

Abstract
Optimal fat mass is necessary for normal gonadotropin levels in adults, and both undernutrition and overnutrition suppress gonadotropins: thus, the gonadotropin response to relative adipose mass is biphasic. Adult obesity is associated with blunted luteinizing hormone (LH) pulse amplitude that is partially attributable to increased LH clearance rate.
Testosterone appears to have a biphasic effect on gonadotropin production in females. Moderate elevations of testosterone appear to stimulate LH production at both the hypothalamic and pituitary level, while very high levels of testosterone suppress LH.
Thus, obesity per se appears to suppress gonadotropin production, and moderate hyperandrogenemia in women appears to stimulate LH. The ordinary hypergonadotropic hyperandrogenism of obese women appears to be an exception to this model because it is usually due to polycystic ovary syndrome (PCOS), a condition in which intrinsic functional ovarian hyperandrogenism and excess adiposity share a common origin that involves insulin-resistant hyperinsulinemia. LH elevation seems to be secondary to hyperandrogenemia and is absent in the most obese cases.
Overweight early pubertal girls have significant blunting of sleep-related LH production, which is the first hormonal change of puberty. The data are compatible with the possibility that excess adiposity may paradoxically subtly suppress hypothalamic-pituitary-gonadal function in early puberty although it is known to contribute to the early onset of puberty.
Keywords: obesity, hyperandrogenism, luteinizing hormone, puberty, polycystic ovary syndrome
1. Introduction
Extrinsic environmental factors are responsible for a widening epidemic of obesity that has serious health consequences after a long asymptomatic period (Franks et al., 2010; Yang et al., 2010). Gonadal dysfunction accounts for perhaps the most common symptoms that bring obesity in adolescents and young adults to medical attention.
Reproductive endocrinologists have long faced a common paradox about the relationship between obesity and gonadal dysfunction. The gonadal dysfunction of obese men is typically hypogonadotropic hypoandrogenism, yet that of obese women is typically hypergonadotropic hyperandrogenism. Recent research sheds light on this paradox.
Accumulating evidence indicates that obesity per se appears to suppress gonadotropin production in both sexes and that moderate hyperandrogenism in women appears to stimulate luteinizing hormone (LH), as modeled in Fig. 1. We argue that the common occurrence of hypergonadotropic hyperandrogenism in obese women is an exception to this model because it is due to polycystic ovary syndrome (PCOS), a condition in which insulin-resistant hyperinsulinism plays a role in both the androgen and adiposity excess. The frequency of this condition, which affects 5–10% of reproductive-age women (Ehrmann, 2005), is one factor that has obscured appreciation of the specific effects of obesity—as distinct from hyperandrogenism—on the neuroendocrine control of reproduction in women. Another obscuring factor has been the evidence that adiposity, indexed by body mass index (BMI), is positively associated with androgen production not only in PCOS, but in women with asymptomatic obesity (Quinkler et al., 2004; Strain et al., 2003; Taponen et al., 2003). In elucidating these relationships, research has revealed a new paradox: excessive adiposity appears to advance the onset of puberty while suppressing neuroendocrine-gonadal axis function thereafter.
Figure 1.
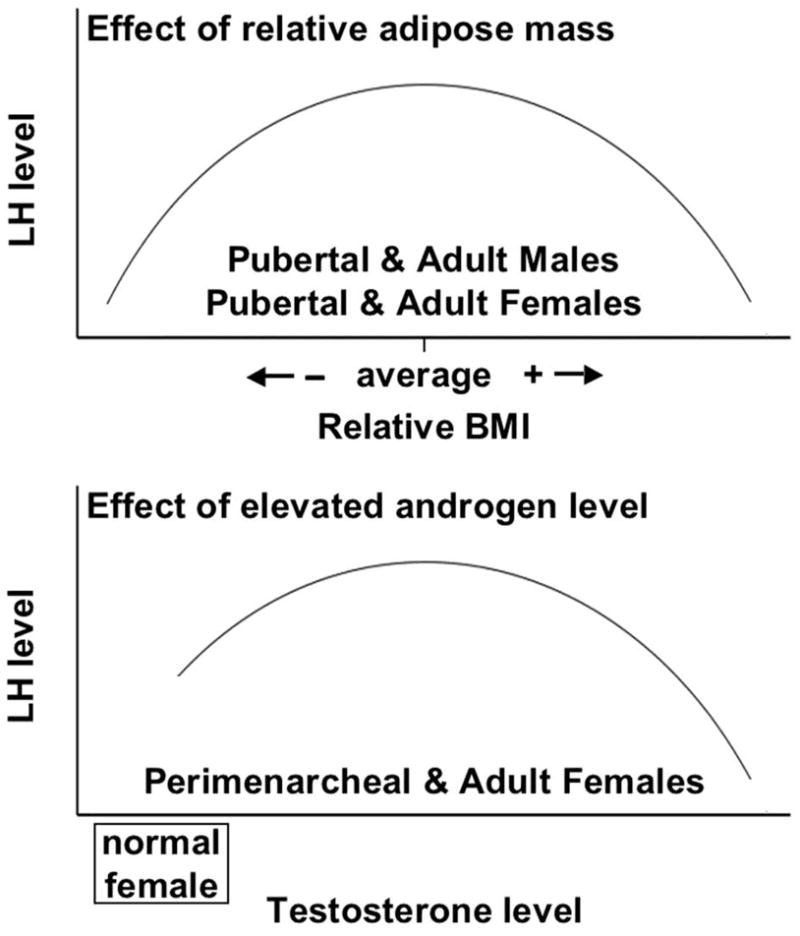
Model depicting the independent, biphasic effects of adiposity and androgen levels on pubertal and adult LH production. Top panel: In both sexes an optimal degree of adiposity is necessary for gonadotropin production, and both undernutrition and overnutrition suppress it. Bottom panel: In females, the moderately elevated levels of androgen typical of hyperandrogenic women with PCOS (approximately 70–150 ng/dl) stimulate LH production, but very high levels of androgen (in the upper male range, >600 ng/dl) have an LH-suppressive effect.
2. Relationship of nutritional status to neuroendocrine function
Undernutrition has long been recognized as a cause of hypogonadism in both men and women (Marshall and Kelch, 1986; Vermeulen, 1993). A mechanistic explanation appeared with the discovery that the hypogonadism is mediated by deficiency of leptin, an adipokine secreted by adipocytes (Rosenbaum and Leibel, 1999; Welt et al., 2004). Leptin is necessary for proper function of the hypothalamic gonadotropin releasing hormone (GnRH) pulse generator. Consequently, undernutrition is characterized by decreased LH pulse frequency and decreased LH pulse amplitude. LH pulse frequency is a surrogate for GnRH pulse frequency, while LH pulse amplitude reflects either GnRH pulse size or the pituitary response to GnRH (Apter et al., 1993).
Obesity has become recognized as a cause of hypoandrogenism in men (Kaufman and Vermeulen, 2005; MacDonald et al., 2010; Vermeulen, 1993; Wu et al., 2008). Obesity also causes low SHBG levels, which results in low total testosterone; normal free testosterone levels are preserved in mild obesity. Free testosterone levels fall in proportion to total testosterone with increasing BMI (Zumoff et al., 1990), and they become subnormal in severely obese men. Increased plasma free estradiol may play a role in preventing a compensatory rise in LH (Strain et al., 2003; Zumoff et al., 2003). LH pulse amplitude is decreased, and serum LH is decreased or normal.
Women with morbid obesity who are eumenorrheic and clinically normoandrogenic were recently reported to have drastically blunted follicular phase LH levels (Jain et al., 2007). This asymptomatic obesity was associated with suppressed LH pulse amplitude. This lowered LH was found to have only a subtle effect on follicular function: estrogen production was normal during the follicular phase of the menstrual cycle, but was significantly depressed at the time of the mid-cycle ovulatory surge, after which corpus luteum insufficiency ensued.
In women with PCOS, excess adiposity influences the blood LH level. Before proceeding, it should be said that the neuroendocrine basis for the LH elevation of PCOS is complex. Spontaneous LH pulse frequency is increased, which is particularly striking in overnight studies since these women typically lack the normal nocturnal slowing of pulse frequency that is the residual effect of ovulation in the previous cycle (McCartney et al., 2002; Taylor et al., 1997). LH pulse amplitude is also increased (Kazer et al., 1987; Waldstreicher et al., 1988), which is reflected in the size of the pulse induced promptly by GnRH (Arroyo et al., 1997) or GnRH agonist administration (Barnes et al., 1989). LH secreted early (30–60 min) in response to GnRH or GnRH agonist injection subcutaneously represents the readily releasable gonadotrope pool of preformed LH (GnRH agonist, being long-acting, then stimulates synthesis and release of a second, slowly releasable gonadotropin pool) (Bremner and Paulsen, 1974; Redding et al., 1972; Rosenfield et al., 1996; Zimmer et al., 2010).
In PCOS, BMI is inversely related to baseline mean LH levels (Fig. 2A) (Holte et al., 1994; Lawson et al., 2008; Mortensen et al., 2009; Taylor et al., 1997). The change in spontaneous LH secretion is attributable to a fall in pulse amplitude, while the high pulse frequency is unchanged. Similarly to LH responsiveness to GnRH(Arroyo et al., 1997; Pagan et al., 2006), the early LH response to GnRH agonist is significantly elevated in PCOS (p<0.01) and inversely related to BMI (Fig. 2B), more-so than to baseline LH. It is higher in non-obese (BMI <30.0) than obese PCOS (p<0.01) (Mortensen et al., 2009). Baseline and stimulated LH are normal in the majority of PCOS patients with BMI >40. These associations are not found in normal women.
Figure 2.

