

Abstract
Heterotrimeric guanine nucleotide–binding protein (G protein)–coupled receptors (GPCRs), which constitute the largest family of membrane proteins, mediate responses to diverse physiological stimuli. The presence of melanocortin 2 receptors (MC2Rs) on the plasma membrane requires the presence of either MC2R accessory protein (MRAP) or MRAP2, which are homologous accessory proteins. Here, we show that, whereas MRAP was essential for activation of MC2R signaling, MRAP2 was an endogenous inhibitor that competed with MRAP for binding to MC2R and decreased the potency of adrenocorticotropic hormone (ACTH), the endogenous agonist for MC2Rs, in stimulating the production of adenosine 3′,5′-monophosphate (cAMP). ACTH bound with high affinity to MC2Rs in the presence of MRAP, but not MRAP2. The ability of MRAP and MRAP2 to influence ligand-binding affinity was specific to MC2R, because these proteins had little effect on the binding of NDP-α-melanocyte–stimulating hormone to MC4R or on its stimulation of cAMP responses. These results demonstrate that the balance of stimulatory and inhibitory accessory proteins can control the sensitivity of a GPCR to its natural agonist.
Introduction
In the classical view, the cellular signal generated by a heterotrimeric guanine nucleotide–binding protein (G protein)–coupled receptor (GPCR) is governed by the affinity and efficacy of its natural agonist. In recent years, this simplistic model has been expanded by studies of biased agonists that differ from one another in their ability to elicit different biological responses (1), the identification of endogenous peptides that act as antagonists or inverse agonists (2–5), and strong evidence that GPCRs form multimeric complexes that contain receptor dimers and sometimes additional proteins (6–8). To pass the rigorous quality control system in the endoplasmic reticulum (ER) and move through the Golgi apparatus to the plasma membrane, GPCRs interact with general chaperones such as calnexin and, in some cases, with accessory proteins that are specific for a given group of receptors. Dopamine receptor–interacting protein 78 (DRIP78) (9), receptor activity–modifying proteins (RAMPs) (10–13), receptor-transporting proteins (RTPs) (14), and melanocortin receptor accessory proteins (MRAPs) (15) are among the accessory proteins that promote the proper localization of receptors at the cell surface (6, 7, 10, 13). In addition to facilitating the trafficking of receptors to the plasma membrane, RAMPs act as switches to control the ligand preferences of two class B GPCRs, calcitonin and calcitonin receptor–like receptors (10–12).
The melanocortin (MC) receptor family consists of five Gs-coupled receptors that control various physiological functions in response to four distinct agonists, adrenocorticotropic hormone (ACTH, also known as corticotrophin) and α, β, and γ melanocyte–stimulating hormone (MSH), which are derived from the proopiomelanocortin precursor protein, and two inverse agonists, agouti and agouti-related proteins (2–5). The MC2 receptor (MC2R) serves as the physiological receptor for ACTH in the adrenal cortex. Recently, the essential role of an accessory protein for the MC2 receptor, MC2R accessory protein (MRAP), was described (15). Loss of either MC2R (16) or MRAP (15) causes severe resistance to ACTH, which results in severe glucocorticoid deficiency that can be fatal (17).
In the absence of an accessory protein, MC2R is trapped in the ER and is unable to signal (15, 18–21). MRAP is required both for the trafficking of MC2R to the cell surface and for its signaling; these functions of MRAP are distinct, because certain mutant MRAP proteins promote receptor trafficking but not signaling (19–21). Knockdown of MRAP blunts signaling by MC2R in adrenocortical cells (22). All mammals have genes that encode two homologous proteins, MRAP and MRAP2. The N-terminal and transmembrane domains of MRAP and MRAP2 are conserved, although MRAP2 lacks a sequence that is essential for the function of MRAP in mediating the signaling of MC2R (19). MRAP and MRAP2 messenger RNA (mRNA) and protein are found in the adrenal cortex, and both MRAPs enable localization of MC2R at the cell surface (15, 19–23). Here, we tested the ability of these two accessory proteins to support MC2R-dependent responses stimulated by ACTH.
Results
Effects of MRAP and MRAP2 on MCR signaling
To compare signal transduction by MC2R in the presence of MRAP or MRAP2, we transfected Chinese hamster ovary (CHO) cells with plasmids encoding MC2R and either one of the two accessory proteins, incubated the cells with different concentrations of ACTH, and measured the accumulation of adenosine 3′,5′-monophosphate (cAMP). In the presence of MRAP, ACTH stimulated a strong MC2R-mediated cAMP response with a median effective concentration (EC50) of 2.4 ± 1.2 nM. In the presence of MRAP2, ACTH (1 μM) caused only a small increase in the amount of cAMP (Fig. 1A). The abundance of MC2R at the plasma membrane was the same in cells that had either MRAP or MRAP2 (Fig. 1B), but was low in cells that lacked either accessory protein. The response profiles show why MC2Rs expressed with MRAP2 gave a substantial cAMP response when ACTH was tested at 1 μM (23) but not at 100 nM (19).
Fig. 1.
