

Abstract
Atherosclerosis, a chronic inflammatory disease of the vascular system, presents significant challenges to developing effective molecular diagnostics and novel therapies. A systems biology approach integrating data from large-scale measurements (e.g. transcriptomics, proteomics and genomics) is successfully contributing to deciphering regulatory networks underlying the response of many different cellular systems to perturbations. Such a network analysis strategy using pathway information and data from multiple measurement platforms, tissues and species is a promising approach to elucidate the mechanistic underpinnings of complex diseases. Here, we present our views on the contributions that a systems approach can bring to the study of atherosclerosis, propose ways to tackle the complexity of the disease in a systems manner and review recent systems-level studies of the disease.
Keywords: atherosclerosis, systems biology, network analysis
Atherosclerosis and systems biology
Atherosclerosis is a complex multifactorial disease characterized by the accumulation of inflammatory cells, lipoproteins and fibrous tissue in the wall of large arteries (Lusis, 2000). It is the primary cause of heart attacks and strokes and thus is the underlying cause of the majority of deaths globally, accounting for approximately 29% of all deaths worldwide. Cardiovascular disease disproportionately affects low and middle income countries and is projected to remain the single leading cause of death worldwide for the next 20 years (World Health Organization, 2009). Despite the enormous economic and social burden of this disease, we lack both a full understanding of its underlying mechanism and the ability to personalize its diagnosis and treatment.
Research over the past several decades has revolutionized our understanding of the pathogenesis of atherosclerosis. Previously atherosclerosis was viewed primarily as a passive process of cholesterol accumulation in the vessel wall, and the clinical manifestations were attributed primarily to the degree of stenosis. We now understand that atherosclerosis is a complex and active process and that the ultimate clinical presentation results from the interaction of multiple cell types and organ systems (Corti et al, 2004; Libby & Theroux, 2005). Because of its underlying complexity, the study and treatment of atherosclerosis presents several fundamental challenges that the emerging discipline of systems biology is uniquely suited to address.
Systems biology is the comprehensive, quantitative analysis of the manner in which all of the components of a biological system interact over time (Zak & Aderem, 2009). To study atherosclerosis, one would for example consider the human body as the biological system and naturally, all the molecules, cells, tissues and organs that play a role in the pathology of this disease are its components. While systems biology is dependent on new ‘omics’-scale technologies, it is not defined by these technologies. Rather, the systems approach involves the integration of the data derived from these measurement tools into comprehensive predictive models.
Systems biology is hypothesis-driven, global, quantitative, iterative, integrative and dynamic. Its practice begins with the acquisition of global sets of biological data from as many hierarchical levels of information as possible [i.e. deoxyribonucleic acid (DNA) sequences, ribonucleic acid (RNA) expression, protein or lipid abundance]. This is the starting point for formulating detailed graphical or mathematical models, which are then iteratively refined through a hypothesis-driven process of system perturbation and data integration. Cycles of this process will result in more accurate models; ultimately, these models will explain the systems-level properties of the biological system of interest. Once the model is sufficiently accurate and detailed, it will allow researchers to accomplish two tasks never before possible: (1) predict the behaviour of the system given any perturbation and (2) redesign or perturb the molecular network to create new emergent properties. Taking the atherosclerosis analogy further, a researcher or clinician could (1) predict the body's response to, for example, a new diet or medication and (2) design an appropriate intervention that prevents atherosclerosis-promoting events or shifts them to anti-atherosclerotic ones. This latter possibility lies at the heart of preventative medicine.
Glossary
Cholesterol
A steroid metabolite found in cell membranes. It is transported in the blood in lipoprotein particles and excess circulating cholesterol is associated with atherosclerosis.
Data-mining
Pattern-finding in a large dataset.
Familial hypercholesterolemia
A genetic disorder usually caused by mutations in the LDL-receptor or in ApoB that results in high levels of LDL and premature atherosclerosis.
Fatty streak
Early atherosclerotic lesion containing mainly cholesterol and macrophages.
Feedback loop
A network motif in which a node (molecule) indirectly (or directly) regulates itself.
Feed-forward loop
A network motif in which a node (molecule) both directly and indirectly regulates a downstream target node.
High density lipoprotein (HDL)
High density lipoprotein (HDL)—‘good cholesterol’, high levels are thought to be protective against atherosclerosis.
Low density lipoprotein (LDL)
Low density lipoprotein (LDL)—‘bad cholesterol’, high circulating levels have been shown to correlate with atherosclerosis.
Plaque
The buildup of cells and cholesterol in the arterial wall. Severe plaque buildup can narrow the arterial lumen interfering with the flow of blood.
Seed
A group of nodes (molecules) used as the starting point in ad hoc network construction.
Stenosis
The abnormal narrowing of an artery.
Tangier disease
A genetic disorder resulting in very low levels of HDL caused by a mutation in the ABCA1 transporter.
Thrombosis
Formation of a blood clot.
The levels of complexity of atherosclerosis
Atherosclerosis involves the interplay among thousands of molecules in multiple interacting cells types including macrophages, endothelial cells and smooth muscle cells (SMCs). The disease occurs in different forms throughout the body (Trogan et al, 2006) and is affected by inputs from multiple organ systems including the vascular system, the endocrine system, adipose tissue, the liver, the gastrointestinal tract and the kidneys (Fig 1). Epidemiologic and treatment studies have also shown that the disease is modulated by a variety of genetic and environmental factors (Yusuf et al, 2004). For example, alteration in the relative abundance of various plasma lipoproteins such as low-density lipoprotein (LDL) and high-density lipoprotein (HDL) has been shown to be of primary importance in the development of the disease. The levels of these lipoproteins are influenced by multiple genetic factors (such as mutations in the LDL receptor gene which cause familial hypercholesterolemia (Brown & Goldstein, 1974) and mutations in the ABCA1 gene which cause Tangier disease (Rust et al, 1999) as well as diet, exercise and medications (Steinberg, 2004, 2005a, b, 2006). Many of the risk factors for atherosclerosis, including dyslipidemia, hypertension, diabetes and obesity, involve the interaction of several organ systems such as the liver, kidneys, gastrointestinal tract and hormonal systems (Assmann et al, 1999). Furthermore, systemic inflammation, which has been shown to be critically involved in both the development and eventual clinical complications of atherosclerosis, involves immune cells and mediators located at the site of plaque formation as well as distal organ systems such as the liver and adipose tissue (Libby et al, 2002; Ross, 1999). Thus, atherosclerosis results from the complex interplay of genetic and environmental risk factors at a whole-organism level (Fig 1).
Figure 1. The pathophysiology of atherosclerosis involves interacting systems at multiple levels.

- Genetic and environmental factors.
- Multiple organs and organ systems are involved in the disease process.
