules are common, it would evidently be helpful to have a minimal model that captures essential aspects of these networks. For example, we can expect that deforming self-assembled or thermally reversible networks will ultimately cause a breakdown of network structure by virtue of the stress-induced disruption of the associations defining the network, with a consequent loss of network stiffness. Once the network has “melted” due to the imposition of such stresses, it should recover its pre-stressed state (eventually) through associations and dissociations occurring under a state of dynamic equilibrium. Next we introduce a minimal model to describe this effect. It is noted that if the network undergoes an instability under deformation, such as fracture or significant plastic flow, before melting then our theory does not apply; network deformation then leads to an irreversible modification of the network.
A. Simple model of the shear modulus T dependence of self-assembling networks
We treat this problem generally by considering simplified models of the emergence of elasticity and self-assembly. Rigidity percolation theory41,42 and effective medium theory indicate that the bulk and shear moduli of lattice structures composed of central force springs connecting the lattice points varies linearly with the fraction Φ of possible connected bonds, provided a sufficient number of bonds (rigidity percolation threshold) for the structure to exist as a solid. The rigidity percolation threshold is notably a greater concentration than the geometrical percolation threshold where infinite network first forms, i.e., greater constraints on the particle motions are generally required for rigidity. The effective medium treatment of rigidity percolation motivates taking the shear modulus to be proportional to the fraction Φ of associating species in the self-assembled state,
| (8) |
where is the limiting modulus (limiting linear regime due to linkages created by self-assembly) in the fully self-assembled state and Φ* corresponds to a critical amount of ruptured associations where rigidity is lost. The approximation in eqn (8) applies when Φ is near unity and has the advantage of not requiring the introduction of a new fit parameter, Φ*.
The extent of self-assembly Φ is the basic order parameter governing self-assembly and there has been much work to calculate this quantity for specific self-assembly models. Expressions for Φ for realistic assembly models often involve rather complex analytic relations that are not generally amenable to closed analytic expression. There is a simplified two-state model for Φ that provides a useful approximation for many practical applications and we adopt this relation here for our purposes. Specifically, we model Φ by the relation,43
| (9) |
where Δh and Δs are the enthalpies and entropies of the assembly process and kBT represents the thermal energy. The variation of Φ of many other more complicated assembly models is mimicked rather well by eqn (9), which can be approximated even further by expanding the free energy of assembly about the self-assembly transition temperature.43 This yields the alternative simplified relation,
| (10) |
where Do describes the temperature breadth of the transition and ζp is a constant (see [43] for the specification of these fit parameters in terms of the energetic variables of the self-assembly model). Eqn (10) has the same mathematical form as the occupation density from statistical mechanics of fermions and this functional form has been denoted the ‘Fermi function’ ΦF in the experimental literature, as discussed below.
We thus have the simple approximation for the T dependence of the shear modulus G0, which is prescribed by eqn (8) where Φ is given by either eqn (9) or the simplified expression eqn (10). Fig. 2 shows the variation of as a function of T for representative values of the energetic parameters Δh and Δs. We see that G0 drops to zero with increasing temperature in a sigmoidal manner as the temperature is increased. This phenomenon is observed in diverse biomaterials44,45 and nano-technology applications,46 where is often fit to the phenomenological Fermi function. (This function is apparently a phenomenological counterpart to the Vogel-Fulcher-Tammann relation for the temperature dependence of the viscosity and structural relaxation time of glass-forming and some self-assembling systems (e.g., see Kumar and Douglas47). Note that “two-state expression” for Φ in eqn (9), and the Fermi approximant defined by eqn (10), are nearly indistinguishable from each other so that either relation seems reasonable for quantifying T dependent changes in the elasticity of network solid materials. The Fermi function has been particularly utilized in food science field in the quantification of the mechanical properties of breakfast cereals,48 and other common food products, and recently this relation has been applied to the description of the T dependence of the elastic moduli of polymeric nanofibers, etc.46 (Recent observations of the formation of a thermally reversible network structure consistent with equilibrium branched polymer network structure in a model glass-forming polymer melt lends some support to the application of our model to glassy materials.49 The common use of the Fermi function in the characterization of network materials motivated our explanation of how the Fermi function can be approximately derived from a molecular model and stimulated our initial modeling of the elastic properties of self-assembled networks.
Fig. 2.
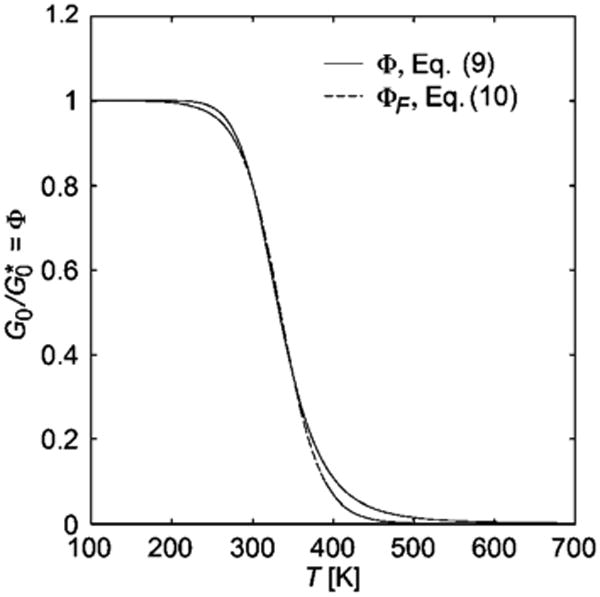
Variation of the self-assembly order parameter Φ or the reduced linear shear modulus with temperature from eqn (9) and (10), respectively. The illustrative curves are for Δh = − 35 kJ/mol and Δs = −105 J/mol K, and for Tp = 335 K and D0 = 25 K. Increasing the magnitude of Δs at fixed Δh causes the “melting’ transition to become progressively sharper, while reducing the transition temperature itself (inflection point in curve).
B. Deformation dependence of the stability of self-assembled networks
To describe the elastic properties of deformed networks formed by self-assembly, we must account for changes in the free energy of self-assembly accompanying the network deformation process. There has been much recent work quantifying the effect of applied deformation on binding constants in ligand-receptor pairs in the context of modeling cellular adhesion. Bell and others (see Bongrand50) have introduced a simplified model for how deformation alters association equilibrium constants. Specifically, Bell et al.51 argued that the free energy of the equilibrium constant governing the association and dissociation of the binding species is modified by simply adding a quadratic term in the extent of deformation with a phenomenological local force constant describing how the free energy of assembly (binary binding of the associating species) becomes modified by deformation. In the spirit of the minimal model introduced above to describe network elasticity in vanishing deformation limit, we introduce the simple Bell model expression for the deformation dependence of the free energy association constant governing the self-assembly process to obtain a deformation dependent expression for Φ,
| (11) |
where kf is the force constant mentioned above. In Fig. 3, we show the variation of Φ(λ) at a fixed T when λ is varied and we see that increasing λ causes Φ (and corresponding the reduced shear modulus, ) to decrease sigmoidally. We also show Φ(T,λ) in Fig. 4 as a function of T at fixed λ values where we see the self-assembly transition temperature (inflection point of Φ curve) simply shifts to lower T as λ increases. Eqn (11) leads us expect a general tendency for self-assembled networks to “melt” upon deformation and a corresponding tendency towards strain softening if deformation is large enough to induce network disordering.
