

Abstract
Single strand breaks (SSBs) are one of the most frequent DNA lesions caused by endogenous and exogenous agents. The most utilized alkaline-based assays for SSB detection frequently give false positive results due to the presence of alkali-labile sites that are converted to SSBs. Methoxyamine, an acidic O-hydroxylamine, has been utilized to measure DNA damage in cells. However, the neutralization of methoxyamine is required prior to usage. Here we developed a convenient, specific SSB assay using alkaline gel electrophoresis (AGE) coupled with a neutral O-hydroxylamine, O-(tetrahydro-2H-pyran-2-yl)hydroxylamine (OTX). OTX stabilizes abasic sites (AP sites) to prevent their alkaline incision while still allowing for strong alkaline DNA denaturation. DNA from DT40 and isogenic polymerase β null cells exposed to methyl methanesulfonate were applied to the OTX-coupled AGE (OTX-AGE) assay. Time-dependent increases in SSBs were detected in each cell line with more extensive SSB formation in the null cells. These findings were supported by an assay that indirectly detects SSBs through measuring NAD(P)H depletion. An ARP-slot blot assay demonstrated a significant time-dependent increase in AP sites in both cell lines by 1 mM MMS compared to control. Furthermore, the Pol β-null cells displayed greater AP site formation than the parental DT40 cells. OTX use represents a facile approach for assessing SSB formation, whose benefits can also be applied to other established SSB assays.
Keywords: Alkylating agent, AP sites, Base excision repair, single strand breaks, O-Hydroxylamine
1. Introduction
Thousands of single strand breaks (SSBs) are believed to be produced in cellular DNA daily from endogenous and exogenous sources as well as during DNA repair processes, such as base excision repair (BER) [1]. If unrepaired, these lesions threaten genetic integrity through their potential conversion to lethal double strand breaks during DNA replication. Therefore, repair of SSBs is critical to the cell. BER is a primary response pathway for the repair of deleterious DNA lesions including non-bulky DNA adducts, apurinic/apyrimidinic (AP) sites, and SSBs. In its simplest form, BER proceeds as follows: 1) removal of a damaged base through N-glycosylic bond cleavage by a mono-functional DNA glycosylase with AP site formation; 2) incision 5’ to the AP site by a Type II AP endonuclease, resulting in a SSB with 3’-hydroxyl and 5’-deoxyribosephosphate (5’-dRp) margins; 3) removal of the 5’-dRp and synthesis of the missing nucleotide by DNA polymerase beta (Pol β); 4) and strand ligation by a ligase [2]. Efficient BER is imperative because of the inadvertent generation of SSBs as pathway intermediates. While SSBs are one of the most frequent DNA lesions, well-characterized assays with a high specificity for SSBs are lacking.
The ability to detect SSBs has generated much interest for assessing genotoxicity. However, many of these approaches can generate confounded SSB data. A specific example is the single cell gel electrophoresis (comet) assay, which has gained much popularity throughout the past decade due to its ease and relative inexpensiveness [3-4]. Similar to other SSB assays, the comet assay usually employs alkaline conditions (pH > 13) to denature DNA. The presence of alkali-labile sites (ALSs), such as AP sites, at high pH can lead to DNA strand cleavage. The resulting overestimation of SSB formation compromises the reliability of data obtained by the comet assay and other alkaline-based SSB assays [3]. The potential for such variability has been observed in intra- and inter- laboratory studies [5-6]. The comet assay performed at pH 12.1-12.5 has been reported to detect SSBs without the contribution of artifactual SSBs from cleaved ALSs; however, this modification has also been reported to reduce the sensitivity of that assay [7].
O-Hydroxylamines have been documented to react with the aldehydic group of AP sites to create very stable complexes that are refractory to β-elimination by enzymatic activity (i.e. AP or dRp lyases) or by high pH [8-10]. Methoxyamine hydrochloride has been used ex vivo as a BER inhibitor [11-13] and in vitro as a stabilizer of AP sites for the detection of damaged bases (e.g. N7-methylpurines) [14, 15] or AP sites [16, 17]. However, the acidic methoxyamine hydrochloride must be precisely neutralized [13, 18-20] before the usage. Furthermore, the neutralization of methoxyamine hydrochloride may lead to high salt contamination. The presence of salt retards DNA migration during electrophoresis, ultimately leading to an underestimate of SSB formation [21,22]. As an alternative to methoxyamine hydrochloride, the O-hydroxylamine, O-(tetrahydro-2Hpyran-2-yl)hydroxylamine (OTX), is available in a form that does not require neutralization, thus avoiding the resulting salt contamination. Here we report the development of a novel OTX-coupled alkaline gel electrophoresis (AGE) method for the specific detection of SSBs. Furthermore, this newly developed OTX-coupled AGE analysis, combined with an aldehyde reactive probe (ARP)-slot-blot analysis and NAD(P)H depletion assay allowed us to detect an accumulation of DNA repair intermediate product during acute exposure of cells to the methylating agent, methyl methanesulfonate (MMS).
2. Materials and Methods
2.1. Cell Culture and Chemical Exposure
DT40 cells and isogenic DT40-derived Pol β-null cells were cultivated as described previously [23]. DT40 and Pol β-null cells were seeded in 10-cm dishes and allowed to divide overnight to reach a density of approximately 1×106 cells/mL. Cells were exposed to a continuous treatment of 1 mM MMS (Aldrich) for up to 4 hours. Control cells received 1X PBS for 0.5 hour. After incubation, cells were harvested, washed with chilled 1X PBS, pelleted, and stored at -80 °C until DNA isolation. Using this protocol, DT40 cells were also incubated in the absence of chemical to obtain a stock of untreated DNA.
2.2. DNA Isolation from Cells
DNA was extracted using a PureGene DNA extraction kit (Gentra Systems, Inc.) with modifications as described previously [24].
