

Abstract
Excitotoxicity is considered to be an important mechanism involved in various neurodegenerative diseases in the central nervous system (CNS) such as Alzheimer's disease (AD). However, the mechanism by which excitotoxicity is implicated in neurodegenerative disorders remains unclear. Kainic acid (KA) is an epileptogenic and neuroexcitotoxic agent by acting on specific kainate receptors (KARs) in the CNS. KA has been extensively used as a specific agonist for ionotrophic glutamate receptors (iGluRs), for example, KARs, to mimic glutamate excitotoxicity in neurodegenerative models as well as to distinguish other iGluRs such as α-amino-3-hydroxy-5-methylisoxazole-4-propionate receptors and N-methyl-D-aspartate receptors. Given the current knowledge of excitotoxicity in neurodegeneration, interventions targeted at modulating excitotoxicity are promising in terms of dealing with neurodegenerative disorders. This paper summarizes the up-to-date knowledge of neurodegenerative studies based on KA-induced animal model, with emphasis on its potentials and limitations.
1. Introduction
Excitotoxicity is involved in a variety of neurodegenerative disorders in the central nervous system (CNS) including Alzheimer's disease (AD), Parkinson disease (PD), amyotrophic lateral sclerosis (ALS), and multiple sclerosis (MS). In the past decades, great achievements have been obtained in elucidating excitatory signaling pathways, yet the mechanism by which glutamate receptors is implicated in neurodegenerative disorders remains unclear. Kainic acid (KA), an analog of excitotoxic glutamate, can elicit selective neuronal death in the brain of rodents, of which the pathological changes partially mimic neurodegeneration in the CNS. Thus, KA-induced neurodegeneration in rodents has been used as a model for exploring the pathogenesis of excitotoxicity in neurodegenerative disorders.
2. Generic Characteristics of KA and KA Receptors (KARs)
KA was originally isolated from the seaweed called “Kainin-sou” or “Makuri”. The molecular formula of KA are C10H15NO4 and the molar mass of KA is 213.23 (Figure 1). Initially, KA was used as an antihelminth compound to remove worms from the gut. Subsequent studies indicated that KA is a nondegradable analog of glutamate and a potent neurotoxin [1]. KA exerts its neuroexcitotoxic and epileptogenic properties by acting on kainate receptors (KARs). Upon binding to KARs, KA induces a number of cellular events, including the influx of cellular Ca2+, production of reactive oxygen species (ROS), and mitochondrial dysfunction leading to neuronal apoptosis and necrosis [2]. KA has been extensively utilized as a specific agonist for ionotropic glutamate receptors (iGluRs); that is, KARs to mimic the effect of glutamate in neurodegenerative models, as well as to distinguish other ionotropic receptors for glutamate. KARs mediate most of kainate-induced seizures and excitotoxic neuronal death, whereby serving as a promising therapeutic target for neurodegeneration.
Figure 1.
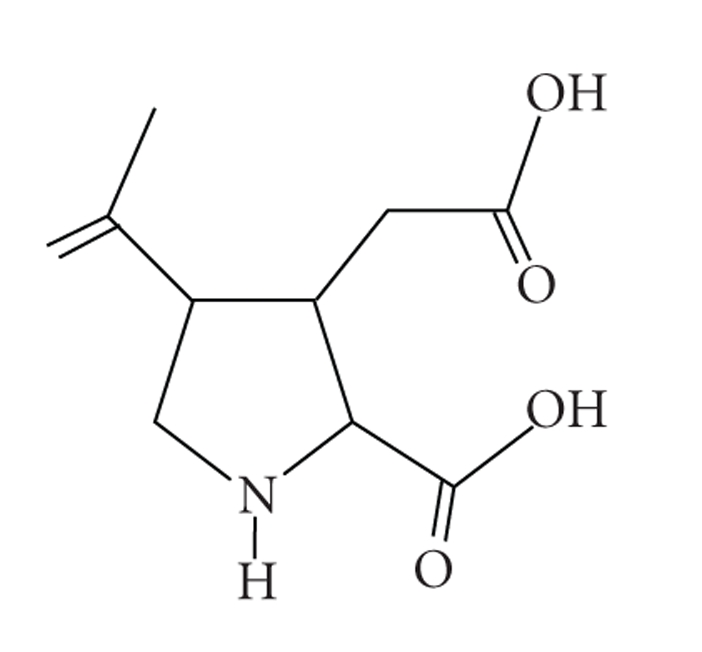
The molecular structure of kainic acid (2-carboxy-4-isopropenylpyrrolidin-3-ylacetic acid, KA). The molecular formula of KA are C10H15NO4 and the molar mass of KA is 213.23 g/mol.
Two broad categories of glutamate receptors (GluRs), that is, iGluRs and metabotropic GluRs (mGluRs), differ with reference to their functions. There are three major subtypes of iGluRs: N-methyl-D-aspartate receptor (NMDARs), α-amino-3-hydroxy-5-methylisoxazole-4-propionatereceptor (AMPARs), and KARs, termed according to the types of synthetic agonists [3]. The latter two subtypes are usually referred to together as non-NMDA receptors. The KARs family is divided into two subfamilies, including GluR5-7 and KAR1-2. A new nomenclature for iGluR family has been recently recommended by the International Union of Pharmacology Committee on Receptor Nomenclature and Drug Classification (NC-IUPHAR) [4]. In view of the popularity of the conventional nomenclature in previous studies, the present paper follows its conventional use.
3. Features of KA-Induced Neurodegenerative Animal Model
Systemic (intravenous or intraperitoneal), intranasal, or local administration of KA results in a series of clinical manifestations and pathological changes in rodents, including recurrent seizures, behavioral changes of rodents, oxidative stress including the generation of ROS and reactive nitrogen species (RNS), hippocampal neuronal death and glial activation, and so forth [2].
3.1. KA Induces Recurrent Seizures and Behavioral Changes in Rodents
In rodents, systemic administration of KA leads to a well-characterized seizure syndrome [5, 6]. During the first 20–30 minutes, the animals show “staring” spells, followed by head nodding. Then, they stand upright and fall down, with wet-dog shakes for around 30 minutes. One hour after KA administration, the animals start to present with recurrent limbic motor seizures. The limbic seizures then develop into status epilepticus and lasted 1-2 hours [7].
After KA administration, rodents show behavioral changes in the water maze, the object exploration tasks, and the open-field test in association with selective damage in the hippocampus. Deficiency in short-term spatial memory and long-term spatial learning, a higher degree of anxiety [8], and depression-like behaviors were also observed [9]. Physical exercise and selective cyclooxygenase (COX)-2 inhibitors like celecoxib may help to enhance the function recovery [10, 11].
3.2. Selective Hippocampal Neurodegeneration in Rodents after KA Challenge
Among the agents used to selectively destroy hippocampal neurons, ibotenic acid (IBO) and N-methyl-D-aspartate (NMDA) are nonspecific in acting on different cell types and usually these toxins do not cause seizures. In contrast, focal injection of KA resulted in hippocampal damage that occurred primarily in the CA3 area [12]. Similar selective vulnerability to KA was also seen when KA is administered via systemic injection. Selective vulnerability of hippocampal neurons was related to the distribution of the AMPARs/KARs in the brain [13]. KARs are most abundant in CA3 region and the activation of KARs can induce the production of ROS and compromise the function of mitochondria in the region [14, 15]. The stimulation of KARs excites CA3 neurons directly and increases glutamate efflux [16, 17]. Systemic injection of KA produced extensive neuronal death, primarily within the hippocampus hilus, CA3, and CA1 areas [18]. The hilar neurons are sensitive to KA-induced excitotoxicity, but neuronal loss in the other hippocampal areas varies between animal species and strains [19, 20]. KA at higher doses can also induce neurotoxicity in the medial amygdaloid nuclei [21].
KA-caused neuronal death involves endoplasmic reticulum (ER) stress, activation of ER sensor proteins, and induction of the fragmentation of the ER membrane [22], as well as Ca2+ overload [2]. C-jun N-terminal kinase (JNK)3 plays a detrimental role whereas other MAPKs like p38 and extracellular signal-regulated kinases 1/2 (ERK 1/2) seem to play a beneficial role [23]. Ikappa B kinase (IκB)/nuclear factor-kappa B- (NFκB-) dependent microglia activation contributes to KA-induced hippocampal neuronal death in vivo through induction of inflammatory mediators like tumor necrosis factor (TNF)-α and interleukin (IL)-1β [24].