Fig. 3.
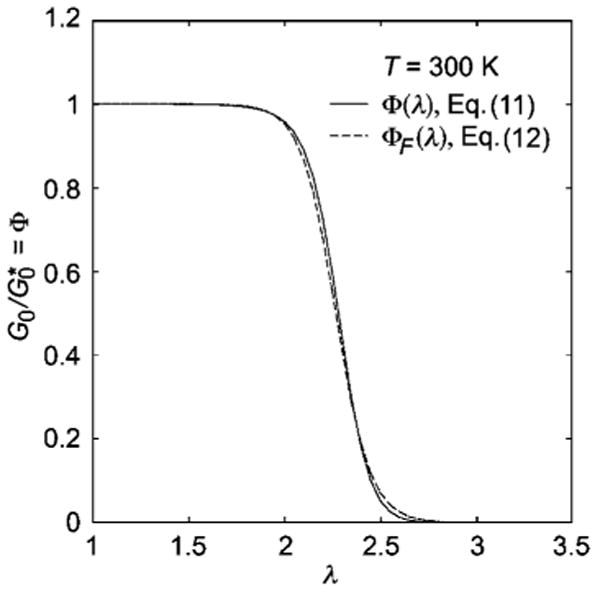
Variation of the self-assembly order parameter Φ or the reduced linear shear modulus with deformation ratio λ at constant temperature from eqn (11) and (12), respectively. The illustrative curves are for Δh = −40.75 kJ/mol, Δs = −70 J/mol K, kf = 12 kJ/mol and ζ = 120, λ* = 2.7, δ = 0.09.
Fig. 4.
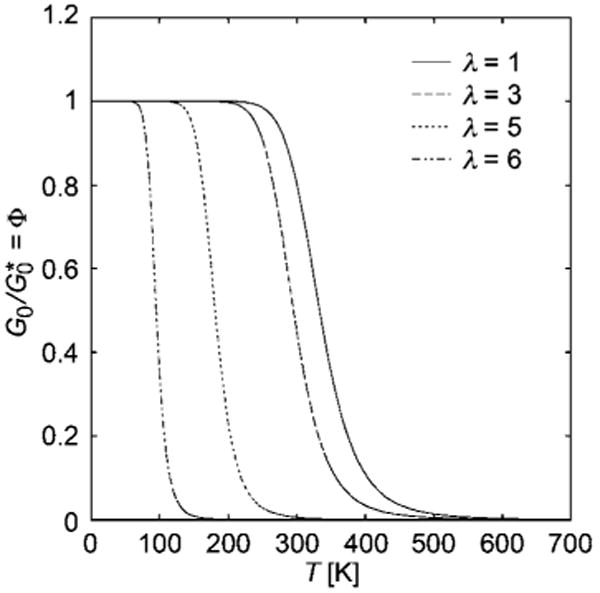
Variation of the self-assembly order parameter Φ and reduced linear shear modulus order with T for different fixed values of the stretch ratio λ from eqn (10). Illustrative curves are for Δh = −35 kJ/mol, Δs = − 105 J/mol K, and kf = 1 kJ/mol.
Eqn (10), in conjunction with eqn (11), provides a minimal model for describing how changes in the free energy of assembly with deformation alter the elastic properties of assembled structures change with deformation. The order parameter for self-assembly Φ(λ) exhibits a sigmoidal variation with increased deformation λ at a fixed temperature and we can similarly define a simple Fermi model approximant for Φ,
| (12) |
by analogy to eqn (10) where ζ and δ are adjustable parameters and λ* is the critical stretch at a fixed T at which softening initiates. As before, the Fermi approximant is most appropriate when the fiber self-assembly transition is sharp (i.e., cooperative).
C. Viscoelasticity of self-assembled networks
The idealized model for the elasticity of self-assembled networks in the previous section neglects relaxation processes associated with varying the rate at which strain is applied. In reality, relaxation processes over time progressively reduce the stress applied to the network when the application of stress is made at progressively lower rates. This is another complexity that must be confronted in modeling the elasticity of networks. Evidence for these strain rate effects have been reported for telechelic polymer networks and gelatin gels by Berret and Serero52 and Groot et al.,53 respectively. In the former system, strain hardening and stress maxima were found to be shear rate dependent. In the latter case, the initial shear modulus (G0), yield stress and yield strain of gelatin all varied with the applied shear rate. Of particular interest is the increase in all three mechanical properties with increasing strain rate. An assumption of the modeling of strain rate effects is that deformation rates remain low enough to remain in a regime where the equilibrium constant governing the self-assembly process remains well defined.
We may again draw upon recent work on modeling the viscoelastic properties of self-assembling fluid to model this kind of viscoelastic effect. Stukalin et al.54 have recently modeled stress relaxation of solutions of self-assembling polymers and have found the stress relaxation function ψ(t) of these solutions to generally have a stretched exponential form,
| (13) |
where 0 < α < 1 and τr is an average structural relaxation time. The exponent α primarily affects the averaging of the relaxation of the individual assembled chains over the size distribution that generally accompanies the self-association by virtue of Boltzmann's law. We adopt eqn (13) as a model of the stress relaxation process of our assembled networks. Further, we take the deformation rate (ω) at which the stress is applied to equal the reciprocal of time in eqn (13) and define t/τr = 1/ωτr by an overall deformation rate parameter, Γ. The relaxation stiffness at a finite dimensionless rate of network deformation Γ for our deformed networks is then taken to equal,
| (14) |
where Ω = exp[−(1/Γ)α]. A factor of this kind has been previously invoked by Berret and Serero52 to describe variable viscoelasticity as a function of Γ in their network elasticity measurements.
Many self-assembling systems recover their unstressed properties following a cessation of the applied stress as the system recovers its quiescent state. This phenomenon has been a particular focus of interest in actin networks subjected to stretching,55,56 a phenomenon that has been suggested to be of great significance in cellular function.57 We may model this recovery process by multiplying Φ in eqn (11) by a corresponding stress recovery function, R(t/τR) = (G0 − Gcessation) [1 − e−(t/τR)α] + Gcessation, where τR is a recovery time for network ‘rejuvenation’ and where Gcessation is the cross-link contribution to the modulus at the time of cessation of the applied stress, a quantity that can be calculated from the model above. Note that this expression implies that R(t) approaches its equilibrium value G0 at long times. In general, we expect recovery time τR to be on the order of the linear viscoelastic relaxation time τr in eqn (13). We emphasize that our modeling does not describe the common situation at very large deformation where the material fractures or suffers some instability or extensive plastic deformation that preempts that network melting process.
Combining eqn (13) and (14) with the entangled rod network model [eqn (7)] provides a simple model for the deformation rate dependence σ of these networks in uniaxial deformation,
| (15) |
Correspondingly, for a network of flexible chains with thermally reversible associations we have,
| (16) |
Many real associating networks form over long timescales and exhibit ‘aging’ behavior in their mechanical properties over long timescales. Sollich58,59 has developed an interesting constituitive modeling approach for ‘glassy’ material systems that is frequently cited as providing qualitative insights into measurements on biological thermoreversible gels. This model should be useful in the complementary case to our work where the polymer network is in a highly non-equilibrium glassy state, a situation that may be operative in the fiber networks in living systems where the network properties are affected by the presence of motor proteins, chemical reactions involving ATP, calcium reaction diffusion waves modulating the fiber assembly, etc.