2.3. AP Site Preparation and Alkali Treatment for Extracted DNA
AP sites were produced in stock DT40 DNA through exposure to heat/acid conditions. DT40 DNA was incubated in sodium citrate buffer (10 mM sodium citrate, 10 mM NaH2PO4, 10 mM NaCl, pH 5) for 5 minutes at 70 °C, followed by rapid chilling on ice to stop the reaction [8]. Depurinated DNA was collected using microcon YM-3 centrifugal filter devices (Amicon Bioseparations). This DNA was then washed several times with HEPES buffer (50 mM, pH 7) supplemented with 1 mM 2,2,6,6-tetramethylpiperidine-N-oxyl (TEMPO). DNA was finally resuspended in HEPES buffer containing 1 mM TEMPO. Heat/acid-treated DT40 DNA was further exposed to various concentrations of NaOH (0.1-250 mM) in HEPES buffer (50 mM) at 37 °C for 15 minutes. The samples were isolated, washed, and resuspended as described above.
2.4. NAD(P)H Depletion Assay
SSB repair capacity was determined by a real-time colorimetric NAD(P)H depletion assay described previously [25,26] using 1 × 105 cells/well . To confirm the activation of PARP-1 during continuous MMS exposure, cells were also co-exposed to PARP inhibitor 3-aminobenzamide (3-AB, 20 mM, Sigma).
2.5. ARP-Slot Blot (ASB) Assay
AP sites were measured using an ARP (Dojindo Molecular Technology, Gaithersburg, MD, USA) -coupled slot blot technique, previously described [8].
2.6. AGE
An AGE protocol [27] with modifications was used to assess SSBs and ALSs in DNA from exposed cells. Agarose gel (0.7%) was prepared in 50 mM NaCl containing 1 mM EDTA, pH 7.0. After casting, the gel was soaked in mild alkaline running buffer (30 mM NaOH, 1 mM EDTA, pH 12.4) for at least 30 minutes. Equal amounts of DNA (3-5 μg) were incubated for 1 hour at 37 °C in the presence or absence of OTX (Aldrich) in 50 mM HEPES buffer (pH 7.0). OTX does not require titration to neutral pH by NaOH before or during incubation with DNA. Following OTX exposure, the samples were denatured through treatment with an alkaline buffer (100 mM NaOH, 30 mM OTX, 50 mM HEPES, pH 12.8) for 20 minutes at 37 °C. Samples were mixed with loading buffer (10 mM NaOH, 95% formamide (Fluka), 0.05% bromophenol blue, 0.05% xylene cyanol) and were loaded onto the agarose gel. The gel was electrophoresed at a constant voltage (30 V) for 16 hours at 4 °C. At the completion of electrophoresis, the gel was placed in neutralization buffer (400 mM Tris, pH 7.5) for 20 minutes at room temperature. Gels were stained with acridine orange (Sigma) (5 μg/mL) for 1 hour or with SYBR Gold (10,000X; Invitrogen) for 40 minutes followed by destaining in deionized water. DNA migration was visualized using an Image Station 440CF system (Kodak). The extent of DNA migration was determined using the AlphaEaseFC (Alpha Innotech Inc.) gel documentation software. A profile of intensity values was generated for each lane with automatic correction for background intensity. In addition, the integrated intensity values from a blank lane were also subtracted as background. DNA remaining in the region ≥ 23.1 kb was considered intact while DNA in the region < 23.1 kb was considered fragmented [28]. The percent fragmented compared to total lane integrated intensity value were determined for each lane.
2.7. SSB Formation at N7-Methylguanine Adducts through Heat/Piperidine Displacement
To visualize the conversion of MMS-induced N7-methylguanine (N7-meG) adducts into SSBs, the AGE assay described above was modified with a piperidine treatment [29]. Equal amounts of DNA (25 μg) were incubated for 1 hour at 37° C in the presence of MMS (0 or 1 mM) and 30 mM OTX (Aldrich) in 50 mM HEPES buffer (pH 7). Treated DNA was washed and collected using microcon YM-3 centrifugal filter devices . The treated DNA was incubated with alkaline buffer (pH ≥ 12.8) containing either 100 mM NaOH or 1 M piperidine for 20 minutes at 37 °C. This DNA was washed, collected by centrifugal filter devices, and the concentration determined by UV. DNA (3-5 μg) from each sample was mixed with loading dye and was loaded onto a 0.7% agarose gel prepared as previously described. Electrophoresis, neutralization, staining, and image analysis were performed as described above.
2.8. Statistical Analyses
A student’s t-test was utilized to determine the significant differences (p<0.05) between groups for absorbance of medium containing formazan dye, extent of NAD(P)H depletion and DNA fragmentation, and the number of AP sites.
3. Results and Discussion
3.1. Affect of pH on Cleavage of AP Sites
The use of alkaline conditions in established SSB assays has been shown to be an efficient means to denature and unwind DNA for the detection of the lesions. However, during DNA denaturation at high pH, DNA is cleaved the sugar/phosphate chain at 3’ the intact AP sites (i.e., AP sites without an adjacent nick) are nicked by β-elimination [30]. Because intact AP sites appear to be one of the major ALSs present in genomic DNA, current AGE analysis overestimate SSB content in genomic DNA, To establish the alkaline denaturation condition for the AGE assay, we used freshly isolated, genomic DNA. The extracted DNA was treated with heat/acid to generate intact AP sites, followed by exposure to various NaOH concentrations ranging in pH from 10 to greater than 13. This alkaline treatment of the AP site containing DNA was performed prior to mild alkaline (pH 12.4) agarose gel electrophoresis. Samples in NaOH concentrations with pH below 11.9 displayed large amounts of DNA just below the loading wells, similar to the control sample with no NaOH exposure prior to electrophoresis (Figure 1A, Lanes 2-4 versus Lane 1). DNA denaturation and unwinding relies on the disruption of hydrogen bonds at pH > 12 [31,32]. This data shows that these concentrations of NaOH (pH < 12) were unable to denature the DNA for efficient electrophoresis. With increasing NaOH concentration (pH > 12.2), the amount of DNA just below the wells decreased due to enhanced DNA migration (Figure 1A, Lanes 5-12). While the increase in DNA migration may be partly due to cleavage of intact AP sites by the increasing NaOH concentration, the decrease in the amount of DNA just below the wells indicates that DNA denaturation was increasingly improved at these NaOH concentrations. Based on these results, we selected 100 mM NaOH (pH 12.8; Figure 1A, Lane 10) for DNA unwinding in subsequent experiments. Under these conditions, freshly isolated, genomic DNA without heat/acid-treatment showed no major DNA fragmentation (data not shown).
