

Abstract
BACKGROUND AND PURPOSE
The arachidonyl-amino acid N-arachidonyl-glycine (NAGly) is an endogenous lipid, generated within the spinal cord and producing spinally mediated analgesia via non-cannabinoid mechanisms. In this study we examined the actions of NAGly on neurons within the superficial dorsal horn, a key site for the actions of many analgesic agents.
EXPERIMENTAL APPROACH
Whole cell patch clamp recordings were made from lamina II neurons in rat spinal cord slices to examine the effect of NAGly on glycinergic and NMDA-mediated synaptic transmission.
KEY RESULTS
N-arachidonyl-glycine prolonged the decay of glycine, but not β-alanine induced inward currents and decreased the amplitude of currents induced by both glycine and β-alanine. NAGly and ALX-1393 (inhibitor of the glycine transporter, GLYT2), but not the GLYT1 inhibitor, ALX-5407, produced a strychnine-sensitive inward current. ALX-5407 and ALX-1393, but not NAGly prolonged the decay phase of glycine receptor-mediated miniature inhibitory postsynaptic currents (IPSCs). NAGly prolonged the decay phase of evoked IPSCs, although to a lesser extent than ALX-5407 and ALX-1393. In the presence of ALX-1393, NAGly shortened the decay phase of evoked IPSCs. ALX-5407 increased and NAGly decreased the amplitude of evoked NMDA-mediated excitatory postsynaptic currents.
CONCLUSIONS AND IMPLICATIONS
Our results suggest that NAGly enhanced inhibitory glycinergic synaptic transmission within the superficial dorsal horn by blocking glycine uptake via GLYT2. In addition, NAGly decreased excitatory NMDA-mediated synaptic transmission. Together, these findings provide a cellular explanation for the spinal analgesic actions of NAGly.
Keywords: synaptic transmission, glycine, transporter, pain, dorsal horn, substantia gelatinosa, endocannabinoid, arachidonate
Introduction
N-arachidonyl-glycine (NAGly) is the carboxylic acid congener of the endocannabinoid anandamide and is generated at high levels in the spinal cord and small intestine, with lower levels in brain, kidney and skin (Huang et al., 2001). A number of studies have implicated NAGly in the modulation of pain and inflammation. Oral and intraplantar NAGly reduce responses to acute noxious thermal stimuli and intraplantar formalin (Burstein et al., 2000; Huang et al., 2001; Barbara et al., 2009). Furthermore, intrathecal NAGly reduces the allodynia and hyperalgesia associated with animal models of chronic inflammatory and neuropathic pain (Succar et al., 2007; Vuong et al., 2008). The spinal actions of NAGly in chronic pain states are not reduced by cannabinoid CB1 and CB2 receptor antagonists (Succar et al., 2007; Vuong et al., 2008) which is consistent with the finding that NAGly, unlike the endocannabinoid arachidonyl ethanolamide (anandamide), has negligible affinity for the cannabinoid CB1 receptor (Sheskin et al., 1997; receptor and channel nomenclature follows Alexander et al., 2009). NAGly also differs from anandamide in that it has low affinity for the vanilloid transient receptor potential channel, TRPV1 (De Petrocellis et al., 2000; Ross et al., 2009), but has moderate affinity for the anandamide degrading enzyme, fatty acid amide hydrolase (FAAH) and cyclooxygenase (COX)-2 (Huang et al., 2001; Prusakiewicz et al., 2002; Cascio et al., 2004; McHugh et al., 2010). Conversely, NAGly has been reported to be a degradation product of anandamide, which is partly mediated via FAAH (Cascio et al., 2004; Bradshaw et al., 2009). NAGly has also been identified as a ligand for T-type Ca2+-channels (Barbara et al., 2009; Ross et al., 2009), GRP18 (Kohno et al., 2006; Yin et al., 2009; McHugh et al., 2010) and possibly other GPCRs (see Oh et al., 2008; Williams et al., 2009; Yin et al., 2009; Parmar and Ho, 2010).
A potential target for the spinal analgesic actions of NAGly is the glycine transporter system which belongs to the Na+/Cl- dependent family of neurotransmitter transporters. There are two subtypes of glycine transporters, GLYT1 which is widely expressed in glial cells throughout the nervous system and GLYT2 which has a more restricted expression pattern on the presynaptic terminals of glycinergic neurons (Zafra et al., 1995; Spike et al., 1997; Zeilhofer et al., 2005). It has recently been demonstrated that NAGly inhibits GLYT2, but has no effect on GLYT1, or the GABA transporter GAT1 (Wiles et al., 2006). Both GLYT1 and GLYT2 are expressed at relatively high levels within the superficial dorsal horn, a major integrative centre of nociception within the CNS (Zafra et al., 1995; Spike et al., 1997). Like NAGly, spinal administration of GLYT1 and GLYT2 inhibitors reduces allodynia and hyperalgesia in a range of chronic pain models (Hermanns et al., 2008; Morita et al., 2008; Tanabe et al., 2008). While the pain relieving actions of both GLYT1 and GLYT2 inhibitors are mediated by glycine receptors, co-activation of NMDA receptors limits the anti-allodynic actions of GLYT1 inhibitors (Morita et al., 2008). Cellular studies have demonstrated that GLYT1 and GLYT2 inhibitors modulate glycinergic and NMDA-mediated synaptic transmission in lamina X of the spinal cord and in spinal cord cultures (Bradaia et al., 2004; Xu et al., 2005; Rousseau et al., 2008). Thus, glycine transport inhibitors may modulate both inhibitory and excitatory synaptic transmission within the dorsal horn via the actions of endogenous glycine at glycine receptors and NMDA ion channels respectively. The cellular actions of NAGly within the spinal cord are, however, unknown as are the actions of glycine transport inhibitors within the superficial dorsal horn. In the present study we examined the effect of NAGly on neurons within lamina II of the superficial dorsal horn, in rats.
Methods
All animal care and experimental procedures followed the guidelines of the ‘NH&MRC Code of Practice for the Care and Use of Animals in Research in Australia’ and were approved by the Royal North Shore Hospital Animal Care and Ethics Committee. We used P14–18 Sprague-Dawley rats. Animals were housed in individually ventilated cages with their littermates and mother, under a 12:12 h light/dark cycle, with environmental enrichment and free access to water and standard rat chow. For the experiments, animals were deeply anaesthetized with isoflurane and decapitated. The dura was incised and the spinal column quickly removed and placed in ice cold artificial cerebrospinal fluid (ACSF) of composition: (mM): NaCl, 126; KCl, 2.5; NaH2PO4, 1.4; MgCl2, 1.2; CaCl2, 2.4; glucose, 11; NaHCO3, 25. Transverse (300 µm) slices of the lumbar spinal cord (L4–6) were cut (vibratome; VT1000S: Leica, Nussloch, Germany) and maintained at 34°C in a submerged chamber containing ACSF equilibrated with 95% O2 and 5% CO2.