3.3. Glial Activation after KA Treatment
KA binds to KARs expressed on neurons and microglia, leading to (1) rapid Ca2+ influx, (2) activation of Ca2+-dependent enzymes and generation of ROS and RNS, (3) excessive Ca2+, ROS and RNS lead to mitochondrial dysfunction, and (4) nuclear condensation and DNA fragmentation. Alternatively, intense Ca2+ overload can directly cause mitochondrial swelling and damage, causing acute neuronal cell death [2]. KA-induced neuronal death is accompanied by the activation of gliocytes, that is, microglia and astrocytes, characterized by the clustering of activated gliocytes in the hippocampal lesions [25, 26].
Microglia are the main effector cells of the inflammatory responses in the CNS, exert their functions as phagocytes, and interact with other gliocytes and neurons [27]. The physiological role of microglia can be partly linked to neuroprotection whereas under pathophysiological conditions, microglia may become activated and secrete a great amount of proinflammatory cytokines, chemokines, complements, and so forth [28]. Activated microglia may play a neuroprotective role in MS and its animal model, experimental autoimmune encephalomyelitis (EAE), by facilitating reparatory and regenerative processes [29]. However, in other neurodegenerative diseases such as AD and PD, microglia may initiate and aggravate the disease process through secreting proinflammatory and cytotoxic factors [30, 31]. In KA-induced excitotoxic neurodegeneration, activated microglia express MHC class I, MHC class II, and costimulatory molecules, produce complements, cytokines (IL-1, IL-6, IL-12, IL-18, TNF-α, etc.), chemokines, ROS, and RNS, and secrete proteases and excitatory amino acids, which contribute to the neuronal death [27, 28, 32] (Figure 2). Stanniocalcin 2 (STC2), the paralog of STC1, exerts neuroprotective actions against excitotoxic insults of KA through direct inhibition of microglial activation [33]. Despite the important role of microglia in KA-induced neurodegeneration, evidence is lacking that microglia participates in initiating the inflammatory and degenerative process. Seemingly, activation of microglia is just a “byproduct” of KA-induced excitotoxicity, but serves as an “accomplice” in this process [34].
Figure 2.

KA-induced microglial activation. Activated microglia express MHC class I and II, costimulatory molecules (CD80 and CD86), chemokine receptors (CCR2, 3, 5, CXCR3, 4, etc.), cytokine receptors (IL-10R, IL-12R, IL-18R, IFNgR, TNFR, TGFβR, etc.), complement receptors (FCγRI-III and CR1, 3, 4, etc.), and prostaglandin receptors, produce complements, prostaglandins, cytokines (IL-1, IL-6, IL-10, IL-12, IL-18, TNF-α, TGF-β, etc.), chemokines (IP-10, MIP-1α, MIP-1β, MCP-1, IL-8, RANTES, etc.), ROS, and RNS, and secrete neutrophins (NGF, BDNF, and NT-3, 4, etc.), proteases, and excitatory amino acids, and so forth, which may either contribute to excitotoxic damage or be protective against KA damage. IL: interleukin; CCR: C-C chemokine receptor; CXCR: CXC chemokine receptor; IFNgR: interferon gamma receptor; TNF: tumor necrosis factor; TGF: transforming growth factor; IP: Interferon gamma-inducible protein; MIP: macrophage inflammatory protein; MCP: monocyte chemotactic protein; RANTES: regulated upon activation, normal T-cell expressed and presumably secreted; NGF: nerve growth factor; BDNF: brain-derived neurotrophic factor; NT: neurotrophin.
Astrocytes were formerly regarded as passive supporters of neurons for decades. However, a new concept of neuron-glial intercommunication that astrocytes play a dynamic role by integrating neuronal inputs and modulating synaptic activity, whereby contributing to neurodegenerative development, has been recently acknowledged [35, 36]. Astrocytes can prevent neurons from death and promote proliferation and differentiation of precursor cells by producing growth factors [37–39]. Moreover, astrocytes express functional receptors for the excitatory neurotransmitters like iGluRs [40]. In pathological conditions, astrocytes may contribute to brain damage by releasing ROS and proinflammatory factors. Failure in water transport also triggers brain edema, reversal of neurotransmitter transporters, and Ca2+-dependent exocytosis. Furthermore, the opening of high-permeability plasmalemmal channels contributes to glutamate excitotoxicity [41]. The expression of glial fibrillary acidic protein (GFAP), a marker for astrogliosis, was shown to steadily increase as early as one day after intrahippocampal or intraperitoneal injection of KA [42, 43]. Aged astrocyte specifically induced substance (OASIS) is known to be expressed on astrocytes and involved in the ER stress [44], which plays an important role in attenuating neuronal damage induced by KA [45]. Upregulated transcription factors in astrocytes, including NF-κB and nuclear factor erythroid-2-related factor 2 (Nrf2) induce the expression of neuroprotective molecules [46, 47].
3.4. KA-Mediated Oxidative Stress
KA receptors have both presynaptic modulatory and postsynaptic excitatory actions [48, 49]. The activation of KA receptors leads to membrane depolarization and increased influx of cellular calcium, which are required to trigger the cascade of neuronal death [50]. ROS/RNS have been implicated in the pathogenesis of various neurodegenerative disorders [51]. KA administration increases the generation of ROS and RNS. The increased formation of thiobarbituric acid-reactive substances (TBARS) and decreased total antioxidant status (TAS) in the cerebral cortex, cerebellum, and brain stem of rats subjected to KA indicate the presence of oxidative stress in excitotoxicity [52]. The exact mechanisms regarding the increased production of nitric oxide (NO) in excitotoxicity are not well established. However, stimulation of KARs induces NO release, which, in turn, modulates glutamate transmission [53]. NO may induce changes in neuronal signaling pathways [54]. Generation of other free radicals also contributes to neuronal damage [55]. KA induces immediate production of COX-2 that is involved in hippocampal neuronal death [56]. COX-2 facilitates the recurrence of hippocampal seizures and stimulates hippocampal neuronal loss after KA administration [57, 58]. PGE(2), the synthesis of which is dependent on COX, is pathologically increased in the brain after KA treatment, in close association with neuronal death [59]. In addition, seizure-induced products of lipid peroxidation, such as F(2)-isoprostanes and isofurans, play critical roles in the initiation and modulation of inflammation and oxidative stress, serving as reliable indices of oxidative stress in vivo [60].
3.5. KA-Enhanced Inflammatory Mediator Production
Neurodegeneration is associated with increased inflammatory mediators in the CNS such as in AD, MS, and so forth [61, 62]. Activated microglia and astrocytes after KA treatment release a large amount of inflammatory mediators such as NO, IL-6, IL-12, TGF-β, TNF-α, IL-18, and IL-1β [28, 63, 64]. Manipulation of inflammatory mediators may affect the outcome with regard to seizure activity, behavioral changes, as well as the neuropathological consequences in KA-induced neurodegeneration [65]. Important inflammatory mediators including NO, IL-6, TGF-β, TNF-α, IL-12, IL-18, and IL-1β are herein summarized.
The production of NO represents one of the principle features of activated macrophage/microglia, and NO is a major effector in the innate immunity [66]. NO can be formed enzymatically from L-arginine by inducible NO synthase (iNOS) in neuroglia [67]. KA administration increases the generation of ROS and RNS by neuroglia. Microglia can produce large amounts of soluable factors like NO [68]. Elevated production of NO by increased activity of iNOS is thought to contribute to KA-induced neuronal damage [69]. iNOS-deficient mice are resistant to KA-induced neuronal death [70]. Similarly, pretreatment with aminoguanidine, a selective iNOS inhibitor, significantly suppressed KA-induced neuronal death in the hippocampal CA3 area with concomitant decrease in iNOS expression and microglial activation [70].