Figure 1. Establishment of OTX-coupled AGE assay.
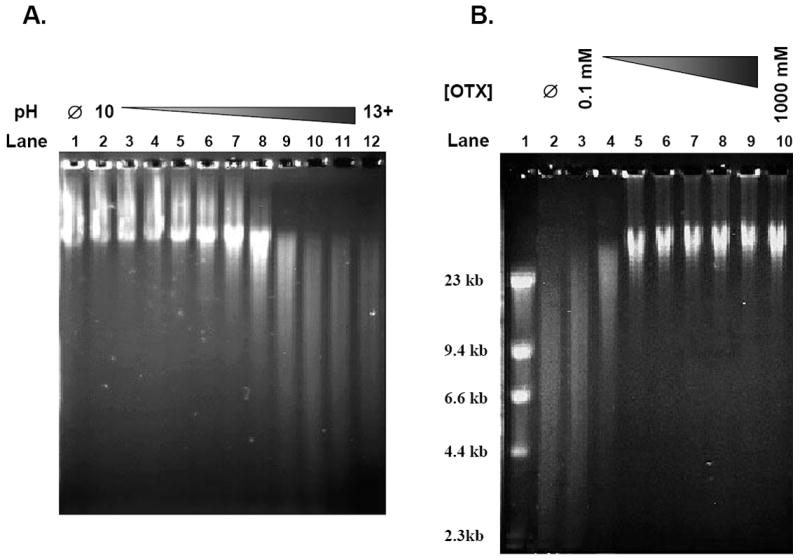
(A) Examining pH effect on DNA alkaline denaturation. Genomic DT40 DNA was exposed to heat/acid conditions for 5 minutes to induce intact AP sites. The DNA was then exposed to various concentrations of NaOH to create a pH gradient (pH 10-13+) prior to being loaded into an agarose gel and electrophoresed under alkaline conditions (pH 12.4). The gels were stained with acridine orange and the image was obtained with a Kodak Image Station 440CF system. Lane 1 – No NaOH (Ø); Lanes 2-4 – pH ≤ 11.9; Lanes 5-8 – pH 12.2-12.6; Lanes 9-12 – pH ≥ 12.7. (B) OTX optimization for the protection of AP sites. Genomic DT40 DNA was exposed to heat/acid conditions for 5 minutes to induce intact AP sites. The DNA was then exposed to a concentration gradient of OTX (0.1-1000 mM) prior to denaturation with 100 mM NaOH (pH 12.8) and electrophoresed under alkaline conditions (pH 12.4) in an agarose gel. The gel was stained with acridine orange and the image was obtained with a Kodak Image Station 440CF system. Lane 1 – Marker; Lane 2 – No OTX (Ø); Lanes 3-10 – Heat/acid DNA + OTX (0.1, 0.3, 1, 3, 10, 30, 100 and 1000 mM).
It has been previously reported that AP sites are stable against alkaline cleavage at pH < 12.6 [31,32]. However, our findings suggest that AP sites generated by heat/acid conditions are stable at pH ≤ 11.9 but may be increasingly cleaved at pH ≥ 12.2. This suggests that any current SSB assay that relies on alkaline conditions at pH ≥ 12.2 for DNA denaturation may be inducing artifactual SSBs through the cleavage of intact AP sites, resulting in an overestimate of SSB formation. As the nearly complete denaturation of DNA requires incubation of DNA at pH ≥12.7 (Figure 1A, Lane 10-12), we could not accomplish DNA denaturation without stabilizing AP sites.
3.2. O-(tetrahydro-2H-pyran-2-yl)hydroxylamine Optimization for Protection of AP Sites
We next assessed the usefulness of stabilizing AP sites prior to complete unwinding at high pH for subsequent gel electrophoresis. It has been well characterized that O-hydroxylamines, such as acidic methoxyamine hydrochloride, efficiently react with AP sites to generate very stable complexes, which are resistant to β-elimination [8]. The acidic O-hydroxylamines need to be titrated to neutral pH before usage, leading to high salt contamination in DNA samples. This high salt contamination may hinder migration of DNA during electrophoresis due to inefficient DNA denaturation. It also affects the electric field in the gel. Of the different O-hydroxylamine compounds, we selected pH neutral OTX, because of its commercial availability at reasonable cost and its simple preparation, which requires no titration to neutral pH, thereby eliminating high salt contamination in DNA samples. To optimize the concentration of OTX, DNA containing intact AP sites was exposed to OTX (0.1 - 1000 mM) at 37 °C for 1 hour prior to denaturation. At concentrations of OTX < 1 mM (Figure 1B, Lanes 3-4), DNA migration was unhindered compared to the unprotected control (Figure 1B, Lane 2). At concentrations of OTX ≥ 1 mM, there was little or no DNA migration beyond the 23.1 kb size marker (Figure 1B, Lanes 5-10). DNA that did not migrate past the 23.1 kb marker was considered to be intact DNA [28]; therefore, 30 mM OTX (Figure 1B, Lane 8) was selected as the optimum concentration to protect AP sites from alkaline cleavage and was utilized for the remainder of the experiments in this study.