The relationship of serum LH to BMI and free testosterone levels in PCOS (n=88). A. The relationship of 2-hr mean baseline LH to BMI. B. The early LH response (average level 30–60 min minus baseline) to GnRH agonist (leuprolide acetate 10 μg/kg) is inversely correlated to BMI. C. The early LH response to GnRH agonist is significantly positively correlated with the plasma free testosterone level, but to a lesser degree than with BMI. Free testosterone tends to correlate with BMI (r=0.188, p=0.08, data not shown). These relationships were not significant in 53 asymptomatic, eumenorrheic volunteers (normal 5-95th percentile shown in hatched areas), about half of whom had a polycystic ovary and 13% of whom had subclinical PCOS by Rotterdam criteria. Data from previously reported groups (Mortensen et al., 2009).
The blunting of LH pulsations in obese PCOS is at least in part due to accelerated metabolism of LH (Srouji et al., 2007). Clearance of gonadotropins from the circulation is related to the sulfonation and sialylation patterns of the component isoforms: sulfonated isoforms are cleared more rapidly than sialylated ones (Wide et al., 2009). Although sialylation of LH molecules is increased in PCOS, the percent of sulfonated LH isoforms is proportional to BMI in PCOS (Wide et al., 2007). Thus, the increase in sulfonated LH isoforms in obese PCOS seems likely to account for their accelerated LH turnover. Since LH turnover seems to be the major determinant of LH bioavailability, this change would be expected to decrease LH in vivo bioactivity (Mi et al., 2008).
The mediator of the obesity effect on LH pulse amplitude is unclear. Since obesity causes insulin resistance and since PCOS patients are more insulin-resistant than BMI-matched controls (Dunaif et al., 1989), insulin resistance is a major candidate mediator. However, studies of the effects of insulin on gonadotropin production by diverse methods have not yielded a consistent picture (Eagleson et al., 2003; Lawson et al., 2008; Mehta et al., 2005).
In summary, these considerations are compatible with a unisex model in which optimal fat mass is necessary for normal gonadotropin levels in adults, and both undernutrition and overnutrition suppress gonadotropins: thus, the gonadotropin response to relative adipose mass is biphasic when controlling for the level of androgen (Fig. 1). In both men and women, the suppressed LH of excess adiposity is associated with gonadal underfunction. While the ultimate effect of nutritional extremes on LH levels is the same, the mechanisms differ: undernutrition suppresses both LH pulse amplitude and frequency, while overnutrition appears to only affect LH pulse amplitude and to act at least in part by causing increased LH clearance rate.
3. The relationship of androgen status to neuroendocrine function
Blood levels of testosterone, the major circulating androgen, are normally one-tenth as high in women as in men (Rosenfield, 2005). This is because the testes form testosterone from precursor androstenedione using a uniquely efficient type of 17β-hydroxysteroid dehydrogenase (17β-HSD, type 3), whereas the ovaries and adrenal glands carry out this reaction by the less active type 5 17β-HSD (17β-HSD5) (Du et al., 2009). The 17β-HSD5 gene is also expressed widely in peripheral tissues, such as fat, liver, and skin. As a result, about half of testosterone production in women arises by direct secretion from the ovaries and adrenal glands, and about half arises by conversion from circulating precursor androstenedione in peripheral tissues. The adipose contribution to androgen production in women appears to take place primarily in subcutaneous depots (Quinkler et al., 2004).
Androgen blood concentrations of women are not tightly controlled in negative feedback fashion by the neuroendocrine system (Ehrmann et al., 1995;Rosenfield, 1999). Estradiol is the gonadotropin-regulated ovarian steroid, and cortisol is the adrenocorticotropin-regulated adrenal steroid. Androgens are formed in the ovary and adrenal glands as metabolic by-products of estradiol and cortisol biosynthesis. Thus, androgen production in women is a by-product of estradiol secretion by the ovaries and cortisol secretion by the adrenal cortices. In the periphery androgens are formed as by-products of the function of specialized tissues, independently of neuroendocrine regulation. For example, 17β-HSD5 expression in adipose tissue varies depending upon the status of fat accumulation: 17β-HSD5 expression and testosterone formation increase as preadipocytes differentiate into mature adipocytes, and 17β-HSD5 expression in subcutaneous fat correlates with BMI and decreases with weight loss (Quinkler et al., 2004). Even the gonadotropic portion of this multifaceted system for androgen production is not efficiently regulated: women’s LH output is less sensitive than follicle-stimulating hormone (FSH) output to negative feedback by estrogen-progestin (Daniels and Berga, 1997; Hemrika et al., 1993). Rather, the ovarian androgenic response to LH appears to be modulated by intraovarian regulatory mechanisms that coordinate thecal androgen formation with granulosa cell estrogen formation, and the ovarian hyperandrogenism of PCOS appears to arise because this system goes awry.
Counter-intuitively, it is now appears that modest elevations of androgens up-regulate LH secretion in sexually mature females. PCOS is the most common and best characterized moderately hyperandrogenic condition in which LH levels are significantly elevated, but it is not the only one. LH baseline and GnRH-stimulated levels are elevated in women with non-classic congenital adrenal hyperplasia (Feldman et al., 1992) and are normal when androgens are suppressed by glucocorticoid replacement treatment (Barnes et al., 1994). LH is similarly elevated in moderately virilizing ovarian thecomas (Dunaif et al., 1984; Givens et al., 1975), though tumors that elaborate testosterone levels in the 600–700 ng/dl range suppress women’s LH (Rosenfield, 1985).
A number of lines of evidence support the concept that modest elevation of plasma testosterone stimulates LH production. For one thing, testosterone is a positive predictor of LH in multivariate models (Lawson et al., 2008). Direct evidence of a testosterone effect on LH has recently been developed. Early short-term (Dewis et al., 1986; Dunaif, 1986)or non -intensively sampled longer-term (Serafini et al., 1986) studies of the effects of testosterone administration showed remarkably little effect on LH or the menstrual cycle other than the suppressive effects of very high levels. However, the effect of testosterone infusion was recently reexamined using deconvolution analysis to quantify the pulsatile properties of LH secretion in a 12-hr overnight study, in which seven normal post-menarcheal and seven PCOS adolescent girls were tested (Ropelato et al., 2009). In normals, when testosterone levels were increased 3-fold (to 120 ng/dl=4.1 nM), LH pulsatile secretion (amount over the 12-hr period) increased 50% (p<0.05), while basal LH secretion over the 12-hr period did not change. When testosterone levels were raised 6-fold (to 245 ng/dl=8.5 nM), mean serum LH fell 22% (p<0.05) due to a fall in basal LH secretion and a return of pulsatile secretion to normal. PCOS adolescents were resistant to testosterone: only when testosterone was raised 4-fold to 300 ng/dl (10.4 nM) were effects seen, and at this level, while testosterone reduced basal LH secretion, it further increased LH pulsatile secretion. The increases in pulsatile secretion are probably accounted for by both increases in pulse mass and pulse frequency, although the study was not powered sufficiently to establish this.
It appears that the effect of testosterone elevation on LH in women results in part from increased LH pulse frequency. The elevated LH pulse frequency of PCOS is relatively resistant to suppression by combined estrogen and progestin administration (Daniels and Berga, 1997; Pastor et al., 1998). This resistance of hyperandrogenic women to estrogen-progesterone negative feedbackis normalized by anti -androgen therapy (Eagleson et al., 2000). Thus, the persistently rapid LH pulse frequency seems to be explained by hyperandrogenemia impairing sensitivity of the hypothalamic GnRH pulse generator to the slowing that normally results from estrogen-progestin negative feedback.
It also seems likely that testosterone excess in females stimulates LH pulse amplitude. This effect appears not to be mediated by interference with the estrogen-progestin negative feedback effect since the above estrogen-progesterone treatment suppresses LH pulse amplitude normally (Daniels and Berga, 1997; Pastor et al., 1998). The significant positive correlation between the baseline free testosterone level and the early LH response to GnRH agonist in PCOS (Fig. 2C) is compatible with a stimulatory effect of testosterone on LH pulse amplitude. Note that the negative correlation of adiposity on the early LH response to GnRH agonist in PCOS (Fig. 2B) is much more significant than the positive correlation of testosterone (Fig. 2C), even though BMI tends to correlate with testosterone. While the correlations between testosterone and LH could indicate the converse, that LH excess stimulates testosterone excess, there is other evidence that testosterone positively affects the LH pulse amplitude, and in addition evidence discussed in the next section argues for LH excess being a minor factor in the genesis of the hyperandrogenism.
Direct evidence for both positive and suppressive effects of testosterone on LH pulse amplitude has been reported in a series of studies in men. In the latest of these, normal men were temporarily “medically castrated” by administering an inhibitor of testosterone biosynthesis, and gonadotropin-deficient men underwent a companion “hypophysiotropic clamp” study in which a fixed regimen of pulsatile GnRH was infused (Pitteloud et al., 2008). In the former group, replacement doses of testosterone (that yielded a blood level of 730 ng/dl) suppressed the mean LH concentration. GnRH pulse frequency was suppressed, but LH pulse amplitude was stimulated. In the latter group, replacement estradiol (average blood level 23 pg/ml) lowered LH pulse amplitude in response to GnRH. It was deduced that androgen-stimulated hypothalamic GnRH pulse amplitude mediated by the androgen receptor was countered by suppression of the pituitary LH response that was mediated by pituitary aromatization to estradiol. Thus, the androgen effect on LH secretion depends upon the balance of signaling through the androgen and estrogen receptors.
There is also a sexual dimorphism in gonadotropin production that indicates a net testosterone stimulatory effect on LH pulse amplitude in response to GnRH (Rosenfield et al., 1996; Zimmer et al., 2010). Normal men have a higher LH response than women within the first hour after a GnRH agonist injection, although baseline LH levels are similar between the sexes; then men’s LH plateaus. However, women’s LH and FSH levels continue to rise, becoming significantly higher than men’s by 3–4 hours. The LH/FSH ratio is higher in men than women at baseline (p=0.01), but not at the 4-hr peak. These data are interpretable in terms of the two-pool model of gonadotropin release discussed above. The data suggest that normal males have a larger readily releasable pool of gonadotropins than females, but females have a greater capacity to synthesize gonadotropins in response to GnRH agonist administration. This ability of women to synthesize a large amount of LH may be what enables them to mount the mid-cycle gonadotropin surge and does not appear to be influenced by moderate androgen excess. Hyperandrogenic women (PCOS) have the high early LH responsiveness to GnRH/agonist of men, but retain the female ability to subsequently mount surge levels of gonadotropins (Mortensen et al., 2009).