Electrophysiological procedures
Individual slices were transferred to a slice chamber (volume 0.5 mL) and superfused continuously (1.8 mL·min−1) with ACSF at 34°C using an inline temperature controller. Superficial dorsal horn neurons located throughout lamina II were visualized using infra-red Dodt-tube optics on an upright microscope (Olympus BX50, Olympus, Sydney, Australia) and whole-cell voltage clamp recordings (holding potential −65 mV, liquid junction potential corrected) were made using an Axopatch 200B (Molecular Devices, Sunnyvale, CA, USA). The internal solution contained (mM): CsCl 140, HEPES 10, EGTA 0.2, MgCl2 1, QX-314 3, MgATP 2 and NaGTP 0.3 (pH 7.3 and osmolality 280–285 mosmol·L−1). This internal solution had an ECl of 0 mV leading to inward, glycine receptor–mediated, currents in neurons at −65 mV. Series resistance (<25 MΩ) was compensated by 80% and continuously monitored during experiments.
All recordings were filtered (1 kHz low-pass filter) and sampled (5 kHz) for on-line and off-line analysis using AxographX (Axograph Scientific, Sydney Australia). Recordings examining the effect of exogenous glycine were made in the presence of the non-NMDA receptor antagonist 6-cyano-2,3-dihdroxy-7-nitro-quinoxaline (CNQX, 5 µM) and the GABAA receptor blocker bicuculline (30 µM). The time course of the membrane current during washout of exogenously applied glycine was measured as the time to reach one decay time constant (τ, time at 36.8% of current at start of washout). Glycine receptor-mediated electrically evoked inhibitory postsynaptic currents (IPSCs) were elicited in neurons via ACSF-filled glass stimulating electrodes placed within lamina II (rate 0.03 s−1, intensity 1–50 V, 0.05–0.2 ms) in the presence of CNQX (5 µM), bicuculline (30 µM) and the NMDA antagonist DL-(-)-2-Amino-5-phosphonopentanoic acid (AP5, 25 µM). Evoked NMDA-mediated excitatory postsynaptic currents (EPSCs) were elicited in the presence of CNQX (5 µM), strychnine (3 µM) and picrotoxin (100 µM) in Mg2+-free ACSF via ACSF-filled glass stimulating electrodes placed on the dorsal rootlets (rate 0.05–0.067 s−1, intensity 1–20 V, 0.05–0.2 ms). Spontaneous miniature IPSCs were recorded in the presence of tetrodotoxin (TTX, 500 nM), CNQX and bicuculline. Miniature IPSCs, 4.5–5.5 standard deviations above baseline noise, were automatically detected by a sliding template algorithm and manually checked. The kinetics of synaptic currents were measured by their width [at 36.8% (1/e) of the peak for IPSCs and at 50% of the peak for EPSCs] and 10–90% rise-time. In addition, the time constant of the decay phase of IPSCs was fit to an exponential using the least square method based on a simplex algorithm (Chery and de Koninck, 1999). Glycine receptor-mediated evoked and miniature IPSCs had mixed decay phase kinetics, with some being best fit by one and others by two exponentials (as assessed by visual inspection and the sums of squared errors of the fits).
Data analysis
Normalized cumulative distribution plots of IPSC characteristics were constructed and compared using the Kolmogorov–Smirnov test. Statistical assessment of drug effects were made using Student's t-test, or Wilcoxon's summed rank test for non-normally distributed data, and comparisons between drug groups were made using a one-way ANOVA with Newman-Keuls adjustment for post hoc tests using Prism (GraphPad Software, La Jolla, USA). Differences were considered significant when P < 0.05 and all numerical data are expressed as mean ± SEmean.
Materials
N-arachidonyl-glycine was obtained from Cayman Chemical Co. (Ann Arbor, MI, USA); dl-2-amino-5-phosphonopentanoic acid (AP5), bicuculline methiodide, 5,7-dichlorokynurenic acid (DCKA), 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX), QX314 and TTX from Ascent Scientific (Bristol, UK). β-alanine, O-[(2-benzyloxyphenyl-3-fluorophenyl) methyl]-L-serine (ALX-1393), N-[3-(4′-Fluorophenyl)-3-(4′-phenylphenoxy)propyl]sarcosine hydrochloride (ALX-5407, NFPS), glycine, picrotoxin, strychnine hydrochloride and all other reagents from Sigma (Sydney, Australia). Stock solutions of NAGly (in ethanol), ALX-1393 and ALX-5407 (both in DMSO) were diluted (1:1000–1:3000) to working concentrations in ACSF immediately before use and applied by superfusion to the slice chamber. NAGly, ALX-1393 and ALX-5407 were applied to one cell per slice, and after each recording the entire system was washed in 30% ethanol.
Results
N-arachidonyl-glycine enhances the effect of exogenously applied glycine
In the presence of CNQX (5 µM) and bicuculline (30 µM), glycine produced an inward current in all lamina II neurons tested which rapidly desensitized at higher concentrations (Figure 1A). The glycine-induced inward current was concentration dependent, had an EC50 of 345 µM (95% confidence interval 226–525 µM) and a Hill slope of 1.2 ± 0.2 (Figure 1C). In addition, β-alanine produced concentration dependent inward currents with an EC50 of 485 µM (95% confidence interval 328–765 µM) and a Hill slope of 1.0 ± 0.1 (Figure 1B and C).
Figure 1.
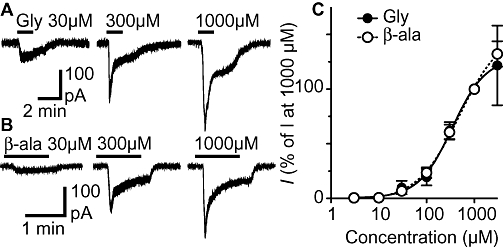
Glycine and β-alanine induce concentration dependent currents in lamina II neurons. Membrane currents induced by (A) glycine (Gly) and (B) β-alanine (β-ala). (C) Concentration response curves for the membrane currents induced by glycine and β-alanine; current values were normalized to those produced at 1000 µM in each neuron. A logistic function was fitted to each curve to determine the EC50 and Hill slope. (A) and (B) are from different neurons.