3.3. Accumulation of Single Strand Break Repair Intermediates in BER-Deficient DT40 Cells Exposed to MMS
The accumulation of SSBs leads to PARP-1 activation and consumption of NAD+. NAD+ can be indirectly monitored in living cells through the measurement of intracellular NAD(P)H depletion as detected by the formation of formazan dye converted from tetrazolium salts (e.g., XTT) during chemical exposure [25]. As with other SSB assays, we could not exclude the contribution of double strand breaks from the results. However, even in highly replicating Saccharomyces cerevisiae deficient in either RAD6 or RAD52, MMS predominantly causes SSBs over double strand breaks [33,34]; therefore, most of the NAD(P)H depletion in wild-type DT40 and Pol β ko cells caused by MMS is likely due to SSBs. While the formazan products measured by absorbance at 450 nm increased in a nearly linear manner with increasing time in DT40 cells exposed to PBS (Figure 2A), DT40 cells exposed to 1 mM MMS for up to 4 hours displayed a slower rate of formation of formazan products compared to the control group (Figure 2A). Formazan formation exhibited greater suppression in Pol β-null cells exposed to MMS than in parental DT40 cells (Figure 2B). Consequently, the Pol β-null cells displayed a time-dependent, more extensive NAD(P)H depletion than parental DT40 cells by 1 mM MMS exposure (Figure 2C). NAD(P)H depletion was prevented by the PARP inhibitor 3-AB (Figure 2C). Therefore, we can conclude that NAD(P)H depletion in DT40 and Pol β-null cells exposed to MMS was due to an accumulation of SSBs. These results are in good agreement with our previous reports [35].
Figure 2. Intracellular NAD(P)H depletion in DT40 and Pol β-null cells exposed to 1 mM MMS or 1x PBS.
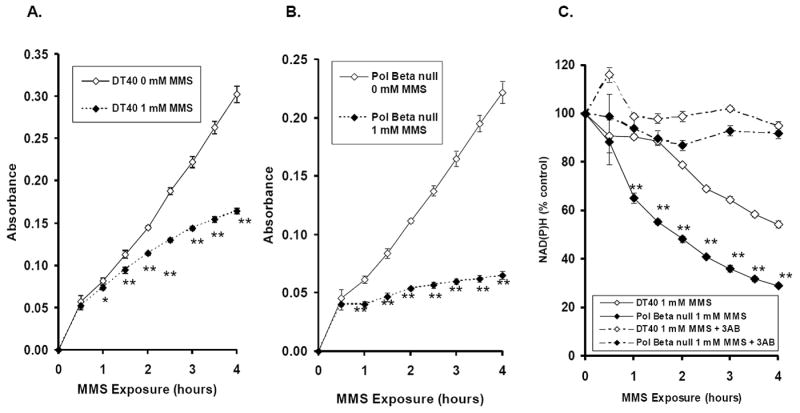
Intracellular NAD(P)H levels were detected by formation of formazan dye converted from tetrazolium salt in cells continuously exposed to 0 or 1 mM MMS for up to 4 hours. The amount of formazan dye in the culture medium was detected by absorbance at 450 nm using a plate reader. The intracellular NAD(P)H depletion is an indicator of PARP activation caused by accumulation of SSBs in the cell lines exposed MMS. (A-B) Absorbance measurements in the DT40 and Pol β-null cells, respectively. (C) DT40 and Pol β-null cells were continuously exposed to 1 mM MMS in the presence or absence of 3-AB (20 mM). Absorbance data plotted as percent control. The mean values represent three independent measurements. Bars indicate SD. Statistical Significance: * p < 0.05, ** p < 0.01, compared to PBS control (A-B) and DT40 cells (C) for the experiments in the absence of 3-AB.
If SSBs are actually being generated during the repair of MMS-induced DNA damage as indicated by the NAD(P)H depletion assay mentioned above, then we should be able to detect the formation of these SSBs using our OTX-AGE assay. We first exposed DT40 cells to PBS (1-4 hours) as a control. No difference in DNA migration in the cells exposed to PBS for 4 hours was found, which indicates that no significant SSB accumulation under 4-hour cultivation conditions (data not shown). To determine the extent to which SSBs were being generated, DNA was isolated from DT40 and Pol β-null cells exposed to 1 mM MMS (1-4 hours) for the direct detection of SSBs by OTX-AGE. Visually, the gel-based assay showed a slight time-dependent increase in DNA migration induced by MMS (1 mM) in DT40 cells (Figure 3A, Lanes 4, 6, 8 and 10). The ratio between the amount of DNA fragmented to less than 23.1 kb and the total amount of DNA for each treated sample was compared to the PBS control value (Figure 3B). The data suggest that an accumulation of BER intermediate product starts even after a 30-min exposure to MMS in DT40 cells.
Figure 3. SSB detection in DT40 and Pol β-null cells exposed continuously to 1 mM MMS for up to 4 hours.