III. Results and discussion
A. Application to flexible chain networks with permanent cross-links
It is well known that the classical theory of rubber elasticity has limited applicability to the description of dry rubbery materials composed of flexible polymer chains, although this model becomes quite a useful description to swollen polymer networks where interchain packing interactions are diminished by swelling.60,61 Many molecular models have been introduced to describe the elasticity of dry rubbery materials, but a recent evaluation of these models by Han, Horkay and McKenna38 indicated that only two models are empirically successful in fitting the elastic behavior of real rubbery materials– the localization model (LM) of Gaylord and Douglas and the Flory-Erman ‘junction fluctuation’ model. (The Flory-Erman model contains a parameter relating to fluctuation in the junction positions that affects the degree to which their displacement is affine under macroscopic deformation.) The LM has the advantage of physical transparency and makes predictions for the model parameters that have been validated by measurement. For example, McKenna et al.62 found that the LM describes torsional rigidity measurements on dry rubbers having a range of cross-linking densities where the classical term is fixed to its classical value (C0 = ½) and Ge was found to have exactly the form predicted by eqn (5), the entanglement contribution to the dry rubber elasticity Ge is linear in Gc and extrapolates to the plateau modulus in the limit of vanishing cross-linking density (See Appendix A). The LM prediction for the dependence of on molecular structure (e.g. chain cross-sectional dimension) also accords rather well with observations on numerous polymers.36 The junction-fluctuation model does not have this predictive capability.
We briefly illustrate the nature of dry rubber elasticity in comparison to the classical rubber elasticity theory in Fig. 5 and 6 (the reduced stress is defined relative to the classical rubber elasticity scaling), where comparison is made to the classical data of Rivlin and Saunders63 and Pak and Flory's64 on the compression and extension of dry rubbers (natural rubber and polydimethylsiloxane, respectively) at moderate cross-linking densities. The data is shown in the form of a reduced stress relative to the classical theory to emphasize the deviation from the classical theory.
Fig. 5.
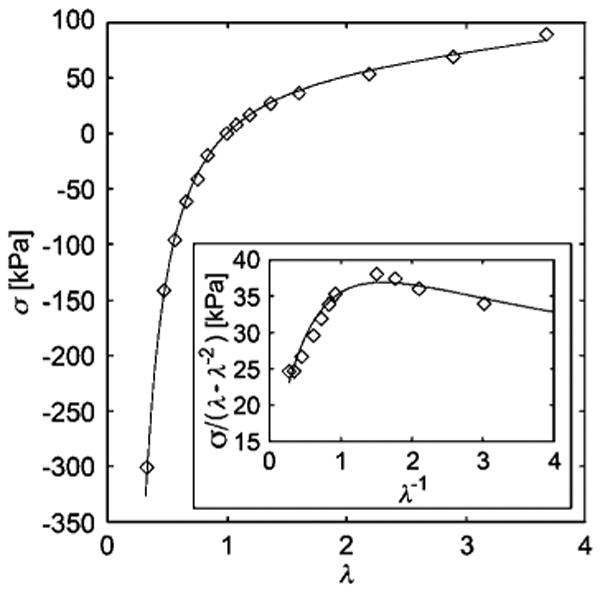
Stress-strain plot of Rivlin and Saunders's data63 for the compression and extension of vulcanized natural rubber. The data is fit with the localization model, with fitting parameters Gc = 12 kPa and Ge = 94 kPa with correlation coefficient R2 = 0.999 (maximum deviation in stress is 6.17 kPa). Scaling errors arising from digitization of the original plots are estimated to be no more than ±10%. Inset: reduced stress vs. inverse deformation or ‘Mooney-Rivlin’ plot.
Fig. 6.
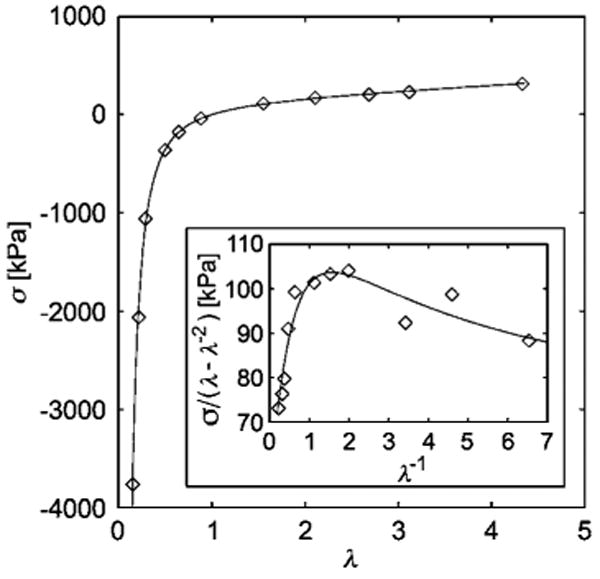
Stress-strain plot of Pak and Flory's data64 for the elongation and compression of polydimethylsiloxane. The data is fit with the localization model, with fitting parameters Gc = 55 kPa and Ge = 184 kPa, and with R2 = 0.999 (maximum deviation in stress is 98.6 kPa). Scaling errors arising from digitization of the original plots are estimated to be no more than ±10%. Inset: reduced stress vs. inverse deformation. The LM also compares well with biaxial extension,20,21 and torsional deformation measurements on dry rubbers62 and network elasticity of rubbers cross-linked in a deformed state.75,76 Torsional deformation62 measurements performed over an wide range of νx, where C0 was fixed to equal ½, yielded a fitted Ge having exactly the form predicted by eqn (5), i.e., Ge is linear in νx with slope γ, where Ge extrapolates to the plateau or transient rubbery modulus of the entangled polymer melt.
The fits of the LM to this rubber elasticity benchmark data reveals that the non-classical contribution (Ge) to the shear modulus is nearly a factor of five times larger than the contribution from the classical network elasticity. This observation underscores the limitations of classical rubber elasticity to the description of dry rubber materials.
B. Application to permanent stiff chain networks
From our development in Sect. II, we many expect networks of cross-liked stiff fibers to exhibit a qualitatively different elasticity. In particular, such networks are predicted theoretically to exhibit strain stiffening rather than strain softening under extension and this phenomenon has recently been receiving significant experimental and theoretical attention,2,52,53,55,65,66 mainly because of its biological importance.
We illustrate this phenomenon with some other widely recognized data in Fig. 7, where we also show comparisons to the rod network model with entanglement interactions included. As noted before, this model is equivalent, in the absence of entanglement interactions, to the phenomenological Fung model. The Fung model is widely successful in describing the load-deformation behavior of various types of soft tissues.4–11 This type of elasticity is common in real ‘gel’ materials and we emphasize how different this behavior is from the strain softening found for flexible polymer chain networks (Fig. 5 and 6). (At very large deformations, flexible polymer networks exhibit a singular strain stiffening associated with the finite extensibility of real flexible chains; Gaussian chains are infinitely extensible). Strain stiffening is also apparent in microscale observations on self-assembled fiber networks. For example, Fig. 8 illustrates the microscale deformation of chicken sternal cartilage determined by the atomic force microscope (AFM). As in the other stiff chain network systems, we are able to obtain a good fit to our network model. Numerous recent studies of the deformation of fiber networks have indicated similar data as described in the representative data shown in Fig. 7 and 8.