This sexual dimorphism in gonadotropin output is discernable in the newborn. During the neonatal “mini-puberty”, serum LH rises to become higher in boys than in girls. This dimorphism has been shown to be dependent upon a functional androgen receptor (Bouvattier et al., 2002): genetic males with complete androgen insensitivity due to inactivating androgen receptor mutations have blunted LH levels at baseline and in response to GnRH. In addition, congenitally virilized girls have the male-type of gonadotropin response to GnRH agonist at puberty and are at risk for PCOS (Barnes et al., 1994; Ghizzoni et al., 1996). Studies in a variety of animal models support the concept that prenatal androgenization programs development of a masculine pattern of LH secretion at puberty by reducing expression of hypothalamic progesterone receptors (Abbott et al., 2005; Foecking et al., 2005; Sarma et al., 2005).
In summary, the data are compatible with a model in which moderate elevations of testosterone appear to act through the androgen receptor to exert stimulatory effects on LH production at both the hypothalamic and pituitary levels, while frankly virilizing levels of testosterone suppress LH. Thus, the data are compatible with a model in which testosterone in females has a biphasic effect on LH production when controlling for adiposity (Fig. 1).
4. The case for PCOS being unique in its relationships among androgens, obesity, and LH
PCOS is the common cause of obesity in young women that becomes symptomatic in the perimenarcheal period. It is currently defined as otherwise unexplained hyperandrogenic anovulation, with a polycystic ovary being a widely accepted alternative to anovulation as a criterion for ovarian dysfunction (Azziz et al., 2009; Rotterdam ESHRE/ASRM-Sponsored PCOS Consensus Workshop Group, 2004; Zawadzki and Dunaif, 1992). However, there is uniform recognition that the PCOS spectrum involves obesity and/or insulin resistance. Metabolic syndrome, the manifestation of insulin resistance interacting with obesity and age, occurs at least two-fold more often than expected for obesity status from adolescence onwards (Leibel et al., 2006).
PCOS appears to arise as a complex trait with contributions from heritable and intrauterine and external environmental factors (Rosenfield, 2007). Polygenic influences appear to account for about 70% of the variance in pathogenesis. Hyperandrogenemia, polycystic ovaries, and the metabolic syndrome have heritable components that seem to be fundamental to the pathogenesis of the disorder (Leibel et al., 2006). In our series of adolescents with PCOS, metabolic syndrome was found in three-quarters of fathers and one-third of mothers. The presence of a polycystic ovary in these girls corresponded to the presence of paternal metabolic syndrome more often than it did to the presence of a maternal polycystic ovary.
There has been considerable debate over whether PCOS is fundamentally a neuroendocrine disorder (in which hyperpulsatility of GnRH is the origin of the problem), an ovarian disorder (in which intrinsic ovarian dysfunction is the origin), or a metabolic disorder (in which insulin resistance is a key element). Our research has led us to favor the concept that the essence of PCOS is intrinsic functional ovarian hyperandrogenism that is closely linked to the metabolic disorder.
The increased LH of PCOS was initially considered the cause of the androgen excess (Yen, 1980). This is reasonable since LH stimulates theca cell development and steroidogenesis and, indeed, is necessary for the expression of gonadal steroidogenic enzymes and sex hormone secretion. However, once the adult steady-state is reached, further LH increase has little further effect on androgen levels because excess LH causes homologous desensitization of theca cells (Ehrmann et al., 1995; Rosenfield, 1999). Desensitization involves down-regulation of LH receptor expression and steroidogenesis. Because steroidogenic down-regulation is primarily exerted on 17,20-lyase activity, which converts 17alpha-hydroxycorticoids to 17-ketosteroids, 17-hydroxyprogesterone levels rise in response to increased LH levels, but there is only a limited rise in androgens (Hirshfeld-Cytron et al., 2009; McCartney et al., 2004; Mortensen et al., 2009).
Our working hypothesis is that PCOS usually results from intrinsic ovarian dysfunction that is due to dysregulation of transcription factors common to the function of a wide variety of tissues (Fig. 3) (Du et al., 2009; Ehrmann et al., 1995; Hirshfeld-Cytron et al., 2009; Nelson et al., 1999; Rosenfield, 1999). The ovarian hyperandrogenism is “functional” because it is gonadotropin-dependent; however, LH excess does not seem to ordinarily be the fundamental stimulus to hyperandrogenism. Rather, PCOS patients typically are hypersensitive to LH stimulation, which is indicated by disproportionate hyper-responsiveness of 17-hydroxyprogesterone relative to other ovarian steroids. This appears to be due to intrinsic dysregulaton of ovarian theca cells, in which most steroidogenic enzymes are constitutively overexpressed and hyper-responsive to cyclic adenosine monophosphate stimulation (Jakimiuk et al., 2001; Nelson et al., 1999; Nelson et al., 2001). Hyperinsulinemia and abnormal paracrine signaling from dysregulated granulosa cells contribute to theca cell dysfunction, causing an “escape” from desensitization, in which the set-point for desensitization to LH rises. Insulin synergizes with gonadotropins to up-regulate androgen formation by counteracting desensitization. Thus, the ovaries function as if sensitive to the effects of insulin on steroidogenesis in a state of peripheral resistance to the effects of insulin on glucose metabolism (Ehrmann et al., 1995);
Figure 3.
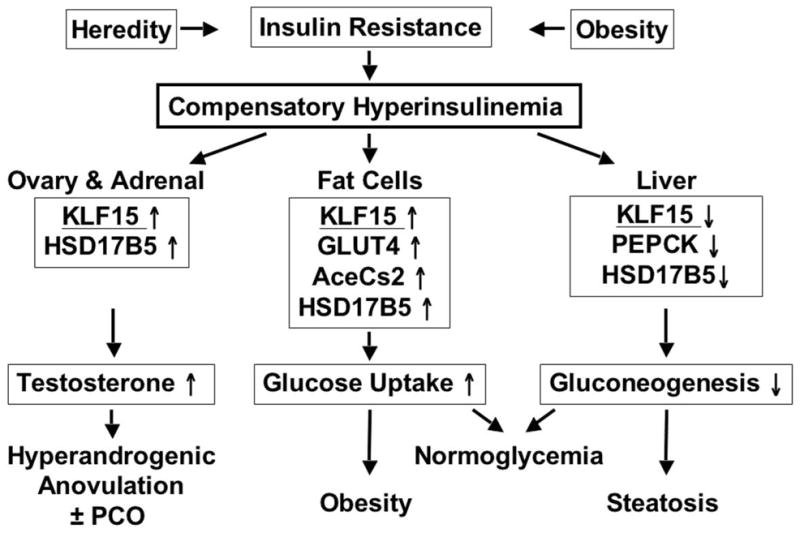
Model of the proposed role of KLF15 protein in the pathogenesis of PCOS. Increased KLF15 expression in response to the compensatory hyperinsulinemia of insulin resistance of any cause restores glucose homeostasis, but at the expense of obesity, hepatic steatosis, and hyperandrogenism. KLF15 influences glucose metabolism and 17β-HSD5 gene (HSD17B5) expression in multiple tissues. In the ovary, the KLF15 response to insulin up-regulates HSD17B5 expression and increases testosterone biosynthesis, contributing to follicular maturation arrest and a polycystic ovary (PCO). In fat and muscle (not shown), KLF15 expression is stimulated by insulin to up-regulate the glucose transporter 4 (GLUT4) and acetyl-CoA synthetase (AceCs2) genes, which increase glucose uptake, causing obesity and muscularity. In liver, KLF15 expression is inhibited by insulin to down-regulate the phosphoenolpyruvate carboxykinase(PEPCK) gene, which decreases hepatic gluconeogenesis, predisposing to fatty liver. Modified and reproduced with permission from Du, et al (Du et al., 2009), copyright 2009, The Endocrine Society.
Adipocyte lipogenesis, analogousto ovarian steroidogenesis, is insulin -sensitive (Corbould and Dunaif, 2007). One action of insulin is to stimulate formation of testosterone by 17β-HSD5. Insulin does so by stimulating expression of KLF15, a Kruppel-like transcription factor that is part of the gene’s proximal promoter co-activator complex (Du et al., 2009). KLF15 is a multifunctional transcriptional regulator that promotes glucose uptake in a wide variety of tissues (Fig. 3). The KLF15 binding site of the 17β-HSD5 gene is unique to humans and orangutans; evolutionarily, it seems to contribute to an anabolic “thrifty phenotype”. In adipocytes, this action stimulates lipogenesis; in liver and muscle, it stimulates gluconeogenesis. Thus, the hyperinsulinemia that is compensatory for the insulin resistance of PCOS would be expected to contribute to both androgen and fat excess.
In summary, PCOS is a unique multifactorial disorder, the essence of which is intrinsic functional ovarian hyperandrogenism. Our hypothesis is that it results from a generalized transcriptional dysregulation that uniformly causes intrinsic ovarian hyperandrogenism and often causes insulin-resistant hyperinsulinism, which contributes to both excess androgen and excess adiposity. While the disorder is gonadotrop in-dependent, gonadotropins being necessary for the expression of gonadal steroidogenic enzymes, LH excess does not seem to ordinarily be the fundamental cause of the hyperandrogenism. LH excess is variable and depends on obesity-androgen balance. When it is present, homologous desensitization limits the androgenic response to it: thus, LH excess does not appear to cause the hyperandrogenism, although it may aggravate it.
5. Obesity and androgen effects on pubertal onset and progression
Important aspects of the effects of adiposity and sex steroids on LH production are different in children than in adults. The mature relationships of each seem to be established at different stages of puberty.