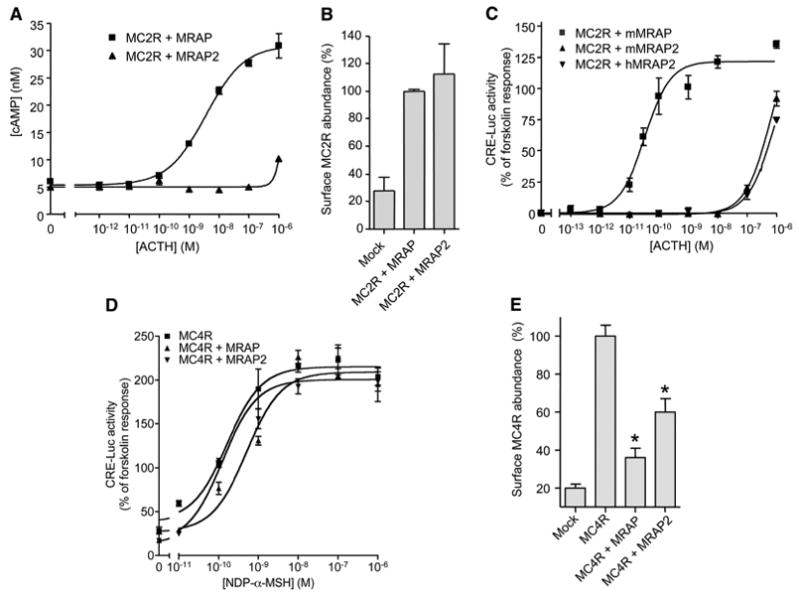
Effects of MRAP and MRAP2 on MC2R and MC4R signaling. (A to E) CHO cells were transfected with plasmids encoding HA-hMC2R or HA-hMC4R and V5-mMRAP-FLAG, V5-mMRAP2-FLAG, or V5-hMRAP2-FLAG and (C and D) CRE-Luc. (A) cAMP accumulation in cells transfected with plasmid encoding HA-hMC2R that were incubated for 20 min with 0.1 mM isobutylmethylxanthine and either vehicle or ACTH (1–39). CRE-Luc activity in cells transfected with plasmid encoding HA-hMC2R (C) or HA-hMC4R (D) that were incubated for 4 hours with vehicle, ACTH(1-39), NDP-α-MSH, or forskolin (20 μM). Data are normalized to the forskolin response. ACTH(1-39) and NDP-α-MSH did not generate any responses in mock-transfected cells. The forskolin response in cells that had mMRAP2 averaged 69 ± 29% higher than that in cells that had mMRAP (n = 11 experiments). (B and E) Surface HA-hMC2Rs or HA-hMC4Rs in nonpermeabilized cells were detected by ELISA with an antibody against the HA tag. In (B), data are normalized to the value with MRAP, and in (E), data are normalized to the value with MC4R alone. Shown are the mean and SE or range from representative experiments performed in triplicate, for (A) through (D), or in duplicate, for (E). *P < 0.05 versus receptor alone.
To confirm the strikingly different effects of the two accessory proteins, we measured ACTH responses with a far more sensitive cAMP reporter assay. In addition to being transfected with plasmids encoding MC2R and either MRAP or MRAP2, CHO cells were transfected with a plasmid encoding luciferase (luc) under the control of a promoter containing four nonpalindromic cAMP-responsive elements (CREs) from the rat insulin promoter (CRE-Luc) (24). This method in which cAMP drives the expression of luc, yielded results consistent with those in which cAMP was measured directly (Fig. 1A). In the more sensitive reporter assay, the EC50 for ACTH was (8.0 ± 2.8) × 10−11 M in cells containing MC2R and MRAP and was greater than 0.1 μM in cells containing MC2R and MRAP2; in these cells the response did not reach a plateau (Fig. 1C). Identical results were obtained with mouse and human MRAP2 (Fig. 1C). No constitutive activity was detected, nor did ACTH inhibit the production of cAMP in the presence of forskolin (a stimulator of adenylyl cyclase) in cells containing MC2R and either MRAP or MRAP2, eliminating the possibility that MRAP2 switched the G protein–coupling preference of MC2R from Gs to Gi.
Although only MC2R requires an accessory protein for proper trafficking, MRAP and MRAP2 coimmunoprecipitate with all five MCRs (23). MRAP2 mRNA is found in the adrenal glands and the brain but not elsewhere, whereas MRAP mRNA is found in multiple tissues (15, 19, 20, 22, 23, 25). To test the specificities of the actions of MRAP and MRAP2 on MC2R, we measured their effects on signaling by MC4R. MC4Rs can be expressed heterologously in cells without the addition of an accessory protein. Neither MRAP nor MRAP2 substantially altered the potency of the agonist NDP-α-MSH to stimulate MC4R-mediated production of cAMP. In the reporter assay, EC50 values averaged (1.1 ± 0.3), (2.7 ± 1.1), and (1.2 ± 0.2) × 10−10 M in cells containing MC4R alone, in cells containing MC4R and MRAP and in cells containing MC4R and MRAP2, respectively (Fig. 1D). Both MRAP and MRAP2 decreased the abundance of MC4R at the cell surface (Fig. 1E), as previously reported (23). The contrast between the powerful effects of MRAP and MRAP2 on MC2R and the minimal effects on MC4R emphasizes the selectivity of these accessory proteins. The lack of effect of MRAPs on MC4R signaling makes it unlikely that these accessory proteins exert major effects on Gs, adenylyl cyclase, or other downstream signaling components.
Because the ACTH-stimulated activation of mitogen-activated protein kinases (MAPKs) is largely independent of cAMP-dependent protein kinase (PKA) (26), we tested the ability of ACTH to stimulate the phosphorylation of the MAPKs extracellular signal–regulated kinases (ERKs) ERK1 and ERK2 in serum-deprived cells transfected with plasmids encoding MC2R and either MRAP or MRAP2 (Fig. 2A). In cells containing both MC2R and MRAP, ACTH strongly stimulated the phosphorylation of ERK1 and ERK2 with an EC50 of 1.1 ± 0.1 nM. In cells containing MC2R and MRAP2, ACTH only weakly activated the MAPK pathway, with an EC50 of (6.8 ± 3.5) × 10−8 M (Fig. 2B). These data indicate that the disparity in MC2R signaling with MRAP and MRAP2 extends to multiple downstream pathways.
Fig. 2.