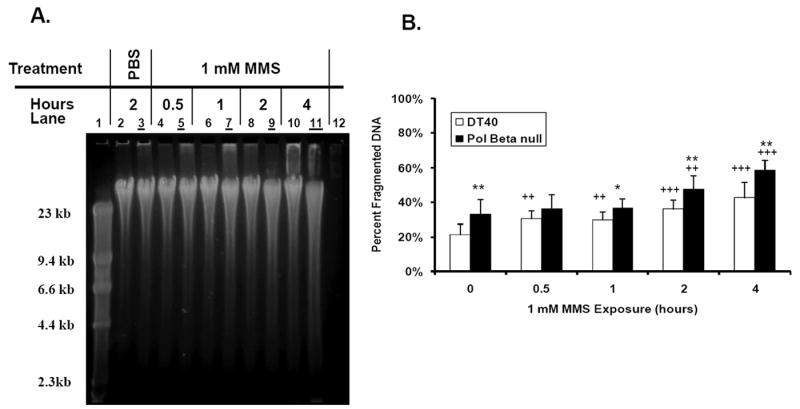
(A) Visualization of SSB formation in the cell lines through OTX-coupled AGE analysis. Images obtained using SYBR Gold and the Kodak Image Station 440CF system. Even lanes - DT40 cells; Odd lanes (with underline) - Pol β-null cells; Lane 1 - Marker; Lanes 2-3 – PBS control; Lanes 4-11 - 1 mM MMS exposure for 0.5-4 hours; Lane 12 - Blank. (B) Graphic representation of fragmented DNA in each sample as determined by the AlphaEaseFC gel documentation software. The mean values represent four independent measurements. Bars indicate SD. Statistical significance: ++ p < 0.01, +++ p < 0.001, compared to PBS control of the cell line; * p < 0.05, ** p < 0.01, compared to the corresponding treatment in the wild-type cell line.
Similarly, the Pol β-null cell line also displayed a time-dependent increase in DNA migration during the 1 mM MMS exposure (Figure 3A, Lanes 5, 7, 9 and 11). Significant differences were observed in accumulation of SSBs in comparison to the PBS control at the 2 and 4 hour time points (Figure 3B). Comparison of the Pol β-null cells to the DT40 wild-type cells showed significant increases in fragmented DNA during exposure to 1 mM MMS indicating greater SSB formation in the repair deficient cell line (Figure 3B). Though the numerical data did not show a significant difference in SSB accumulation at the shorter MMS exposure times compared to PBS control, this could be partly explained by the presence of a higher background of endogenous SSBs in the Pol β-null cells than in parental DT40 cells (Figure 3A and B, PBS control). Overall, there is a good agreement between the NAD(P)H depletion assay and OTX-AGE analysis. We also conclude that the combination of a robust NAD(P)H depletion assay and the specific, direct OTX-AGE analysis provides a more precise demonstration of SSB formation.
3.4. AP Site Formation in BER-Deficient DT40 Cells during MMS Exposure
BER of alkylated bases leads to the formation of AP sites as pathway intermediates. The accumulation of AP sites during acute MMS exposure was examined in DNA not exposed to OTX, which was extracted from DT40 and Pol β-null cells using the ASB assay. Both cell lines displayed a significant time-dependent increase in AP sites by 1 mM MMS compared to control. Furthermore, the Pol β-null cells displayed greater AP site formation than the parental DT40 cells (Figure 4). While the number of AP sites significantly increased by 1 mM MMS treatment for 1 hour in this study, our previous study showed no increase in the number of AP sites in mouse embryonic fibroblasts deficient or proficient in Pol β after 1-hour exposure to 1 mM MMS [36]. This discrepancy can be explained by an improvement in DNA extraction through the use of the free radical scavenger TEMPO [24]. TEMPO reduces the artifactual induction of aldehydic sites in DNA (e.g., AP sites) that are detected by the ASB assay. This discrepancy may also be explained by inherent differences between mammalian and avian cell lines. Interestingly, there was an increase in the number of AP sites induced by 1 mM MMS in Pol β-null cells as compared to parental DT40 cells in the present study; whereas, we recently reported that 1 mM MMS induced similar numbers of AP sites in DT40 cells and their PARP-1 null cells [25]. This difference in the extent of AP site accumulation could be explained by a greater ability of the PARP-1 null cells than the Pol β-null cells to remove 5’-dRp.
Figure 4.

AP site detection in DT40 and Pol β-null cells exposed continuously to 1 mM MMS for up to 4 hours. The number of AP sites was determined by the ASB assay. The mean values represent four independent measurements. Bars indicate SD. Statistical significance: ** p < 0.01, compared to DT40 cells.
N7-meG, which is considered an ALS, is the predominate lesion formed by SN2 methylating agents, such as MMS [37]. Therefore, NaOH treatment might contribute SSB formation derived from alkaline-labile N7-meG during AGE. To assess this phenomenon, we simultaneously exposed isolated genomic DNA to MMS (0 or 1 mM) and 30 mM OTX for 1 hour at 37°C to induce N7-meG formation and protect pre-existing or endogenous aldehydic sites from alkaline strand scission during subsequent AGE. As a positive demonstration of the contribution of ALSs to the SSBs detected, we incubated treated DNA with piperidine instead of NaOH during DNA denaturation. The introduction of heat/piperidine and resulting alkaline pH induced SSBs at N7-meG adducts through a series of chemical events involving imidazole ring opening of N7-meG, displacement of the resulting formamido-pyrimidine from the deoxyribose backbone with AP site formation, and β-elimination of the AP site with concurrent SSB formation [29]. In addition, Greenberg and his colleagues beautifully demonstrated that FAPy-G sites in 2‘-deoxyoligonucleotides at a defined site in oligonucleotides are only partially cleaved by exposure to 1 M piperidine at 37 °C for 20 min at 6.15% [38]. When the MMS-treated DNA received piperidine treatment, a notable increase in DNA migration was observed (data not shown) due to the presence of the N7-meG lesions in the sample. However, the increase in DNA migration was not seen in the PBS control that also received piperidine treatment (data not shown). In MMS-treated and control DNA subjected to NaOH-induced DNA denaturation there was no difference in the amount of DNA migration (data not shown). In contrast to piperidine treatment, the lack of migration observed in the MMS-exposed DNA treated with NaOH indicates that this denaturation condition cleaves the N7-meG lesions only sparingly; thus, these lesions did not contribute to artifactual SSB detection.