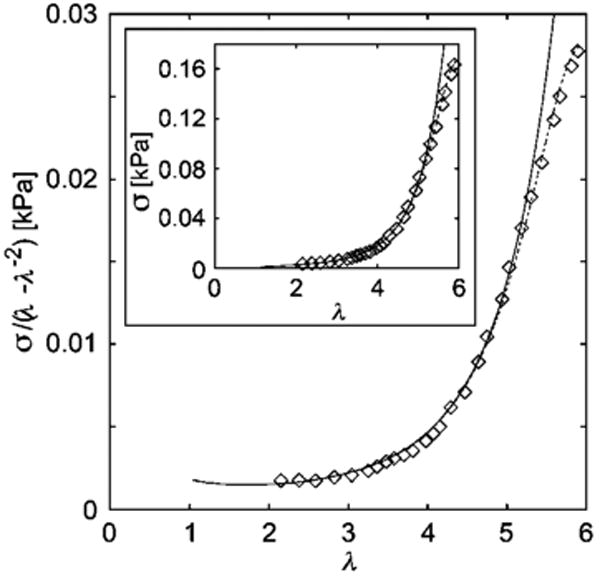
Fig. 7.
Reduced stress vs. stretch plot of the shear deformation of a neurofilament gel.66 The data is fit with the rod chain network model, both with (solid curve) and without (dashed curve) applying the Fermi approximation, eqn (11) as a factor, with fitting parameters: Gc = 750 kPa, Ge = 1000 kPa, b = 0.125, ζ = 0.9, λ* = 6, and δ = 0.3. Inset: stress vs. deformation, with R2 = 0.998 for the fit with the simple Fermi approximant (maximum deviation in stress is 0.004 kPa).
Fig. 8.
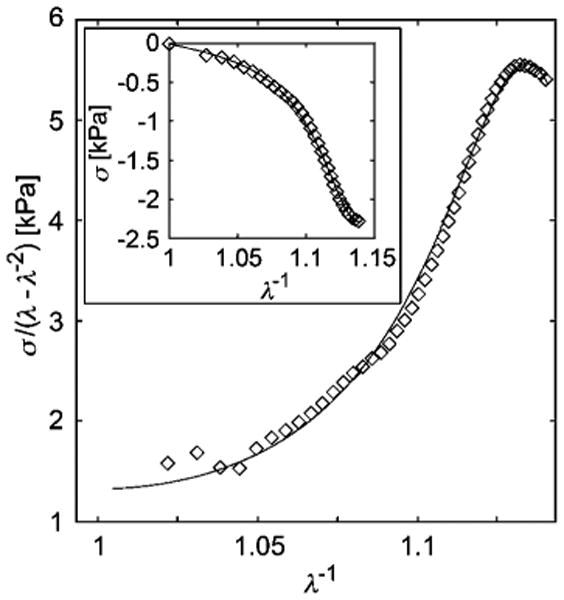
Reduced stress vs. inverse stretch plot of the microindentation of chicken sternal cartilage. The data is fit with the rod chain + localization model in which the additional Fermi approximation, eqn (11), is applied: fitting parameters Gc = 0.9 kPa, Ge = 0.8 kPa, b = 45, ζ = 1, λ* = 0.8745, and δ = − 0.008. Uncertainties in measured stresses are mainly attributed to sample surface roughness and inclination (estimated to be on the order of several percent due to the relatively large size of the probe), and to errors in measuring the probe radius (estimated to be no greater than ±5%). Inset: stress vs. inverse deformation where R2 = 0.998 (maximum deviation in stress is 0.17 kPa).
C. Application to self-assembled fiber networks
It is apparent from Fig. 7 that there is a tendency for the strain stiffening to weaken with increasing deformation, leading a slight turnover in the data for large λ. Indeed, this tendency is normal and progressively increases with increasing deformation. To address this general phenomenon, we recognize that the fibers composing the gel form by the dynamic process of molecular self-assembly and that the associations governing network connectivity eventually break down under stress, which for systems formed by equilibrium self-assembly should be perfectly reversible given enough time for recovery (which can be a very long time!). We illustrate this general phenomenon - stiffening followed by abrupt softening - in Fig. 9 in the case of a gelatin gel under deformation. Similar observations have recently been reported for actin55,56 (where reversible fiber buckling was invoked to rationalize the facile recovery of the elastic properties of the undeformed system) and recently in thermally reversible gels of block copolymers67).
Fig. 9.
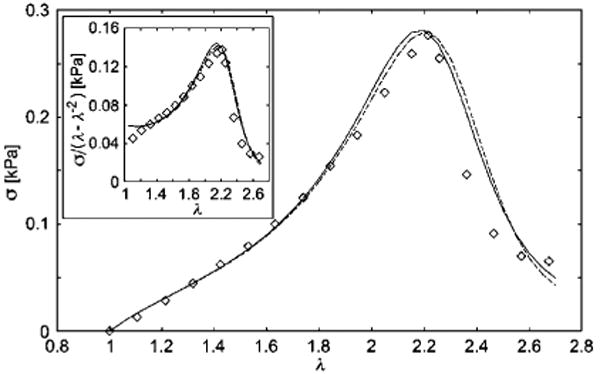
Stress vs. stretch plot of the shear deformation of a gelatin gel.53 The data is fit with two forms of the rod chain network model: one curve indicates the simple Fermi function description of the deformation dependence of the shear modulus (solid curve; fitting parameters Gc = 56 Pa, Ge = 12 Pa, b = 0.4, ζ = 210, λ* = 2.85, and δ = 0.1; R2 = 0.952, maximum deviation in stress is 0.06 kPa) and the other curve indicates a comparison where the full expression for the order parameter is utilized [eqn (11)] (dashed curve; fitting parameters Gc = 56 Pa, Ge = 12 Pa, b = 0.4, Δh = −38.1 kJ/mol, Δs = −75.0 J/mol K, and kf = 9.0 kJ/mol; R2 = 0.948, maximum deviation in stress is 0.06 kPa). Inset: reduced stress vs. deformation.
We next compare our model of self-assembling stiff networks subject to a deformation dependent change in the stability of network. Fig. 9 shows that this simple model is able to capture the stiffening and subsequent softening in an effective manner when we use the simple Fermi approximation for the deformation dependence of the linear shear modulus. The self-assembly energetic parameters obtained from a fit to this data (Δh = −38 kJ/mol and Δs = −77.6 J/mol K) are similar to values observed in many self-assembling systems (e.g., Δh and Δs for the equilibrium polymerization of α-methylstyrene have been found to equal Δh = −35 kJ/mol and Δs = −105 J/mol K). We expect our model of the elasticity of self-assembled stiff chain networks to be widely applicable to diverse biological and synthetic networks.
Another basic aspect of networks of dynamically associating species is a sensitivity of the elastic response to the rate of straining the sample. We illustrate this expected effect schematically in Fig. 10 for a range of dimensionless strain rates Γ where we take α = 0.8, as in recent measurements on telechelic polymer networks.52 Our main point here is that we can understand the qualitative effect of varying strain rate on the elasticity of this class of gels within our network model. We look forward to comparing our model to other data over a range of thermodynamic conditions to further test the model in the future.
Fig. 10.