To determine whether the currents induced by exogenous glycine and β-alanine were affected by NAGly, we repeatedly superfused sub-EC50 concentrations of glycine (30–100 µM) for a 1.5–2 min period every 5–7 min (Figure 2A and C). When glycine was superfused alone, it induced an inward current which rapidly reversed following washout and gradually reduced in amplitude with each successive application (Figure 2A and B). We next examined whether the glycine-induced current was affected by addition of NAGly (10–60 µM) at concentrations which produce maximal inhibition of the glycine transport GLYT2 (Wiles et al., 2006). When NAGly was added following the third application of glycine it led to a reduction in the amplitude of the glycine current and a prolongation of its decay following washout (Figure 2C and D).
Figure 2.
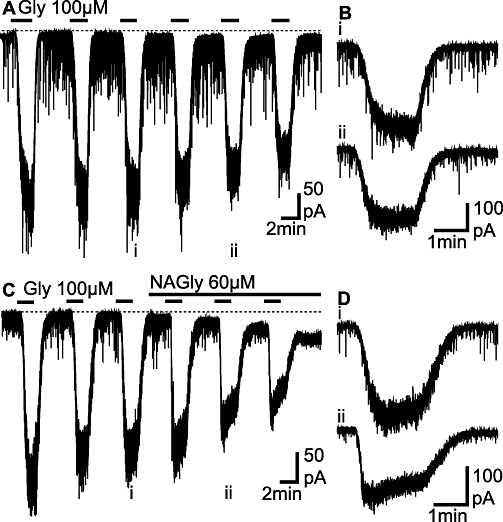
N-arachidonyl-glycine (NAGly) prolongs the decay phase of currents induced by exogenously applied glycine. (A, C) Membrane currents induced by repeated, brief applications of a sub-maximal concentration of glycine (Gly, 100 µM). In (C) NAGly (60 µM) was applied following the third application of glycine. (B) and (D) show expanded traces of the membrane currents during the (i) third and (ii) fifth applications of glycine from the traces in (A) and (C) respectively. (A, B) and (C, D) are from two different neurons.
To quantify the effect of NAGly we compared the amplitude and decay times for the third and fifth applications of glycine and β-alanine, for neurons in which NAGly was, or was not added to the superfusate. The decay time of the glycine-induced current was increased from 15 ± 2 s to 29 ± 5 s (P = 0.0005, n = 17) when NAGly was added to the superfusate (Figure 3). The change in decay times of the glycine-induced current was similar at all concentrations of NAGly tested (10 µM = 188 ± 44, 30 µM = 173 ± 15, 60 µM = 212 ± 38% of pre-NAGly, n = 3, 7, 7, P = 0.7) and it might be noted that NAGly (10 µM) has no effect at TRPV1 channels (De Petrocellis et al., 2000; Ross et al., 2009). By contrast, there was no significant change in decay time of the β-alanine (pre = 15 ± 2 s; NAGly = 15 ± 2 s, P = 0.8, n = 10) induced current when NAGly was added to the superfusate (Figure 3). There was also no significant change in decay times of the glycine (pre = 19 ± 3 s; NAGly = 21 ± 4 s, p = 0.2, n = 13) and β-alanine (pre = 11 ± 2 s; NAGly = 12 ± 2 s, p = 0.3, n = 10) induced currents when NAGly was not added to the superfusate (Figure 3). The amplitudes of the glycine- and β-alanine-induced currents were reduced from 216 ± 48 pA to 144 ± 25 pA (P = 0.008, n = 17) and from 262 ± 80 pA to 198 ± 62 pA (P = 0.02, n = 10) when NAGly was added to the superfusate (Figure 3). Similarly, the amplitudes of the glycine- and β-alanine-induced currents were reduced from 221 ± 32 pA to 175 ± 24 pA (P = 0.03, n = 13) and from 200 ± 26 pA to 179 ± 30 pA (P = 0.03, n = 10) when NAGly was not added to the superfusate (Figure 3). These observations suggest that the prolongation of the decay phase of the glycine-induced current was due to inhibition of glycine transport (Wiles et al., 2006).
Figure 3.
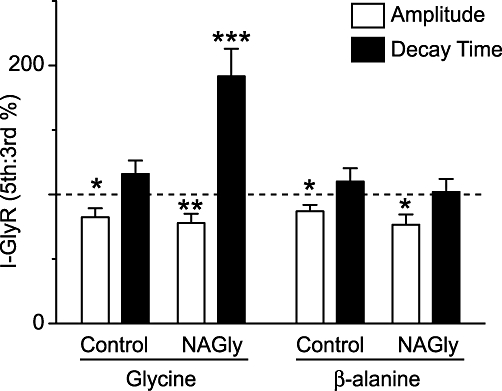
N-arachidonyl-glycine (NAGly) prolongs the decay phase of currents induced by exogenous glycine, but not β-alanine. Bar chart of the amplitude and decay time of the membrane currents produced by exogenous application of glycine (100 µM) and β-alanine (100 µM). The data are expressed as a percentage ratio of the 5th : 3rd application of glycine, or β-alanine. NAGly (10–60 µM), or control ACSF (control) was added continuously after the 3rd application of glycine, or β-alanine (as in Figure 2). *P < 0.05, **P < 0.01, ***P < 0.001. ACSF, artificial cerebrospinal fluid.
N-arachidonyl-glycine enhances the effect of endogenous glycine
We next examined whether NAGly enhanced the actions of endogenously released glycine. In the presence of TTX (300 nM), CNQX (5 µM) and bicuculline (30 µM), addition of NAGly (30 µM) induced a slowly developing inward current and an increase in the variance of the inward current, both of which did not reverse upon washout of NAGly, but were abolished by the addition of strychnine (3 µM) (Figure 4A–C). When averaged across all neurons the strychnine-sensitive component of the inward current and increase in variance produced by NAGly were significant (Figure 5A and B, P = 0.004, 0.004, n = 9). We also examined the effect of the GLYT1 and GLYT2 inhibitors ALX-5407 and ALX-1393 (Atkinson et al., 2001; Aubrey and Vandenberg, 2001; Dohi et al., 2009). ALX-1393 (1 µM), but not ALX-5407 (1 µM) produced an inward current (P = 0.03, 0.06) and an increase in the variance of the inward current (P = 0.03, 0.06) which was reversed by strychnine (3 µM) (Figure 5A and B, n = 6 each).
Figure 4.