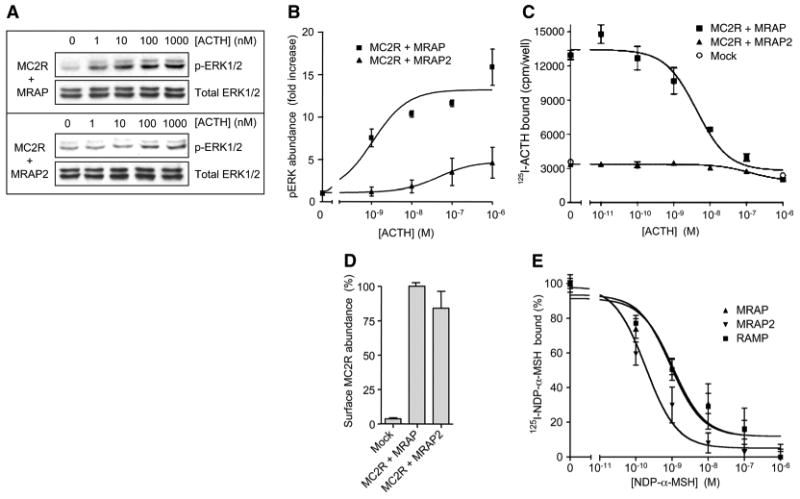
Effects of MRAP and MRAP2 on agonist binding and MAPK signaling. (A) MC2R-mediated phosphorylation of ERK1/2. HEK 293 cells transfected with plasmids encoding HA-hMC2R and either mMRAP or mMRAP2 were serum-deprived overnight and incubated with vehicle or ACTH(1-39) for 5 min. Phosphorylated and total ERK1/2 proteins were identified by Western blotting. (B) Densitometric analysis of the abundance of pERK. (C) Analysis of 125I-labeled ACTH binding. CHO cells transfected with plasmid encoding HA-hMC2R and either mMRAP or mMRAP2 were incubated for 1 hour at 37°C with 125I-labeled ACTH and unlabeled ACTH(1–39). (D) The abundance of HA-hMC2R at the cell surface was measured under the same conditions as those used for measurement of the binding of 125I-labeled ACTH. Data are normalized to the value with MRAP. (E) Analysis of 125I-labeled NDP-α-MSH binding. CHO cells transfected with plasmids encoding HA-hMC4R and either mMRAP or mMRAP2 were incubated for 2 hours at 37°C with 125I-labeled NDP-α-MSH and unlabeled NDP-α-MSH. IC50 values for NDP-α-MSH were 0.92, 1.1, and 0.19 nM and maximal binding was 100, 65, and 165% in cells transfected with plasmid encoding RAMP3 (control), mMRAP, and mMRAP2, respectively. There was no specific binding of 125I-labeled NDP-α-MSH to mock-transfected cells or to cells transfected with plasmids encoding MRAPs alone. Shown are the mean and SE from representative experiments performed in triplicate, for (A) through (D), or pooled data from three experiments performed in duplicate or triplicate for (E).
Effects of MRAP and MRAP2 on the binding affinities of MCRs
The simplest explanation for our results is that the affinity of MC2R for ACTH is highly dependent on the accessory protein present. We measured the binding of 125I-labeled ACTH to cells transfected with plasmids encoding MC2R and either MRAP or MRAP2 (Fig. 2C). In cells containing MRAP, MC2Rs bound to 125I-labeled ACTH with high affinity and specificity; the median inhibitory concentration (IC50) for displacement with unlabeled ACTH was 5.7 ± 0.3 nM. The abundance of receptor at the surface was measured under the conditions used for radioligand-binding and cAMP assays. When MRAP2 was present, the density of receptors at the cell surface was nearly the same as in cells that contained MRAP (Fig. 2D), but no specific binding was detected, indicating that the MC2R-MRAP2 complex had extremely low affinity for ACTH, which explained the decrease in the potency of ACTH in all of the signaling pathways analyzed. It was not clear whether MRAP acted as a co-receptor that was physically involved in binding to ACTH or acted indirectly to induce MC2R to adopt a conformation that had a higher affinity for ACTH. In contrast to their dramatically different effects on the binding of 125I-labeled ACTH to MC2Rs, MRAP and MRAP2 exerted relatively small effects on the affinity of MC4R for 125I-labeled NDP-α-MSH (Fig. 2E).
Topology of MRAP-MRAP2 dimers
MRAP and MRAP2 have single membrane-spanning helical domains. We have shown that MRAP forms antiparallel homodimers; that is, the protein is inserted into the membrane in opposing orientations (18). MRAP is the only eukaryotic protein thus far known to display this dual topology. Coimmunoprecipitation assays with V5- and FLAG-tagged MRAP and MRAP2 have shown that MRAP and MRAP2 form homodimers and heterodimers in cell lysates (18, 19, 22), but the orientation of MRAP-MRAP2 dimers is not known. To examine the arrangement of MRAP-MRAP2 heterodimers, we performed bimolecular fluorescence complementation experiments (27). Briefly, yellow fluorescent protein (YFP) can be split into two fragments that are not fluorescent on their own. If these fragments are fused to proteins that interact closely, then fluorescent YFP can be reconstituted. We expressed two proteins in cells. One was a fusion protein of MRAP or MRAP2 with a fragment of YFP (YFP-F1) at the N terminus and the other was a fusion protein with the complementary fragment (YFP-F2) at the C terminus of MRAP or MRAP2. In this case, molecular complementation of YFP could only occur if the two proteins were inserted across the membrane in opposite directions and in close proximity to one another (Fig. 3). As a control, we performed the same experiment with RAMP3, a single transmembrane protein that forms parallel dimers with an extracellular N terminus (10, 12).
Fig. 3.
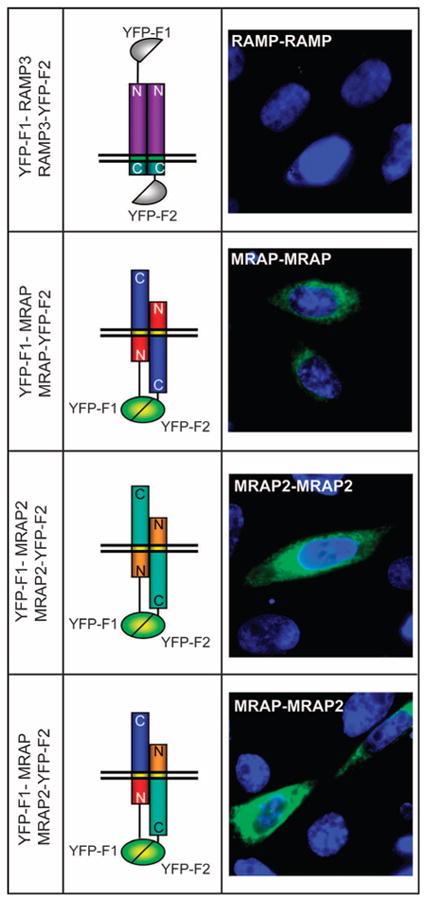
Homodimers and heterodimers of MRAP and MRAP2 are arranged in an antiparallel manner. YFP fluorescence imaging was performed in live CHO cells transfected with plasmids encoding YFP-F1-RAMP3 and RAMP3-YFP-F2, YFP-F1-mMRAP and mMRAP-YFP-F2, YFP-F1-mMRAP2 and mMRAP2-YFP-F2, or YFP-F1-mMRAP and mMRAP2-YFP-F2. Nuclei are counterstained in blue.