- Risk factors that are influenced by genetic and environmental inputs and that involve multiple organ systems contribute to the development and progression of atherosclerosis.
- Atherosclerosis progresses through several stages, each of which involves the interaction of multiple cell types and molecular processes.
Atherosclerosis progresses through multiple stages from early fatty streaks, to advanced lesions to plaque rupture, with each stage being characterized by different cellular and molecular components (Fig 1D). Atherosclerotic plaque development begins with endothelial cell activation, including overexpression of leukocyte adhesion proteins such as vascular cell adhesion molecule 1 VCAM-1 (Cybulsky & Gimbrone, 1991). Chemoattractants such as monocyte chemoattractant protein 1 (MCP-1) then promote migration of leukocytes into the intima (Boring et al, 1998), where macrophage colony stimulating factor (CSF1) promotes the differentiation of monocytes into macrophages (Rajavashisth et al, 1990). These macrophages express scavenger receptors that allow them to engulf and modify lipoproteins and become foam cells which secrete inflammatory mediators [such as interleukin-1 (IL-1), tumour-necrosis factor-α (TNF-α), nitric oxide and endothelin] (Hansson et al, 2006), that amplify inflammation in the vessel wall and can contribute to additional leukocyte accumulation, SMC proliferation and extracellular matrix remodelling (Brown & Goldstein, 1983; Greaves & Gordon, 2009). Multiple other leukocytes are recruited to the lesion and have been demonstrated to play a critical role in disease development (Weber et al, 2008). Whereas foam cell accumulation characterizes fatty streaks, deposition of fibrous tissue defines the more advanced atherosclerotic lesion. SMCs synthesize the bulk of the extracellular matrix that characterizes this phase of plaque evolution (Raines & Ferri, 2005). Plaque rupture resulting from inflammatory activation and the ensuing thrombosis commonly cause the most acute complications of atherosclerosis such as myocardial infarctions or stroke (Fuster et al, 2005).
There are multiple challenges facing clinicians in the treatment of atherosclerotic vascular disease. Atherosclerosis is a chronic condition that develops silently over decades, presenting with clinical manifestations only very late in the course of the disease. Current strategies to detect early disease rely heavily on population-based risk factor assessment but lack the ability to individualize these risk assessments. Currently available diagnostic tools are only able to detect advanced disease. Furthermore, while many pharmacologic interventions directed at reducing correlative risk factors have been shown to reduce the population-based cardiovascular mortality rate, no methods are currently available to track the vascular response in an individual patient and therefore to predict that patient's risk of future events. Thus, current strategies do not holistically address the multiple factors that contribute to the observed pathology.
Applying network analysis to atherosclerosis questions
Given the complexities of atherosclerosis, a systems biology approach that samples multiple levels of hierarchical data and then integrates the results into coherent network models offers many advantages. Complex biological networks are organized around sub-networks of gene modules that contribute to the robustness of the entire system. From a systems-level perspective, disease states represent perturbations, from genetic or environmental factors, on complex networks of interacting components on multiple scales (molecules, macromolecules, organelles, cells, tissues and organs).
The recent development of global measurement and analysis technologies, and their integration under the aegis of systems biology, offer an unprecedented opportunity to overcome the difficulties inherent in atherosclerosis research and treatment. The complex spatial and temporal relationships involved in the disease need to be understood in the context of a dynamic interaction network. Because atherosclerotic plaques evolve over time from simple fatty streaks to advanced lesions prone to plaque rupture, a useful model of the disease process must be able to accommodate changes in molecular composition and interactions over time. Below, we describe the use of interaction networks in systems biology and their application to atherosclerosis research.
A network is a framework that represents the relationships among the features that make up complex biological systems. Biological networks are made up of nodes, which represent molecular entities (such as DNA variations, RNA, proteins and metabolites), edges that represent the relationships between these entities, and network properties that represent the state of the molecular entities over time (Fig 2). The network topology represents all of the interactions involved in a given biological system. A cornerstone of the systems biology approach is the construction of a network representing the disease process, using a collection of methods that together can be called network analysis. Network analysis can help identify feedback mechanisms and network regulatory motifs that capture the emergent properties of the system, such as robustness to perturbation, multistability or homeostatic control.
Figure 2. Conceptualizing the disease process as a network.
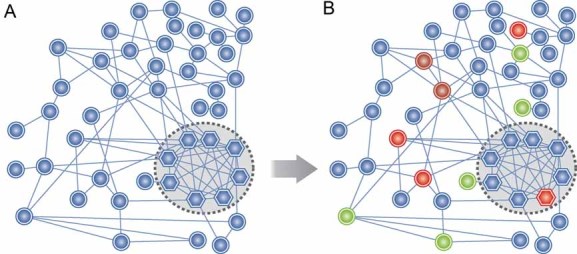
- In a systems biology approach, the goal is to uncover a network of molecular interactions whose altered state can be correlated with disease progression. In this simple conceptual network diagram, a node (biomolecule) is represented by a circle or hexagon, and an interaction between two nodes (edges) is represented by a line between them. A highly interconnected subnetwork of molecules (hexagons, inside dotted circle region) is shown.
- Colored nodes denote changes in the network during disease progression (a red node denotes increased expression and green denotes decreased expression).
Network analysis can be divided into two approaches, ad hoc network construction and pathway analysis. Both are useful for analysing expression data pertaining to a complex disease such as atherosclerosis, and they have complementary advantages and limitations. The pathway approach is more straightforward to interpret, but it is limited to only those biological functions and processes that are represented in a pathway database. Arbitrary divisions between canonical pathways can also limit the effectiveness of the pathway approach. On the other hand, the ad hoc approach has greater potential to reveal novel molecular connections within the data, but the networks generated in the ad hoc approach can be challenging to interpret. We outline a procedure and relevant resources for each approach below.
The pathway approach
In the pathway approach a network diagram is organized around curated lists of interactions known to be involved in a specific molecular process, for example, ‘eicosanoid biosynthesis’ or ‘toll-like receptor signalling’. These pathway-oriented interaction networks can be obtained from freely accessible pathway repositories (Table 1) and from commercial pathway databases such as Ingenuity Pathways Analysis (IPA, Ingenuity), MetaCore (GeneGo) and Pathway Studio (Ariadne). There are also several freely available software tools that can mine multiple pathway databases (see Table 1). Beyond analysis in terms of separate pathways, an atherosclerosis dataset can also be analysed across multiple pathways to identify molecules or genes that operate within more than one atherosclerosis-associated pathway (e.g. see Ghazalpour et al, 2004).
Table 1.
Public molecular pathway databases and software tools to access them
 |
For each database, a link to the main website and a list of compatible data-mining software tools is provided. The estimated number of non-redundant mammalian pathways in each database is shown.