Body fat and sex steroids are among the somatic stimuli to skeletal growth and maturation that determinethe onset of puberty (Divall et al., 2010; Flor-Cisneros et al., 2004). The hypothesis that attainment of a critical amount of body fat is necessary for the onset of normal pubertal development in humans and lowerspecies (Frisch, 1984) is now well-accepted. Low fat stores are associated with delayed puberty and, when severe, gonadotropin deficiency (Marshall and Kelch, 1986). Conversely, excessadiposity is associated with advanced onset of puberty in girls (Rosenfield et al., 2009). In obese boys the most robust evidence also indicates that puberty begins slightly early (Sorensen et al., 2010), though an association with delayed puberty has been suspected (Kelch et al., 1980; Lee et al., 2010). Leptin appears to be an essential mediator of the effect of fat stores on the onset of puberty, signaling the central nervous system that energy stores are sufficient to support the high energy demands of reproduction, and thus facilitating the onset of substantial pulsatile GnRH secretion (Ahima et al., 1997).
Puberty begins when the hypothalamus begins to release substantial amounts of GnRH in a pulsatile manner during sleep (Grumbach et al., 1974; Knobil, 1980). The first hormonal change of puberty is consequently the onset of sleep-related LH release (Boyar et al., 1972). No studies have been designed to examine whether this occurs early in girls with excessive adiposity. However, a secondary analysis of our data in prepubertal girls suggests that the onset of sleep-related LH secretion occurs earlier in overweight than in normal weight prepubertal girls (Fig. 4A) (Bordini et al., 2009), a finding consistent with the concept that a fat-related signal such as leptin facilitates the activation of hypothalamic GnRH release.
Figure 4.

Overnight LH levels and pulse characteristics of normal weight (NW) and overweight (OW, average BMI 95th percentile) prepubertal and early pubertal girls. A. Secondary analysis of data on prepubertal girls 6–9 years old. Primary analysis showed the two groups to have similar mean levels awake and similar mean levels asleep, so the data of the two groups were pooled for analysis; this analysis showed a small, but significant sleep-related rise in LH in these prepubertal girls (p <0.005) (Bordini et al., 2009). Secondary analysis showed that this rise was attributable to the OW group, according to nominal P values (Pu). None of the prepubertal girls had detectable LH pulses awake, and only 2 of 9 NW and 5 of 11 OW prepubertal girls had detectable LH pulses during sleep; the latter sufficed to yield nominally significant sleep-related rises in pulse amplitude and frequency in the OW prepubertal group. Light dotted line shows LH assay limit of sensitivity. B. Primary analysis of data on early pubertal girls 9–13 years old (premenarcheal, average breast stage 3) (Bordini et al., 2009). Unlike normal NW girls, OW pubertal girls did not have significant sleep -related rises in mean LH or FSH, nor did they have a significant sleep-related rise in LH pulse amplitude. Pulse frequency was also significantly lower during sleep in the OW pubertal girls. P values corrected for multiple comparisons.
GnRH secretion is extremely sensitive to sex steroid negative feedback during early puberty, and this sensitivity wanes as the reproductive axis matures, permitting increasing GnRH and gonadotropin secretion (Grumbach et al., 1974; Rosenfield and Fang, 1974). However, androgen or estrogen exposure that is sufficient to suppress gonadotropin production during early puberty (Kelch et al., 1985; Kulin et al., 1969) paradoxically advances the onset of puberty (Foster et al., 1984; Pescovitz et al., 1984). This has appeared to result from sex steroids synchronously influencing both somatic and central nervous system developmental maturation. It now seems that this sex hormone effect does not end with attainment of adulthood. The hypogonadotropic state of about ten percent of men with congenital hypogonadotropic hypogonadism remits after the discontinuation of sex hormone replacement therapy (Raivio et al., 2007). Similar neuroendocrine recovery occurs after sex steroid treatment in most men and women with inactivating mutationsof the hypothalamic neurokinin B signaling system (Gianetti et al., 2010). Thus, it appears as if, contrary to their negative feedback effect on hypothalamic GnRH secretion, sex steroids have the ability to enhance the onset of GnRH pulse generation in both children and adults.
The onset of PCOS in the perimenarcheal period and its association with obesity has led to the postulate that obesity predisposes to the development of PCOS by causing androgen-mediated premenarcheal LH excess (McCartney et al., 2007; McCartney et al., 2009). This concept is in line with the effects of obesity and androgen excess to accelerate the onset of puberty in girls. To test this concept, we surveyed the hypothalamic-pituitary-ovarian function of asymptomatic pubertal volunteers with normal and elevated BMI (Bordini et al., 2009). Contrary to the hypothesis, however, we found that the sleep-related onset of LH secretion, the earliest hormonal change of puberty, was blunted in asymptomatic overweight early pubertal female volunteers (premenarcheal with average BMI 95th percentile, breast stage 3) who had no evidence of androgen excess.
In contrast to normal weight volunteers of similar early pubertal stage, these overweight girls had insignificantsleep -related rises in mean LH and FSH (Fig. 4B). Their nocturnal LH, determined at 20-min intervals by continuous blood withdrawal, was characterized by both a significantly lower rise in LH pulse amplitude and a significantly lower LH pulse frequency during sleep than was that of normal weight girls. These alterations occurred in the absence of significantly different sex steroid levels. While the LH, FSH, and estradiol responses to GnRH agonist testing did not differ significantly between the overweight and normal-weight groups, the overweight girls had significantly lower estradiol responses to GnRH agonist as the peak LH responses rose, compatible with lower LH bioactivity in the overweight group.
These observations confirm and extend the similar findings of McCartney, et al about nocturnal LH pulsatility that were obtained in a small group of peripubertal girls (Tanner stage 1–2) by overnight sampling at 10-min intervals (McCartney et al., 2009). Thus, there is agreement that nocturnal LH pulse frequency and amplitude are subnormal in overweight early pubertal girls. This blunted LH pulse frequency suggests a less vigorous hypothalamic drive to puberty than in normal-weight girls. The transient gonadotropin deficiency that has been reported in obese boys (Kelch et al., 1980)may be related to these findings.
However, there is controversy about if, when, and how simple obesity may cause LH excess in relation to menarche. McCartney, et al have published a series of studies that have led them to conclude that hyperandrogenism resulting from obesity predisposes to the elevated LH secretion of PCOS commencing during breast stage 3 of puberty (McCartney et al., 2006; McCartney et al., 2007; McCartney et al., 2009). This is at odds with our above data in healthy overweight volunteers (Fig. 4B) (Bordini et al., 2009). Where data has been provided for premenarcheal stage 3 hyperandrogenic adolescents in the studies of McCartney, et al, the bone age is advanced, suggesting that the inclusion of hyperandrogenic adolescents with primary amenorrhea due to PCOS is confounding their conclusions.
To test the hypothesis that the combination of excessive adiposity and hyperandrogenism peripubertally are risk factors for early pubertal LH excess, we characterized a group of 6–8 year-old overweight girls (average BMI 97th percentile) who had slowly progressive sexual precocity (average breast stage 3) preceded by the mild hyperandrogenism of premature adrenarche (Bordini et al., 2010). These girls had suppressed sleep-related LH secretion, like the above healthy overweight early pubertal girls. Though they had pubertal LH responses to GnRH agonist testing, they differed from asymptomatic overweight early pubertal girls in having significantly lower gonadotropin levels awake and in response to GnRH agonist. Their estradiol levels were lower during sleep, but otherwise estradiol and testosterone levels were normal for pubertal stage. These results argue against the mild prepubertal hyperandrogenism of excess adiposity and premature adrenarche up-regulating LH secretion during early puberty. However, our study group was not hyperandrogenic for their stage of pubertal maturation, so it remains unclear at what point in pubertal development, and at what level, hyperandrogenemia becomes capable of stimulating pulsatile LH secretion, as it does after menarche. Rather, the results are consistent with the possibility that early pubertal girls are particularly sensitive to the gonadotropin-suppressive effects of excess fat mass, even in the presence of mild hyperandrogenism.
The suppression of gonadotropins during sleep in early puberty would be expected to cause slow progression of puberty. Thus, we speculate that obesity may underlie the precocity of girls in whom, like these, the onset of puberty is only mildly premature and puberty is subsequently slowly progressive. Many sexually precocious girls in this 6–8 year age range do not require treatment to preserve height potential (Carel et al., 2009).
Our data suggest that the effects of excess adiposity on gonadotropin production during puberty are distinct from those on the onset of puberty, analogous to the paradoxical effects of androgen on GnRH secretion pre-and post -pubertally. The obesity effect on gonadotropin production during early puberty is suppressive; while comparable to the effect of excess adiposity on gonadotropin production in adults, the decrease in LH pulse frequency in overweight girls during early puberty suggests a suppressive effect on GnRH secretion that is not seen in adults. However, our conclusions must be considered preliminary and warrant further study. They are limited by lack of polysomnographic documentation of sleep stage and integrity. We cannot be certain that the apparent reduction in LH pulse frequency in overweight pubertal girls is real, since it may be an artifact of reduction of LH pulse amplitude to levels below assay sensitivity. It is also possible that blunting of LH pulse amplitude during sleep in overweight girls is compensated by a shift in diurnal LH rhythm, such that 24-hr LH secretion is not altered.
Reversal of diurnal LH rhythm has been reported in post-menarcheal adolescents with PCOS, (Zumoff et al., 1983). The two subsequent studies of LH in adolescents over a 24-hr period were consistent with this finding (Apter et al., 1994; Porcu et al., 1987). Originally, this abnormality was considered evidence that PCOS resulted from a primary “chronobiologic” abnormality of neuroendocrine function. However, in view of recent evidence that the neuroendocrine abnormalities of PCOS are secondary, as discussed in the preceding section, the explanation must be reconsidered. Thus, hyperandrogenemia--or obesity, for which two of the three studies were not controlled--must be considered as a possible cause of a shift in diurnal rhythmicity.