No fluorescence was visible in live CHO cells that contained YFP-F1-RAMP3 and RAMP3-YFP-F2, because the complementary fragments of YFP were on different sides of the membrane (Fig. 3). As expected (19), fluorescence was detected in cells that contained YFP-F1-MRAP and MRAP-YFP-F2, demonstrating the presence of antiparallel homodimers of MRAP. Fluorescence was also strong in cells that contained YFP-F1-MRAP2 and MRAP2-YFP-F2 or YFP-F1-MRAP and MRAP2-YFP-F2, showing that MRAP2 homodimers and MRAP-MRAP2 heterodimers were antiparallel in arrangement.
Dominant-negative actions of MRAP2
That MRAP2 does not promote efficient MC2R signaling even though it supports trafficking of MC2R to the plasma membrane raised the question of the physiological role of MRAP2. We hypothesized that MRAP2 was an inhibitory accessory protein and that the relative concentrations of MRAP and MRAP2 in the cell could determine its responsiveness to ACTH. To test this idea, we measured the effect of different amounts of MRAP2 on cAMP signaling in CHO cells that contained MC2R and MRAP (Fig. 4A). As before, the abundance of MC2R at the cell surface was not decreased by the addition of MRAP2. At the same time, downstream signaling in these cells was dramatically lowered compared to that in cells containing MC2R and MRAP. Increasing the amount of MRAP2 protein in the presence of a fixed amount of MRAP led to a substantial shift in the ACTH dose-response curve to the right, consistent with a dominant-negative role for MRAP2. These results strongly suggested that MRAP2 represented an inhibitory accessory protein capable of antagonizing the actions of MRAP and lowering the sensitivity of MC2R for its natural agonist ACTH.
Fig. 4.
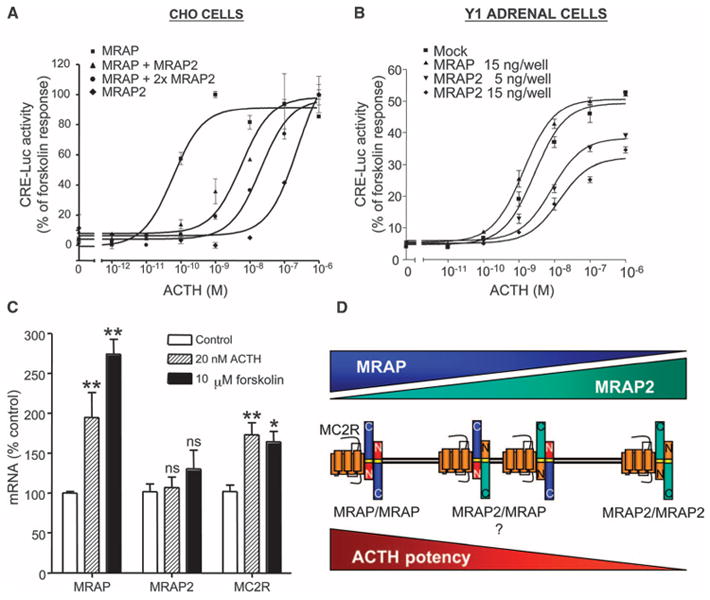
Competition between MRAP and MRAP2 affects MC2R signaling. (A) Responses to ACTH(1–39) in CHO cells transfected with plasmids encoding CRE-Luc and HA-hMC2R and mMRAP (3 ng), mMRAP2 (3 ng), or mMRAP (3 ng) and mMRAP2 (15 or 30 ng). (B) Responses of Y1 adrenal cells transfected with plasmids encoding CRE-Luc and mMRAP (15 ng) or mMRAP2 (5 or 15 ng) to ACTH(1–39). (C) Y1 cells were incubated for 18 hour alone or with ACTH(1–39) (20 nM) or forskolin (10 μM). Relative amounts of mRNA were estimated by real-time quantitative RT-PCR. *P < 0.05; **P < 0.01; ns, not significant, versus control. (D) Possible dimers of MRAP and MRAP2. The effects of the MRAP-MRAP2 heterodimers on MC2R signaling are hypothetical. Shown are the mean and SE from representative experiments performed in triplicate for (A) and (B) or pooled data from three experiments performed in duplicate or triplicate for (C).
The clonal adrenal cell line Y1 contains MC2R, MRAP, and MRAP2 mRNAs (20), demonstrating that these proteins can be found in the same cell. To test whether MRAP2 inhibited signaling by endogenous MC2R, we used the CRE-Luc reporter assay system to measure ACTH-dependent responses in Y1 cells transfected with a plasmid encoding CRE-luc; in this system, only successfully transfected cells were studied (Fig. 4B). ACTH elicited a strong cAMP response in Y1 cells transfected with the reporter plasmid alone (EC50 = 1.95 ± 1.00 nM). Cotransfection of cells with the plasmid encoding MRAP2 caused a dose-dependent decrease in cAMP signaling and a shift in the ACTH dose-response curve to the right, consistent with the concept that MRAP2 competes with MRAP for binding to MC2R and inhibits ACTH-induced signaling. In contrast, cotransfection of cells with the plasmid encoding MRAP did not cause a substantial change in the response to ACTH, which suggested that the amount of endogenous MRAP in these cells was not limiting. This result also implied that the abundance of endogenous MRAP2 was not high enough to inhibit MC2R signaling substantially; if it were, then the increased abundance of MRAP in transfected cells would compete with endogenous MRAP2 and increase the responsiveness of these cells to ACTH.