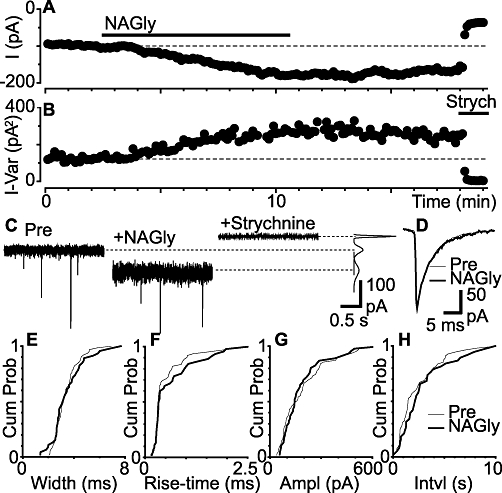
N-arachidonyl-glycine (NAGly) produces a glycine receptor-mediated inward current, but has no effect on miniature IPSCs. Time plots of the (A) membrane current and (B) variance of the membrane current during application of NAGly (30 µM) and strychnine (3 µM). (C) Raw current traces of the membrane current for epochs before (Pre) and during NAGly and strychnine. Inset on the right shows the variance of the membrane current (taken from periods where no spontaneous miniature IPSCs were observed). (D) Averaged traces of spontaneous miniature IPSCs before (Pre) and during superfusion of NAGly. Cumulative probability distribution plots of miniature IPSC (E) width (F) rise-time (G) amplitude and (H) inter-event interval, before and during superfusion of NAGly (Kolmogorov–Smirnov test P = 0.9, 0.5, 0.7, 0.2). (A)–(H) are from the same neuron. IPSCs, inhibitory postsynaptic currents.
Figure 5.
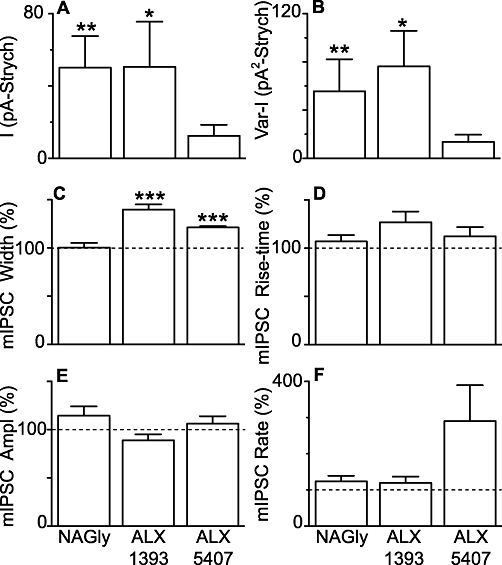
ALX-1393 and ALX-5407, but not NAGly increase the decay time of miniature IPSCs. Bar charts showing the mean (A) inward membrane current and (B) increase in the variance of the membrane current induced by NAGly (30 µM), ALX-1393 (1 µM) and ALX-5407 (1 µM) measured relative to the values obtained after addition of strychnine. Bar charts showing the effect of NAGly (30 µM), ALX-1393 (1 µM) and ALX-5407 (1 µM) on the (C) width (D) rise-time (E) amplitude and (F) rate of glycine receptor-mediated spontaneous miniature IPSCs, expressed as a percentage of the pre-drug value. *P < 0.05, **P < 0.01, ***P < 0.001. ALX-1393, O-[(2-benzyloxyphenyl-3- fluorophenyl) methyl]-L-serine; ALX-5407 (NFPS), N-[3-(4′-fluorophenyl)-3-(4′-phenylphenoxy)propyl]sarcosine hydrochloride; IPSCs, inhibitory postsynaptic currents; NAGly, N-arachidonyl-glycine.
In these recordings, strychnine (3 µM)-sensitive spontaneous miniature IPSCs were observed which, on average, had an amplitude of 76 ± 9 pA, 10–90% rise-time of 0.52 ± 0.05 ms, and width at 36.8% of peak of 4.5 ± 0.3 ms (Figure 4C and D, n = 21). In most neurons, the decay phase of the miniature IPSCs was best fitted by a monoexponential curve (mean time constant = 4.2 ± 0.3 ms, n = 18/21 neurons). NAGly (30 µM) had no effect on the kinetics of miniature IPSCs in most neurons, as assessed by their width and rise-time (Figure 4D). Correspondingly, NAGly (30 µM) did not alter the cumulative probability distributions of miniature IPSC width and rise-time in most neurons (Figure 4E and F, P > 0.05 for 8/9 and 8/9 neurons respectively). In addition, NAGly (30 µM) had no effect on the cumulative probability distributions of the amplitude and inter-event intervals of miniature IPSCs in most neurons (Figure 4G and H, P > 0.05 for 8/9 and 7/9 neurons respectively). When averaged across all neurons, NAGly (30 µM) had no effect on the mean width, rise-time, amplitude and rate of miniature IPSCs (Figure 5C–F, P = 0.9, 0.3, 0.2, 0.2, n = 9).
Unlike NAGly, both ALX-1393 and ALX-5407 produced an increase in the width of miniature IPSCs (P = 0.0006, <0.0001), but had no effect on their rise-times (P = 0.06, 0.3) (Figure 5C and D, n = 6 each). This suggests that ALX-1393 and ALX-5407 altered the kinetics of miniature IPSCs largely by prolonging the decay phase. ALX-1393 and ALX-5407 had no effect on the amplitude of miniature IPSCs (Figure 5E, P = 0.1, 0.5). ALX-1393 had no effect on miniature IPSC rate (Figure 5F, P = 0.3). ALX-5407 had a variable effect on miniature IPSC rate (Figure 5F, range = 78–750% of pre-ALX-5407, P = 0.1), producing an increase in a subset of neurons (n = 3/6). This is partly consistent with earlier studies, some of which have observed presynaptic facilitation of transmitter release (Turecek and Trussell, 2001; Bradaia et al., 2004; Xu et al., 2005), and possibly reflects the diverse phenotypes of dorsal horn neurons.
Effect of N-arachidonyl-glycine on evoked glycinergic synaptic transmission
We next examined the effect of NAGly, ALX-1393 and ALX-5407 on action potential evoked IPSCs. In the presence of CNQX (5 µM), AP5 (25 µM) and bicuculline (30 µM), electrical stimulation elicited IPSCs which were abolished by strychnine (3 µM; Figure 6A–D). These glycine receptor-mediated evoked IPSCs had an average amplitude of 580 ± 62 pA and width of 8.1 ± 0.5 ms (n = 29). Unlike miniature IPSCs, the decay phase of the evoked IPSCs in most recordings was best fitted by two exponentials (n = 25/29 neurons, mean time constants = 4.7 ± 0.2 ms and 28.9 ± 1.5 ms). Superfusion of NAGly (30 µM) produced a significant increase in the width of evoked IPSCs, but had no effect on the evoked IPSC amplitude (Figure 6A and E, P = 0.02 and 0.4, respectively, n = 11). Both ALX-1393 (1 µM) and ALX-5407 (1 µM) produced an increase in the width of evoked IPSCs (Figure 6B,C and E, P = 0.004, 0.0006, n = 9 each) which was greater than for NAGly (P < 0.05). ALX-1393, but not ALX-5407 produced a reduction in the amplitude of evoked IPSCs (Figure 6E, P = 0.02, 0.3).