The ad hoc approach
As opposed to the pathway approach, the ad hoc approach is unbiased. Here, differentially expressed molecules [ideally identified using a significance threshold that accounts for multiple hypothesis tests (Storey & Tibshirani, 2003)] are grouped into sub-networks that are highly interconnected in an interaction dataset. Often, differentially expressed molecules may share an interacting partner that is not itself differentially expressed, such as a common regulator, substrate, etc. To reveal such indirect connections, molecules can be added to the network that, by virtue of their interactions, enhance the network's connectedness [e.g. its clustering coefficient (Watts & Strogatz, 1998)]. The ad hoc network construction approach does not rely on curated pathway information, but instead is applied using large databases of molecular interactions or associations. Most commonly used are databases of protein–protein interactions (PPI), protein–DNA interactions and protein–metabolite interactions. Interaction databases fall into two categories, large-scale interaction repositories, which may aggregate interactions from high-throughput PPI screens from various species and tissue types, and interaction databases that are focused on a specific class of molecules or functions. A listing of commonly used, publicly accessible interaction databases is given in Table 2. An important limitation of many of these databases is that they aggregate findings from a variety of model cell types, and thus the interaction data are not necessarily derived from atherosclerosis-relevant tissues or models. Another caveat is that literature-based interaction databases necessarily provide more information on better-studied molecules, which may introduce bias into the network model.
Table 2.
Public molecular interaction databases
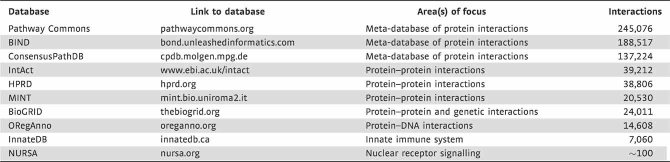 |
For each database, the Web address of the main portal for the database is given, along with a description of the database's area(s) of emphasis. The ‘Interactions’ column provides an estimate of the number of human protein–protein interactions within each database (except for ORegAnno, where it lists the number of transcription factor binding sites in the database).
Typically, ad hoc network analysis begins with a network constructed from a ‘seed’ collection of molecules identified in an expression study and any direct interactions between them. To this seed network are then added molecules that have high connectivity to the seed network, thus growing a highly interconnected molecular network in an iterative fashion. The resulting network can be analysed for enrichment of functional annotations to gain insight into its specific biological functions. Several software tools are available that can perform ad hoc network construction and analysis, as well as statistical and bioinformatic analysis of high-throughput data (Table 3). Integrating data from different high-throughput measurement platforms (e.g. transcriptomic and proteomic) can be particularly useful to comprehensively detect disease-associated genes, and statistical approaches have been specifically developed to iteratively expand a molecular network using multiple data types (Hwang et al, 2005).
Table 3.
Freely available software tools that can be used for data integration, network construction and network analysis
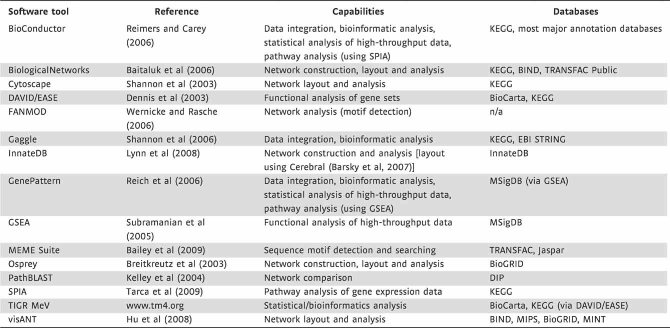 |
Several commercial tools are also available for network construction and analysis, such as Ingenuity Pathways Analysis, Pathway Studio, MetaCore and TRANSFAC Professional (BioBase). The ‘databases’ column indicates interaction or pathway databases that the software tool can directly query or interact with.
Analysing large-scale expression measurements in the context of molecular pathways or interaction networks can reveal key regulatory molecules and functional modules involved in the disease process and suggest hypotheses regarding the system response to perturbation. Originally used in the context of model organisms such as yeast and bacteria, ad hoc network analysis has more recently been applied to the study of mammalian systems. Particularly relevant to atherosclerosis, several studies have examined networks involved in the inflammatory response in macrophages. Gilchrist et al combined transcriptional profiling and analysis of promoter sequences to identify activating transcription factor 3 ATF3 as a regulator of macrophage response to the bacterial endotoxin lipopolysaccharide (Gilchrist et al, 2006). Based on the network analysis, ATF3 was predicted to act as a negative regulator of Toll-like receptor 4 TLR4-induced expression of key pro-inflammatory genes such as Il6 and Il12b, and this prediction was validated both in vitro and in vivo. A second transcriptomic study analysed the dynamic transcriptional response of macrophages to stimulation with various Toll-like receptor TLR agonists. Using a probabilistic framework, transcriptomic data were integrated with promoter sequence scanning (scanning for cis-regulatory motifs from the TRANSFAC database) to predict transcription factors that regulate clusters of TLR-responsive genes. TGFB-induced factor homeobox 1 TGIF1 was identified as a potential novel transcriptional regulator of a cluster containing the cytokines Csf2 and Gm1960 (Ramsey et al, 2008).
Taking network analysis further
Network analysis can enhance the utility of even simple transcriptomic studies, as we illustrate in Fig 3. Expression data-mining based solely on gene functional annotations is limited by incomplete annotations and the fact that key regulators of disease progression may not be differentially expressed. Furthermore, within a disease-associated functional module, only a small fraction of molecular species may be differentially expressed. Network analysis can extend beyond gene annotation enrichment analysis by taking into account interactions between the molecules in the expression dataset (and intermediaries). Among other advantages, this enables the identification of regulatory molecules that may not be differentially expressed (Fig 3A). A differentially expressed molecule can also be identified as a candidate regulator based on its proximity to disease-associated molecules in the interaction network (Fig 3B). Moreover, the sub-network involving a differentially expressed molecule may indicate its significance, for example, if it resides in a feedback loop regulating the level of a disease-associated protein or metabolite. As an example of this network-oriented approach, we analysed transcriptomic data from murine macrophages treated with oxidatively modified LDL (oxLDL), a stimulus that is associated with foam cell formation. The analysis and the resulting network, which are detailed in Box 1 and Fig 4, show how a potential regulator of the response (p65/Rela) can be identified even though it is not differentially expressed. This simple example also reveals the potential for network analysis to provide a more complete picture of the cellular response than would analysing the annotations of only the differentially expressed genes. Moreover, network analysis can uncover network regulatory motifs [e.g. feedback loops, feed-forward loops, etc. (Alon, 2007)] controlling the response.
Figure 3. Regulatory molecules implicated by network analysis.