ACTH increases the abundance of MC2R mRNA in mouse and human adrenal cells and this response can be mimicked by forskolin, which increases the concentration of intracellular cAMP by directly stimulating adenylyl cyclase (28, 29). To determine whether ACTH also regulated the abundance of endogenous MRAP and MRAP2 mRNAs, we incubated Y1 cells with ACTH (20 nM) or forskolin (10 μM) overnight, conditions used previously to identify genes regulated by ACTH in Y1 cells. In agreement with Schimmer et al. (29), we found that ACTH and forskolin increased the abundance of MC2R mRNA (Fig. 4C). ACTH and forskolin stimulated an even larger increase in the abundance of MRAP mRNA, but had no substantial effect on that of MRAP2 mRNA (Fig. 4C). These findings are consistent with a mechanism whereby ACTH, or its second messenger cAMP, sensitizes Y1 adrenal cells to ACTH by increasing in tandem the abundance of its receptor MC2R and its stimulatory accessory protein MRAP without altering the abundance of the inhibitory accessory protein MRAP2.
Discussion
This study describes a previously uncharacterized mechanism of regulation of GPCR signaling by identifying the first inhibitory accessory protein, MRAP2. MRAP2 prevents the binding of ACTH to MC2R and, consequently, receptor signaling, by competing with the obligatory MC2R accessory protein MRAP (Fig. 4D). The finding that ACTH is almost 10,000-fold less potent in stimulating the MC2R-MRAP2 complex than it is at stimulating the MC2R-MRAP complex may explain why patients with familial glucocorticoid deficiency type II, which is caused by inactivating mutations in MRAP, are resistant to ACTH even though they presumably have adrenal MRAP2 (15). Plasma concentrations of ACTH vary diurnally, rise during stress, and are increased in a setting of adrenal insufficiency, but ACTH would not reach the micromolar concentrations that are apparently required to activate MC2Rs that are found with MRAP2 alone.
Surface localization of the MC2 receptor is facilitated by either MRAP or MRAP2. These accessory proteins may promote folding, posttranslational modification, or trafficking of the receptor from the ER to the plasma membrane. MRAP and MRAP2 may not be equivalent, however, because MRAP appears to facilitate the surface localization of glycosylation mutants of the MC2R more efficiently than does MRAP2 (30). Based on their studies of a series of MC2-MC4 receptor chimeras, Fridmanis et al. (31) proposed that several regions of the MC2R function as intracellular retention signals and that the MRAP accessory proteins mask these signals to enable localization of the receptor at the plasma membrane.
Although both MRAP and MRAP2 allow MC2R to reach the cell surface, only MRAP enables the receptor to signal effectively. The molecular mechanisms for this specificity have not been identified. RAMPs have been proposed to act as co-receptors (10, 12) and a co-receptor model is consistent with the available data for MRAP, but there is no evidence that MRAP interacts directly with ACTH. It is also conceivable that MRAP, similar to RAMPs, changes ligand specificity, but there is no evidence that any ligand other than ACTH binds to MC2R. An alternative model, whereby MRAP causes a conformational change in the receptor that opens an ACTH-binding pocket, is supported by studies of MC2-MC4 receptor chimeras (31).
Why has MRAP2 evolved to escort functionally inactive MC2Rs to the plasma membrane? In a setting where MRAP and MRAP2 are both present with the MC2R, the dominant-negative MRAP2 protein, acting in MRAP2-MRAP2 homodimers, or perhaps in MRAP-MRAP2 heterodimers, may provide an additional means of controlling the response of cells to ACTH. The ratio of the abundance of MRAP to MRAP2 could tune the affinity of MC2R for ACTH and regulate the ACTH-response profile of the cells (Fig. 4D). MRAP2 likely acts as a natural, dominant-negative form of MRAP. Roy et al. (32) reported that the EC50 value for ACTH is fourfold higher in cells that contain MC2R and the β-form of human MRAP than in cells that contain MC2R and the α-form of MRAP, which suggests that the ratio of the abundance of MRAP splice variants may also contribute to regulating the responsiveness of cells to ACTH. Selective effects of MRAP and MRAP2 may play a physiologic role at other receptors in the MCR family or at GPCRs not currently known to interact with these accessory proteins.
The MCR family has evolved several unusual features. Whereas most GPCRs appear to be regulated by one or more natural agonists, several of the MCRs have appreciable constitutive activity and are regulated in both directions by physiological agonists and inverse agonists (2–5). Indeed, mutations that reduce constitutive, but not agonist-dependent, signaling by the MC4R cause an obesity phenotype in humans (33). In the presence of agouti or agouti-related peptide (AGRP), the dose-response curve for signaling by MC1Rs or MC4Rs is shifted to the right. Analogously, in the presence of MRAP2, the dose-response curve for signaling by the MC2R is shifted to the right. These entirely distinct mechanisms both serve to regulate the sensitivity of MCRs to their agonists. It seems likely that other GPCRs will be found to undergo similarly complex modes of regulation. Competition between active and inhibitory accessory proteins also provides an opportunity for drug design.
Materials and Methods
Reagents
Plasmids encoding hMC2R and hMC4R with three N-terminal hemagglutinin (HA) tags and plasmid encoding RAMP3 were obtained from the Missouri S&T cDNA Resource Center. Plasmids encoding YFP-F1 and YFP-F2 were obtained from C. Berlot (Weis Center for Research, Geisinger Clinic, Danville, PA) (34), and plasmid encoding RIP CRE-luc was obtained from G. Holz (State University of New York Upstate Medical Center, Syracuse, NY) (24). Constructs for split YFP fusion proteins were prepared by polymerase chain reaction (PCR) and verified by sequencing as described (19). Primer sequences are available on request. Unless noted, all MRAP constructs were from mouse and MC2R and MC4R were from human. Monoclonal antibody against the V5 tag was from AbDSerotec; monoclonal antibody against the HA tag (HA11) was from Covance; monoclonal antibody against the FLAG tag was obtained from Sigma; horseradish peroxidase (HRP)–conjugated antibodies against mouse and rabbit immunoglobulins were from Biorad; and antibodies against total ERK1/2 and phosphorylated ERK1/2 were from Cell Signaling Technology. ACTH(1–39) and NDP-α-MSH were purchased from Phoenix Pharmaceuticals.