IL-6, which is secreted by macrophages, dendritic cells, T cells, and so forth, bears both pro- and anti-inflammatory functions. IL-6 was initially categorized into a T helper (Th) 1 cytokine [71]. However, IL-6 can induce IL-4 production by naïve Th0 cells and their differentiation into effector Th2 cells [72]. IL-6 appears to be a critical factor in early phases of CNS insults, taking part in the orchestration of attempts for tissue repair [69]. Levels of IL-6 are increased in the cerebrospinal fluid (CSF) in humans after tonic-clonic seizures [73]. IL-6 mRNA is increased in the hippocampus, cortex, amygdale, and meninges, and IL-6 receptor is upregulated in the hippocampus in the rat brain after KA-induced status epilepticus [73]. Neuronal death is more pronounced when IL-6 is produced in limbic seizures induced by KA [26]. Upregulated expression of IL-6 represented an endogenous neuroprotective mechanism against NMDAR-mediated injury in cerebral ischemia [74]. IL-6 knockout mice exhibit significantly higher seizure susceptibility to NMDA, AMPA, and KA, and the excitatory amino acid system seems more active in the CNS of IL-6 deficient mice [75]. Specifically, IL-6 deficiency increases neuronal injury and impaired the inflammatory response after KA treatment, characterized by reduced reactive astrogliosis and microgliosis versus increased morphological hippocampal damage, oxidative stress, iNOS expression, and apoptotic neuronal death [76].
IL-12 consists of p35 and p40 subunits, which form the bioactive IL-12p70 when combined together. IL-12 is required for the induction of IFN-γ production, critical for the polarization of the Th1 immune response. In EAE, the proinflammatory cytokines IL-12, IFN-γ, TNF-α, IL-1β, IL-6, and IL-18 are involved in the initiation and development of the local immune responses in the CNS [77]. IL-12 can be produced by activated microglia in response to KA, and IL-12p35 deficiency may alleviate hippocampal injury upon KA challenge, indicating a critical role of IL-12 in excitotoxin-induced brain injury [65]. Moreover, expression IL-12 was increased in IL-18 deficient mice as compared with controls in association with increased microglial activation, suggesting that the null role of IL-18 in excitotoxic injury may be compensated by the upregulated levels of IL-12 [78].
TGF-β bears anti-inflammatory, immunosuppressive, and neuroprotective features. It can downregulate (micro)glial cytokine production, for example, IL-1 and TNF-α [69]. TGF-β1 mRNA in the regenerating neurons is correlated with glial activation [79]. Exogenous administration of TGF-β1 protects excitotoxic lesions and neuronal death [80]. TGF-β1 mRNA is also increased in the hippocampus after KA-induced seizures, particularly in the areas of neurodegeneration [81]. TGF-β3 is upregulated in response to KA, and exogenous TGF-β3 significantly attenuates KA-induced seizures and neuronal loss in a dose-dependent manner, indicating a protective role of TGF-β3 [82].
TNF-α is mainly produced by microglia and astrocytes in the CNS. KA-activated microglia expressed high levels of TNF-α mRNA and protein. As with many other cytokines, TNF-α bears neuroprotective properties in contrast to its well-known deleterious role as a proinflammatory cytokine, which implies an intricate biological balance in immune and inflammatory responses mediated by TNF-α [83]. The mechanisms by which TNF reduces brain injury may involve the upregulation of proteins, such as neuronal apoptosis inhibitor protein (NAIP), which maintains calcium homeostasis or reduces free radical generation [84]. The protective roles of TNF-α in KA-induced neurodegeneration can also be mediated via TNFR2 signaling pathway [85]. However, inconsistent findings suggested that TNF-α derived from KA-activated microglia can increase the excitotoxicity of hippocampal neurons and can induce neuronal apoptosis in vitro and in vivo [86].
IL-18 is a proinflammatory cytokine which can enhance postsynaptic AMPAR responses in CA1 pyramidal neurons via the release of glutamate, thereby facilitating basal hippocampal synaptic transmissions [87]. IL-18 knockout mice showed a diminished microglial activation and reduced dopaminergic neuronal loss after acute 1-methy-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) treatment [88]. In KA-induced excitotoxicity, IL-18 immunoreactivity was found in the hippocampal microglia of mice whereas IL-18R immunoreactivity was observed in astrocytes. Levels of IL-18 and IL-18R in the hippocampus were upregulated progressively from day 1 and reached the peak on day 3 post-KA treatment [89]. We found that exogenous administration of IL-18 aggravated KA-induced injuries in C57BL/6 mice, and IL-12 could compensate the function of IL-18 and worsen seizure severity as well as hippocampal neurodegeneration in IL-18 deficient mice [78].
IL-1β, a proinflammatory cytokine produced in abundance by activated microglia, acts as a regulatory factor in the multiple connections among the immune, nervous, and endocrine systems. IL-1β exerts its effects through IL-1 receptor type I (IL-1RI) and IL-1 receptor accessory protein (IL-1RAcP) [90]. IL-1β plays a pivotal role in the neuroinflammation associated with certain forms of neurodegeneration, including cerebral ischemia and excitotoxic brain injury [91]. Compelling evidence in experimental neurodegenerative models indicates that IL-1β is pivotal in the excitotoxicity leading to neuronal death [92]. Primary cultured microglia are significantly activated by KA with increased IL-1β levels [93]. IL-1β can mediate KA-induced excitotoxic injuries to hippocampal neurons in vitro and in vivo [93].
4. Neurodegenerative Animal Model Induced by Intranasal Administration of KA: Our Experience
C57BL/6 mice are commonly used for transgenic studies, while they are resistant to systemic administration of KA (intravenous or intraperitoneal). However, Chen et al. in our group, developed a model of KA-induced neurodegeneration by intranasal administration of KA into C57BL/6 mice [94]. This model provides a valuable tool to explore the role of excitotoxicity in neurodegeneration using transgenic mice with C57BL/6 background. Within 15 minutes after KA administration, C57BL/6 mice are catatonic and staring, followed by myoclonic twitching, frequent rearing, and repeated falling. Approximately 30–40 minutes after administration, the mice present with status epilepticus, which lasts 1–5 hours. Some serious cases die from seizures. Then the mice assume a hunched posture and remain immobile for additional few hours [78]. As to behavioral changes, intranasal administration of KA induced elevated levels of spontaneous activity in the open-field test [94].
The mechanism as regards how KA induces excitotoxic neurodegeneration through intranasal administration is underclarified whereas we have several speculations. Firstly, KA may reach the hippocampus via passive diffusion, a route utilized extensively in both clinical practice and laboratory investigations [95]. Secondly, KARs were found in olfactory nerves and the olfactory bulb [96]. Protracted postsynaptic stimulation induced by KA can be relayed through projections to the hippocampus and thus result in neuronal damage. Thirdly, KA may directly enter the CSF via the nasal cavity, since the CSF has direct connection with the olfactory bulb [97]. Additionally, drugs with a molecular weight of less than 20 kDa can be directly transported from the nasal cavity to the CSF [98]. Finally, KA may reach the hippocampus via the blood circulation [99].
5. Potentials of Antiglutamate Excitotoxicity as a Therapeutic Target
Considering the importance of oxidative stress in KA-induced excitotoxic damage, antioxidant and anti-inflammatory treatments may attenuate or prevent the neurodegenerative disorders involved glutamate excitotoxicity [100].
COX-2 inhibitors as a therapeutic drug for the neuronal loss after KA treatment have been studied [57]. Selective COX-2 inhibitors, for example, celecoxib, rofecoxib, NS398, and SC58125, can suppress the synthesis of PGE(2), whereby blocking hippocampal neuronal death [10, 58]. Free radical scavengers (FRSs) are known to prevent neuronal loss induced by excitotoxins [101, 102]. Edaravone (Ed), a newly developed FRS [103], and melatonin, the pineal secretory product [104], were potent to attenuate KA-induced neuronal death, suppress lipid peroxidation, and inhibit microglial activation. Sinapic acid (SA), a GABA (A) receptor agonist, can attenuate KA-induced hippocampal damage in mice via its anticonvulsive activity through GABA (A) receptor activation and radical scavenging activity [102]. Tetramethylpyrazine (TMP), a principal ingredient of Ligusticum wallichi Franchat (a Chinese herb), can function as a reductant/antioxidant to quench ROS, block lipid peroxidation, and protect enzymatic antioxidants such as glutathione peroxidase and glutathione reductase [105]. TMP may prevent and rescue KA-induced neuronal loss via the preservation of the structural and functional integrity of mitochondria, evidenced by maintaining the mitochondrial membrane potential, ATP production, and complex I and III activities [106].