3.5. Comprehensive analyses for characterization of an accumulation of BER intermediate product
BER pathways consist of a series of steps, leading to the generation of repair intermediate products within the pathway, such as AP sites and SSBs. These BER intermediates are more toxic and mutagenic than some of the initial base damage (e.g., N7-meG). Therefore, it is important to characterize the extent of the accumulation of BER intermediate product with high specificity and sensitivity in cells and tissues. Our comprehensive analyses using a combination of the newly developed OTX-AGE assay, the previously developed NAD(P)H depletion analysis along with the ASB for AP sites allow us to better characterize an accumulation of BER intermediate product in cells exposed to MMS. The NAD(P)H depletion assay provides indirect evidence of PARP-1/2 activation with very high sensitivity and reproducibility, but somewhat low specificity. This assay has been utilized in cultured cell experiments to understand both when and to what extent DNA damaging agents cause SSBs in a real-time scenario [25,26,39-41]. Once we identify the starting point of PARP activation, we have a much better chance to understand the accumulation of BER intermediate product using more specific biomarkers. Based on the results obtained by the NAD(P)H depletion assay, we selected a 1 mM MMS exposure for 0.5 to 4 hours. The ASB assay can measure the number of AP sites in genomic DNA. In the present study, a significant increase in AP sites was detected slightly earlier than PARP activation as detected by the NAD(P)H depletion assay. Furthermore, the Pol β-null cells showed greater AP site formation than the parental DT40 cells. These results suggest that, in the absence of Pol β, 5’-dRp may accumulate in the cells. An alternative possibility is that APE1 may inefficiently incise DNA strand at AP site generated from N7-meG and N3-methyladenine in DT40 cells deficient in Pol β. Previously, during in vitro experiments, Pol β has been shown to displace APE1 from the damaged site during BER [42]. Therefore, in Pol β-null cells, the efficiency of APE1 may be expected to decrease as less APE1 is released from the site of repair. The resulting product inhibition of APE1 could further lead to an increase in AP sites in the Pol β-null cells.
3.6. Advantages of the OTX-Coupled AGE Assay and Application to Other SSB Assays
In addition to being a specific means to detect SSBs, the OTX-AGE assay offers a number of advantages. This approach is a relatively easy, time efficient assay based on common electrophoresis techniques. Because it is an electrophoretic method, the equipment used in the assay is often available in laboratories and the chemicals are readily available from commercial sources. Since the OTX-AGE assay uses very little DNA (3-5 μg per sample), this method can be used in situations where DNA sample amounts are limited. This assay is also versatile because samples analyzed by OTX-AGE may represent either DNA isolated from various tissues or cells after in vivo exposures, or DNA purified after in vitro exposures to genotoxicants; this latter experimental design would not be applicable to the comet assay. The simple method of OTX-AGE analysis is also very attractive. This method serves as a valuable means to screen the ability of exogenous and endogenous agents to induce SSBs prior to assessing additional measures of DNA damage.
The use of OTX may also benefit other established alkaline-based SSB assays, such as the comet assay. Inclusion of OTX in the cell washing, lysis, and denaturation buffers for the comet assay should protect AP sites within cells during processing and electrophoresis. Lysis of the cells followed by complete DNA denaturation could then be achieved with high alkaline conditions without a reduction in assay sensitivity or specificity.
Acknowledgments
The authors thank Dr. Bruna P. Brylawski and John Ridpath for their critical reading of this manuscript. This study was supported in part by National Institute of Environmental Health Science grants (T32-ES07126, P42-ES05948, P30-ES10126); National Cancer Institute (T32-CA72319); and Environmental Protection Agency Science to Achieve Results Graduate Fellowship (FP-91643601 to B. F. P.).
Footnotes
Conflict of Interest statement The authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Caldecott KW. Mammalian single-strand break repair: mechanisms and links with chromatin. DNA Repair. 2007;6:443–453. doi: 10.1016/j.dnarep.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 2.Fortini P, Dogliotti E. Base damage and single-strand break repair: mechanisms and functional significance of short- and long-patch repair subpathways. DNA Repair. 2007;6:398–409. doi: 10.1016/j.dnarep.2006.10.008. [DOI] [PubMed] [Google Scholar]
- 3.Anderson D, Plewa MJ. The international comet assay workshop. Mutagenesis. 1998;13:67–73. doi: 10.1093/mutage/13.1.67. [DOI] [PubMed] [Google Scholar]