Cloning of hMRAP2 and mMRAP2
The cloning of mMRAP2 from mouse adrenal glands was previously described (19). hMRAP2 was cloned from the human cell line SHSY5Y with the same approach and the following primers: 5 -TATATATATAGCTAGCGCCACCATGGGTAAGCCTATCCCCAACCCTCTGCTCGGTCTTGATTCTACTTCCGCCCAGAGGTTAATTTC-3 (forward) and 5 -TATATATAACGCGTATCCAGGTCTTTGTGTGAGGTCTG-3 (reverse). The amplicon was then inserted into the pciNeo expression plasmid between the Nhe I and Mlu I sites flanked by the coding region for the V5 and 3X FLAG epitopes. Untagged hMRAP2 was cloned and inserted into the pcDNA3.1 expression plasmid between the Bam HI and Eco RI sites with the following primers: 5 -TATATATAGGATCCGCCACCATGTCCGCCCAGAGGTTAATTTC-3 (forward) and 5 -TATATATAGAATTCTCAATCCAGGTCTTTGTGTGAGGTCTG-3 (reverse). Cells transfected with plasmids encoding V5-mMRAP2-FLAG or untagged hMRAP2 yielded virtually identical results in functional assays. Unless otherwise noted, mouse MRAP and MRAP2 constructs were tested, and these appeared to be functionally equivalent to the corresponding human MRAPs.
Cell culture and transfections
CHO, Y1, SHSY5Y, and human embryonic kidney (HEK) 293 cells were grown in Dulbecco's modified Eagle's medium (DMEM)–F-12 medium supplemented with 5% fetal bovine serum. Cells were transfected with the appropriate plasmids with Fugene HD (Roche) 24 to 48 hours before experiments were performed. Total plasmid concentrations were kept identical for all transfections in each experiment by the addition of empty vector.
cAMP and CRE-Luc assays
CHO cells were plated in white 96-well plates, transfected the next day with plasmids encoding MC2R and accessory proteins, as noted, and used in experiments the following morning. Cells were challenged with vehicle or peptide in DMEM–F-12 supplemented with 0.1% bovine serum albumin (BSA). Concentrations of cAMP were measured with the FRET-based LANCE cAMP assay kit (Perkin Elmer). Cells were incubated with 0.1 mM isobutylmethylxanthine and vehicle or peptide for 20 min at 37°C before the assay. In the luciferase reporter assay, cells were transfected with a CRE-luc reporter plasmid (24) and then incubated with vehicle, peptide, or forskolin (20 μM) for 4 hours at 37°C. The medium was removed and 100 μl of One-Step luciferase assay reagent (Nanolight Technology) was added and the dish was kept in the dark for 10 to 30 min at room temperature. Luminescence was then measured with a Victor plate reader (Perkin Elmer).
Radioligand binding assays
Cells in 12-well dishes were incubated for 20 min in binding buffer [Dulbecco's phosphate-buffered saline (PBS) with 20 mM Hepes (pH 7.5), 0.1% BSA] at 37°C, and were then incubated for 1 hour in binding buffer (0.4 ml per well) containing between 100,000 and 250,000 cpm of 125I-labeled ACTH (ACTH(1–39)23Tyr[125I]) or 125I-labeled NDP-α-MSH (New England Nuclear, 2000 Ci/mmol). Dishes were placed on ice and cells were washed three times with cold PBS, solubilized in 0.1% SDS, and quantified in a Beckman gamma counter.
Detection of surface epitopes
Epitopes on the extracellular side of the plasma membrane were measured by fixed-cell enzyme-linked immunosorbent assay (ELISA) on nonpermeabilized cells. Cells were washed with PBS, fixed for 10 min with 2% paraformaldehyde in PBS, washed, blocked in 5% milk in PBS, and processed for ELISA as described previously (35, 36) with a 1:5000 dilution of monoclonal antibody against the HA tag. In the absence of MRAP or MRAP2, the amount of MC2R at the cell surface was negligible. ELISA assays were not saturated under the conditions used.
Imaging of live cells
CHO cells on glass coverslips were transfected 16 to 18 hours before experiments and incubated with Hoechst 33342 (3 μg/ml) for 5 min at 37°C before imaging. A Nikon Diaphot inverted microscope with 100×/1.3 numerical aperture (NA) oil objective, a Photometrics CoolSNAP ES camera, and the appropriate filter sets from Chroma were used. Images were captured with Metamorph software from Universal Imaging. Micrographs displayed were exposed and processed identically.
Detection of activated MAPKs
HEK 293 cells were plated in 24-well dishes coated with poly-l-lysine and transfected with the appropriate plasmids as noted earlier. The next day, the culture medium was removed and cells were incubated with serum-free media overnight and then treated with vehicle or agonist for 5 min at 37°C. Dishes were then placed on ice and cells were scraped in sample buffer. Proteins were resolved by SDS–polyacrylamide gel electrophoresis (SDS-PAGE), and total ERK1/2 and phosphorylated ERK1/2 proteins were identified by Western blotting, performed as described previously (36), with the appropriate antibodies at a 1:1000 dilution by chemiluminescence. HEK 293 cells rather than CHO cells were used because they gave a lower background in MAPK assays.
Quantitative real-time reverse transcription (RT)-PCR
Y1 cells grown in six-well dishes were treated as described, and the medium was aspirated and RNA isolated with the RNeasy kit (Qiagen). Complementary DNA (cDNA) was prepared with the Power SybrGreen Cells to CT kit (Ambion). Real-time PCR was carried out on an ABI Prism 7000 real-time PCR cycler in 20-μl reactions, each containing 2 to 4 μl of cDNA, 4 pmol of primers, and 10 μl of Invitrogen SYBRGreen ER qPCR Supermix with MC2R from Origene. Relative concentrations of mRNAs were calculated by the 2ΔΔCt method (37) with glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as standard. Ct values for GAPDH were not affected by ACTH or forskolin.