Preclinical data have suggested KARs as an attractive target for drug development. Tezampanel, a nonselective KAR inhibitor, can reduce migraine pain and other symptoms in a clinical phase II study. This suppressive effect is probably attributable to the inhibition of KARs. Another KAR antagonist, NS1209, can alleviate refractory status epilepticus and neuropathic pain in phase II trials, but further research on this agent has been suspended [107]. Tat-GluR6-9c, a GluR6 C-terminus peptide conjugated to a Tat peptide, confers neuroprotection against neuronal death induced by KA-induced excitotoxic brain injury without perturbing KAR-mediated currents. This is mediated by its inhibition of JNK activation [15, 108, 109].
Expression of p27, a cyclin-dependent kinase inhibitor which arrests cell cycle at G1-S phase, was downregulated in the neuronal cell death caused by several reasons like glutamate toxicity [110]. It is demonstrated that reduced expression of endogenous p27 induced cell death in cultured cortical neurons by transfection of p27 small interfering RNA (siRNA) [111]. p27 may alleviate KA-induced seizure and hippocampal degeneration [112]. Early calpain activation is involved in neurotoxicity in the hippocampus after KA insults. Treatment with the calpain inhibitor MDL 28170 significantly suppressed the neurodegeneration induced by KA [113]. The roles of the activation of caspases and p21 in the process of neuronal death caused by KA insult have been reported in several studies. However, it is still unclear whether caspases and p21 are involved in KA-induced neurodegeneration, as evidence by negative and conflicting findings [114, 115].
5.1. Limitations of KA-Induced Neurodegenerative Model
Notwithstanding the advantages of KA-induced model in neurodegenerative studies, potential limitations should be recognized. KA-induced neurodegeneration in animals is an acute monophasic disorder, which differs from chronic neurodegenerative disorders in clinic. An alternative way to induce the chronic model, that is, long-term administration of KA at a lower dose, cannot mimic the pathogenesis of neurodegeneration adequately. This is evidenced by the fact that monosodium glutamate, a frequently added flavor enhancer, does not increase the incidence of neurodegenerative disorders. Despite a widespread belief that glutamate may elicit Chinese Restaurant Syndrome (CRS), migraine headache, and asthma, literature reviewing does not suggest that individuals may be uniquely sensitive to glutamate [116]. This might be partially explained by the dose-dependent effects of excitatory amino acids in the CNS. The implications of excitatory amino acid transmitters are complex. It may only represents one of several pathways involved in pathological cell death, but not necessarily the predominant or decisive one. Interventions targeted at glutamate excitotoxicity are promising, yet to be with caution, because iGluRs represent one of the major excitatory neurotransmitter pathways in the physiological functions of the brain, and the blockade of iGluRs might be disastrous to the CNS functions.
Moreover, mouse strains vary significantly in their sensitivity to KA-induced neurodegeneration [117]. In general, the C57BL/6, C57BL/10, and F1 C57BL/6∗CBA/J strains of mice are resistant to systemic administration of KA, while the FVB/N, ICR and DBA/2 J strains are vulnerable [19]. Since intranasal administration can break the tolerance to KA in C57BL/6 mice, as demonstrated by Chen et al. [94], administration routes should be carefully selected when resistant strains are used. Thus, pilot studies are always indispensable; otherwise, the results might be confounded by the resistance.
Despite the theoretical or preclinical significance of KARs, concerns regarding the clinical prospects of drugs targeting AMPARs and KARs prevail. This is due to the fact that interference with excitotoxicity is accompanied unduly with effects on synaptic transmission. The recent discovery of proteins that anchor and interact with iGluRs promises a new strategy to cytoprotective therapy without interfering with synaptic transmission. The reason why iGluRs interactors appear more suitable as therapeutic targets than the receptors per se is that they might interfere specifically with excitotoxic pathways while leaving synaptic transmission untouched [118]. Also, the relationship between the structure and physiological functions of KARs is difficult to study in the CNS because of the large number of subunits of iGluRs, their widespread distribution, and neuronal heterogeneity [119, 120].
6. Concluding Remarks
Excitotoxicity is considered to be a major mechanism involved in cell death of various CNS diseases including neurodegenerative disorders. In the past decades, great progress has been made in elucidating excitatory signaling pathways whereas the mechanism by which excitatory amino acids and their receptors are implicated in neurodegenerative diseases remains little known. KA-mediated excitotoxicity has long been used as a model for elucidating mechanisms underlying oxidative stress and inflammation in neurodegenerative disorders. However, the routes of KA administration should be taken into account, especially for those strains resistant to conventional ways of administration.
Acknowledgments
This work was supported by grants from the China Scholarship Council, Stiftelsen för Gamla Tjänarinnor, Swedish Medicine Association, Gun och Bertil Stohnes foundation and Swedish National Board of Health and Welfare. X.-Y. Zheng and H.-L. Zhang contributed equally to this work.
References
- 1.Vincent P, Mulle C. Kainate receptors in epilepsy and excitotoxicity. Neuroscience. 2009;158(1):309–323. doi: 10.1016/j.neuroscience.2008.02.066. [DOI] [PubMed] [Google Scholar]
- 2.Wang Q, Yu S, Simonyi A, Sun GY, Sun AY. Kainic acid-mediated excitotoxicity as a model for neurodegeneration. Molecular Neurobiology. 2005;31(1–3):3–16. doi: 10.1385/MN:31:1-3:003. [DOI] [PubMed] [Google Scholar]
- 3.Kew JNC, Kemp JA. Ionotropic and metabotropic glutamate receptor structure and pharmacology. Psychopharmacology. 2005;179(1):4–29. doi: 10.1007/s00213-005-2200-z. [DOI] [PubMed] [Google Scholar]
- 4.Collingridge GL, Olsen RW, Peters J, Spedding M. A nomenclature for ligand-gated ion channels. Neuropharmacology. 2009;56(1):2–5. doi: 10.1016/j.neuropharm.2008.06.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ben-Ari Y. Limbic seizure and brain damage produced by kainic acid: mechanisms and relevance to human temporal lobe epilepsy. Neuroscience. 1985;14(2):375–403. doi: 10.1016/0306-4522(85)90299-4. [DOI] [PubMed] [Google Scholar]
- 6.Mulle C, Sailer A, Pérez-Otaño I, et al. Altered synaptic physiology and reduced susceptibility to kainate- induced secures in GluR6-deficient mice. Nature. 1998;392(6676):601–605. doi: 10.1038/33408. [DOI] [PubMed] [Google Scholar]
- 7.Chuang Y-C, Chang AYW, Lin J-W, Hsu S-P, Chan SHH. Mitochondrial dysfunction and ultrastructural damage in the hippocampus during kainic acid-induced status epilepticus in the rat. Epilepsia. 2004;45(10):1202–1209. doi: 10.1111/j.0013-9580.2004.18204.x. [DOI] [PubMed] [Google Scholar]
- 8.Ratté S, Lacaille JC. Selective degeneration and synaptic reorganization of hippocampal interneurons in a chronic model of temporal lobe epilepsy. Advances in Neurology. 2006;97:69–76. [PubMed] [Google Scholar]