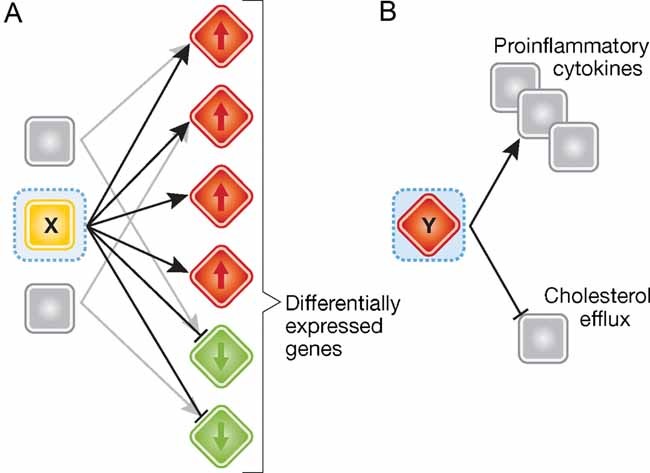
- A regulatory molecule ‘X’ that is not differentially expressed can be implicated by its connectivity to differentially expressed molecules, in the interaction network.
- A differentially expressed molecule ‘Y’ can be identified as a key regulator through its connections to key disease-associated molecules and biological processes. Red/green colours denote up/downregulation of mRNA.
Box 1: Network analysis of macrophage response oxLDL
As an example of network analysis, we analysed transcriptomic data from murine macrophages treated with oxLDL, a stimulus that is associated with foam cell formation. Using the PLIER algorithm (Affymetrix, 2005), 542 genes were identified as differentially expressed. Ad hoc network analysis was performed on the 542 genes using MetaCore, yielding a network (Fig 4) associated with three biological processes relevant to foam cell formation: ‘regulation of foam cell differentiation’ (P < 10−10), ‘regulation of macrophage differentiation’ (P < 10−9) and ‘lipoprotein catabolic process’ (P < 10−9). A functional enrichment test of only the differentially expressed genes in the network fails to detect these enrichments, because only a fraction of the molecules in the network are differentially expressed. The interaction network has multiple interactions with sphingolipids, consistent with the findings of Wheelock et al (2009) based on the data from Kleemann et al (2007).
Figure 4. Network involving macrophage genes with altered expression in response to oxLDL.

The network includes 20 oxLDL-responsive genes. Nodes (molecules) are arranged based on subcellular location. A red (blue) circle on the upper right-hand side of the molecule indicates upregulation (downregulation) when cells are treated for 24 h. Each node is designated by a glyph that indicates the type of molecule (receptor, kinase/phosphatase, scaffold, etc.) (Kleemann et al, 2007). Each edge indicates an interaction, with an arrowhead indicating the flow of control in the specific interaction (if known).
The application of both pathway (Cagnin et al, 2009) and ad hoc (Skogsberg et al, 2008) network analysis has facilitated extraction of biologically meaningful information from microarray messenger RNA (mRNA) studies of atherosclerotic plaques. Such studies are critical in that they analyse disease-relevant tissue [e.g. whole mouse aorta (Skogsberg et al, 2008) or human coronary and carotid arteries (Cagnin et al, 2009)] but they pose numerous analysis challenges. For example, the lesion samples contain a mix of cell types obtained at a fixed time point. These studies allow the construction of a ‘parts list’ of molecules that may participate in the process—which is extremely useful in the interpretation and analysis of complementary in vitro studies. However, to go beyond such lists requires the application of network analysis. By doing this type of analysis the authors provided insights into disease pathogenesis that would not otherwise have been apparent. For example, network analysis of transcriptional data from lesions suggested a small group of cholesterol-responsive genes whose functional annotations were suggestive of involvement in lipid uptake or metabolism (Skogsberg et al, 2008). Screening this gene network with siRNA in an in vitro macrophage cholesterol accumulation assay showed that no single intervention ablates foam cell formation. These findings are consistent with the viewpoint that foam cell formation in vivo is likely to be resistant to targeting a single molecule, and instead may require a combined therapeutic approach.
Both pathway and ad hoc network analysis depend on the quality and comprehensiveness of the underlying interaction database. One approach to extend beyond the available databases is to construct networks using molecular associations based on semantic mining of relevant scientific literature. This has the advantage of enabling an investigator to explore networks organized around relevant biological search terms such as ‘atherosclerosis’, ‘foam cell’ or ‘cardiovascular disease’. The interactions in these networks are based on co-occurrence of molecule names within abstracts of disease-associated articles, with the molecule names separated by a keyword that is suggestive of molecular interaction (e.g. ‘binds’, ‘modifies’, ‘phosphorylates’, etc.). This literature network approach has been used in the analysis mRNA profiles of human atherosclerotic tissue (Ashley et al, 2006; King et al, 2005). Due to limitations inherent in human studies, these investigations could not identify transcriptional markers of early-stage disease, and they identified a relatively modest number of differentially expressed genes compared to controlled transcriptomic studies of lesions in mice. Integrating data from both human and model systems maximizes the probability of obtaining robust, physiologically relevant findings. Tabibiazar et al leveraged transcriptional data from mouse aortas and human coronary arteries to identify atherosclerosis-related genes that are predictive of disease severity in mouse and lesion grade in human (Tabibiazar et al, 2005). Their analysis confirmed many genes whose transcript levels are known to correlate with disease severity and identified functional classes of genes that are novel in the context of atherosclerosis (such as RUNX transcription factors and histone deacetylases). Such a dual-species approach has the benefit of ensuring that the transcriptional correlates of disease severity identified in mice are relevant in human.
Integrating transcriptomic, proteomic and metabolomic measurements into network analysis can yield a more complete picture of changes responsible for the initiation of atherosclerotic vascular disease than can be obtained by analysing a single measurement type. Applying such an approach in a mouse model of atherosclerosis has demonstrated the importance of transcriptional and metabolic reprogramming of the liver as a key driver of the inflammatory process underlying atherogenesis (Clish et al, 2004; Kleemann et al, 2007).
Examining a combination of genetic and transcriptomic measurements in the context of molecular pathways and biological processes associated with atherosclerosis has also been used as a novel method to uncover disease biomarkers (Hagg et al, 2008; Torkamani et al, 2008). For example, this approach identified insulin receptor substrate 2 (IRS2), whose expression is higher in macrophages from individuals with atherosclerosis than from control subjects. Through genetic association analysis of a larger cohort, a single nucleotide polymorphism SNP in the IRS2 promoter was identified that results in higher IRS2 gene expression and increased risk of coronary heart disease (Hagg et al, 2008). Thus, this approach utilizing a tiered study design has identified a potential novel biomarker for the development of coronary heart disease.