- 4.Burlinson B, Tice RR, Speit G, Agurell E, Brendler-Schwaab SY, Collins AR, Escobar P, Honma M, Kumaravel TS, Nakajima M, Sasaki YF, Thybaud V, Uno Y, Vasques M, Hartmann A. In Vivo Comet Assay Workgroup, part of the Fourth International Workgroup on Genotoxicity Testing. Fourth international workgroup on genotoxicity testing: results of the in vivo Comet assay workgroup. Mutat Res. 2007;627:31–35. doi: 10.1016/j.mrgentox.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 5.European Standards Committee on Oxidative DNA Damage. Measurement of DNA oxidation in human cells by chromatographic and enzymic methods. Free Radical Biol Med. 2003;34:1089–1099. doi: 10.1016/s0891-5849(03)00041-8. [DOI] [PubMed] [Google Scholar]
- 6.Gedik CM, Collins A. European Standards Committee on Oxidative DNA Damage. Establishing the background level of base oxidation in human lymphocyte DNA: results of an interlaboratory validation study. FASEB J. 2005;19:82–84. doi: 10.1096/fj.04-1767fje. [DOI] [PubMed] [Google Scholar]
- 7.Speit G, Schutz P, Bonzheim I, Trenz K, Hoffmann H. Sensitivity of the FPG protein towards alkylation damage in the comet assay. Toxicol Lett. 2004;146:151–158. doi: 10.1016/j.toxlet.2003.09.010. [DOI] [PubMed] [Google Scholar]
- 8.Nakamura J, Walker VE, Upton PB, Chiang S-Y, Kow YW, Swenberg JA. Highly sensitive apurinic/apyrimidinic site assay can detect spontaneous and chemically induced depurination under physiological conditions. Cancer Res. 1998;58:222–225. [PubMed] [Google Scholar]
- 9.Nakamura J, Swenberg JA. Endogenous apurinic/apyrimidinic sites in genomic DNA of mammalian tissues. Cancer Res. 1999;59:2522–2526. [PubMed] [Google Scholar]
- 10.Horton JK, Prasad R, Hou E, Wilson SH. Protection against methylation-induced cytotoxicity by DNA polymerase beta-dependent long patch base excision repair. J Biol Chem. 2000;275:2211–2218. doi: 10.1074/jbc.275.3.2211. [DOI] [PubMed] [Google Scholar]
- 11.Buschfort C, Muller MR, Seeber S, Rajewsky MF, Thomale J. DNA excision repair profiles of normal and leukemic human lymphocytes: functional analysis at the single-cell level. Cancer Res. 1997;57:651–658. [PubMed] [Google Scholar]
- 12.Ukai A, Maruyama T, Mochizuki S, Ouchida R, Masuda K, Kawamura K, Tagawa M, Kinoshita K, Sakamoto A, Tokuhisa T, O-Wang J. Role of DNA polymerase theta in tolerance of endogenous and exogenous DNA damage in mouse B cells. Genes Cells. 2006;11:111–121. doi: 10.1111/j.1365-2443.2006.00922.x. [DOI] [PubMed] [Google Scholar]
- 13.Vermeulen C, Verwijs-Janssen M, Cramers P, Begg AC, Vens C. Role for DNA polymerase beta in response to ionizing radiation. DNA Repair. 2007;6:202–212. doi: 10.1016/j.dnarep.2006.09.011. [DOI] [PubMed] [Google Scholar]
- 14.Scicchitano DA, Hanawalt PC. Repair of N-methylpurines in specific DNA sequences in chinese hamster ovary cells: absence of strand specificity in the dihydrofolate reductase gene. Proc Natl Acad Sci USA. 1989;86:3050–3054. doi: 10.1073/pnas.86.9.3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bartlett JD, Scicchitano DA, Robison SH. Two expressed human genes sustain slightly more DNA damage after alkylating agent treatment than an inactive gene. Mutat Res. 1991;255:247–256. doi: 10.1016/0921-8777(91)90028-n. [DOI] [PubMed] [Google Scholar]
- 16.Talpaert-Borlé M. Formation, detection and repair of AP sites. Mutat Res. 1987;181:45–56. doi: 10.1016/0027-5107(87)90286-7. [DOI] [PubMed] [Google Scholar]
- 17.Fortini P, Bignami M, Dogliotti E. Evidence for AP site formation related to DNA-oxygen alkylation in CHO cells treated with ethylating agents. Mutat Res. 1990;236:129–137. doi: 10.1016/0921-8777(90)90040-c. [DOI] [PubMed] [Google Scholar]
- 18.Cashmore AR, Brown DM, Smith JD. Selective reaction of methoxyamine with cytosine bases in tyrosine transfer ribonucleic acid. J Mol Biol. 1971;59:359–373. doi: 10.1016/0022-2836(71)90056-8. [DOI] [PubMed] [Google Scholar]
- 19.Liuzzi M, Weinfeld M, Paterson MC. Selective inhibition by methoxyamine of the apurinic/apyrimidinic endonuclease activity associated with pyrimidine dimer-DNA glycosylases from Micrococcus luteus and bacteriophage T4. Biochemistry. 1987;26:3315–3321. doi: 10.1021/bi00386a011. [DOI] [PubMed] [Google Scholar]
- 20.Wang Y, Liu L, Wu C, Bulgar A, Somoza E, Zhu W, Gerson SL. Direct detection and quantification of abasic sites for in vivo studies of DNA damage and repair. Nucl Med Biol. 2009;36:975–983. doi: 10.1016/j.nucmedbio.2009.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Olive PL, Wlodek D, Durand RE, Banath JP. Factors influencing DNA migration from individual cells subjected to gel electrophoresis. Exp Cell Res. 1992;198:259–267. doi: 10.1016/0014-4827(92)90378-l. [DOI] [PubMed] [Google Scholar]
- 22.Fairbairn DW, Olive PL, O’Neill KL. The comet assay: a comprehensive review. Mutat Res. 1995;339:37–59. doi: 10.1016/0165-1110(94)00013-3. [DOI] [PubMed] [Google Scholar]
- 23.Ridpath JR, Nakamura A, Tano K, Luke AM, Sonoda E, Arakawa H, Buerstedde J-M, Gillespie DAF, Sale JE, Yamazoe M, Bishop DK, Takata M, Takeda S, Watanabe M, Swenberg JA, Nakamura J. Cells deficient in the FANC/BRCA pathway are hypersensitive to plasma levels of formaldehyde. Cancer Res. 2007;76:11117–11122. doi: 10.1158/0008-5472.CAN-07-3028. [DOI] [PubMed] [Google Scholar]