- 9.Gröticke I, Hoffmann K, Löscher W. Behavioral alterations in a mouse model of temporal lobe epilepsy induced by intrahippocampal injection of kainate. Experimental Neurology. 2008;213(1):71–83. doi: 10.1016/j.expneurol.2008.04.036. [DOI] [PubMed] [Google Scholar]
- 10.Gobbo OL, O’Mara SM. Post-treatment, but not pre-treatment, with the selective cyclooxygenase-2 inhibitor celecoxib markedly enhances functional recovery from kainic acid-induced neurodegeneration. Neuroscience. 2004;125(2):317–327. doi: 10.1016/j.neuroscience.2004.01.045. [DOI] [PubMed] [Google Scholar]
- 11.Gobbo OL, O'Mara SM. Exercise, but not environmental enrichment, improves learning after kainic acid-induced hippocampal neurodegeneration in association with an increase in brain-derived neurotrophic factor. Behavioural Brain Research. 2005;159(1):21–26. doi: 10.1016/j.bbr.2004.09.021. [DOI] [PubMed] [Google Scholar]
- 12.Jarrard LE. Use of excitotoxins to lesion the hippocampus: update. Hippocampus. 2002;12(3):405–414. doi: 10.1002/hipo.10054. [DOI] [PubMed] [Google Scholar]
- 13.Bloss EB, Hunter RG. Hippocampal kainate receptors. Vitamins and Hormones. 2010;82:167–184. doi: 10.1016/S0083-6729(10)82009-6. [DOI] [PubMed] [Google Scholar]
- 14.Carriedo SG, Sensi SL, Yin HZ, Weiss JH. AMPA exposures induce mitochondrial Ca2+ overload and ROS generation in spinal motor neurons in vitro. Journal of Neuroscience. 2000;20(1):240–250. doi: 10.1523/JNEUROSCI.20-01-00240.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lauri SE, Bortolotto ZA, Bleakman D, et al. A critical role of a facilitatory presynaptic kainate receptor in mossy fiber LTP. Neuron. 2001;32(4):697–709. doi: 10.1016/s0896-6273(01)00511-6. [DOI] [PubMed] [Google Scholar]
- 16.Contractor A, Sailer AW, Darstein M, et al. Loss of kainate receptor-mediated heterosynaptic facilitation of mossy-fiber synapses in KA2-/- mice. Journal of Neuroscience. 2003;23(2):422–429. doi: 10.1523/JNEUROSCI.23-02-00422.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rodríguez-Moreno A, Sihra TS. Presynaptic kainate receptor facilitation of glutamate release involves protein kinase A in the rat hippocampus. Journal of Physiology. 2004;557(3):733–745. doi: 10.1113/jphysiol.2004.065029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee HN, Jeon GS, Kim DW, Cho I-H, Cho SS. Expression of adenomatous polyposis coli protein in reactive astrocytes in hippocampus of kainic acid-induced rat. Neurochemical Research. 2009;35(1):114–121. 1–8. doi: 10.1007/s11064-009-0036-3. [DOI] [PubMed] [Google Scholar]
- 19.McLin JP, Steward O. Comparison of seizure phenotype and neurodegeneration induced by systemic kainic acid in inbred, outbred, and hybrid mouse strains. European Journal of Neuroscience. 2006;24(8):2191–2202. doi: 10.1111/j.1460-9568.2006.05111.x. [DOI] [PubMed] [Google Scholar]
- 20.Kasugai M, Akaike K, Imamura S-I, et al. Differences in two mice strains on kainic acid-induced amygdalar seizures. Biochemical and Biophysical Research Communications. 2007;357(4):1078–1083. doi: 10.1016/j.bbrc.2007.04.067. [DOI] [PubMed] [Google Scholar]
- 21.Pereno GL, Beltramino CA. Differential role of gonadal hormones on kainic acid-induced neurodegeneration in medial amygdaloid nucleus of female and male rats. Neuroscience. 2009;163(3):952–963. doi: 10.1016/j.neuroscience.2009.06.059. [DOI] [PubMed] [Google Scholar]
- 22.Sokka A-L, Putkonen N, Mudo G, et al. Endoplasmic reticulum stress inhibition protects against excitotoxic neuronal injury in the rat brain. Journal of Neuroscience. 2007;27(4):901–908. doi: 10.1523/JNEUROSCI.4289-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.De Lemos L, Junyent F, Verdaguer E, et al. Differences in activation of ERK1/2 and p38 kinase in Jnk3 null mice following KA treatment. Journal of Neurochemistry. 2010;114(5):1315–1322. doi: 10.1111/j.1471-4159.2010.06853.x. [DOI] [PubMed] [Google Scholar]
- 24.Cho I-H, Hong J, Suh EC, et al. Role of microglial IKKβ in kainic acid-induced hippocampal neuronal cell death. Brain. 2008;131(11):3019–3033. doi: 10.1093/brain/awn230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen Z, Duan R-S, Quezada HC, et al. Increased microglial activation and astrogliosis after intranasal administration of kainic acid in C57BL/6 mice. Journal of Neurobiology. 2005;62(2):207–218. doi: 10.1002/neu.20099. [DOI] [PubMed] [Google Scholar]
- 26.Ravizza T, Rizzi M, Perego C, et al. Inflammatory response and glia activation in developing rat hippocampus after status epilepticus. Epilepsia. 2005;46(supplement 5):113–117. doi: 10.1111/j.1528-1167.2005.01006.x. [DOI] [PubMed] [Google Scholar]
- 27.Mack CL, Vanderlugt-Castaneda CL, Neville KL, Miller SD. Microglia are activated to become competent antigen presenting and effector cells in the inflammatory environment of the Theiler’s virus model of multiple sclerosis. Journal of Neuroimmunology. 2003;144(1-2):68–79. doi: 10.1016/j.jneuroim.2003.08.032. [DOI] [PubMed] [Google Scholar]
- 28.Kato H, Walz W. The initiation of the microglial response. Brain Pathology. 2000;10(1):137–143. doi: 10.1111/j.1750-3639.2000.tb00250.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Napoli I, Neumann H. Protective effects of microglia in multiple sclerosis. Experimental Neurology. 2009;225(1):24–28. doi: 10.1016/j.expneurol.2009.04.024. [DOI] [PubMed] [Google Scholar]
- 30.Marinova-Mutafchieva L, Sadeghian M, Broom L, Davis JB, Medhurst AD, Dexter DT. Relationship between microglial activation and dopaminergic neuronal loss in the substantia nigra: a time course study in a 6-hydroxydopamine model of Parkinson’s disease. Journal of Neurochemistry. 2009;110(3):966–975. doi: 10.1111/j.1471-4159.2009.06189.x. [DOI] [PubMed] [Google Scholar]
- 31.Venneti S, Wiley CA, Kofler J. Imaging microglial activation during neuroinflammation and Alzheimer’s disease. Journal of Neuroimmune Pharmacology. 2009;4(2):227–243. doi: 10.1007/s11481-008-9142-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Penkowa M, Molinero A, Carrasco J, Hidalgo J. Interleukin-6 deficiency reduces the brain inflammatory response and increases oxidative stress and neurodegeneration after kainic acid-induced seizures. Neuroscience. 2001;102(4):805–818. doi: 10.1016/s0306-4522(00)00515-7. [DOI] [PubMed] [Google Scholar]
- 33.Byun J-S, Lee J-W, Kim SY, et al. Neuroprotective effects of stanniocalcin 2 following kainic acid-induced hippocampal degeneration in ICR mice. Peptides. 2010;31(11):2094–2099. doi: 10.1016/j.peptides.2010.08.002. [DOI] [PubMed] [Google Scholar]
- 34.Giaume C, Kirchhoff F, Matute C, Reichenbach A, Verkhratsky A. Glia: the fulcrum of brain diseases. Cell Death and Differentiation. 2007;14(7):1324–1335. doi: 10.1038/sj.cdd.4402144. [DOI] [PubMed] [Google Scholar]
- 35.Gill SS, Hou Y, Ghane T, Pulido OM. Regional susceptibility to domoic acid in primary astrocyte cells cultured from the brain stem and hippocampus. Marine Drugs. 2008;6(1):25–38. [PMC free article] [PubMed] [Google Scholar]
- 36.Vesce S, Rossi D, Brambilla L, Volterra A. Glutamate release from astrocytes in physiological conditions and in neurodegenerative disorders characterized by neuroinflammation. International Review of Neurobiology. 2007;82:57–71. doi: 10.1016/S0074-7742(07)82003-4. [DOI] [PubMed] [Google Scholar]