In spite of the possible effect of hyperandrogenism on LH rhythmicity and its significant effect on GnRH pulse frequency in post-menarcheal adolescents, it appears to have no effect on the diurnal LH rhythm before menarche. We have serendipitously observed preservation of a sleep-related LH rise in an asymptomatic premenarcheal girl with PCOS (Fig. 5) (Bordini et al., 2009), albeit with elevated LH levels, and a hyperandrogenic premenarcheal girl studied because of hirsutism had similar findings during a 24-hr study (Apter et al., 1994).
Figure 5.
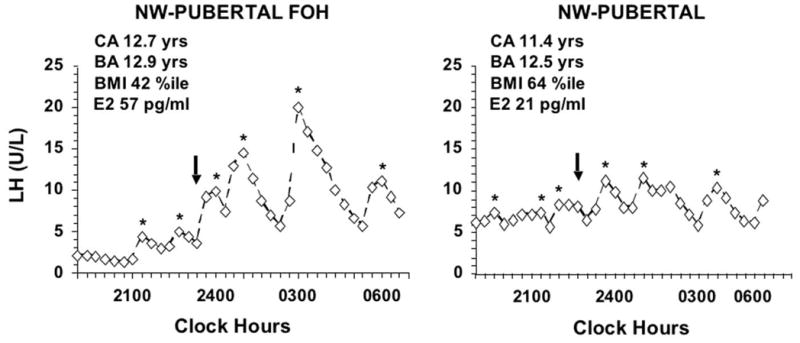
Overnight LH profiles in a premenarcheal, normal-BMI (NW) girl with PCOS-like functional ovarian hyperandrogenism (FOH) in comparison to a NW control, both with stage 3 breast development. The FOH was detected because of outlier hormone levels during a study of normal volunteers (Bordini et al., 2009): it was manifest as an elevated plasma free testosterone level (15 pg/ml) and 17-hydroxyprogesterone peak response to GnRHag (256 ng/dl) in the absence of a polycystic ovary; peak sleep LH was 3 S.D. above normal (7.7 ± 4.1, SD, U/L) with a significant sleep-related rise in mean LH. CA = chronologic age, BA = bone age. Arrows denote sleep onset, asterisks denote significant LH pulses.
It remains unclear at what point in pubertal development, and at what level, hyperandrogenemia becomes capable of affecting pulsatile LH secretion, as it does after menarche. The resistance to estrogen -progestin negative feedback of hyperandrogenemia, while significant, occurs in less than half of adolescents, less consistently than in adults, almost all of whom are resistant (Blank et al., 2009). This discrepancy between adolescents and adults suggests to us that this resistance to negative feedback is not inherent to the disorder. Rather, it suggests that resistance only becomes apparent when the immature high sensitivity to sex steroid negative feedback fully wanes, i.e., when the activation of the GnRH pulse generator is fully mature.
In summary, while excess adiposity appears to accelerate the onset of puberty by facilitating the onset of pubertal GnRH and gonadotropin secretion during sleep, overweight early pubertal girls have significant blunting of sleep-related LH production. This suggests that excess adiposity, in the absence of substantial sex steroid excess, may subtly suppress hypothalamic-pituitary-gonadal function in early pubertal girls, although it is known to accelerate the onset of puberty. LH pulse amplitude during early puberty is diminished to such an extent that pulse frequency also seems to be affected, raising the possibility of a hypothalamic effect that has not been discerned in studies of adults. The data are compatible with the possibility that excess adiposity may contribute to both the early onset and the slow progression of puberty. However, we cannot rule out the possibility that obesity or hyperandrogenism affect the pubertal diurnal rhythm of LH.
6. Conclusions
Accumulating evidence suggests that obesity per se suppresses gonadotropin production in both sexes and that moderate hyperandrogenism in women stimulates it. We review the evidence that the common symptomatic hyperandrogenism of young obese women does not fit this paradigm of hypogonadism in obesity because it is due to PCOS, a condition in which excess androgen and excess adiposity share a common origin that involves insulin excess. While LH is necessary for steroidogenesis, LH elevation appears to be secondary to hyperandrogenemia and is absent in the most obese cases; it does not seem to cause the hyperandrogenism, although it may aggravate it.
Overweight early pubertal girls have significant blunting of sleep-related LH production. These data are compatible with the possibility that excess adiposity may subtly suppress hypothalamic-pituitary-gonadal function in early puberty as it does in adults and thereby slow progression of puberty although, paradoxically, it is known to contribute to the early onset of puberty. However, the possibility that obesity or hyperandrogenism affect the pubertal 24-hr diurnal rhythm of LH cannot be excluded.
Acknowledgments
This research was supported in part by the Eunice Kennedy Shriver NICHD/NIH through cooperative agreement [U54-041859] as part of the Specialized Cooperative Centers Program in Reproduction and Infertility Research and HD-39267 (RLR) and RR-00055 and UL1RR024999 from the National Center For Research Resources.
Abbreviations
- BMI
Body mass index
- FSH
Follicle-stimulating hormone
- GnRH
Gonadotropin releasing hormone
- 17β-HSD5
17β-hydroxsteroid dehydrogenase type 5
- KLF
Kruppel-like factor
- LH
Luteinizing hormone
- PCOS
Polycystic ovary syndrome
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abbott DH, Barnett DK, Bruns CM, Dumesic DA. Androgen excess fetal programming of female reproduction: a developmental aetiology for polycystic ovary syndrome? Hum Reprod Update. 2005;11:357–74. doi: 10.1093/humupd/dmi013. [DOI] [PubMed] [Google Scholar]
- Ahima RS, Dushay J, Flier SN, Prabakaran D, Flier JS. Leptin accelerates the onset of puberty in normal female mice. J Clin Invest. 1997;99:391–5. doi: 10.1172/JCI119172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apter D, Butzow TL, Laughlin GA, Yen SS. Gonadotropin-releasing hormone pulse generator activity during pubertal transition in girls: pulsatile and diurnal patterns of circulating gonadotropins. J Clin Endocrinol Metab. 1993;76:940–949. doi: 10.1210/jcem.76.4.8473410. [DOI] [PubMed] [Google Scholar]
- Apter D, Bützow T, Laughlin G, Yen S. Accelerated 24-hour luteinizing hormone pulsatile activity in adolescent girls with ovarian hyperandrogenism: relevance to the developmental phase of polycystic ovarian syndrome. J Clin Endocrinol Metab. 1994;79:119–125. doi: 10.1210/jcem.79.1.8027216. [DOI] [PubMed] [Google Scholar]
- Arroyo A, Laughlin GA, Morales AJ, Yen SSC. Inappropriate gonadotropin secretion in polycystic ovary syndrome: influence of adiposity. J Clin Endocrinol Metab. 1997;82:3728–33. doi: 10.1210/jcem.82.11.4377. [DOI] [PubMed] [Google Scholar]
- Azziz R, Carmina E, Dewailly D, Diamanti-Kandarakis E, Escobar-Morreale HF, Futterweit W, Janssen OE, Legro RS, Norman RJ, Taylor AE, Witchel SF. The Androgen Excess and PCOS Society criteria for the polycystic ovary syndrome: the complete task force report. Fertil Steril. 2009;91:456–88. doi: 10.1016/j.fertnstert.2008.06.035. [DOI] [PubMed] [Google Scholar]
- Barnes RB, Rosenfield RL, Burstein S, Ehrmann DA. Pituitary-ovarian responses to nafarelin testing in the polycystic ovary syndrome. N Engl J Med. 1989;320:559–565. doi: 10.1056/NEJM198903023200904. [DOI] [PubMed] [Google Scholar]
- Barnes RB, Rosenfield RL, Ehrmann DA, Cara JF, Cuttler L, Levitsky LL, Rosenthal IM. Ovarian hyperandrogenism as a result of congenital adrenal virilizing disorders: Evidence for perinatal masculinization of neuroendocrine function in women. J Clin Endocrinol Metab. 1994;79:1328–1333. doi: 10.1210/jcem.79.5.7962325. [DOI] [PubMed] [Google Scholar]
- Blank SK, McCartney CR, Chhabra S, Helm KD, Eagleson CA, Chang RJ, Marshall JC. Modulation of GnRH pulse generator sensitivity to progesterone inhibition in hyperandrogenic adolescent girls - Implications for regulation of pubertal maturation. J Clin Endocrinol Metab. 2009;94:2360–66. doi: 10.1210/jc.2008-2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordini BD, Littlejohn EE, Rosenfeld RL. Blunted sleep-related LH rise in healthy premenarcheal pubertal girls with elevated body mass index. J Clin Endocrinol Metab. 2009;94:1168–1175. doi: 10.1210/jc.2008-1655. [Comment in: J Clin Endocrinol Metab. 2009 Apr;94:10946] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordini BD, Littlejohn EE, Rosenfield RL. LH dynamics in overweight girls with premature adrenarche and slowly progressive sexual precocity. Intl J Pediatr Endocrinol. 2010;2010 doi: 10.1155/2010/724696. 724696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouvattier C, Carel JC, Lecointre C, David A, Sultan C, Bertrand AM, Morel Y, Chaussain JL. Postnatal changes of T, LH, and FSH in 46, XY infants with mutations in the AR gene. J Clin Endocrinol Metab. 2002;87:29–32. doi: 10.1210/jcem.87.1.7923. [DOI] [PubMed] [Google Scholar]