Figure 6.
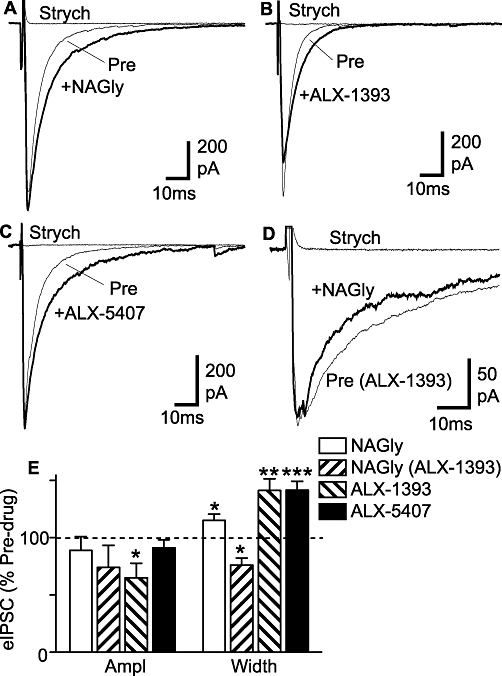
NAGly, ALX-1393 and ALX-5407 increase the decay time of glycine receptor-mediated evoked IPSCs. Averaged evoked IPSCs before (Pre) and during application of (A) NAGly (30 µM) (B) ALX-1393 (1 µM) and (C) ALX-5407 (1 µM), then during addition of strychnine (Strych, 3 µM). (D) Averaged evoked IPSCs in a neuron pre-incubated in ALX-1393 (1 µM) and then during addition of NAGly (30 µM), followed by strychnine. (D) Bar chart showing the mean amplitude and width of evoked IPSCs (eIPSC) in the presence of NAGly (30 µM), ALX-1393 (1 µM) and ALX-5407 (1 µM), expressed as a percentage of the pre-drug value [pre-drug was either control ACSF, or ALX-1393 (1 µM) for the NAGly (ALX-1393) group]. *P < 0.05, **P < 0.01, ***P < 0.001. Traces in (A)–(D) are from different neurons. ACSF, artificial cerebrospinal fluid; ALX-1393, O-[(2-benzyloxyphenyl-3- fluorophenyl) methyl]-L-serine; ALX-5407 (NFPS), N-[3-(4′-fluorophenyl)-3-(4′-phenylphenoxy)propyl]sarcosine hydrochloride; IPSCs, inhibitory postsynaptic currents; NAGly, N-arachidonyl-glycine.
The lesser effect of NAGly on evoked IPSC width compared with ALX-1393 and ALX-5407 could have been due to poor penetration into the slice. Alternatively, it might have been some other mechanism which functionally opposed the prolongation of the evoked IPSC decay phase resulting from GLYT blockade. In this regard, NAGly is a low affinity glycine receptor antagonist (Yang et al., 2008), similar to SR95331, which shortens the evoked IPSC decay phase if there is sufficient spill-over of glycine (Beato, 2008; Balakrishnan et al., 2009). We therefore examined the effect of NAGly on glycine receptor-mediated evoked IPSCs when GLYT2 was already inhibited by pre-incubating slices in ALX-1393. In the presence of ALX-1393 (1 µM), superfusion of NAGly (30 µM) produced a decrease in the width of evoked IPSCs, but had no effect on the evoked IPSC amplitude (Figure 6D and E, P = 0.01 and 0.2, respectively, n = 6).
Effect of N-arachidonyl-glycine on NMDA-mediated synaptic transmission
Endogenous glycine is also required as a co-activator for excitatory, NMDA receptor-mediated, synaptic transmission. In the presence of Mg2+-free ACSF which contained strychnine (3 µM), picrotoxin (100 µM) and CNQX (5 µM), electrical stimulation of the dorsal rootlets elicited EPSCs which were abolished by AP5 (25–50 µM, n = 12) (Figure 7A and B) and were reduced by the NMDA receptor glycine binding site antagonist DCKA (30 µM, n = 5). These evoked EPSCs had an amplitude of 151 ± 24 pA (range = 54–337 pA), 10–90% rise-time of 15.1 ± 3.4 ms (range = 4.8–45.3 ms) and half-width of 133 ± 24 ms (range = 48–292 ms) (n = 12). Superfusion of NAGly (30 µM) produced a decrease in the amplitude of evoked EPSCs (Figure 7A and C, P = 0.01, n = 6). By contrast, ALX-5407 (1 µM) produced an increase in the amplitude of evoked EPSCs (Figure 7B and C, P = 0.04, n = 6). NAGly and ALX-5407 had no effect on the half-width of evoked EPSCs (Figure 7A–C, P = 0.2 and 0.4 respectively). When nothing was added to the superfusate, the amplitude and half-width of evoked EPSCs were not significantly different to baseline levels when measured over similar time periods (Figure 7C, P = 0.9 and 0.6, respectively, n = 5).
Figure 7.
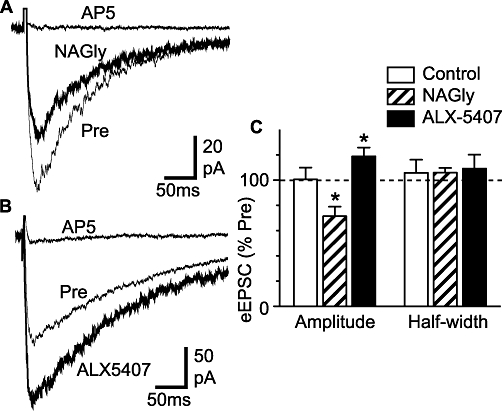
ALX-5407 increases and NAGly decreases NMDA-mediated evoked EPSCs. Averaged evoked EPSCs before (Pre) and during application of (A) NAGly (30 µM), and (B) ALX-5407 (1 µM), then during addition of AP5 (25–30 µM). (C) Bar chart showing the mean amplitude and half-width of evoked EPSCs (eEPSC) after an 8–10 min application of NAGly (30 µM), ALX-5407 (1 µM) or control ACSF (control), expressed as a percentage of the pre-application value. *P < 0.05. Traces in (A) and (B) are from different neurons. ACSF, artificial cerebrospinal fluid; ALX-5407 (NFPS), N-[3-(4′-fluorophenyl)-3-(4′-phenylphenoxy)propyl]sarcosine hydrochloride; AP5, DL-(-)-2-amino-5-phosphonopentanoic acid; EPSCs, excitatory postsynaptic currents; NAGly, N-arachidonyl-glycine.