Data analysis
All experiments were repeated a minimum of two times on different days in separate experiments. Data points in ELISA assays are the mean and range of duplicate or mean and SEM of three to six determinations. EC50 values were determined with Prism software. The statistical significance of differences was analyzed by unpaired Student's t test or analysis of variance (ANOVA) with Dunnett's posttest analysis. EC50 values are the means ± SEM from three to five experiments.
Acknowledgments
The authors appreciate the valuable contributions of A. Jakubowski.
Funding: This work was supported by NIH grant DK19974.
Footnotes
Author contributions: Both authors made contributions to all aspects of this study.
Competing interests: The authors declare no financial conflicts of interest. The University of Rochester Technology Transfer Office requires that a materials transfer agreement (MTA) be signed before the plasmids used in this study can be provided to other researchers.
References and Notes
- 1.Kohout TA, Nicholas SL, Perry SJ, Reinhart G, Junger S, Struthers RS. Differential desensitization, receptor phosphorylation, β-arrestin recruitment, and ERK1/2 activation by the two endogenous ligands for the CC chemokine receptor 7. J Biol Chem. 2004;279:23214–23222. doi: 10.1074/jbc.M402125200. [DOI] [PubMed] [Google Scholar]
- 2.Haskell-Luevano C, Monck EK. Agouti-related protein functions as an inverse agonist at a constitutively active brain melanocortin-4 receptor. Regul Pept. 2001;99:1–7. doi: 10.1016/s0167-0115(01)00234-8. [DOI] [PubMed] [Google Scholar]
- 3.Nijenhuis WA, Oosterom J, Adan RA. AgRP(83–132) acts as an inverse agonist on the human-melanocortin-4 receptor. Mol Endocrinol. 2001;15:164–171. doi: 10.1210/mend.15.1.0578. [DOI] [PubMed] [Google Scholar]
- 4.Lu D, Willard D, Patel IR, Kadwell S, Overton L, Kost T, Luther M, Chen W, Woychik RP, Wilkison WO, Cone RD. Agouti protein is an antagonist of the melanocyte-stimulating-hormone receptor. Nature. 1994;371:799–802. doi: 10.1038/371799a0. [DOI] [PubMed] [Google Scholar]
- 5.Cone RD. Studies on the physiological functions of the melanocortin system. Endocr Rev. 2006;27:736–749. doi: 10.1210/er.2006-0034. [DOI] [PubMed] [Google Scholar]
- 6.Bernier V, Bichet DG, Bouvier M. Pharmacological chaperone action on G-protein-coupled receptors. Curr Opin Pharmacol. 2004;4:528–533. doi: 10.1016/j.coph.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 7.Bockaert J, Fagni L, Dumuis A, Marin P. GPCR interacting proteins (GIP) Pharmacol Ther. 2004;103:203–221. doi: 10.1016/j.pharmthera.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 8.Bulenger S, Marullo S, Bouvier M. Emerging role of homo- and heterodimerization in G-protein-coupled receptor biosynthesis and maturation. Trends Pharmacol Sci. 2005;26:131–137. doi: 10.1016/j.tips.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 9.Bermak JC, Li M, Bullock C, Zhou QY. Regulation of transport of the dopamine D1 receptor by a new membrane-associated ER protein. Nat Cell Biol. 2001;3:492–498. doi: 10.1038/35074561. [DOI] [PubMed] [Google Scholar]
- 10.Hay DL, Poyner DR, Sexton PM. GPCR modulation by RAMPs. Pharmacol Ther. 2006;109:173–197. doi: 10.1016/j.pharmthera.2005.06.015. [DOI] [PubMed] [Google Scholar]
- 11.McLatchie LM, Fraser NJ, Main MJ, Wise A, Brown J, Thompson N, Solari R, Lee MG, Foord SM. RAMPs regulate the transport and ligand specificity of the calcitonin-receptor-like receptor. Nature. 1998;393:333–339. doi: 10.1038/30666. [DOI] [PubMed] [Google Scholar]
- 12.Parameswaran N, Spielman WS. RAMPs: The past, present and future. Trends Biochem Sci. 2006;31:631–638. doi: 10.1016/j.tibs.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 13.Bouschet T, Martin S, Henley JM. Receptor-activity-modifying proteins are required for forward trafficking of the calcium-sensing receptor to the plasma membrane. J Cell Sci. 2005;118:4709–4720. doi: 10.1242/jcs.02598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saito H, Kubota M, Roberts RW, Chi Q, Matsunami H. RTP family members induce functional expression of mammalian odorant receptors. Cell. 2004;119:679–691. doi: 10.1016/j.cell.2004.11.021. [DOI] [PubMed] [Google Scholar]
- 15.Metherell LA, Chapple JP, Cooray S, David A, Becker C, Ruschendorf F, Naville D, Begeot M, Khoo B, Nurnberg P, Huebner A, Cheetham ME, Clark AJ. Mutations in MRAP, encoding a new interacting partner of the ACTH receptor, cause familial glucocorticoid deficiency type 2. Nat Genet. 2005;37:166–170. doi: 10.1038/ng1501. [DOI] [PubMed] [Google Scholar]
- 16.Clark AJ, McLoughlin L, Grossman A. Familial glucocorticoid deficiency associated with point mutation in the adrenocorticotropin receptor. Lancet. 1993;341:461–462. doi: 10.1016/0140-6736(93)90208-x. [DOI] [PubMed] [Google Scholar]
- 17.Chan LF, Clark AJ, Metherell LA. Familial glucocorticoid deficiency: Advances in the molecular understanding of ACTH action. Horm Res. 2008;69:75–82. doi: 10.1159/000111810. [DOI] [PubMed] [Google Scholar]