- 37.Braun A, Dang J, Johann S, Beyer C, Kipp M. Selective regulation of growth factor expression in cultured cortical astrocytes by neuro-pathological toxins. Neurochemistry International. 2009;55(7):610–618. doi: 10.1016/j.neuint.2009.06.004. [DOI] [PubMed] [Google Scholar]
- 38.Dakubo GD, Beug ST, Mazerolle CJ, Thurig S, Wang Y, Wallace VA. Control of glial precursor cell development in the mouse optic nerve by sonic hedgehog from retinal ganglion cells. Brain Research. 2008;1228(C):27–42. doi: 10.1016/j.brainres.2008.06.058. [DOI] [PubMed] [Google Scholar]
- 39.Sandhu JK, Gardaneh M, Iwasiow R, et al. Astrocyte-secreted GDNF and glutathione antioxidant system protect neurons against 6OHDA cytotoxicity. Neurobiology of Disease. 2009;33(3):405–414. doi: 10.1016/j.nbd.2008.11.016. [DOI] [PubMed] [Google Scholar]
- 40.Brand-Schieber E, Werner P. AMPA/kainate receptors in mouse spinal cord cell-specific display of receptor subunits by oligodendrocytes and astrocytes and at the nodes of Ranvier. GLIA. 2003;42(1):12–24. doi: 10.1002/glia.10136. [DOI] [PubMed] [Google Scholar]
- 41.Heneka MT, Rodríguez JJ, Verkhratsky A. Neuroglia in neurodegeneration. Brain Research Reviews. 2010;63(1-2):189–211. doi: 10.1016/j.brainresrev.2009.11.004. [DOI] [PubMed] [Google Scholar]
- 42.Bendotti C, Guglielmetti F, Tortarolo M, Samanin R, Hirst WD. Differential expression of S100β and glial fibrillary acidic protein in the hippocampus after kainic acid-induced lesions and mossy fiber sprouting in adult rat. Experimental Neurology. 2000;161(1):317–329. doi: 10.1006/exnr.1999.7262. [DOI] [PubMed] [Google Scholar]
- 43.Ding M, Haglid KG, Hamberger A. Quantitative immunochemistry on neuronal loss, reactive gliosis and BBB damage in cortex/striatum and hippocampus/amygdala after systemic kainic acid administration. Neurochemistry International. 2000;36(4-5):313–318. doi: 10.1016/s0197-0186(99)00139-4. [DOI] [PubMed] [Google Scholar]
- 44.Saito A, Hino S-I, Murakami T, Kondo S, Imaizumi K. A novel ER stress transducer, OASIS, expressed in astrocytes. Antioxidants and Redox Signaling. 2007;9(5):563–571. doi: 10.1089/ars.2006.1520. [DOI] [PubMed] [Google Scholar]
- 45.Chihara K, Saito A, Murakami T, et al. Increased vulnerability of hippocampal pyramidal neurons to the toxicity of kainic acid in OASIS-deficient mice. Journal of Neurochemistry. 2009;110(3):956–965. doi: 10.1111/j.1471-4159.2009.06188.x. [DOI] [PubMed] [Google Scholar]
- 46.Vargas MR, Johnson DA, Sirkis DW, Messing A, Johnson JA. Nrf2 activation in astrocytes protects against neurodegeneration in mouse models of familial amyotrophic lateral sclerosis. Journal of Neuroscience. 2008;28(50):13574–13581. doi: 10.1523/JNEUROSCI.4099-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vargas MR, Johnson JA. The Nrf2-ARE cytoprotective pathway in astrocytes. Expert Reviews in Molecular Medicine. 2009;11, article no. e17 doi: 10.1017/S1462399409001094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Campbell SL, Mathew SS, Hablitz JJ. Pre- and postsynaptic effects of kainate on layer II/III pyramidal cells in rat neocortex. Neuropharmacology. 2007;53(1):37–47. doi: 10.1016/j.neuropharm.2007.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Youn D-H, Randić M. Modulation of excitatory synaptic transmission in the spinal substantia gelatinosa of mice deficient in the kainate receptor GluR5 and/or GluR6 subunit. Journal of Physiology. 2004;555(3):683–698. doi: 10.1113/jphysiol.2003.057570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brorson JR, Manzolillo PA, Miller RJ. Ca2+ entry via AMPA/KA receptors and excitotoxicity in cultured cerebellar Purkinje cells. Journal of Neuroscience. 1994;14(1):187–197. doi: 10.1523/JNEUROSCI.14-01-00187.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Frantseva MV, Perez Velazquez JL, Tsoraklidis G, et al. Oxidative stress is involved in seizure-induced neurodegeneration in the kindling model of epilepsy. Neuroscience. 2000;97(3):431–435. doi: 10.1016/s0306-4522(00)00041-5. [DOI] [PubMed] [Google Scholar]
- 52.Swamy M, Sirajudeen KNS, Chandran G. Nitric oxide (NO), citrullineNO cycle enzymes, glutamine synthetase, and oxidative status in kainic acid-mediated excitotoxicity in rat brain. Drug and Chemical Toxicology. 2009;32(4):326–331. doi: 10.1080/01480540903130641. [DOI] [PubMed] [Google Scholar]
- 53.Nakaki T, Mishima A, Suzuki E, Shintani F, Fujii T. Glufosinate ammonium stimulates nitric oxide production through N-methyl D-aspartate receptors in rat cerebellum. Neuroscience Letters. 2000;290(3):209–212. doi: 10.1016/s0304-3940(00)01363-x. [DOI] [PubMed] [Google Scholar]
- 54.Prast H, Philippu A. Nitric oxide as modulator of neuronal function. Progress in Neurobiology. 2001;64(1):51–68. doi: 10.1016/s0301-0082(00)00044-7. [DOI] [PubMed] [Google Scholar]
- 55.Ueda Y, Yokoyama H, Nakajima A, Tokumaru J, Doi T, Mitsuyama Y. Glutamate excess and free radical formation during and following kainic acid-induced status epilepticus. Experimental Brain Research. 2002;147(2):219–226. doi: 10.1007/s00221-002-1224-4. [DOI] [PubMed] [Google Scholar]
- 56.Kim EJ, Lee JE, Kwon KJ, Lee SH, Moon C-H, Baik EJ. Differential roles of cyclooxygenase isoforms after kainic acid-induced prostaglandin E2 production and neurodegeneration in cortical and hippocampal cell cultures. Brain Research. 2001;908(1):1–9. doi: 10.1016/s0006-8993(01)02432-5. [DOI] [PubMed] [Google Scholar]
- 57.Takemiya T, Matsumura K, Yamagata K. Roles of prostaglandin synthesis in excitotoxic brain diseases. Neurochemistry International. 2007;51(2–4):112–120. doi: 10.1016/j.neuint.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 58.Takemiya T, Maehara M, Matsumura K, Yasuda S, Sugiura H, Yamagata K. Prostaglandin E2 produced by late induced COX-2 stimulates hippocampal neuron loss after seizure in the CA3 region. Neuroscience Research. 2006;56(1):103–110. doi: 10.1016/j.neures.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 59.Kawaguchi K, Hickey RW, Rose ME, Zhu L, Chen J, Graham SH. Cyclooxygenase-2 expression is induced in rat brain after kainate-induced seizures and promotes neuronal death in CA3 hippocampus. Brain Research. 2005;1050(1-2):130–137. doi: 10.1016/j.brainres.2005.05.038. [DOI] [PubMed] [Google Scholar]
- 60.Patel M, Liang L-P, Hou H, et al. Seizure-induced formation of isofurans: novel products of lipid peroxidation whose formation is positively modulated by oxygen tension. Journal of Neurochemistry. 2008;104(1):264–270. doi: 10.1111/j.1471-4159.2007.04974.x. [DOI] [PubMed] [Google Scholar]
- 61.Rojo LE, Fernández JA, Maccioni AA, Jimenez JM, Maccioni RB. Neuroinflammation: implications for the pathogenesis and molecular diagnosis of Alzheimer’s disease. Archives of Medical Research. 2008;39(1):1–16. doi: 10.1016/j.arcmed.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 62.Bennett JL, Stüve O. Update on inflammation, neurodegeneration, and immunoregulation in multiple sclerosis: therapeutic implications. Clinical Neuropharmacology. 2009;32(3):121–132. doi: 10.1097/WNF.0b013e3181880359. [DOI] [PubMed] [Google Scholar]
- 63.Colton CA, Wilcock DM. Assessing activation states in microglia. CNS and Neurological Disorders. 2010;9(2):174–191. doi: 10.2174/187152710791012053. [DOI] [PubMed] [Google Scholar]
- 64.Kerschensteiner M, Meinl E, Hohlfeld R. Neuro-immune crosstalk in CNS diseases. Neuroscience. 2009;158(3):1122–1132. doi: 10.1016/j.neuroscience.2008.09.009. [DOI] [PubMed] [Google Scholar]