Discussion
The findings of the present study indicate that the endogenous arachidonyl amino acid NAGly enhances glycinergic synaptic transmission within the lumbar superficial dorsal horn, a major centre for the integration of ascending pain information in the CNS. The enhancement of endogenous glycinergic tone and synaptic transmission was mediated by inhibition of glycine uptake via GLYT2, although this was likely to be limited by direct, low-affinity, glycine receptor antagonism. In addition, NAGly inhibited excitatory NMDA-mediated synaptic transmission. Together, these actions provide multiple cellular substrates for the analgesic actions of NAGly within the spinal cord.
In the present study NAGly prolonged the current induced by exogenous application of glycine, but not by the glycine receptor agonist β-alanine, the latter of which is a poor substrate for glycine transporters. In addition, NAGly produced a strychnine-sensitive inward current and an increase in the variance of this current which was mimicked by the GLYT2 inhibitor ALX-1393, but not by the GLYT1 inhibitor ALX-5407. These observations suggest that NAGly prolongs the actions of exogenous glycine and enhances ‘tonic’ endogenous glycine levels within the lumbar superficial dorsal horn, both of which are consistent with our previous finding that NAGly inhibits the cloned glycine transporter GLYT2, but not GLYT1 (Wiles et al., 2006). The difference between NAGly/ALX-1393 and ALX-5407 is also consistent with GLYT2 having a greater driving force for glycine uptake than GLYT1, and is a consequence of their differing stoichiometries (Roux and Supplisson, 2000). It might be noted that our observations, however, differ from those in lamina X of the spinal cord where both GLYT1 and GLYT2 inhibitors produce glycine receptor-mediated currents (Bradaia et al., 2004) and in cultured spinal dorsal horn neurons where GLYT1 and two inhibitors do not produce ‘tonic’ currents (Xu et al., 2005). This suggests that the actions of transporters are region-specific, even within the spinal cord, and that they vary between ex vivo slices and cultured spinal systems, as the latter might have a lower density of synaptic zones and transporters.
In the present study ALX-1393, but not ALX-5407 reduced the amplitude of evoked glycine receptor-mediated synaptic transmission, as observed earlier using GLYT1/2 knockout and pharmacological blockade (Gomeza et al., 2003a; Bradaia et al., 2004). These observations are consistent with the idea that GLYT1 regulates glycinergic neurotransmission by terminating the actions of glycine within the synaptic cleft, while GLYT2 maintains glycinergic transmission by transporting glycine into presynaptic glycinergic terminals where it becomes available for vesicle replenishment (Gomeza et al., 2003a,b; Aubrey et al., 2007; Rousseau et al., 2008). The observations are also consistent with the anatomical evidence that GLYT1 is expressed in glial cells immediately adjacent to the glycinergic synaptic cleft and GLYT2 is expressed more remotely in the perisynaptic region of the presynaptic terminals of glycinergic neurons in the dorsal horn (Zafra et al., 1995; Spike et al., 1997). In contrast to ALX-1393, NAGly had no effect on the amplitude of evoked and miniature IPSCs suggesting that NAGly is a less efficacious GLYT2 inhibitor than ALX-1393. It is also possible that GLYT2 inhibition only reduces glycinergic IPSC amplitude after prolonged periods of application, or during intense stimulation, as observed in earlier spinal cord studies (Bradaia et al., 2004; Xu et al., 2005; Aubrey et al., 2007). This is consistent with our finding that ALX-1393 reduced the amplitude of evoked, but not miniature IPSCs. The lack of effect of NAGly on IPSC amplitude (and on glycine receptor-mediated currents induced by exogenous glycine and β-alanine) was also surprising given that we have previously reported that NAGly is an antagonist at some glycine receptor subtypes (Yang et al., 2008; but see below).
In the present study ALX-1393 and ALX-5407 prolonged the decay phase of evoked and miniature glycine receptor-mediated synaptic transmission, as observed previously in lamina X with other GLYT1 and two inhibitors (Bradaia et al., 2004). We also found that NAGly prolonged the decay phase of evoked IPSC, although to a lesser extent than ALX-1393, and had no effect on miniature IPSCs. These observations are not consistent with other studies which have shown that GLYT1, but not GLYT2 knockout and pharmacological blockade prolongs the decay phase of glycinergic IPSC kinetics in hypoglossal motoneurons and cultured spinal cord neurons (Singer et al., 1998; Gomeza et al., 2003b; Xu et al., 2005; Rousseau et al., 2008). The decay phase of glycine receptor-mediated miniature IPSCs in lamina II neurons was best fitted by a single exponential with a short time constant, as observed previously in various spinal cord preparations (Chery and de Koninck, 1999; Graham et al., 2003; Katsurabayashi et al., 2004; Rousseau et al., 2008). By contrast, we found that the decay phase of evoked IPSCs was best fitted by two exponentials, the slower of which was likely to reflect glycine spill-over from neighbouring synapses and/or diffusion onto distal perisynaptic receptors during synchronous evoked release (see Beato, 2008; Balakrishnan et al., 2009). The differential effects of NAGly on evoked and miniature IPSCs suggest that NAGly inhibits a transporter which is engaged during synchronous release and spill-over associated with evoked IPSCs, but not during asynchronous release associated with miniature IPSCs. Low affinity glycine receptor antagonists, such as SR-95531, shorten evoked glycine receptor-mediated IPSCs under conditions in which there is significant spill-over by preventing rebinding as the concentration of glycine drops during the later phase of IPSCs (Beato, 2008; Balakrishnan et al., 2009). In the present study, it was observed that after occlusion of GLYT2 mediated blockade, NAGly shortened the decay phase of evoked IPSCs. This is consistent with our recent finding that NAGly can act as a low affinity glycine receptor antagonist (Yang et al., 2008) which would partially counteract the prolongation of IPSC decay phase resulting from NAGly-induced inhibition of GLYT2.
The transmission of nociceptive information through the superficial dorsal horn is influenced by both excitatory and inhibitory neurons (Narikawa et al., 2000; Santos et al., 2007). We found that NAGly decreased the amplitude of evoked NMDA-mediated EPSCs. This was surprising because glycine transport blockade and the subsequent spill-over would be expected to increase NMDA-mediated EPSCs given that it is also an NMDA receptor co-agonist (e.g. Johnson and Ascher, 1987). Indeed, ALX-5407 increased the amplitude of evoked NMDA-mediated EPSCs, as observed previously for other GLYT1 inhibitors in the brain and lamina X of the spinal cord (e.g. Chen et al., 2003; Bradaia et al., 2004; Martina et al., 2004). This suggests that endogenous glycine levels are high enough to co-activate, but do not saturate the NMDA receptor glycine binding site in our slice preparation. The NAGly-induced inhibition of evoked NMDA-mediated EPSCs was unlikely to be due to reduced glycine spill-over because NAGly had no effect on the amplitude of glycine receptor-mediated IPSCs, unlike that previously reported for nocistatin in lamina II neurons (Ahmadi et al., 2003). Our findings suggest that NAGly acts directly on NMDA receptors as a competitive antagonist at the glycine binding site, although confirmation of this requires examination of NAGly–glycine interactions on NMDA-mediated currents in isolated dorsal horn neurons.