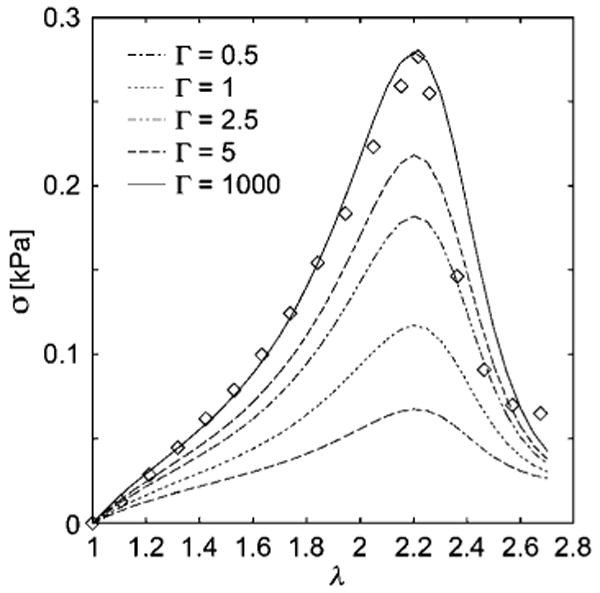
The data and fit using the order parameter form of the rod polymer network model from Fig. 9 are reproduced, along with fits in which the deformation rate Γ is progressively decreased. The tendency of σ to plateau at large λ in measurements of this kind is often due to the onset of gel fracture which preempts the gel melting process,67 an effect that is not described by our simple stress-induced network disassembly model. Our model should only be applied when the material can recover its initial network properties, at least approximately, after network breakdown under stress.
D. Application to self-assembled flexible chain networks
Gelation may also arise from the association of flexible chains and the effect of large deformation on the elasticity of these gels has obvious practical interest. For example, we can expect this situation to apply to nanoparticles (NP) dispersed in a polymer matrix in cases where the association of the NP with the polymer chains creates the equivalent of dynamic cross-links. There is a large literature showing a strong softening of NP-filled networks where the effect is temperature dependent and where the effect is observed following a small extent of deformation. The Payne effect is often attributed to the breakdown of particle-polymer associations within the network,68 as in our model, or the irreversible breakup of NP clusters within the network. The breakup of the physical associations should be reversible while this is not the case for the breakup of the NP clusters. Our model only considers the first mechanism of strain softening and does not address the irreversible NP breakup mechanism of strain-softening, which no doubt arises in some nanocomposites.
We consider the consequences of deformation on flexible polymer networks based on the same model for the deformation dependence of G0 used in our model of a self-assembled stiff network. In our illustrative calculations, we restrict attention to unentangled flexible polymer networks where we can appropriately neglect the entanglement contribution (i.e., Ge = 0). This approximation is probably more generally applicable for polymer nanocomposites at low NP concentrations since the effect of deformation on the entanglement contribution to the network elasticity (Mullin's effect) normally occurs at relatively large extents of deformation in comparison to the Payne effect.
Fig. 11 illustrates the result of these approximations for our highly idealized model of a NP filled unentangled flexible chain network. We see from Fig. 11 that the strain softening can be a large effect at small deformations and that effect becomes progressively larger upon cooling. These general trends in measurements are observed in real nanocomposites, where the strain softening effect vanishes at high temperature where the cross-links ‘melt’. We have chosen the transition temperature for network dissociation (inflection point temperature of Φ in Fig. 2) to be 120 °C which is close to the value that seems to apply to the NP measurements of Kalfus and Jancar.68 More quantitative comparisons between our model and experiments are needed, but our model clearly captures correct physical trends in this practically important class of thermally reversible gels. The effects of entanglement interactions on the Payne effect, appropriate to the situation where the polymer chains are long, can be incorporated by simply including a Ge contribution into our model. However, this generalization does not change the qualitative nature of strain softening found for the NP filled polymer melts, unless the extent of deformation is so large as to significantly modifies the entanglement contribution to the rubber elasticity. Next, we consider the deformation rate dependence on the entanglement contribution to flexible polymer networks composed of long flexible polymer chains. This Mullins effect is a non-trivial effect even in the absence of added NP.
Fig. 11.
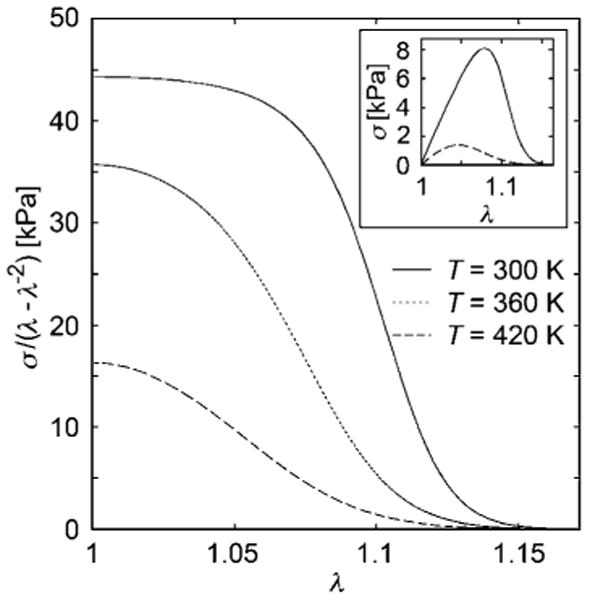
Reduced stress plot of a flexible unentangled chain network that conforms to the classical model with Gc = 45 kPa, showing the effect of temperature on strain softening behavior. Thermodynamic parameters: Δh = − 40kJ/mol, Δs = −100 J/molK, and kf= 1000 kJ/mol. Inset shows the stress-stretch relationship at room temperature and at elevated temperature. In the context of carbon black and nanoparticle filled rubbers, this type of strongly temperature dependent strain softening is called the “Payne effect”.68,78 Note that the stiffness in these nanoparticle cross-linked rubber networks increases upon cooling, which is opposite from classical theory of rubber elasticity with permanent cross-links.
In these illustrative computations, we assume that the network connectivity contribution to the network elasticity of the flexible polymer network is negligible in comparison to the entanglement contribution, which physically corresponds to a low cross-link density in the melt state (just enough cross-links for the network to be in a percolated solid state). In this model, the only contribution to the stress under steady deformation in the localization model equals Ge (1 − λ−3/2). Applying the same model as described above for the rate of relaxation of this entanglement contribution then provides a model of the strain rate Γ and strain dependence of entanglement contribution to the elasticity, Ge(1 − λ−3/2) exp[(−1/Γe)3/5], where Γe = ω τer is the product of the deformation rate ω and an entanglement recovery time τer. This recovery time should be on the order of the terminal stress relaxation time τT ∼ M3.4 of the polymer melt.24 Recovery after a large deformation can also be modeled through the introduction of corresponding stress recovery function R(t/τer) in a fashion described above for the recovery of the elastic properties of network whose structure is broken down under stress where τer governs the rate of recovery to the entangled state. We plan to study this recovery phenomena in future work. At a higher level of development, we could incorporate NP into entangled polymer melts where the effects of the associative or permanent network junctions are also considered. Application to such complex systems, however, will require validation of the model in simpler situations where there are not so many effects in play.
E. Observations on biological fiber network gels
Storm and colleagues66 have recently emphasized the characteristic strain stiffening properties of many biopolymer networks and the neurofilament gel reproduced in Fig. 7 provides a good illustrative example. Applying the full rod network model, along with the strain dependence of the shear modulus leads to a better fit of the data, as illustrated in Fig. 7. The effect of deformation on the thermal stability of the network is evident in this data.