- Boyar R, Finkelstein J, Roffwarg H, Kapen S, Weitzman L. Synchronization of augmented luteinizing hormone secretion with sleep during puberty. N Engl J Med. 1972;287:582–586. doi: 10.1056/NEJM197209212871203. [DOI] [PubMed] [Google Scholar]
- Bremner WJ, Paulsen CA. Two pools of luteinizing hormone in the human pituitary: evidence from constant administration of luteinizing hormone-releasing hormone. J Clin Endocrinol Metab. 1974;39:811–5. doi: 10.1210/jcem-39-5-811. [DOI] [PubMed] [Google Scholar]
- Carel JC, Eugster EA, Rogol A, Ghizzoni L, Palmert MR, Antoniazzi F, Berenbaum S, Bourguignon JP, Chrousos GP, Coste J, Deal S, de Vries L, Foster C, Heger S, Holland J, Jahnukainen K, Juul A, Kaplowitz P, Lahlou N, Lee MM, Lee P, Merke DP, Neely EK, Oostdijk W, Phillip M, Rosenfield RL, Shulman D, Styne D, Tauber M, Wit JM. Consensus statement on the use of gonadotropin-releasing hormone analogs in children. Pediatrics. 2009;123:e752–62. doi: 10.1542/peds.2008-1783. [DOI] [PubMed] [Google Scholar]
- Corbould A, Dunaif A. The adipose cell lineage is not intrinsically insulin resistant in polycystic ovary syndrome. Metabolism. 2007;56:716–22. doi: 10.1016/j.metabol.2006.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels T, Berga S. Resistance of gonadotropin releasing hormone drive to sex steroid-induced suppression in hyperandrogenic anovulation. J Clin Endocrinol Metab. 1997;82:4179–4183. doi: 10.1210/jcem.82.12.4402. [DOI] [PubMed] [Google Scholar]
- Dewis P, Newman M, Ratcliffe WA, Anderson DC. Does testosterone affect the normal menstrual cycle? Clin Endocrinol (Oxf) 1986;24:515–21. doi: 10.1111/j.1365-2265.1986.tb03280.x. [DOI] [PubMed] [Google Scholar]
- Divall SA, Williams TR, Carver SE, Koch L, Bruning JC, Kahn CR, Wondisford F, Radovick S, Wolfe A. Divergent roles of growth factors in the GnRH regulation of puberty in mice. J Clin Invest. 2010;120:2900–9. doi: 10.1172/JCI41069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du X, Rosenfield RL, Qin K. KLF15 is a transcriptional regulator of the human 17β-hydroxysteroid dehydrogenase type 5 gene. A potential link between regulation of testosterone production and fat stores in women. J Clin Endocrinol Metab. 2009;94:2594–601. doi: 10.1210/jc.2009-0139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunaif A, Scully RE, Andersen RN, Chapin DS, Crowley WF., Jr The effects of continuous androgen secretion on the hypothalamic-pituitary axis in woman: evidence from a luteinized thecoma of the ovary. J Clin Endocrinol Metab. 1984;59:389–93. doi: 10.1210/jcem-59-3-389. [DOI] [PubMed] [Google Scholar]
- Dunaif A. Do androgens directly regulate gonadotropin secretion in the polycystic ovary syndrome? J Clin Endocrinol Metab. 1986;63:215–21. doi: 10.1210/jcem-63-1-215. [DOI] [PubMed] [Google Scholar]
- Dunaif A, Segal KR, Futterweit W, Dobrjansky A. Profound peripheral insulin resistance, independent of obesity, in polycystic ovary syndrome. Diabetes. 1989;38:1165–1174. doi: 10.2337/diab.38.9.1165. [DOI] [PubMed] [Google Scholar]
- Eagleson CA, Gingrich MB, Pastor CL, Arora TK, Burt CM, Evans WS, Marshall JC. Polycystic ovarian syndrome: evidence that flutamide restores sensitivity of the gonadotropin-releasing hormone pulse generator to inhibition by estradiol and progesterone. J Clin Endocrinol Metab. 2000;85:4047–52. doi: 10.1210/jcem.85.11.6992. [DOI] [PubMed] [Google Scholar]
- Eagleson CA, Bellows AB, Hu K, Gingrich MB, Marshall JC. Obese patients with polycystic ovary syndrome: evidence that metformin does not restore sensitivity of the gonadotropin-releasing hormone pulse generator to inhibition by ovarian steroids. J Clin Endocrinol Metab. 2003;88:5158–62. doi: 10.1210/jc.2003-030167. [DOI] [PubMed] [Google Scholar]
- Ehrmann DA, Barnes RB, Rosenfield RL. Polycystic ovary syndrome as a form of functional ovarian hyperandrogenism due to dysregulation of androgen secretion. Endocrin Rev. 1995;16:322–353. doi: 10.1210/edrv-16-3-322. [DOI] [PubMed] [Google Scholar]
- Ehrmann DA. Medical progress: polycystic ovary syndrome. N Engl J Med. 2005;352:1223–36. doi: 10.1056/NEJMra041536. [DOI] [PubMed] [Google Scholar]
- Feldman S, Billaud L, Thalabard J-C, Raux-Demay MC, Mowszowicz I, Kuttenn F, Mauvais-Jarvis P. Fertility in women with late-onset adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab. 1992;74:635–639. doi: 10.1210/jcem.74.3.1310999. [DOI] [PubMed] [Google Scholar]
- Flor-Cisneros A, Leschek EW, Merke DP, Barnes KM, Coco M, Cutler GB, Jr, Baron J. In boys with abnormal developmental tempo, maturation of the skeleton and the hypothalamic-pituitary-gonadal axis remains synchronous. J Clin Endocrinol Metab. 2004;89:236–41. doi: 10.1210/jc.2002-021954. [DOI] [PubMed] [Google Scholar]
- Foecking EM, Szabo M, Schwartz NB, Levine JE. Neuroendocrine consequences of prenatal androgen exposure in the female rat: absence of luteinizing hormone surges, suppression of progesterone receptor gene expression, and acceleration of the gonadotropin-releasing hormone pulse generator. Biol Reprod. 2005;72:1475–83. doi: 10.1095/biolreprod.105.039800. [DOI] [PubMed] [Google Scholar]
- Foster C, Comite F, Pescovitz O, Ross J, Loriaux D, Cutler CJ. Variable response to a long-acting agonist of luteinizing hormone-releasing hormone in girls with McCune-Albright syndrome. J Clin Endocrinol Metab. 1984;59:801. doi: 10.1210/jcem-59-4-801. [DOI] [PubMed] [Google Scholar]
- Franks PW, Hanson RL, Knowler WC, Sievers ML, Bennett PH, Looker HC. Childhood obesity, other cardiovascular risk factors, and premature death. N Engl J Med. 2010;362:485–93. doi: 10.1056/NEJMoa0904130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch R. Body fat, puberty, and fertility. Biol Rev Camb Philos Soc. 1984;59:161–88. doi: 10.1111/j.1469-185x.1984.tb00406.x. [DOI] [PubMed] [Google Scholar]
- Ghizzoni L, Virdis R, Vottero A, Cappa M, Street ME, Zampolli M, Ibanez L, Bernasconi S. Pituitary-ovarian responses to leuprolide acetate testing in patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab. 1996;81:601–606. doi: 10.1210/jcem.81.2.8636275. [DOI] [PubMed] [Google Scholar]
- Gianetti E, Tusset C, Noel SD, Au MG, Dwyer AA, Hughes VA, Abreu AP, Carroll J, Trarbach E, Silveira LF, Costa EM, de Mendonca BB, de Castro M, Lofrano A, Hall JE, Bolu E, Ozata M, Quinton R, Amory JK, Stewart SE, Arlt W, Cole TR, Crowley WF, Kaiser UB, Latronico AC, Seminara SB. TAC3/TACR3 mutations reveal preferential activation of gonadotropin-releasing hormone release by neurokinin B in neonatal life followed by reversal in adulthood. J Clin Endocrinol Metab. 2010;95:2857–67. doi: 10.1210/jc.2009-2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Givens JR, Andersen RN, Wiser WL, Donelson AJ, Coleman SA. A testosterone-secreting, gonadotropin-responsive pure thecoma and polycystic ovarian disease. J Clin Endocrinol Metab. 1975;41:845–53. doi: 10.1210/jcem-41-5-845. [DOI] [PubMed] [Google Scholar]
- Grumbach M, Roth J, Kaplan S, Kelch R. Hypothalamic-pituitary regulation of puberty in man: Evidence and concepts derived from clinical research. In: Grumbach M, Grave C, Mayer F, editors. The Control of the Onset of Puberty. John Wiley & Sons; New York: 1974. pp. 115–207. [Google Scholar]
- Hemrika DJ, Slaats EH, Kennedy JC, de Vries Robles-Korsen TJ, Schoemaker J. Pulsatile luteinizing hormone patterns in long term oral contraceptive users. J Clin Endocrinol Metab. 1993;77:420–6. doi: 10.1210/jcem.77.2.8345046. [DOI] [PubMed] [Google Scholar]
- Hirshfeld-Cytron J, Barnes RB, Ehrmann DA, Caruso A, Mortensen MM, Rosenfield RL. Characterization of functionally typical and atypical types of polycystic ovary syndrome. J Clin Endocrinol Metab. 2009;94:1587–94. doi: 10.1210/jc.2008-2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holte J, Bergh T, Gennarelli G, Wide L. The independent effects of polycystic ovary syndrome and obesity on serum concentrations of gonadotrophins and sex steroids in premenopausal women. Clin Endocrinol (Oxf) 1994;41:473–81. doi: 10.1111/j.1365-2265.1994.tb02578.x. [DOI] [PubMed] [Google Scholar]
- Jain A, Polotsky AJ, Rochester D, Berga SL, Loucks T, Zeitlian G, Gibbs K, Polotsky HN, Feng S, Isaac B, Santoro N. Pulsatile luteinizing hormone amplitude and progesterone metabolite excretion are reduced in obese women. J Clin Endocrinol Metab. 2007;92:2468–73. doi: 10.1210/jc.2006-2274. [DOI] [PubMed] [Google Scholar]