- 65.Chen Z, Duan R-S, Concha QH, et al. IL-12p35 deficiency alleviates kainic acid-induced hippocampal neurodegeneration in C57BL/6 mice. Neurobiology of Disease. 2004;17(2):171–178. doi: 10.1016/j.nbd.2004.07.018. [DOI] [PubMed] [Google Scholar]
- 66.Stuehr DJ, Cho HJ, Kwon NS, Weise MF, Nathan CF. Purification and characterization of the cytokine-induced macrophage nitric oxide synthase: an FAD- and FMN-containing flavoprotein. Proceedings of the National Academy of Sciences of the United States of America. 1991;88(17):7773–7777. doi: 10.1073/pnas.88.17.7773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wiesinger H. Arginine metabolism and the synthesis of nitric oxide in the nervous system. Progress in Neurobiology. 2001;64(4):365–391. doi: 10.1016/s0301-0082(00)00056-3. [DOI] [PubMed] [Google Scholar]
- 68.Hanisch U-K. Microglia as a source and target of cytokines. GLIA. 2002;40(2):140–155. doi: 10.1002/glia.10161. [DOI] [PubMed] [Google Scholar]
- 69.Amor S, Puentes F, Baker D, Van Der Valk P. Inflammation in neurodegenerative diseases. Immunology. 2010;129(2):154–169. doi: 10.1111/j.1365-2567.2009.03225.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Byun J-S, Lee S-H, Jeon S-H, et al. Kainic acid-induced neuronal death is attenuated by aminoguanidine but aggravated by L-NAME in mouse hippocampus. Korean Journal of Physiology and Pharmacology. 2009;13(4):265–271. doi: 10.4196/kjpp.2009.13.4.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yamamoto M, Yoshizaki K, Kishimoto T, Ito H. IL-6 is required for the development of Th1 cell-mediated murine colitis. Journal of Immunology. 2000;164(9):4878–4882. doi: 10.4049/jimmunol.164.9.4878. [DOI] [PubMed] [Google Scholar]
- 72.Diehl S, Rincón M. The two faces of IL-6 on Th1/Th2 differentiation. Molecular Immunology. 2002;39(9):531–536. doi: 10.1016/s0161-5890(02)00210-9. [DOI] [PubMed] [Google Scholar]
- 73.Lehtimäki KA, Peltola J, Koskikallio E, Keränen T, Honkaniemi J. Expression of cytokines and cytokine receptors in the rat brain after kainic acid-induced seizures. Molecular Brain Research. 2003;110(2):253–260. doi: 10.1016/s0169-328x(02)00654-x. [DOI] [PubMed] [Google Scholar]
- 74.All C, Nicole O, Docagne F, et al. Ischemia-induced interleukin-6 as a potential endogenous neuroprotective cytokine against NMDA receptor-mediated excitoxicity in the brain. Journal of Cerebral Blood Flow and Metabolism. 2000;20(6):956–966. doi: 10.1097/00004647-200006000-00008. [DOI] [PubMed] [Google Scholar]
- 75.De Sarro G, Russo E, Ferreri G, et al. Seizure susceptibility to various convulsant stimuli of knockout interleukin-6 mice. Pharmacology Biochemistry and Behavior. 2004;77(4):761–766. doi: 10.1016/j.pbb.2004.01.012. [DOI] [PubMed] [Google Scholar]
- 76.Penkowa M, Molinero A, Carrasco J, Hidalgo J. Interleukin-6 deficiency reduces the brain inflammatory response and increases oxidative stress and neurodegeneration after kainic acid-induced seizures. Neuroscience. 2001;102(4):805–818. doi: 10.1016/s0306-4522(00)00515-7. [DOI] [PubMed] [Google Scholar]
- 77.Stoll G, Jander S, Schroeter M. Cytokines in CNS disorders: neurotoxicity versus neuroprotection. Journal of Neural Transmission. 2000;(59, supplement):81–89. doi: 10.1007/978-3-7091-6781-6_11. [DOI] [PubMed] [Google Scholar]
- 78.Zhang X-M, Duan R-S, Chen Z, et al. IL-18 deficiency aggravates kainic acid-induced hippocampal neurodegeneration in C57BL/6 mice due to an overcompensation by IL-12. Experimental Neurology. 2007;205(1):64–73. doi: 10.1016/j.expneurol.2007.01.019. [DOI] [PubMed] [Google Scholar]
- 79.Kiefer R, Streit WJ, Toyka KV, Kreutzberg GW, Hartung H-P. Transforming growth factor-β1: a lesion-associated cytokine of the nervous system. International Journal of Developmental Neuroscience. 1995;13(3-4):331–339. doi: 10.1016/0736-5748(94)00074-d. [DOI] [PubMed] [Google Scholar]
- 80.Boche D, Cunningham C, Gauldie J, Perry VH. Transforming growth factor-β1-mediated neuroprotection against excitotoxic injury in vivo. Journal of Cerebral Blood Flow and Metabolism. 2003;23(10):1174–1182. doi: 10.1097/01.WCB.0000090080.64176.44. [DOI] [PubMed] [Google Scholar]
- 81.Morgan TE, Nichols NR, Pasinetti GM, Finch CE. TGF-β1 mRNA increases in macrophage/microglial cells of the hippocampus in response to deafferentation and kainic acid-induced neurodegeneration. Experimental Neurology. 1993;120(2):291–301. doi: 10.1006/exnr.1993.1063. [DOI] [PubMed] [Google Scholar]
- 82.Kim H-C, Bing G, Kim S-J, et al. Kainate treatment alters TGF-β3 gene expression in the rat hippocampus. Molecular Brain Research. 2002;108(1-2):60–70. doi: 10.1016/s0169-328x(02)00514-4. [DOI] [PubMed] [Google Scholar]
- 83.Lu M-O, Zhang X-M, Mix E, et al. TNF-α receptor 1 deficiency enhances kainic acid-induced hippocampal injury in mice. Journal of Neuroscience Research. 2008;86(7):1608–1614. doi: 10.1002/jnr.21600. [DOI] [PubMed] [Google Scholar]
- 84.Thompson C, Gary D, Mattson M, Mackenzie A, Robertson GS. Kainic acid-induced naip expression in the hippocampus is blocked in mice lacking TNF receptors. Molecular Brain Research. 2004;123(1-2):126–131. doi: 10.1016/j.molbrainres.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 85.Balosso S, Ravizza T, Perego C, et al. Tumor necrosis factor-α inhibits seizures in mice via p75 receptors. Annals of Neurology. 2005;57(6):804–812. doi: 10.1002/ana.20480. [DOI] [PubMed] [Google Scholar]
- 86.Zhu W, Zheng H, Shao X, Wang W, Yao Q, Li Z. Excitotoxicity of TNFα derived from KA activated microglia on hippocampal neurons in vitro and in vivo. Journal of Neurochemistry. 2010;114(2):386–396. doi: 10.1111/j.1471-4159.2010.06763.x. [DOI] [PubMed] [Google Scholar]
- 87.Kanno T, Nagata T, Yamamoto S, Okamura H, Nishizaki T. Interleukin-18 stimulates synaptically released glutamate and enhances postsynaptic AMPA receptor responses in the CA1 region of mouse hippocampal slices. Brain Research. 2004;1012(1-2):190–193. doi: 10.1016/j.brainres.2004.03.065. [DOI] [PubMed] [Google Scholar]
- 88.Sugama S, Wirz SA, Barr AM, Conti B, Bartfai T, Shibasaki T. Interleukin-18 null mice show diminished microglial activation and reduced dopaminergic neuron loss following acute 1-methyl-4-phenyl-1,2,3,6- tetrahydropyridine treatment. Neuroscience. 2004;128(2):451–458. doi: 10.1016/j.neuroscience.2004.07.020. [DOI] [PubMed] [Google Scholar]
- 89.Jeon GS, Park SK, Park SW, Kim DW, Chung CK, Cho SS. Glial expression of interleukin-18 and its receptor after excitotoxic damage in the mouse hippocampus. Neurochemical Research. 2008;33(1):179–184. doi: 10.1007/s11064-007-9434-6. [DOI] [PubMed] [Google Scholar]
- 90.Wang D, Zhang S, Li L, Liu X, Mei K, Wang X. Structural insights into the assembly and activation of IL-1β 2 with its receptors. Nature Immunology. 2010;11(10):905–911. doi: 10.1038/ni.1925. [DOI] [PubMed] [Google Scholar]
- 91.Oprica M, Eriksson C, Schultzberg M. Inflammatory mechanisms associated with brain damage induced by kainic acid with special reference to the interleukin-1 system. Journal of Cellular and Molecular Medicine. 2003;7(2):127–140. doi: 10.1111/j.1582-4934.2003.tb00211.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Liu B, Hong J-S. Role of microglia in inflammation-mediated neurodegenerative diseases: mechanisms and strategies for therapeutic intervention. Journal of Pharmacology and Experimental Therapeutics. 2003;304(1):1–7. doi: 10.1124/jpet.102.035048. [DOI] [PubMed] [Google Scholar]