The extracellular matrix (ECM) of cartilage provides another good example of such a rigid chain biological network. Along with charged glycosaminoglycans, the meshwork of collagen fibers provides tissue with its strength and compressive resistance. In compression (or indentation, which is essentially a compressive process69), strain stiffening is dominant, as illustrated by our measurements shown in Fig. 8. The rod network model capably describes the observed elasticity very well until the onset of strain softening, which is often observed to be much more dramatic than found in Fig. 7.
Networks comprised of two distinct components having very different flexibility can also exhibit strong softening. For example, the addition of platelet cells to fibrin gels65 produces dramatic change in gel elasticity. When the highly deformable platelets are added the network it behaves more like a flexible chain network where the rod-like protein filaments play the role of effective cross-links.
IV. Conclusions
We have developed a series of simple models of the elasticity of network materials that address essential physical aspects of this broad class of materials. Building on former successes for flexible polymer networks in the dry state (rubbery materials) we have extended the former modeling to include networks of stiff polymers. Some classical observations on the elasticity of rubbery materials are compared with our models to illustrate their effectiveness and to provide a counterpoint for our new modeling of the elasticity of stiff polymer networks that exhibit a dramatically different elasticity (e.g., strain stiffening versus strain softening at low network deformations).
After showing that our molecular model of stiff polymer networks can reproduce essential trends in the elasticity of stiff polymer networks, we generalize our modeling to address the fact that many gels exhibit cross-links that form by self-assembly or dynamic association. We introduce simple models of the temperature and strain dependence of the elasticity associated with changing the free energy of assembly with deformation and that predict many of the observed elasticity trends seen in real networks, both stiff and flexible chain varieties. Further experimental measurements and modeling are no doubt required to refine the models, but these simple network models seem to capture many of the essential characteristics of complex gels and provide at least qualitative insights into observed trends in these complex materials. Future work should particularly focus on the temperature dependence of the shear modulus to characterize the energetic parameters governing the self-assembly process and the quantification of the rate of strain effects in stiff and flexible chain gels.
Acknowledgments
This work was supported by the Intramural Research Program of the NIH/NICHD. We greatly acknowledge Kendra Erk and Ken Shull of Northwestern University and Richard Gaylord for their critical reading of our manuscript and constructive advice. We thank Rob Riggleman for providing Fig. 1.
Appendix A. The elasticity of dry rubbers
Calculations of the stress-strain relations for dry rubber are then remarkably simple for the LM.20,21 Under uniaxial deformation (λx = λ; λy = λ−1/2; λz = λ−1/2) of an incompressible material in three dimensions, we have the normal stress σ,
| (A.1) |
where V0 is the dry rubber volume. Using the FLM expression for Fnetwork from eqn (A1) implies a remarkably simple relation for σ,
| (A.2) |
The stress relative to its classical deformation variation (λ − λ−2) defines a reduced stress:
| (A.3) |
For large deformations (λ → ∞), this expression reduces to an asymptotic Mooney-Rivlin relation,70,71
| (A.4) |
so that Gc and Ge can be identified approximately with the Mooney parameters 2C1 and 2C2 that are normally considered in characterizing dry rubbers under extension. Blokland72 has provided an extensive tabulation of C2 for a variety of rubbers at relatively high cross-link densities and we tentatively suggest the phenomenological relation γ ≈ 1/3 [see eqn (5)] as a useful rough estimate of the entanglement contribution to rubber elasticity in the absence of direct measurement.
The reduced stress in the LM can be generalized to d-dimensions:
| (A.5) |
which reduces to the classical–like expression σ = Gc[λ − λ−1] in high dimensions73 and an expression applicable to the elasticity of membranes (e.g., polymerized Langmuir films) for d = 2.74
Previous comparisons of the LM to experiment show that this simple model compares well with uniaxial compression and extension measurements, as well as biaxial extension,20,21 and torsional deformation measurements on dry rubbers.62 Moreover, the LM adequately describes network elasticity of rubbers cross-linked in a deformed state.75,76 The torsional deformation measurements of McKenna et al.62 are particularly notable in this type of test of the molecular model since these were performed over a relatively large range of νx. In particular, a fit of these measurements (where C0 was fixed to ½) to the localization model yielded a Ge relation having the form predicted by eqn (5), i.e., Ge is linear in and extrapolates to , the transient shear modulus of the polymer melt. This is reassuring test of the physical soundness of the LM. One of the most challenging tests of a theory of rubber elasticity is the prediction of the changes in the elastic properties of the network upon swelling, which tends to diminish the effects of interchain interaction. In Appendix B, the predictions for the LM are shown to be in broad accord with classical experimental studies on the effect of network swelling on rubber network elasticity. This broad accord of the LM model with these network observations is again encouraging for the physical basis of the model.
Appendix B. The elasticity of swollen rubbers
The theoretical prediction of the elasticity of swollen flexible polymer networks (‘rubber’) from information about the dry rubber is a challenging problem. Before addressing this problem, we note some insightful comments and classic observations made by Gumbrell et al.19 on this topic:
“The change in C2 [non-classical contribution to rubber elasticity; see eqn (A.4)] with volume swelling can be associated with the finite volume of the rubber molecules. This leads to a reduction in the number of possible configurations as two molecules cannot occupy the same space at the same time nor can they pass through one another. The reduction of configurations from this cause would naturally be less in the swollen than in the dry state and in highly swollen rubbers deviations from ideal behavior due to this cause will be small.”
“The value of C2 is found to be large in dry rubbers and decreases to zero at high degrees of swelling.”
The first issue that we must address, in a manner consistent with the former formulation of the LM, is the concentration dependence of the plateau modulus of a polymer melt. In a melt of high molecular mass polymers, the chains are only transiently localized into their local tube environments on the time scale of the stress relaxation time, the terminal time. The system thus responds elastically when perturbed at relatively high frequency measurements, while the system flows over long time scales. Cross-linking locks chains into a permanently localized state- an amorphous solidification transition.77
In the limit νd → 0 (high molecular mass polymer melt), the entanglement contribution Ge is simply due to chain localization as a result of inter-chain interactions:21,24,75 where . If we imagine the network as being comprised of one single molecule (tube) of length N that fills space, then the change in system volume upon swelling V0 → V implies the change in the correlation length ξ ∼ ξ0 ϕ−1/(d−1) and where cross-link density becomes, νd = ϕνd0, where ϕ is the polymer volume fraction. These relations then imply that the plateau modulus of the diluted melt GN (ϕ) scales as,
| (B.1) |
Notably this scaling relation has nothing to do with the fractal character of the chains. This scaling relation is simply a consequence of chain packing. The same packing argument implies that scales in inverse proportionality to the chain cross-sectional area A since ξ2 ∼ A.24 The entanglement contribution to the elasticity of polymer networks, and even polymer melts, should then be reduced for entangled polymer chains with bulky sidegroups.