- Jakimiuk AJ, Weitsman SR, Navab A, Magoffin DA. Luteinizing hormone receptor, steroidogenesis acute regulatory protein, and steroidogenic enzyme messenger ribonucleic acids are overexpressed in thecal and granulosa cells from polycystic ovaries. J Clin Endocrinol Metab. 2001;86:1318–23. doi: 10.1210/jcem.86.3.7318. [DOI] [PubMed] [Google Scholar]
- Kaufman JM, Vermeulen A. The decline of androgen levels in elderly men and its clinical and therapeutic implications. Endocr Rev. 2005;26:833–76. doi: 10.1210/er.2004-0013. [DOI] [PubMed] [Google Scholar]
- Kazer R, Kessel B, Yen S. Circulating luteinizing hormone pulse frequency in women with polycystic ovary syndrome. J Clin Endocrinol Metab. 1987;65:233–236. doi: 10.1210/jcem-65-2-233. [DOI] [PubMed] [Google Scholar]
- Kelch R, Hopwood N, Sauder S, Marshall J. Evidence for decreased secretion of gonadotropin-releasing hormone in pubertal boys during short-term testosterone treatment. Pediatr Res. 1985;19:112–7. doi: 10.1203/00006450-198501000-00030. [DOI] [PubMed] [Google Scholar]
- Kelch RP, Hopwood NJ, Marshall JC. Diagnosis of gonadotropin deficiency in adolescents: limited usefulness of a standard gonadotropin-releasing hormone test in obese boys. J Pediatr. 1980;97:820–4. doi: 10.1016/s0022-3476(80)80279-4. [DOI] [PubMed] [Google Scholar]
- Knobil E. The neuroendocrine control of the menstrual cycle. Recent Prog Horm Res. 1980;36:53. doi: 10.1016/b978-0-12-571136-4.50008-5. [DOI] [PubMed] [Google Scholar]
- Kulin HE, Grumbach MM, Kaplan SL. Changing sensitivity of the pubertal gonadal hypothalamic feedback mechanism in man. Science. 1969;166:1012–3. doi: 10.1126/science.166.3908.1012. [DOI] [PubMed] [Google Scholar]
- Lawson MA, Jain S, Sun S, Patel K, Malcolm PJ, Chang RJ. Evidence for insulin suppression of baseline luteinizing hormone in women with polycystic ovarian syndrome and normal women. J Clin Endocrinol Metab. 2008;93:2089–96. doi: 10.1210/jc.2007-2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee jM, Jacuritu N, Appuglies D, Corwyn RF, Bradley RH, Lumeng JC. Body mass index and timing of pubertal initiation in boys. Arch Pediatr Adolesc Med. 2010;164:139–44. doi: 10.1001/archpediatrics.2009.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leibel NI, Baumann EE, Kocherginsky M, Rosenfield RL. Relationship of adolescent polycystic ovary syndrome to parental metabolic syndrome. J Clin Endocrinol Metab. 2006;91:1275–1283. doi: 10.1210/jc.2005-1707. [DOI] [PubMed] [Google Scholar]
- MacDonald AA, Herbison GP, Showell M, Farquhar CM. The impact of body mass index on semen parameters and reproductive hormones in human males: a systematic review with meta-analysis. Hum Reprod Update. 2010;16:293–311. doi: 10.1093/humupd/dmp047. [DOI] [PubMed] [Google Scholar]
- Marshall J, Kelch R. Gonadotropin-releasing hormone: Role of pulsatile secretion in the regulation of reproduction. N Engl J Med. 1986;315:1459. doi: 10.1056/NEJM198612043152306. [DOI] [PubMed] [Google Scholar]
- McCartney CR, Gingrich MB, Hu Y, Evans WS, Marshall JC. Hypothalamic regulation of cyclic ovulation: evidence that the increase in gonadotropin-releasing hormone pulse frequency during the follicular phase reflects the gradual loss of the restraining effects of progesterone. J Clin Endocrinol Metab. 2002;87:2194–200. doi: 10.1210/jcem.87.5.8484. [DOI] [PubMed] [Google Scholar]
- McCartney CR, Bellows AB, Gingrich MB, Hu Y, Evans WS, Marshall JC, Veldhuis JD. Exaggerated 17-hydroxyprogesterone response to intravenous infusions of recombinant human LH in women with polycystic ovary syndrome. Am J Physiol Endocrinol Metab. 2004;286:E9028. doi: 10.1152/ajpendo.00415.2003. [DOI] [PubMed] [Google Scholar]
- McCartney CR, Prendergast KA, Chhabra S, Eagleson CA, Yoo R, Chang RJ, Foster CM, Marshall JC. The association of obesity and hyperandrogenemia during the pubertal transition in girls: obesity as a potential factor in the genesis of postpubertal hyperandrogenism. J Clin Endocrinol Metab. 2006;91:1714–22. doi: 10.1210/jc.2005-1852. [DOI] [PubMed] [Google Scholar]
- McCartney CR, Blank SK, Prendergast KA, Chhabra S, Eagleson CA, Helm KD, Yoo R, Chang RJ, Foster CM, Caprio S, Marshall JC. Obesity and sex steroid changes across puberty: evidence for marked hyperandrogenemia in pre- and early pubertal obese girls. J Clin Endocrinol Metab. 2007;92:430–6. doi: 10.1210/jc.2006-2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCartney CR, Prendergast KA, Blank SK, Helm KD, Chhabra S, Marshall JC. Maturation of luteinizing hormone (gonadotropin-releasing hormone) secretion across puberty: evidence for altered regulation in obese peripubertal girls. J Clin Endocrinol Metab. 2009;94:56–66. doi: 10.1210/jc.2008-1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta RV, Patel KS, Coffler MS, Dahan MH, Yoo RY, Archer JS, Malcom PJ, Chang RJ. Luteinizing hormone secretion is not influenced by insulin infusion in women with polycystic ovary syndrome despite improved insulin sensitivity during pioglitazone treatment. J Clin Endocrinol Metab. 2005;90:2136–41. doi: 10.1210/jc.2004-1040. [DOI] [PubMed] [Google Scholar]
- Mi Y, Fiete D, Baenziger JU. Ablation of GalNAc-4-sulfotransferase-1 enhances reproduction by altering the carbohydrate structures of luteinizing hormone in mice. J Clin Invest. 2008;118:1815–24. doi: 10.1172/JCI32467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortensen M, Ehrmann DA, Littlejohn E, Rosenfield RL. Asymptomatic volunteers with a polycystic ovary are a functionally distinct but heterogeneous population. J Clin Endocrinol Metab. 2009;94:1579–86. doi: 10.1210/jc.2008-2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson VL, Legro RS, Strauss JF, 3rd, McAllister JM. Augmented androgen production is a stable steroidogenic phenotype of propagated theca cells from polycystic ovaries. Mol Endocrinol. 1999;13:946–57. doi: 10.1210/mend.13.6.0311. [DOI] [PubMed] [Google Scholar]
- Nelson VL, Qin Kn K, Rosenfield RL, Wood JR, Penning TM, Legro RS, Strauss JF, 3rd, McAllister JM. The biochemical basis for increased testosterone production in theca cells propagated from patients with polycystic ovary syndrome. J Clin Endocrinol Metab. 2001;86:5925–5933. doi: 10.1210/jcem.86.12.8088. [DOI] [PubMed] [Google Scholar]
- Pagan YL, Srouji SS, Jimenez Y, Emerson A, Gill S, Hall JE. Inverse relationship between luteinizing hormone and body mass index in polycystic ovarian syndrome: investigation of hypothalamic and pituitary contributions. J Clin Endocrinol Metab. 2006;91:1309–16. doi: 10.1210/jc.2005-2099. [DOI] [PubMed] [Google Scholar]
- Pastor CL, Griffin-Korf ML, Aloi JA, Evans WS, Marshall JC. Polycystic ovary syndrome: evidence for reduced sensitivity of the gonadotropin-releasing hormone pulse generator to inhibition by estradiol and progesterone. J Clin Endocrinol Metab. 1998;83:582–90. doi: 10.1210/jcem.83.2.4604. [DOI] [PubMed] [Google Scholar]
- Pescovitz OH, Comite F, Cassorla F, Dwyer AJ, Poth MA, Sperling MA, Hench K, McNemar A, Skerda M, Loriaux DL. True precocious puberty complicating congenital adrenal hyperplasia: Treatment with a luteinizing hormone-releasing hormone analog. J Clin Endocrinol Metab. 1984;58:857. doi: 10.1210/jcem-58-5-857. [DOI] [PubMed] [Google Scholar]
- Pitteloud N, Dwyer AA, DeCruz S, Lee H, Boepple PA, Crowley WF, Jr, Hayes FJ. Inhibition of luteinizing hormone secretion by testosterone in men requires aromatization for its pituitary but not its hypothalamic effects: evidence from the tandem study of normal and gonadotropin-releasing hormone-deficient men. J Clin Endocrinol Metab. 2008;93:784–91. doi: 10.1210/jc.2007-2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porcu E, Venturoli S, Magrini O, Bolzani R, Gabbi D, Paradisi R, Fabbri R, Flamigni C. Circadian variations of luteinizing hormone can have two different profiles in adolescent anovulation. J Clin Endocrinol Metab. 1987;65:488–493. doi: 10.1210/jcem-65-3-488. [DOI] [PubMed] [Google Scholar]
- Quinkler M, Sinha B, Tomlinson JW, Bujalska IJ, Stewart PM, Arlt W. Androgen generation in adipose tissue in women with simple obesity -a site -specific role for 17beta-hydroxysteroid dehydrogenase type 5. J Endocrinol. 2004;183:331–42. doi: 10.1677/joe.1.05762. [DOI] [PubMed] [Google Scholar]
- Raivio T, Falardeau J, Dwyer A, Quinton R, Hayes FJ, Hughes VA, Cole LW, Pearce SH, Lee H, Boepple P, Crowley WF, Jr, Pitteloud N. Reversal of idiopathic hypogonadotropic hypogonadism. N Engl J Med. 2007;357:863–73. doi: 10.1056/NEJMoa066494. [DOI] [PubMed] [Google Scholar]