Glycinergic inhibitory mechanisms within the spinal cord have a significant role in pain processing, with a reduction in inhibition (disinhibition) contributing to pathological chronic pain states (see Zeilhofer, 2005; Dohi et al., 2009). Like NAGly, spinal delivery of GLYT1 and GLYT2 inhibitors reduces the abnormal pain sensations associated with a range of animal models of chronic inflammatory and neuropathic pain (Succar et al., 2007; Hermanns et al., 2008; Morita et al., 2008; Tanabe et al., 2008; Vuong et al., 2008). Our finding that NAGly enhances the decay phase of synchronous evoked IPSCs, but not asynchronous spontaneous miniature IPSCs suggests that it may preferentially enhance inhibitory glycinergic transmission under conditions in which spinal activity is elevated, such as in chronic pain states. In this regard, the glycine receptor α3 subtype has been proposed to play an important role in chronic pain (Harvey et al., 2004) and mediates the effects of GLYT inhibitors in various models of chronic pain (Morita et al., 2008). It is interesting to note that, unlike GLYT2 inhibitors, intrathecally delivered GLYT1 inhibitors also produce hyperalgesia and have a slower onset of anti-allodynia in chronic pain models, both of which are mediated by NMDA receptors (Morita et al., 2008). Our findings suggest that NAGly might also reduce the pro-algesic NMDA-mediated actions associated with GLYT1 inhibitors. It is unclear whether endogenous NAGly might produce similar effects because the concentration of NAGly used in the present study was likely to be above the gross endogenous levels reported in the spinal cord (Huang et al., 2001). While many aspects of endogenous NAGly signalling remains to be determined, the concentration of NAGly near its site of release may be much higher, as reported for anandamide (Wilson and Nicoll, 2001), and endogenous levels of NAGly may be altered in chronic pain states.
In conclusion, the present results suggest that within lamina II of the spinal cord, the transporter GLYT2 has a role in both vesicle replenishment with glycine and terminating glycinergic transmission by uptake of glycine from the synaptic cleft. The endogenous ligand NAGly differs in some respects from GLYT2 inhibitors and could produce fine tuning of GLYT2 function to enhance the inhibitory glycinergic neurotransmission, while suppressing NMDA transmission. Overall, NAGly may modulate nociceptive transmission within the superficial dorsal horn via multiple mechanisms, including glycine transporters and possibly NMDA receptors, in addition to T-type Ca2+-channels which have been reported to mediate it's peripheral analgesic actions (Barbara et al., 2009; Ross et al., 2009). Together, the pleiotropic nature of NAGly provides novel combinations of endogenous targets for future analgesic strategies.
Acknowledgments
This work was supported by National Health & Medical Research Council of Australia Grant 402564 (CWV and RJV).
Glossary
Abbreviations
- ACSF
artificial cerebrospinal fluid
- ALX-1393
O-[(2-benzyloxyphenyl-3-fluorophenyl) methyl]-L-serine
- ALX-5407 (NFPS)
N-[3-(4′-fluorophenyl)-3-(4′-phenylphenoxy)propyl]sarcosine hydrochloride
- AP5
DL-(-)-2-amino-5-phosphonopentanoic acid
- CNQX
6-cyano-2,3-dihdroxy-7-nitro-quinoxaline
- DCKA
5,7-dichlorokynurenic acid
- EPSC
excitatory postsynaptic current
- FAAH
fatty acid amide hydrolase
- IPSC
inhibitory postsynaptic current
- NAGly
N-arachidonyl-glycine
- TTX
tetrodotoxin
Conflict of interest
None.
Supplemental material
References
- Ahmadi S, Muth-Selbach U, Lauterbach A, Lipfert P, Neuhuber WL, Zeilhofer HU. Facilitation of spinal NMDA receptor currents by spillover of synaptically released glycine. Science. 2003;300:2094–2097. doi: 10.1126/science.1083970. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC) Br J Pharmacol. 2009;158(Suppl. 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson BN, Bell SC, De Vivo M, Kowalski LR, Lechner SM, Ognyanov VI, et al. ALX 5407: a potent, selective inhibitor of the hGIyT1 glycine transporter. Mol Pharmacol. 2001;60:1414–1420. doi: 10.1124/mol.60.6.1414. [DOI] [PubMed] [Google Scholar]
- Aubrey KR, Vandenberg RJ. N[3-(4′-fluorophenyl)-3-(4′-phenylphenoxy)propyl]sarcosine (NFPS) is a selective persistent inhibitor of glycine transport. Br J Pharmacol. 2001;134:1429–1436. doi: 10.1038/sj.bjp.0704381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubrey KR, Rossi FM, Ruivo R, Alboni S, Bellenchi GC, Le Goff A, et al. The transporters GlyT2 and VIAAT cooperate to determine the vesicular glycinergic phenotype. J Neurosci. 2007;27:6273–6281. doi: 10.1523/JNEUROSCI.1024-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balakrishnan V, Kuo SP, Roberts PD, Trussell LO. Slow glycinergic transmission mediated by transmitter pooling. Nat Neurosci. 2009;12:286–294. doi: 10.1038/nn.2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbara G, Alloui A, Nargeot J, Lory P, Eschalier A, Bourinet E, et al. T-type calcium channel inhibition underlies the analgesic effects of the endogenous lipoamino acids. J Neurosci. 2009;29:13106–13114. doi: 10.1523/JNEUROSCI.2919-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beato M. The time course of transmitter at glycinergic synapses onto motoneurons. J Neurosci. 2008;28:7412–7425. doi: 10.1523/JNEUROSCI.0581-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradaia A, Schlichter R, Trouslard J. Role of glial and neuronal glycine transporters in the control of glycinergic and glutamatergic synaptic transmission in lamina X of the rat spinal cord. J Physiol. 2004;559:169–186. doi: 10.1113/jphysiol.2004.068858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradshaw HB, Rimmerman N, Hu SS, Benton VM, Stuart JM, Masuda K, et al. The endocannabinoid anandamide is a precursor for the signaling lipid N-arachidonoyl glycine by two distinct pathways. BMC Biochem. 2009;10:14. doi: 10.1186/1471-2091-10-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burstein SH, Rossetti RG, Yagen B, Zurier RB. Oxidative metabolism of anandamide. Prostaglandins Other Lipid Mediat. 2000;61:29–41. doi: 10.1016/s0090-6980(00)00053-8. [DOI] [PubMed] [Google Scholar]