- 18.Sebag JA, Hinkle PM. Melanocortin-2 receptor accessory protein MRAP forms antiparallel homodimers. Proc Natl Acad Sci USA. 2007;104:20244–20249. doi: 10.1073/pnas.0708916105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sebag JA, Hinkle PM. Regions of melanocortin 2 (MC2) receptor accessory protein necessary for dual topology and MC2 receptor trafficking and signaling. J Biol Chem. 2009;284:610–618. doi: 10.1074/jbc.M804413200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sebag JA, Hinkle PM. Opposite effects of the melanocortin-2 (MC2) receptor accessory protein MRAP on MC2 and MC5 receptor dimerization and trafficking. J Biol Chem. 2009;284:22641–22648. doi: 10.1074/jbc.M109.022400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Webb TR, Chan L, Cooray SN, Cheetham ME, Chapple JP, Clark AJ. Distinct melanocortin 2 receptor accessory protein domains are required for melanocortin 2 receptor interaction and promotion of receptor trafficking. Endocrinology. 2009;150:720–726. doi: 10.1210/en.2008-0941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cooray SN, Almiro Do Vale I, Leung KY, Webb TR, Chapple JP, Egertova M, Cheetham ME, Elphick MR, Clark AJ. The melanocortin 2 receptor accessory protein exists as a homodimer and is essential for the function of the melanocortin 2 receptor in the mouse Y1 cell line. Endocrinology. 2008;149:1935–1941. doi: 10.1210/en.2007-1463. [DOI] [PubMed] [Google Scholar]
- 23.Chan LF, Webb TR, Chung TT, Meimaridou E, Cooray SN, Guasti L, Chapple JP, Egertova M, Elphick MR, Cheetham ME, Metherell LA, Clark AJ. MRAP and MRAP2 are bidirectional regulators of the melanocortin receptor family. Proc Natl Acad Sci USA. 2009;106:6146–6151. doi: 10.1073/pnas.0809918106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chepurny OG, Holz GG. A novel cyclic adenosine monophosphate–responsive luciferase reporter incorporating a nonpalindromic cyclic adenosine monophosphate response element provides optimal performance for use in G protein–coupled receptor drug discovery efforts. J Biomol Screen. 2007;12:740–746. doi: 10.1177/1087057107301856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu A, Choi KL, Wang Y, Permana PA, Xu LY, Bogardus C, Cooper GJ. Identification of novel putative membrane proteins selectively expressed during adipose conversion of 3T3-L1 cells. Biochem Biophys Res Commun. 2002;293:1161–1167. doi: 10.1016/S0006-291X(02)00354-6. [DOI] [PubMed] [Google Scholar]
- 26.Le T, Schimmer BP. The regulation of MAPKs in Y1 mouse adrenocortical tumor cells. Endocrinology. 2001;142:4282–4287. doi: 10.1210/endo.142.10.8441. [DOI] [PubMed] [Google Scholar]
- 27.Hu CD, Chinenov Y, Kerppola TK. Visualization of interactions among bZIP and Rel family proteins in living cells using bimolecular fluorescence complementation. Mol Cell. 2002;9:789–798. doi: 10.1016/s1097-2765(02)00496-3. [DOI] [PubMed] [Google Scholar]
- 28.Mountjoy KG, Bird IM, Rainey WE, Cone RD. ACTH induces up-regulation of ACTH receptor mRNA in mouse and human adrenocortical cell lines. Mol Cell Endocrinol. 1994;99:R17–R20. doi: 10.1016/0303-7207(94)90160-0. [DOI] [PubMed] [Google Scholar]
- 29.Schimmer BP, Cordova M, Cheng H, Tsao A, Goryachev AB, Schimmer AD, Morris Q. Global profiles of gene expression induced by adrenocorticotropin in Y1 mouse adrenal cells. Endocrinology. 2006;147:2357–2367. doi: 10.1210/en.2005-1526. [DOI] [PubMed] [Google Scholar]
- 30.Roy S, Perron B, Gallo-Payet N. Role of asparagine-linked glycosylation in cell surface expression and function of the human adrenocorticotropin receptor (melanocortin 2 receptor) in 293/FRT cells. Endocrinology. 2010;151:660–670. doi: 10.1210/en.2009-0826. [DOI] [PubMed] [Google Scholar]
- 31.Fridmanis D, Petrovska R, Kalnina I, Slaidina M, Peculis R, Schiöth HB, Klovins J. Identification of domains responsible for specific membrane transport and ligand specificity of the ACTH receptor (MC2R) Mol Cell Endocrinol. doi: 10.1016/j.mce.2010.02.032. in press. [DOI] [PubMed] [Google Scholar]
- 32.Roy S, Rached M, Gallo-Payet N. Differential regulation of the human adrenocorticotropin receptor [melanocortin-2 receptor (MC2R)] by human MC2R accessory protein isoforms α and β in isogenic human embryonic kidney 293 cells. Mol Endocrinol. 2007;21:1656–1669. doi: 10.1210/me.2007-0041. [DOI] [PubMed] [Google Scholar]
- 33.Srinivasan S, Lubrano-Berthelier C, Govaerts C, Picard F, Santiago P, Conklin BR, Vaisse C. Constitutive activity of the melanocortin-4 receptor is maintained by its N-terminal domain and plays a role in energy homeostasis in humans. J Clin Invest. 2004;114:1158–1164. doi: 10.1172/JCI21927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mervine SM, Yost EA, Sabo JL, Hynes TR, Berlot CH. Analysis of G protein βγ dimer formation in live cells using multicolor bimolecular fluorescence complementation demonstrates preferences of β1 for particular γ subunits. Mol Pharmacol. 2006;70:194–205. doi: 10.1124/mol.106.022616. [DOI] [PubMed] [Google Scholar]
- 35.Jones BW, Hinkle PM. β-Arrestin mediates desensitization and internalization but does not affect dephosphorylation of the thyrotropin-releasing hormone receptor. J Biol Chem. 2005;280:38346–38354. doi: 10.1074/jbc.M502918200. [DOI] [PubMed] [Google Scholar]
- 36.Jones BW, Song GJ, Greuber EK, Hinkle PM. Phosphorylation of the endogenous thyrotropin-releasing hormone receptor in pituitary GH3 cells and pituitary tissue revealed by phosphosite-specific antibodies. J Biol Chem. 2007;282:12893–12906. doi: 10.1074/jbc.M610854200. [DOI] [PubMed] [Google Scholar]
- 37.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]