- 24.Nakamura J, La DK, Swenberg JA. 5’Nicked apurinic/apyrimidinic sites are resistant to β-elimination by β-polymerase and are persistent in human cultured cells after oxidative stress. J Biol Chem. 2000;275:5323–5328. doi: 10.1074/jbc.275.8.5323. [DOI] [PubMed] [Google Scholar]
- 25.Pachkowski BF, Tano K, Afonin V, Elder RH, Takeda S, Watanabe M, Swenberg JA, Nakamura J. Cells deficient in PARP-1 show an accelerated accumulation of DNA single strand breaks, but not AP sites, over the PARP-1-proficient cells exposed to MMS. Mutat Res. 2009;671:93–99. doi: 10.1016/j.mrfmmm.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakamura J, Asakura S, Hester SD, de Murcia G, Caldecott KW, Swenberg JA. Quantitation of intracellular NAD(P) H can monitor an imbalance of DNA single strand break repair in base excision repair deficient cells in real time. Nucleic Acids Res. 2003;31:e104. doi: 10.1093/nar/gng105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Drouin R, Gao S, Homquist GP. Agarose gel electrophoresis for DNA damage analysis. In: Pfeifer GP, editor. Technologies for Detection of DNA Damage and Mutations. Plenum Press; New York, NY: 1996. pp. 37–43. [Google Scholar]
- 28.Georgiou C, Papapostolou I, Grizntzalis K. Protocol for the quantitative assessment of DNA concentration and damage (fragmentation and nicks) Nature Protocols. 2009;4:125–131. doi: 10.1038/nprot.2008.222. [DOI] [PubMed] [Google Scholar]
- 29.Mattes WB, Hartley JA, Kohn KW. Mechanism of DNA strand breakage by piperidine at sites of N7-alkylguanines. Biochim Biophys Acta. 1986;868:71–76. doi: 10.1016/0167-4781(86)90088-6. [DOI] [PubMed] [Google Scholar]
- 30.Friedberg EC, Walker GC, Wolfram S, Wood RD, Schultz RA, Ellenberger T. DNA Repair and Mutagenesis. 2. ASM Press; Washington, D. C: 2006. DNA damage; pp. 9–69. [Google Scholar]
- 31.Kohn KW. Principles and practices of DNA filter elution. Pharmac Ther. 1991;49:55–77. doi: 10.1016/0163-7258(91)90022-e. [DOI] [PubMed] [Google Scholar]
- 32.Tice RR, Agurell E, Anderson D, Burlinson B, Hartmann A, Kobayashi H, Miyamae Y, Rojas E, Ryu JC, Sasaki YF. Single cell gel/comet assay: guidelines for in vitro and in vivo genetic toxicology testing. Environ Mol Mutagen. 2000;35:206–221. doi: 10.1002/(sici)1098-2280(2000)35:3<206::aid-em8>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 33.Chlebowicz E, Jachymczyk WJ. Repair of MMS-induced DNA double-strand breaks in haploid cells of Saccharomyces cerevisiae, which requires the presence of a duplicate genome. Mol Gen Genet. 1979;167:279–286. doi: 10.1007/BF00267420. [DOI] [PubMed] [Google Scholar]
- 34.Di Primio C, Galli A, Cervelli T, Zoppe M, Rainaldi G. Potentiation of gene targeting in human cells by expression of Saccharomyces cerevisiae Rad52. Nucleic Acids Res. 2005;33:4639–4648. doi: 10.1093/nar/gki778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yoshimura M, Kohzaki M, Nakamura J, Asagoshi K, Sonoda E, Hou E, Prasad R, Wilson SH, Tano K, Yasui A, Lan L, Seki M, Wood RD, Arakawa H, Buerstedde JM, Hochegger H, Okada T, Hiraoka M, Takeda S. Vertebrate POLQ and POLbeta cooperate in base excision repair of oxidative DNA damage. Mol Cell. 2006;24:115–125. doi: 10.1016/j.molcel.2006.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sobol RW, Watson DE, Nakamura J, Yakes FM, Hou E, Horton JK, Ladapo J, Van Houten B, Swenberg JA, Tindall KR, Samson LD, Wilson SH. Mutations associated with base excision repair deficiency and methylation-induced genotoxic stress. Proc Natl Acad Sci USA. 2002;99:6860–6865. doi: 10.1073/pnas.092662499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wyatt MD, Pittman DL. Methylating agents and DNA repair responses: methylated bases and sources of strand breaks. Chem Res Toxicol. 2006;19:1580–1594. doi: 10.1021/tx060164e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Haraguchi K, Delaney MO, Wiederholt CJ, Sambandam A, Hantosi Z, Greenberg MM. Synthesis and characterization of oligodeoxynucleotides containing formamidopyrimidine lesions and nonhydrolyzable analogues. J Am Chem Soc. 2002;124:3263–3269. doi: 10.1021/ja012135q. [DOI] [PubMed] [Google Scholar]
- 39.Bryant HE, Petermann E, Schultz N, Jemth AS, Loseva O, Issaeva N, Johansson F, Fernandez S, McGlynn P, Helleday T. PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination. EMBO J. 2009;28:2601–2615. doi: 10.1038/emboj.2009.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang S, Gong Z, Chen R, Liu Y, Li A, Li G, Zhou J. JWA regulates XRCC1 and functions as a novel base excision repair protein in oxidative-stress-induced DNA single-strand breaks. Nucleic Acids Res. 2009;37:1936–1950. doi: 10.1093/nar/gkp054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chou WC, Wang HC, Wong FH, Ding SL, Wu PE, Shieh SY, Shen CY. Chk2-dependent phosphorylation of XRCC1 in the DNA damage response promotes base excision repair. EMBO J. 2008;27:3140–3150. doi: 10.1038/emboj.2008.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wong D, Demple B. Modulation of the 5’-deoxyrobose-5-phosphate lyase and DNA synthesis activities of mammalian DNA polymerase b by apurinic/apyrimidinic endonuclease 1. J Biol Chem. 2004;279:25268–25275. doi: 10.1074/jbc.M400804200. [DOI] [PubMed] [Google Scholar]