A number of excellent genome-wide association studies (GWAS) have uncovered multiple genetic loci that are associated with the development of atherosclerosis (Assimes et al, 2008; Aulchenko et al, 2009; Erdmann et al, 2009; Jarinova et al, 2009; McPherson et al, 2007; Tregouet et al, 2009), the subsequent clinical complications of atherosclerosis such as myocardial infarction (Kathiresan et al, 2009a) as well as risk factors for atherosclerosis such as hypertension (Newton-Cheh et al, 2009), dyslipidemia (Kathiresan et al, 2007, 2008a, b, 2009b) and obesity (Lindgren et al, 2009; Willer et al, 2009). These studies demonstrate the power of current high-throughput technologies as they have revealed multiple loci that would not have been uncovered with more traditional hypothesis-based methods.
However, the utility of these studies is limited by the fact that GWAS does not necessarily directly indicate the causal gene and does not establish the biological context in which the causal gene operates. The integration of GWAS data with studies that examine the downstream changes in the RNA, protein and metabolite state has the potential to reveal the perturbations in molecular networks that are associated with disease. Additional discovery power can be achieved through integration of QTL and transcriptional profiling analysis of strain-intercrossed mice with varying susceptibility to atherosclerosis (Smith et al, 2006a, b).
The identification of the macrophage-enriched metabolic network (MEMN) is an excellent example of the power of such integrated approaches. This network was constructed by integrating multiple data types including genetic studies and expression data from human and mouse liver and adipose tissue (Chen et al, 2008; Emilsson et al, 2008). The MEMN was strongly associated with obesity, diabetes and heart disease and this was confirmed experimentally in studies that indicate complex feedback control within the network (Mehrabian et al, 2005; Schadt et al, 2008; Yang et al, 2009). These studies identified several genes, including a newly discovered phosphatase gene Ppm1l, that were associated with multiple cardiovascular risk factors including weight, glucose tolerance, levels of free fatty acids and blood pressure. Additionally, these studies suggested that the macrophage not only plays a key role at the local level in the plaque but that it is also a driver of many complex metabolic diseases that are associated with atherosclerosis (Schadt, 2009).
This body of literature has made substantial contributions to the study of atherosclerosis. By increasing our ability to extract biologically meaningful information from high-throughput data sets, network analysis has allowed the identification of potential novel therapeutic targets and diagnostic markers. Moreover, studies that publish complete high-throughput data sets are particularly valuable to the research community because they enable other investigators to mine the data and formulate novel testable hypotheses.
Toward a systems-level disease model
The network analysis tools and methods described above can be extended and refined to accommodate complex study designs spanning multiple tissues. This is particularly relevant to atherosclerosis, where available models of plaque formation and leukocyte infiltration necessarily involve a trade-off between ease of expression profiling and physiological relevance. Network analysis can be performed on data from multiple expression studies, for example, from studies using different models or using different high-throughput measurement platforms. We briefly mention two possible strategies. (i) Differential expression data across multiple studies can be clustered and the clusters used as ‘seed’ lists for ad hoc network construction. This approach is predicated on the hypothesis that clusters derived from data from multiple complementary expression studies (which will have different model artefacts and ‘blind spots’) will enable the construction of a more physiologically relevant network than would be possible using a single expression study. (ii) Present/absent detection calls for gene expression from more physiological models can be used to constrain the list of possible molecules for network construction. We believe this is a particularly promising strategy for extracting maximal information from in vitro expression studies using in vivo-derived expression data.
Although the application of systems biology to the study of complex diseases is in its early stages, these studies are already providing novel insights into atherosclerosis and powerful tools to continue to decipher the intricacies of this disease. The promise of a systems approach includes disease prediction and prevention as well as personalized medicine.
Acknowledgments
We thank Alan Diercks for helpful comments. The authors acknowledge support from NIH contract HHSN272200700038C (National Institute for Allergy and Infectious Diseases). S.A.R. was supported by Award Number K25HL098807 from the National Heart, Lung and Blood Institute.
The authors declare that they have no conflict of interest.
For more information
American Heart Association:
European Atherosclerosis Society:
United States National Heart Lung and Blood Institute:
International Atherosclerosis Society:
Systems Immunology website:
http://www.systemsimmunology.org
Lipid MAPS project website:
Systems Biology portal:
Author's website
References
- Affymetrix I. Guide to Probe Logarithmic Intensity Error (PLIER) Estimation. Technical Note. Santa Clara, CA: Affymetrix; 2005. [Google Scholar]
- Alon U. An Introduction to Systems Biology. Boca Raton, FL: Chapman & Hall/CRC; 2007. [Google Scholar]
- Ashley EA, Ferrara R, King JY, Vailaya A, Kuchinsky A, He X, Byers B, Gerckens U, Oblin S, Tsalenko A, et al. Network analysis of human in-stent restenosis. Circulation. 2006;114:2644–2654. doi: 10.1161/CIRCULATIONAHA.106.637025. [DOI] [PubMed] [Google Scholar]