- Redding TW, Schally AV, Arimura A, Matsuo H. Stimulation of release and synthesis of luteinizing hormone(LH) and follicle stimulating hormone(FSH) in tissue culture of rat pituitaries in response to natural and synthetic LH and FSH releasing hormone. Endocrinology. 1972;90:764–70. doi: 10.1210/endo-90-3-764. [DOI] [PubMed] [Google Scholar]
- Ropelato MG, Rudaz MC, Escobar ME, Bengolea SV, Calcagno ML, Veldhuis JD, Barontini M. Acute effects of testosterone infusion on the serum luteinizing hormone profile in eumenorrheic and polycystic ovary syndrome adolescents. J Clin Endocrinol Metab. 2009;94:3602–10. doi: 10.1210/jc.2009-0402. [DOI] [PubMed] [Google Scholar]
- Rosenbaum M, Leibel RL. The role of leptin in human physiology [editorial; comment] N Engl J Med. 1999;341:913–5. doi: 10.1056/NEJM199909163411211. [DOI] [PubMed] [Google Scholar]
- Rosenfield RL, Fang VS. The effects of prolonged physiologic estradiol therapy on the maturation of hypogonadal teenagers. J Pediatr. 1974;85:830–837. doi: 10.1016/s0022-3476(74)80355-0. [DOI] [PubMed] [Google Scholar]
- Rosenfield RL. Congenital adrenal hyperplasia and reproductive function in female. In: Flamigni C, Venturoli S, Givens J, editors. Adolescence in Females. Year Book Medical Publishers Inc; Chicago: 1985. pp. 373–387. [Google Scholar]
- Rosenfield RL, Perovic N, Ehrmann DA, Barnes RB. Acute hormonal responses to the gonadotropin releasing hormone agonist leuprolide: dose-response studies and comparison to nafarelin. J Clin Endocrinol Metab. 1996;81:3408–3411. doi: 10.1210/jcem.81.9.8784105. [DOI] [PubMed] [Google Scholar]
- Rosenfield RL. Ovarian and adrenal function in polycystic ovary syndrome. Endocrinol Metab Clin N Am. 1999;28:265–293. doi: 10.1016/s0889-8529(05)70070-0. [DOI] [PubMed] [Google Scholar]
- Rosenfield RL. Clinical practice. Hirsutism [Comment in: N Engl J Med. 2006 Apr 6;354(14):1533–5; author reply 1533–5] N Engl J Med. 2005;353:2578–88. doi: 10.1056/NEJMcp033496. [DOI] [PubMed] [Google Scholar]
- Rosenfield RL. Identifying children at risk of polycystic ovary syndrome. J Clin Endocrinol Metab. 2007;92:787–796. doi: 10.1210/jc.2006-2012. [DOI] [PubMed] [Google Scholar]
- Rosenfield RL, Lipton RB, Drum ML. Thelarche, pubarche, and menarche attainment in children with normal and elevated body mass index. Pediatrics. 2009;123:84–8. doi: 10.1542/peds.2008-0146. [Erratum in:Pediatrics. 2009 Apr;123(4):1255] [DOI] [PubMed] [Google Scholar]
- Rotterdam ESHRE/ASRM-Sponsored PCOS Consensus Workshop Group. Revised 2003 consensus on diagnostic criteria and long-term health risks related to polycystic ovary syndrome. Fertil Steril. 2004;81:19–25. doi: 10.1016/j.fertnstert.2003.10.004. [DOI] [PubMed] [Google Scholar]
- Sarma HN, Manikkam M, Herkimer C, Dell’Orco J, Welch KB, Foster DL, Padmanabhan V. Fetal programming: excess prenatal testosterone reduces postnatal luteinizing hormone, but not follicle-stimulating hormone responsiveness, to estradiol negative feedback in the female. Endocrinology. 2005;146:4281–91. doi: 10.1210/en.2005-0322. [DOI] [PubMed] [Google Scholar]
- Serafini P, Silva P, Paulson R, Elkind-Hirsch K, Hernandez M, Lobo R. Acute modulation of the hypothalamic-pituitary axis by intravenous testosterone in normal women. Am J Obstet Gynecol. 1986;155:1288–1292. doi: 10.1016/0002-9378(86)90161-4. [DOI] [PubMed] [Google Scholar]
- Sorensen K, Aksglaede L, Petersen JH, Juul A. Recent changes in pubertal timing in healthy Danish boys: associations with body mass index. J Clin Endocrinol Metab. 2010;95:263–70. doi: 10.1210/jc.2009-1478. [DOI] [PubMed] [Google Scholar]
- Srouji SS, Pagan YL, D’Amato F, Dabela A, Jimenez Y, Supko JG, Hall JE. Pharmacokinetic factors contribute to the inverse relationship between luteinizing hormone and body mass index in polycystic ovarian syndrome. J Clin Endocrinol Metab. 2007;92:1347–52. doi: 10.1210/jc.2006-2716. [DOI] [PubMed] [Google Scholar]
- Strain GW, Zumoff B, Miller LK, Rosner W. Sex difference in the effect of obesity on 24-hour mean serum gonadotropin levels. Horm Metab Res. 2003;35:362–6. doi: 10.1055/s-2003-41358. [DOI] [PubMed] [Google Scholar]
- Taponen S, Martikainen H, Jarvelin MR, Laitinen J, Pouta A, Hartikainen AL, Sovio U, McCarthy MI, Franks S, Ruokonen A. Hormonal profile of women with self-reported symptoms of oligomenorrhea and/or hirsutism: Northern Finland birth cohort 1966 study. J Clin Endocrinol Metab. 2003;88:141–7. doi: 10.1210/jc.2002-020982. [DOI] [PubMed] [Google Scholar]
- Taylor AE, McCourt B, Martin KA, Anderson EJ, Adams JM, Schoenfeld D, Hall JE. Determinants of abnormal gonadotropin secretion in clinically defined women with polycystic ovary syndrome. J Clin Endocrinol Metab. 1997;82:2248–2256. doi: 10.1210/jcem.82.7.4105. [DOI] [PubMed] [Google Scholar]
- Vermeulen A. Environment, human reproduction, menopause, and andropause. Environ Health Perspect. 1993;101(Suppl 2):91–100. doi: 10.1289/ehp.93101s291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldstreicher J, Santoro NF, Hall JE, Filicori M, Crowley WF., Jr Hyperfunction of the hypothalamic-pituitary axis in women with polycystic ovarian disease: indirect evidence forpartial gonadotroph desensitization. J Clin Endocrinol Metab. 1988;66:165–172. doi: 10.1210/jcem-66-1-165. [DOI] [PubMed] [Google Scholar]
- Welt CK, Chan JL, Bullen J, Murphy R, Smith P, DePaoli AM, Karalis A, Mantzoros CS. Recombinant human leptin in women with hypothalamic amenorrhea. N Engl J Med. 2004;351:987–97. doi: 10.1056/NEJMoa040388. [DOI] [PubMed] [Google Scholar]
- Wide L, Naessen T, Sundstrom-Poromaa I, Eriksson K. Sulfonation and sialylation of gonadotropins in women during the menstrual cycle, after menopause, and with polycystic ovarian syndrome and in men. J Clin Endocrinol Metab. 2007;92:4410–7. doi: 10.1210/jc.2007-1342. [DOI] [PubMed] [Google Scholar]
- Wide L, Eriksson K, Sluss PM, Hall JE. Serum half-life of pituitary gonadotropins is decreased by sulfonation and increased by sialylation in women. J Clin Endocrinol Metab. 2009;94:958–64. doi: 10.1210/jc.2008-2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu FC, Tajar A, Pye SR, Silman AJ, Finn JD, O’Neill TW, Bartfai G, Casanueva F, Forti G, Giwercman A, Huhtaniemi IT, Kula K, Punab M, Boonen S, Vanderschueren D. Hypothalamic-pituitary-testicular axis disruptions in older men are differentially linked to age and modifiable risk factors: the European Male Aging Study. J Clin Endocrinol Metab. 2008;93:2737–45. doi: 10.1210/jc.2007-1972. [DOI] [PubMed] [Google Scholar]
- Yang W, Lu J, Weng J, Jia W, Ji L, Xiao J, Shan Z, Liu J, Tian H, Ji Q, Zhu D, Ge J, Lin L, Chen L, Guo X, Zhao Z, Li Q, Zhou Z, Shan G, He J. Prevalence of diabetes among men and women in China. N Engl J Med. 2010;362:1090–1101. doi: 10.1056/NEJMoa0908292. [DOI] [PubMed] [Google Scholar]
- Yen S. The polycystic ovary syndrome. Clin Endocrinol. 1980;12:177–208. doi: 10.1111/j.1365-2265.1980.tb02132.x. [DOI] [PubMed] [Google Scholar]
- Zawadzki J, Dunaif A. Diagnostic criteria for polycystic ovary syndrome: towards a rational approach. In: Polycystic Ovary Syndrome. In: Dunaif A, Givens J, Haseltine F, Merriam G, editors. Current Issues in Endocrinology and Metabolism. Vol. 4. Blackwell Scientific Publications; Cambridge, MA: 1992. pp. 377–384. [Google Scholar]
- Zimmer CA, Ehrmann DA, Rosenfield RL. Potential diagnostic utility of intermittent short-acting GnRH agonist administration in gonadotropin deficiency. Fertil Steril. 2010 doi: 10.1016/j.fertnstert.2010.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zumoff B, Freeman R, Coupey S, Saenger P, Markowitz M, Kream J. A chronobiologic abnormality in luteinizing hormone secretion in teenage girls with the polycysticovary syndrome. N Engl J Med. 1983;309:1206–1209. doi: 10.1056/NEJM198311173092002. [DOI] [PubMed] [Google Scholar]
- Zumoff B, Strain GW, Miller LK, Rosner W, Senie R, Seres DS, Rosenfeld RS. Plasma free and non-sex-hormone-binding-globulin-bound testosterone are decreased in obese men in proportion to their degree of obesity. J Clin Endocrinol Metab. 1990;71:929–31. doi: 10.1210/jcem-71-4-929. [DOI] [PubMed] [Google Scholar]
- Zumoff B, Miller LK, Strain GW. Reversal of the hypogonadotropic hypogonadism of obese men by administration of the aromatase inhibitor testolactone. Metabolism. 2003;52:1126–8. doi: 10.1016/s0026-0495(03)00186-0. [DOI] [PubMed] [Google Scholar]