To calculate the tension of a swollen rubber subjected to deformation,20 we define a swelling factor to describe the first swelling part of the deformation , where λs is the swelling factor. This deformation is followed, for example, by a uniaxial deformation λx = α;λy = α−1/2;λz = α−1/2 so that the tension σ (α) is equal to σ = d[ΔFnetwork/V]/dα or explicitly the LM predicts,
| (B.2) |
The concentration dependent reduced stress I(α,ϕ) ≡ σ/(α − 1/α2)ϕ1/3 then equals,
| (B.3) |
In the large extension limit (α → ∞) eqn (B.3) reduces as before to the Mooney-Rivlin form,
| (B.4) |
where Gc ≡ 2C1 and Ge(ϕ) ≈ 2C2(ϕ) and the ‘Mooney parameter’ C2 exhibits the non-trivial concentration scaling,
| (B.5) |
Although C2 generally vanishes upon swelling, as noted by Gumbrell et al.,19 there is a qualitative difference in the concentration dependence of the entanglement contribution to the elasticity of rubbery materials having relatively high and low cross-linking densities. For lightly cross-linked materials where the chain localization term related to the plateau modulus dominates, we predict that the Mooney parameter should drop off rapidly with concentration,
| (B.6) |
while for highly cross-linked rubbers the scaling becomes like that of classical rubber elasticity theory,
| (B.7) |
Numerous observations on lightly cross-linked rubbers are consistent with eqn (B.5), specifically C2(ϕ) for lightly cross-linked dry rubbers often exhibits a concentration power law19,78,79 near 1 or 4/3, the precise value predicted by the LM. On the other hand, Douglas and McKenna22 have shown that the scaling relation eqn (B.7) holds rather well for relatively highly cross-linked natural rubber. The accord of the LM with these qualitative changes in the elasticity of rubbers, depending on the cross-linking density, is again encouraging.
Evidently, the non-ideal contribution to rubber elasticity can be large in unswollen rubbers, and can even be larger than the contribution arising from chain cross-links. The localization model attributes this non-ideal contribution to strong inter-chain interactions that influence the chain entropy. Comparison of localization model to dry rubber deformation data provides a reasonable description of the elasticity of rubbers in all modes of deformation considered so far. The LM predicts that elasticity of lightly and highly cross-linked rubbers changes in a qualitatively different fashion with network dilution. In view of the simplicity of the analytic form of the model, the physically sensible nature of the parameters derived from it, and its success in capturing qualitative aspects of rubber elasticity in both dry and swollen rubbers, we conclude that the model is a useful working tool in modeling rubbery materials.
In a previous review of the experimental literature to swollen rubbers,38 it was concluded that the constrained junction fluctuation model of Flory and Erman30 provided the “best” empirical description of elasticity of swollen rubbers under different deformation conditions where the fitting parameters of this model were freely adjusted. However, this model offers no predictions about how the fitting parameters should vary with molecular structure or the extent of swelling. A comparison was also made to the localization model in this work where the basic localization variable Ge was not allowed to vary with the concentration, however, so that the predictive capability of the LM to describe the elasticity of swollen rubbers, based on dry state measurements, was not made evident. In view of the empirical success and simple and physically transparent nature of the LM, we believe that this model has significant advantages over other available models to describe the elasticity of both dry and swollen rubbers.
Appendix C. Expression of the shear stress under uniaxial deformation in the rod network model
We deduce the stress-strain relationship for a uniaxial network deformation from eqn (6) as,
| (C.1) |
In simple shear, where the shearing force is applied along the x direction and results in a shear strain γ, the principal stretch ratios are λx = λ, λy = 1, and λz = λ−1. The extent of shear equals, γ = λ − λ−1, and the shear stress S is then defined as,
| (C.2) |
Substituting dλ/dγ = (dγ/dλ)−1 and performing the differentiation, we obtain the desired explicit relation for S,
| (C.3) |
References
- 1.Douglas JF. In: Elastomeric Polymer Networks. Mark JE, Erman B, editors. Prentice Hall; Englewood Cliffs, N.J.: 1992. [Google Scholar]
- 2.Lin YC, Yao NY, Broedersz C, Herrmann H, MacKintosh FC, Weitz DA. Phys Rev Lett. 2010;104:058101. doi: 10.1103/PhysRevLett.104.058101. [DOI] [PubMed] [Google Scholar]
- 3.Kong HJ, Wong E, Mooney DJ. Macromolecules. 2003;36:4582. [Google Scholar]
- 4.Wen Q, Basu A, Winer JP, Yodh A, Janmey PA. New J Phys. 2007;9:428. [Google Scholar]
- 5.Delfino A, Stergiopulos N, Moore JE, Meister JJ. J Biomech. 1997;30:777. doi: 10.1016/s0021-9290(97)00025-0. [DOI] [PubMed] [Google Scholar]
- 6.Fung YC. Am J Physiol. 1967;213:1532. doi: 10.1152/ajplegacy.1967.213.6.1532. [DOI] [PubMed] [Google Scholar]
- 7.Fung YC, Fronek K, Patitucci P. Am J Physiol Heart Circ Physiol. 1979;237:H620. doi: 10.1152/ajpheart.1979.237.5.H620. [DOI] [PubMed] [Google Scholar]
- 8.Horgan CO, Saccomandi G. Biomech Model Mechanobiol. 2003;1:251. doi: 10.1007/s10237-002-0022-z. [DOI] [PubMed] [Google Scholar]
- 9.Pandit A, Lu X, Wang C, Kassab GS. Am J Physiol: Heart Circ Physiol. 2005;288:H2581. doi: 10.1152/ajpheart.00648.2004. [DOI] [PubMed] [Google Scholar]
- 10.Schulze-Bauer CA, Regitnig P, Holzapfel GA. Am J Physiol Heart Circ Physiol. 2002;282:H2427. doi: 10.1152/ajpheart.00397.2001. [DOI] [PubMed] [Google Scholar]
- 11.Zhou J, Fung YC. Proc Natl Acad Sci U S A. 1997;94:14255. doi: 10.1073/pnas.94.26.14255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wall FT, Flory PJ. J Chem Phys. 1951;19:1435. [Google Scholar]
- 13.James HM, Guth E. J Chem Phys. 1943;11:455. [Google Scholar]
- 14.Deam RT, Edwards SF. Philos Trans R Soc London, Ser A. 1976;280:317. [Google Scholar]
- 15.Busse WF. J Phys Chem. 1932;32:2862. [Google Scholar]
- 16.Ferry JD. Polymer. 1979;20:1343. [Google Scholar]