- Assimes TL, Knowles JW, Basu A, Iribarren C, Southwick A, Tang H, Absher D, Li J, Fair JM, Rubin GD, et al. Susceptibility locus for clinical and subclinical coronary artery disease at chromosome 9p21 in the multi-ethnic ADVANCE study. Hum Mol Genet. 2008;17:2320–2328. doi: 10.1093/hmg/ddn132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assmann G, Cullen P, Jossa F, Lewis B, Mancini M. Coronary heart disease: reducing the risk: the scientific background to primary and secondary prevention of coronary heart disease. A worldwide view. International Task force for the Prevention of Coronary Heart disease. Arterioscler Thromb Vasc Biol. 1999;19:1819–1824. doi: 10.1161/01.atv.19.8.1819. [DOI] [PubMed] [Google Scholar]
- Aulchenko YS, Ripatti S, Lindqvist I, Boomsma D, Heid IM, Pramstaller PP, Penninx BW, Janssens AC, Wilson JF, Spector T, et al. Loci influencing lipid levels and coronary heart disease risk in 16 European population cohorts. Nat Genet. 2009;41:47–55. doi: 10.1038/ng.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009;37:W202–W208. doi: 10.1093/nar/gkp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baitaluk M, Sedova M, Ray A, Gupta A. BiologicalNetworks: visualization and analysis tool for systems biology. Nucleic Acids Res. 2006;34:W466–W471. doi: 10.1093/nar/gkl308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barsky A, Gardy JL, Hancock RE, Munzner T. Cerebral: a Cytoscape plugin for layout of and interaction with biological networks using subcellular localization annotation. Bioinformatics. 2007;23:1040–1042. doi: 10.1093/bioinformatics/btm057. [DOI] [PubMed] [Google Scholar]
- Boring L, Gosling J, Cleary M, Charo IF. Decreased lesion formation in CCR2-/- mice reveals a role for chemokines in the initiation of atherosclerosis. Nature. 1998;394:894–897. doi: 10.1038/29788. [DOI] [PubMed] [Google Scholar]
- Breitkreutz BJ, Stark C, Tyers M. Osprey: a network visualization system. Genome Biol. 2003;4:R22. doi: 10.1186/gb-2003-4-3-r22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MS, Goldstein JL. Expression of the familial hypercholesterolemia gene in heterozygotes: mechanism for a dominant disorder in man. Science. 1974;185:61–63. doi: 10.1126/science.185.4145.61. [DOI] [PubMed] [Google Scholar]
- Brown MS, Goldstein JL. Lipoprotein metabolism in the macrophage: implications for cholesterol deposition in atherosclerosis. Annu Rev Biochem. 1983;52:223–261. doi: 10.1146/annurev.bi.52.070183.001255. [DOI] [PubMed] [Google Scholar]
- Cagnin S, Biscuola M, Patuzzo C, Trabetti E, Pasquali A, Laveder P, Faggian G, Iafrancesco M, Mazzucco A, Pignatti PF, et al. Reconstruction and functional analysis of altered molecular pathways in human atherosclerotic arteries. BMC Genomics. 2009;10:13. doi: 10.1186/1471-2164-10-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Zhu J, Lum PY, Yang X, Pinto S, MacNeil DJ, Zhang C, Lamb J, Edwards S, Sieberts SK, et al. Variations in DNA elucidate molecular networks that cause disease. Nature. 2008;452:429–435. doi: 10.1038/nature06757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clish CB, Davidov E, Oresic M, Plasterer TN, Lavine G, Londo T, Meys M, Snell P, Stochaj W, Adourian A, et al. Integrative biological analysis of the APOE*3-leiden transgenic mouse. Omics. 2004;8:3–13. doi: 10.1089/153623104773547453. [DOI] [PubMed] [Google Scholar]
- Corti R, Hutter R, Badimon JJ, Fuster V. Evolving concepts in the triad of atherosclerosis, inflammation and thrombosis. J Thromb Thrombolysis. 2004;17:35–44. doi: 10.1023/B:THRO.0000036027.39353.70. [DOI] [PubMed] [Google Scholar]
- Cybulsky MI, Gimbrone MA., Jr Endothelial expression of a mononuclear leukocyte adhesion molecule during atherogenesis. Science. 1991;251:788–791. doi: 10.1126/science.1990440. [DOI] [PubMed] [Google Scholar]
- Dennis GJ, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: database for annotation, visualization, and integrated discovery. Genome Biol. 2003;4:P3. [PubMed] [Google Scholar]
- Emilsson V, Thorleifsson G, Zhang B, Leonardson AS, Zink F, Zhu J, Carlson S, Helgason A, Walters GB, Gunnarsdottir S, et al. Genetics of gene expression and its effect on disease. Nature. 2008;452:423–428. doi: 10.1038/nature06758. [DOI] [PubMed] [Google Scholar]
- Erdmann J, Grosshennig A, Braund PS, Konig IR, Hengstenberg C, Hall AS, Linsel-Nitschke P, Kathiresan S, Wright B, Tregouet DA, et al. New susceptibility locus for coronary artery disease on chromosome 3q22.3. Nat Genet. 2009;41:280–282. doi: 10.1038/ng.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuster V, Moreno PR, Fayad ZA, Corti R, Badimon JJ. Atherothrombosis and high-risk plaque: part I: evolving concepts. J Am Coll Cardiol. 2005;46:937–954. doi: 10.1016/j.jacc.2005.03.074. [DOI] [PubMed] [Google Scholar]
- Ghazalpour A, Doss S, Yang X, Aten J, Toomey EM, Van Nas A, Wang S, Drake TA, Lusis AJ. Thematic review series: the pathogenesis of atherosclerosis. Toward a biological network for atherosclerosis. J Lipid Res. 2004;45:1793–1805. doi: 10.1194/jlr.R400006-JLR200. [DOI] [PubMed] [Google Scholar]
- Gilchrist M, Thorsson V, Li B, Rust AG, Korb M, Kennedy K, Hai T, Bolouri H, Aderem A. Systems biology approaches identify ATF3 as a negative regulator of Toll-like receptor 4. Nature. 2006;441:173–178. doi: 10.1038/nature04768. [DOI] [PubMed] [Google Scholar]
- Greaves DR, Gordon S. The macrophage scavenger receptor at 30 years of age: current knowledge and future challenges. J Lipid Res. 2009;50:S282–S286. doi: 10.1194/jlr.R800066-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagg DA, Jernas M, Wiklund O, Thelle DS, Fagerberg B, Eriksson P, Hamsten A, Olsson B, Carlsson B, Carlsson LMS, et al. Expression profiling of macrophages from subjects with atherosclerosis to identify novel susceptibility genes. Int J Mol Med. 2008;21:697–704. [PubMed] [Google Scholar]
- Hansson GK, Robertson AK, Soderberg-Naucler C. Inflammation and atherosclerosis. Annu Rev Pathol. 2006;1:297–329. doi: 10.1146/annurev.pathol.1.110304.100100. [DOI] [PubMed] [Google Scholar]
- Hu Z, Snitkin ES, DeLisi C. VisANT: an integrative framework for networks in systems biology. Brief Bioinform. 2008;9:317–325. doi: 10.1093/bib/bbn020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang D, Rust AG, Ramsey S, Smith JJ, Leslie DM, Weston AD, de Atauri P, Aitchison JD, Hood L, Siegel AF, et al. A data integration methodology for systems biology. Proc Natl Acad Sci USA. 2005;102:17296–17301. doi: 10.1073/pnas.0508647102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarinova O, Stewart AF, Roberts R, Wells G, Lau P, Naing T, Buerki C, McLean BW, Cook RC, Parker JS, et al. Functional analysis of the chromosome 9p21.3 coronary artery disease risk locus. Arterioscler Thromb Vasc Biol. 2009;29:1671–1677. doi: 10.1161/ATVBAHA.109.189522. [DOI] [PubMed] [Google Scholar]