- 93.Zheng H, Zhu W, Zhao H, Wang X, Wang W, Li Z. Kainic acid-activated microglia mediate increased excitability of rat hippocampal neurons in vitro and in vivo: crucial role of interleukin-1beta. NeuroImmunoModulation. 2009;17(1):31–38. doi: 10.1159/000243083. [DOI] [PubMed] [Google Scholar]
- 94.Chen Z, Ljunggren H-G, Bogdanovic N, Nennesmo I, Winblad B, Zhu J. Excitotoxic neurodegeneration induced by intranasal administration of kainic acid in C57BL/6 mice. Brain Research. 2002;931(2):135–145. doi: 10.1016/s0006-8993(02)02268-0. [DOI] [PubMed] [Google Scholar]
- 95.Illum L. Transport of drugs from the nasal cavity to the central nervous system. European Journal of Pharmaceutical Sciences. 2000;11(1):1–18. doi: 10.1016/s0928-0987(00)00087-7. [DOI] [PubMed] [Google Scholar]
- 96.Montague AA, Greer CA. Differential distribution of ionotropic glutamate receptor subunits in the rat olfactory bulb. Journal of Comparative Neurology. 1999;405(2):233–246. doi: 10.1002/(sici)1096-9861(19990308)405:2<233::aid-cne7>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 97.Mathison S, Nagilla R, Kompella UB. Nasal route for direct delivery of solutes to the central nervous system: fact or fiction? Journal of Drug Targeting. 1998;5(6):415–441. doi: 10.3109/10611869808997870. [DOI] [PubMed] [Google Scholar]
- 98.Sakane T, Akizuki M, Taki Y, Yamashita S, Sezaki H, Nadai T. Direct drug transport from the rat nasal cavity to the cerebrospinal fluid: the relation to the molecular weight of drugs. Journal of Pharmacy and Pharmacology. 1995;47(5):379–381. doi: 10.1111/j.2042-7158.1995.tb05814.x. [DOI] [PubMed] [Google Scholar]
- 99.Genter MB, Kendig EL, Knutson MD. Uptake of materials from the nasal cavity into the blood and brain: are we finally beginning to understand these processes at the molecular level. Annals of the New York Academy of Sciences. 2009;1170(1):623–628. doi: 10.1111/j.1749-6632.2009.03877.x. [DOI] [PubMed] [Google Scholar]
- 100.Johnson KA, Conn PJ, Niswender CM. Glutamate receptors as therapeutic targets for Parkinson's disease. CNS and Neurological Disorders. 2009;8(6):475–491. doi: 10.2174/187152709789824606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ilieva H, Polymenidou M, Cleveland DW. Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. Journal of Cell Biology. 2009;187(6):761–772. doi: 10.1083/jcb.200908164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kim DH, Yoon BH, Jung WY, et al. Sinapic acid attenuates kainic acid-induced hippocampal neuronal damage in mice. Neuropharmacology. 2010;59(1-2):20–30. doi: 10.1016/j.neuropharm.2010.03.012. [DOI] [PubMed] [Google Scholar]
- 103.Miyamoto R, Shimakawa S, Suzuki S, Ogihara T, Tamai H. Edaravone prevents kainic acid-induced neuronal death. Brain Research. 2008;1209(1):85–91. doi: 10.1016/j.brainres.2008.02.064. [DOI] [PubMed] [Google Scholar]
- 104.Chung S-Y, Han S-H. Melatonin attenuates kainic acid-induced hippocampal neurodegeneration and oxidative stress through microglial inhibition. Journal of Pineal Research. 2003;34(2):95–102. doi: 10.1034/j.1600-079x.2003.00010.x. [DOI] [PubMed] [Google Scholar]
- 105.Zhu X-L, Xiong L-Z, Wang Q, et al. Therapeutic time window and mechanism of tetramethylpyrazine on transient focal cerebral ischemia/reperfusion injury in rats. Neuroscience Letters. 2009;449(1):24–27. doi: 10.1016/j.neulet.2008.09.007. [DOI] [PubMed] [Google Scholar]
- 106.Li S-Y, Jia Y-H, Sun W-G, et al. Stabilization of mitochondrial function by tetramethylpyrazine protects against kainate-induced oxidative lesions in the rat hippocampus. Free Radical Biology and Medicine. 2010;48(4):597–608. doi: 10.1016/j.freeradbiomed.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 107.Swanson GT. Targeting AMPA and kainate receptors in neurological disease: therapies on the horizon? Neuropsychopharmacology. 2009;34(1):249–250. doi: 10.1038/npp.2008.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Liu X-M, Pei D-S, Guan Q-H, et al. Neuroprotection of Tat-GluR6-9c against neuronal death induced by kainate in rat hippocampus via nuclear and non-nuclear pathways. Journal of Biological Chemistry. 2006;281(25):17432–17445. doi: 10.1074/jbc.M513490200. [DOI] [PubMed] [Google Scholar]
- 109.Pei D-S, Wang X-T, Liu Y, et al. Neuroprotection against ischaemic brain injury by a GluR6-9c peptide containing the TAT protein transduction sequence. Brain. 2006;129(2):465–479. doi: 10.1093/brain/awh700. [DOI] [PubMed] [Google Scholar]
- 110.Akashiba H, Matsuki N, Nishiyama N. Calpain activation is required for glutamate-induced p27 down-regulation in cultured cortical neurons. Journal of Neurochemistry. 2006;99(3):733–744. doi: 10.1111/j.1471-4159.2006.04100.x. [DOI] [PubMed] [Google Scholar]
- 111.Akashiba H, Matsuki N, Nishiyama N. p27 small interfering RNA induces cell death through elevating cell cycle activity in cultured cortical neurons: a proof-of-concept study. Cellular and Molecular Life Sciences. 2006;63(19-20):2397–2404. doi: 10.1007/s00018-006-6194-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ueyama C, Akashiba H, Nakayama K, Nakayama KI, Nishiyama N, Matsuki N. Ablation of p27 enhance kainate-induced seizure and hippocampal degeneration. NeuroReport. 2007;18(17):1781–1785. doi: 10.1097/WNR.0b013e3282f16df6. [DOI] [PubMed] [Google Scholar]
- 113.Araújo IM, Gil JM, Carreira BP, et al. Calpain activation is involved in early caspase-independent neurodegeneration in the hippocampus following status epilepticus. Journal of Neurochemistry. 2008;105(3):666–676. doi: 10.1111/j.1471-4159.2007.05181.x. [DOI] [PubMed] [Google Scholar]
- 114.Verdaguer E, Jordà EG, Canudas AM, et al. p21WAF1/Cip1 is not involved in kainic acid-induced apoptosis in murine cerebellar granule cells. Brain Research. 2004;1030(2):297–302. doi: 10.1016/j.brainres.2004.09.052. [DOI] [PubMed] [Google Scholar]
- 115.Verdaguer E, García-Jordà E, Jiménez A, et al. Kainic acid-induced neuronal cell death in cerebellar granule cells is not prevented by caspase inhibitors. British Journal of Pharmacology. 2002;135(5):1297–1307. doi: 10.1038/sj.bjp.0704581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Jinap S, Hajeb P. Glutamate. Its applications in food and contribution to health. Appetite. 2010;55(1):1–10. doi: 10.1016/j.appet.2010.05.002. [DOI] [PubMed] [Google Scholar]
- 117.Yang J, Houk B, Shah J, et al. Genetic background regulates semaphorin gene expression and epileptogenesis in mouse brain after kainic acid status epilepticus. Neuroscience. 2005;131(4):853–869. doi: 10.1016/j.neuroscience.2004.09.064. [DOI] [PubMed] [Google Scholar]
- 118.Aarts MM, Tymianski M. Novel treatment of excitotoxicity: targeted disruption of intracellular signalling from glutamate receptors. Biochemical Pharmacology. 2003;66(6):877–886. doi: 10.1016/s0006-2952(03)00297-1. [DOI] [PubMed] [Google Scholar]
- 119.Thukral V, Chikaraishi D, Hunter DD, Wang JKT. Expression of non-N-methyl-D-aspartate glutamate receptor subunits in the olfactory epithelium. Neuroscience. 1997;79(2):411–424. doi: 10.1016/s0306-4522(96)00699-9. [DOI] [PubMed] [Google Scholar]
- 120.Schauwecker PE. Neuroprotection by glutamate receptor antagonists against seizure-induced excitotoxic cell death in the aging brain. Experimental Neurology. 2010;224(1):207–218. doi: 10.1016/j.expneurol.2010.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]