- 17.Kramer O, Violeta T, Ferry JD. Proc Natl Acad Sci U S A. 1972;69:2216. doi: 10.1073/pnas.69.8.2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Langley NR. Macromolecules. 1968;1:348. [Google Scholar]
- 19.Gumbrell SM, Mullins L, Rivlin RS. Trans Faraday Soc. 1953;49:1495. [Google Scholar]
- 20.Gaylord RJ, Douglas JF. Polym Bull. 1987;18:347. [Google Scholar]
- 21.Gaylord RJ, Douglas JF. Polym Bull. 1990;23:529. [Google Scholar]
- 22.Douglas JF, McKenna GB. Macromolecules. 1993;26:3282. [Google Scholar]
- 23.Edwards SF. Proc Phys Soc. 1967;92:9. [Google Scholar]
- 24.Douglas JF, Hubbard JB. Macromolecules. 1991;24:3163. [Google Scholar]
- 25.De Gennes PG. J Phys, Lett. 1974;35:133. [Google Scholar]
- 26.Heinrich G, Straube E, Helmis G. Adv Polym Sci. 1988;85:33. [Google Scholar]
- 27.Read DJ, McLeish TCB. Phys Rev Lett. 1997;79:87. [Google Scholar]
- 28.McLeish TCB. Adv Phys. 2002;51:1379. [Google Scholar]
- 29.Bent J, Hutchings LR, Richards RW, Gough T, Spares R, Coates PD, Grillo I, Harlen OG, Read DJ, Graham RS, Likhtman AE, Groves DJ, Nicholson TM, McLeish TCB. Science. 2003;301:1691. doi: 10.1126/science.1086952. [DOI] [PubMed] [Google Scholar]
- 30.Flory PJ, Erman B. Macromolecules. 1982;15:800. [Google Scholar]
- 31.Flory PJ. Polym J. 1985;17:1. [Google Scholar]
- 32.Rubinstein M. Macromolecules. 1997;30:8036. [Google Scholar]
- 33.Flory PJ, Gordon M, McCrum NG. Proc R Soc London, Ser A. 1976;351:351. [Google Scholar]
- 34.Dossin LM, Graessley WW. Macromolecules. 1979;12:123. [Google Scholar]
- 35.Fetters LJ, Lohse DJ, Richter D, Witten TA, Zirkel A. Macromolecules. 1994;27:4639. [Google Scholar]
- 36.Wang S. Macromolecules. 2007;40:8684. [Google Scholar]
- 37.Valanis KC, Landel RF. J Appl Phys. 1967;38:2997. [Google Scholar]
- 38.Han WH, Horkay F, McKenna GB. Math Mech Solids. 1999;4:139. [Google Scholar]
- 39.Vilgis TA, Boue F, Edwards SF. In: Moelcular Basis of Polymer Network 1988: Workshop Proceedings. Baumgartner A, Picot CE, editors. Springer-Verlag; Berlin: 1989. [Google Scholar]
- 40.Joly-Duhamel C, Hellio D, Ajdari A, Djabourov M. Langmuir. 2002;18:7158. [Google Scholar]
- 41.Feng S, Thorpe MF. Phys Rev B. 1985;31:276. doi: 10.1103/physrevb.31.276. [DOI] [PubMed] [Google Scholar]
- 42.Thorpe MF. J Non-Cryst Solids. 1985;76:109. [Google Scholar]
- 43.Douglas JF, Dudowicz J, Freed KF. J Chem Phys. 2008;128:224901. doi: 10.1063/1.2909195. [DOI] [PubMed] [Google Scholar]
- 44.Peleg M. Rheol Acta. 1993;32:575. [Google Scholar]
- 45.Peleg M. Cereal Chem. 1996;73:712. [Google Scholar]
- 46.Nakanishi S, Yoshikawa H, Shoji S, Sekkat Z, Kawata S. J Phys Chem B. 2008;112:3586. doi: 10.1021/jp800453p. [DOI] [PubMed] [Google Scholar]
- 47.Kumar SK, Douglas JF. Phys Rev Lett. 2001;87:188301. [Google Scholar]
- 48.Peleg M. J Texture Stud. 1994;25:403. [Google Scholar]
- 49.Riggleman RE, Douglas JF, De Pablo JJ. Soft Matter. 2010;6:292. [Google Scholar]
- 50.Bongrand P. Rep Prog Phys. 1999;62:921. [Google Scholar]
- 51.Bell GI, Dembo M, Bongrand P. Biophys J. 1984;45:1051. doi: 10.1016/S0006-3495(84)84252-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Berret JF, Serero Y. Phys Rev Lett. 2001;87:048303. doi: 10.1103/PhysRevLett.87.048303. [DOI] [PubMed] [Google Scholar]
- 53.Groot RD, Bot A, Agterof WGM. J Chem Phys. 1996;104:9202. [Google Scholar]
- 54.Stukalin EB, Douglas JF, Freed KF. J Chem Phys. 2008;129:094901. doi: 10.1063/1.2976341. [DOI] [PubMed] [Google Scholar]
- 55.Chaudhuri O, Parekh SH, Fletcher DA. Nature. 2007;445:295. doi: 10.1038/nature05459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Trepat X, Deng L, An SS, Navajas D, Tschumperlin DJ, Gerthoffer WT, Butler JP, Fredberg JJ. Nature. 2007;447:592. doi: 10.1038/nature05824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gardel ML, Nakamura F, Hartwig JH, Crocker JC, Stossel TP, Weitz DA. Proc Natl Acad Sci U S A. 2006;103:1762. doi: 10.1073/pnas.0504777103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sollich P, Lequeux F, Hebraud P, Cates ME. Phys Rev Lett. 1997;78:2020. [Google Scholar]
- 59.Sollich P. Phys Rev E. 1998;58:738. [Google Scholar]
- 60.Horkay F, Nagy M. Polym Bull. 1980;3:457. [Google Scholar]
- 61.Horkay F, Zrinyi M. Macromolecules. 1982;15:1306. [Google Scholar]
- 62.McKenna GB, Douglas JF, Flynn KM, Chen Y. Polymer. 1991;32:2128. [Google Scholar]
- 63.Rivlin RS, Saunders DW. Philos Trans R Soc London, Ser A. 1951;243:251. [Google Scholar]
- 64.Pak H, Flory PJ. J Polym Sci B. 1979;17:1845. [Google Scholar]
- 65.Shah JV, Janmey PA. Rheol Acta. 1997;36:262. [Google Scholar]
- 66.Storm C, Pastore JJ, MacKintosh FC, Lebensky TC, Janmey PA. Nature. 2005;435:191. doi: 10.1038/nature03521. [DOI] [PubMed] [Google Scholar]
- 67.Seitz ME, Martina D, Baumberger T, Krishnan VR, Hui CY, Shull KR. Soft Matter. 2009;5:447. Seitz et al. confine themselves to the relatively small deformation regime where strain stiffening is observed, but more recent work by Kendra Erk (unpublished) from Ken Shull's group has examined the strain softening regime for the same block copolymer system. [Google Scholar]
- 68.Kalfus J, Jancar J. Polym Compos. 2007;28:743. [Google Scholar]
- 69.Johnson KL. Contact Mechanics. Cambridge University Press; Cambridge: 1985. [Google Scholar]
- 70.Mooney M. J Appl Phys. 1948;19:434. [Google Scholar]
- 71.Rivlin RS. J Appl Phys. 1947;18:444. [Google Scholar]
- 72.Blokland R. Elasticity and Structure of Polyurethane Networks. Rotterdam University Press; Rotterdam: 1968. [Google Scholar]
- 73.Pelzer H. Monatsh Chem. 1937;71:444. [Google Scholar]
- 74.Rehage H, Veyssie M. Angew Chem, Int Ed Engl. 1990;29:439. [Google Scholar]
- 75.Gaylord RJ, Twardowski TE, Douglas JF. Polym Bull. 1988;20:305. [Google Scholar]
- 76.Twardowski TE, Gaylord RJ. Polym Bull. 1989;21:393. [Google Scholar]
- 77.Goldenfeld N, Goldbart P. Phys Rev E. 1992;45:R5343. doi: 10.1103/physreva.45.r5343. [DOI] [PubMed] [Google Scholar]
- 78.Allen G, Kirkham MJ, Padget J, Price C. Trans Faraday Soc. 1971;67:1278. [Google Scholar]
- 79.Price C, Wolf FP. Proc R Soc London, Ser A. 1976;351:331. [Google Scholar]
- 80.Sternstein SS, Zhu A. Macromolecules. 2002;35:7262. [Google Scholar]