- Kathiresan S, Manning AK, Demissie S, D'Agostino RB, Surti A, Guiducci C, Gianniny L, Burtt NP, Melander O, Orho-Melander M, et al. A genome-wide association study for blood lipid phenotypes in the Framingham Heart Study. BMC Med Genet. 2007;1:S17. doi: 10.1186/1471-2350-8-S1-S17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kathiresan S, Melander O, Anevski D, Guiducci C, Burtt NP, Roos C, Hirschhorn JN, Berglund G, Hedblad B, Groop L, et al. Polymorphisms associated with cholesterol and risk of cardiovascular events. N Engl J Med. 2008a;358:1240–1249. doi: 10.1056/NEJMoa0706728. [DOI] [PubMed] [Google Scholar]
- Kathiresan S, Melander O, Guiducci C, Surti A, Burtt NP, Rieder MJ, Cooper GM, Roos C, Voight BF, Havulinna AS, et al. Six new loci associated with blood low-density lipoprotein cholesterol, high-density lipoprotein cholesterol or triglycerides in humans. Nat Genet. 2008b;40:189–197. doi: 10.1038/ng.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kathiresan S, Voight BF, Purcell S, Musunuru K, Ardissino D, Mannucci PM, Anand S, Engert JC, Samani NJ, Schunkert H, et al. Genome-wide association of early-onset myocardial infarction with single nucleotide polymorphisms and copy number variants. Nat Genet. 2009a;41:334–341. doi: 10.1038/ng.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kathiresan S, Willer CJ, Peloso GM, Demissie S, Musunuru K, Schadt EE, Kaplan L, Bennett D, Li Y, Tanaka T, et al. Common variants at 30 loci contribute to polygenic dyslipidemia. Nat Genet. 2009b;41:56–65. doi: 10.1038/ng.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley BP, Yuan B, Lewitter F, Sharan R, Stockwell BR, Ideker T. PathBLAST: a tool for alignment of protein interaction networks. Nucleic Acids Res. 2004;32:W83–W88. doi: 10.1093/nar/gkh411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King JY, Ferrara R, Tabibiazar R, Spin JM, Chen MM, Kuchinsky A, Vailaya A, Kincaid R, Tsalenko A, Deng DX-F, et al. Pathway analysis of coronary atherosclerosis. Physiol Genomics. 2005;23:103–118. doi: 10.1152/physiolgenomics.00101.2005. [DOI] [PubMed] [Google Scholar]
- Kleemann R, Verschuren L, van Erk MJ, Nikolsky Y, Cnubben NHP, Verheij ER, Smilde AK, Hendriks HFJ, Zadelaar S, Smith GJ, et al. Atherosclerosis and liver inflammation induced by increased dietary cholesterol intake: a combined transcriptomics and metabolomics analysis. Genome Biol. 2007;8:R200. doi: 10.1186/gb-2007-8-9-r200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libby P, Theroux P. Pathophysiology of coronary artery disease. Circulation. 2005;111:3481–3488. doi: 10.1161/CIRCULATIONAHA.105.537878. [DOI] [PubMed] [Google Scholar]
- Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation. 2002;105:1135–1143. doi: 10.1161/hc0902.104353. [DOI] [PubMed] [Google Scholar]
- Lindgren CM, Heid IM, Randall JC, Lamina C, Steinthorsdottir V, Qi L, Speliotes EK, Thorleifsson G, Willer CJ, Herrera BM, et al. Genome-wide association scan meta-analysis identifies three Loci influencing adiposity and fat distribution. PLoS Genet. 2009;5:e1000508. doi: 10.1371/journal.pgen.1000508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lusis AJ. Atherosclerosis. Nature. 2000;407:233–241. doi: 10.1038/35025203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynn DJ, Winsor GL, Chan C, Richard N, Laird MR, Barsky A, Gardy JL, Roche FM, Chan TH, Shah N, et al. InnateDB: facilitating systems-level analyses of the mammalian innate immune response. Mol Syst Biol. 2008;4:218. doi: 10.1038/msb.2008.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPherson R, Pertsemlidis A, Kavaslar N, Stewart A, Roberts R, Cox DR, Hinds DA, Pennacchio LA, Tybjaerg-Hansen A, Folsom AR, et al. A common allele on chromosome 9 associated with coronary heart disease. Science. 2007;316:1488–1491. doi: 10.1126/science.1142447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehrabian M, Allayee H, Stockton J, Lum PY, Drake TA, Castellani LW, Suh M, Armour C, Edwards S, Lamb J, et al. Integrating genotypic and expression data in a segregating mouse population to identify 5-lipoxygenase as a susceptibility gene for obesity and bone traits. Nat Genet. 2005;37:1224–1233. doi: 10.1038/ng1619. [DOI] [PubMed] [Google Scholar]
- Newton-Cheh C, Johnson T, Gateva V, Tobin MD, Bochud M, Coin L, Najjar SS, Zhao JH, Heath SC, Eyheramendy S, et al. Genome-wide association study identifies eight loci associated with blood pressure. Nat Genet. 2009;41:666–676. doi: 10.1038/ng.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raines EW, Ferri N. Thematic review series: the immune system and atherogenesis. Cytokines affecting endothelial and smooth muscle cells in vascular disease. J Lipid Res. 2005;46:1081–1092. doi: 10.1194/jlr.R500004-JLR200. [DOI] [PubMed] [Google Scholar]
- Rajavashisth TB, Andalibi A, Territo MC, Berliner JA, Navab M, Fogelman AM, Lusis AJ. Induction of endothelial cell expression of granulocyte and macrophage colony-stimulating factors by modified low-density lipoproteins. Nature. 1990;344:254–257. doi: 10.1038/344254a0. [DOI] [PubMed] [Google Scholar]
- Ramsey SA, Klemm SL, Zak DE, Kennedy KA, Thorsson V, Li B, Gilchrist M, Gold ES, Johnson CD, Litvak V, et al. Uncovering a macrophage transcriptional program by integrating evidence from motif scanning and expression dynamics. PLoS Comput Biol. 2008;4:e1000021. doi: 10.1371/journal.pcbi.1000021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich M, Liefeld T, Gould J, Lerner J, Tamayo P, Mesirov JP. GenePattern 2.0. Nat Genet. 2006;38:500–501. doi: 10.1038/ng0506-500. [DOI] [PubMed] [Google Scholar]
- Reimers M, Carey VJ. Bioconductor: an open source framework for bioinformatics and computational biology. Methods Enzymol. 2006;411:119–134. doi: 10.1016/S0076-6879(06)11008-3. [DOI] [PubMed] [Google Scholar]
- Ross R. Atherosclerosis—an inflammatory disease. N Engl J Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- Rust S, Rosier M, Funke H, Real J, Amoura Z, Piette JC, Deleuze JF, Brewer HB, Duverger N, Denefle P, et al. Tangier disease is caused by mutations in the gene encoding ATP-binding cassette transporter 1. Nat Genet. 1999;22:352–355. doi: 10.1038/11921. [DOI] [PubMed] [Google Scholar]
- Schadt EE. Molecular networks as sensors and drivers of common human diseases. Nature. 2009;461:218–223. doi: 10.1038/nature08454. [DOI] [PubMed] [Google Scholar]
- Schadt EE, Molony C, Chudin E, Hao K, Yang X, Lum PY, Kasarskis A, Zhang B, Wang S, Suver C, et al. Mapping the genetic architecture of gene expression in human liver. PLoS Biol. 2008;6:e107. doi: 10.1371/journal.pbio.0060107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon PT, Reiss DJ, Bonneau R, Baliga NS. The Gaggle: an open-source software system for integrating bioinformatics software and data sources. BMC Bioinformatics. 2006;7:176. doi: 10.1186/1471-2105-7-176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skogsberg J, Lundstrom J, Kovacs A, Nilsson R, Noori P, Maleki S, Kohler M, Hamsten A, Tegner J, Bjorkegren J. Transcriptional profiling uncovers a network of cholesterol-responsive atherosclerosis target genes. PLoS Genet. 2008;4:e1000036. doi: 10.1371/journal.pgen.1000036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JD, Bhasin JM, Baglione J, Settle M, Xu Y, Barnard J. Atherosclerosis susceptibility loci identified from a strain intercross of apolipoprotein E-deficient mice via a high-density genome scan. Arterioscler Thromb Vasc Biol. 2006a;26:597–603. doi: 10.1161/01.ATV.0000201044.33220.5c. [DOI] [PubMed] [Google Scholar]
- Smith JD, Peng D-Q, Dansky HM, Settle M, Baglione J, Le Goff W, Chakrabarti E, Xu Y, Peng X. Transcriptome profile of macrophages from atherosclerosis-sensitive and atherosclerosis-resistant mice. Mamm Genome. 2006b;17:220–229. doi: 10.1007/s00335-005-0099-7. [DOI] [PubMed] [Google Scholar]
- Steinberg D. Thematic review series: the pathogenesis of atherosclerosis. An interpretive history of the cholesterol controversy: part I. J Lipid Res. 2004;45:1583–1593. doi: 10.1194/jlr.R400003-JLR200. [DOI] [PubMed] [Google Scholar]
- Steinberg D. Thematic review series: the pathogenesis of atherosclerosis: an interpretive history of the cholesterol controversy, part III: mechanistically defining the role of hyperlipidemia. J Lipid Res. 2005a;46:2037–2051. doi: 10.1194/jlr.R500010-JLR200. [DOI] [PubMed] [Google Scholar]
- Steinberg D. Thematic review series: the pathogenesis of atherosclerosis. An interpretive history of the cholesterol controversy: part II: the early evidence linking hypercholesterolemia to coronary disease in humans. J Lipid Res. 2005b;46:179–190. doi: 10.1194/jlr.R400012-JLR200. [DOI] [PubMed] [Google Scholar]
- Steinberg D. Thematic review series: the pathogenesis of atherosclerosis. An interpretive history of the cholesterol controversy, part V: the discovery of the statins and the end of the controversy. J Lipid Res. 2006;47:1339–1351. doi: 10.1194/jlr.R600009-JLR200. [DOI] [PubMed] [Google Scholar]
- Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci USA. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabibiazar R, Wagner RA, Ashley EA, King JY, Ferrara R, Spin JM, Sanan DA, Narasimhan B, Tibshirani R, Tsao PS, et al. Signature patterns of gene expression in mouse atherosclerosis and their correlation to human coronary disease. Physiol Genomics. 2005;22:213–226. doi: 10.1152/physiolgenomics.00001.2005. [DOI] [PubMed] [Google Scholar]
- Tarca AL, Draghici S, Khatri P, Hassan SS, Mittal P, Kim JS, Kim CJ, Kusanovic JP, Romero R. A novel signaling pathway impact analysis. Bioinformatics. 2009;25:75–82. doi: 10.1093/bioinformatics/btn577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torkamani A, Topol EJ, Schork NJ. Pathway analysis of seven common diseases assessed by genome-wide association. Genomics. 2008;92:265–272. doi: 10.1016/j.ygeno.2008.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tregouet DA, Konig IR, Erdmann J, Munteanu A, Braund PS, Hall AS, Grosshennig A, Linsel-Nitschke P, Perret C, DeSuremain M, et al. Genome-wide haplotype association study identifies the SLC22A3-LPAL2-LPA gene cluster as a risk locus for coronary artery disease. Nat Genet. 2009;41:283–285. doi: 10.1038/ng.314. [DOI] [PubMed] [Google Scholar]
- Trogan E, Feig JE, Dogan S, Rothblat GH, Angeli V, Tacke F, Randolph GJ, Fisher EA. Gene expression changes in foam cells and the role of chemokine receptor CCR7 during atherosclerosis regression in ApoE-deficient mice. Proc Natl Acad Sci USA. 2006;103:3781–3786. doi: 10.1073/pnas.0511043103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts DJ, Strogatz SH. Collective dynamics of 'small-world' networks. Nature. 1998;393:440–442. doi: 10.1038/30918. [DOI] [PubMed] [Google Scholar]
- Weber C, Zernecke A, Libby P. The multifaceted contributions of leukocyte subsets to atherosclerosis: lessons from mouse models. Nat Rev Immunol. 2008;8:802–815. doi: 10.1038/nri2415. [DOI] [PubMed] [Google Scholar]
- Wernicke S, Rasche F. FANMOD: a tool for fast network motif detection. Bioinformatics. 2006;22:1152–1153. doi: 10.1093/bioinformatics/btl038. [DOI] [PubMed] [Google Scholar]
- Wheelock CE, Wheelock AM, Kawashima S, Diez D, Kanehisa M, van Erk M, Kleemann R, Haeggström JZ, Goto S. Systems biology approaches and pathway tools for investigating cardiovascular disease. Mol Biosyst. 2009;5:588–602. doi: 10.1039/b902356a. [DOI] [PubMed] [Google Scholar]
- Willer CJ, Speliotes EK, Loos RJ, Li S, Lindgren CM, Heid IM, Berndt SI, Elliott AL, Jackson AU, Lamina C, et al. Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nat Genet. 2009;41:25–34. doi: 10.1038/ng.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization. 2009. Fact Sheet #317—Cardiovascular Disease.
- Yang X, Deignan JL, Qi H, Zhu J, Qian S, Zhong J, Torosyan G, Majid S, Falkard B, Kleinhanz RR, et al. Validation of candidate causal genes for obesity that affect shared metabolic pathways and networks. Nat Genet. 2009;41:415–423. doi: 10.1038/ng.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusuf S, Hawken S, Ounpuu S, Dans T, Avezum A, Lanas F, McQueen M, Budaj A, Pais P, Varigos J, et al. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): case-control study. Lancet. 2004;364:937–952. doi: 10.1016/S0140-6736(04)17018-9. [DOI] [PubMed] [Google Scholar]
- Zak DE, Aderem A. Systems biology of innate immunity. Immunol Rev. 2009;227:264–282. doi: 10.1111/j.1600-065X.2008.00721.x. [DOI] [PMC free article] [PubMed] [Google Scholar]