- Cascio MG, Minassi A, Ligresti A, Appendino G, Burstein S, Di Marzo V. A structure-activity relationship study on N-arachidonoyl-amino acids as possible endogenous inhibitors of fatty acid amide hydrolase. Biochem Biophys Res Commun. 2004;314:192–196. doi: 10.1016/j.bbrc.2003.12.075. [DOI] [PubMed] [Google Scholar]
- Chen L, Muhlhauser M, Yang CR. Glycine tranporter-1 blockade potentiates NMDA-mediated responses in rat prefrontal cortical neurons in vitro and in vivo. J Neurophysiol. 2003;89:691–703. doi: 10.1152/jn.00680.2002. [DOI] [PubMed] [Google Scholar]
- Chery N, de Koninck Y. Junctional versus extrajunctional glycine and GABA(A) receptor-mediated IPSCs in identified lamina I neurons of the adult rat spinal cord. J Neurosci. 1999;19:7342–7355. doi: 10.1523/JNEUROSCI.19-17-07342.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Petrocellis L, Bisogno T, Davis JB, Pertwee RG, Di Marzo V. Overlap between the ligand recognition properties of the anandamide transporter and the VR1 vanilloid receptor: inhibitors of anandamide uptake with negligible capsaicin-like activity. FEBS Lett. 2000;483:52–56. doi: 10.1016/s0014-5793(00)02082-2. [DOI] [PubMed] [Google Scholar]
- Dohi T, Morita K, Kitayama T, Motoyama N, Morioka N. Glycine transporter inhibitors as a novel drug discovery strategy for neuropathic pain. Pharmacol Ther. 2009;123:54–79. doi: 10.1016/j.pharmthera.2009.03.018. [DOI] [PubMed] [Google Scholar]
- Gomeza J, Hulsmann S, Ohno K, Eulenburg V, Szoke K, Richter D, et al. Inactivation of the glycine transporter 1 gene discloses vital role of glial glycine uptake in glycinergic inhibition. Neuron. 2003a;40:785–796. doi: 10.1016/s0896-6273(03)00672-x. [DOI] [PubMed] [Google Scholar]
- Gomeza J, Ohno K, Hulsmann S, Armsen W, Eulenburg V, Richter DW, et al. Deletion of the mouse glycine transporter 2 results in a hyperekplexia phenotype and postnatal lethality. Neuron. 2003b;40:797–806. doi: 10.1016/s0896-6273(03)00673-1. [DOI] [PubMed] [Google Scholar]
- Graham BA, Schofield PR, Sah P, Callister RJ. Altered inhibitory synaptic transmission in superficial dorsal horn neurones in spastic and oscillator mice. J Physiol. 2003;551:905–916. doi: 10.1113/jphysiol.2003.049064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey RJ, Depner UB, Wassle H, Ahmadi S, Heindl C, Reinold H, et al. GlyR alpha3: an essential target for spinal PGE2-mediated inflammatory pain sensitization. Science. 2004;304:884–887. doi: 10.1126/science.1094925. [DOI] [PubMed] [Google Scholar]
- Hermanns H, Muth-Selbach U, Williams R, Krug S, Lipfert P, Werdehausen R, et al. Differential effects of spinally applied glycine transporter inhibitors on nociception in a rat model of neuropathic pain. Neurosci Lett. 2008;445:214–219. doi: 10.1016/j.neulet.2008.09.012. [DOI] [PubMed] [Google Scholar]
- Huang SM, Bisogno T, Petros TJ, Chang SY, Zavitsanos PA, Zipkin RE, et al. Identification of a new class of molecules, the arachidonyl amino acids, and characterization of one member that inhibits pain. J Biol Chem. 2001;276:42639–42644. doi: 10.1074/jbc.M107351200. [DOI] [PubMed] [Google Scholar]
- Johnson JW, Ascher P. Glycine potentiates the NMDA response in cultured mouse brain neurons. Nature. 1987;325:529–531. doi: 10.1038/325529a0. [DOI] [PubMed] [Google Scholar]
- Katsurabayashi S, Kubota H, Higashi H, Akaike N, Ito Y. Distinct profiles of refilling of inhibitory neurotransmitters into presynaptic terminals projecting to spinal neurones in immature rats. J Physiol. 2004;560:469–478. doi: 10.1113/jphysiol.2004.067017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohno M, Hasegawa H, Inoue A, Muraoka M, Miyazaki T, Oka K, et al. Identification of N-arachidonylglycine as the endogenous ligand for orphan G-protein-coupled receptor GPR18. Biochem Biophys Res Commun. 2006;347:827–832. doi: 10.1016/j.bbrc.2006.06.175. [DOI] [PubMed] [Google Scholar]
- Martina M, Gorfinkel Y, Halman S, Lowe JA, Periyalwar P, Schmidt CJ, et al. Glycine transporter type 1 blockade changes NMDA receptor-mediated responses and LTP in hippocampal CA1 pyramidal cells by altering extracellular glycine levels. J Physiol. 2004;557:489–500. doi: 10.1113/jphysiol.2004.063321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHugh D, Hu SS, Rimmerman N, Juknat A, Vogil Z, Walker JM, et al. N-arachidonoyl glycine, an abundant endogenous lipid, potently drives directed cellular migration through GPR18, the putative abnormal cannabidiol receptor. BMC Neurosci. 2010;11:44. doi: 10.1186/1471-2202-11-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita K, Motoyama N, Kitayama T, Morioka N, Kifune K, Dohi T. Spinal antiallodynia action of glycine transporter inhibitors in neuropathic pain models in mice. J Pharmacol Exp Ther. 2008;326:633–645. doi: 10.1124/jpet.108.136267. [DOI] [PubMed] [Google Scholar]
- Narikawa K, Furue H, Kumamoto E, Yoshimura M. In vivo patch-clamp analysis of IPSCs evoked in rat substantia gelatinosa neurons by cutaneous mechanical stimulation. J Neurophysiol. 2000;84:2171–2174. doi: 10.1152/jn.2000.84.4.2171. [DOI] [PubMed] [Google Scholar]
- Oh DY, Yoon JM, Moon MJ, Hwang JI, Choe H, Lee JY, et al. Identification of farnesyl pyrophosphate and N-arachidonylglycine as endogenous ligands for GPR92. J Biol Chem. 2008;283:21054–21064. doi: 10.1074/jbc.M708908200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parmar N, Ho WS. N-arachidonoyl glycine, an endogenous lipid that acts as a vasorelaxant via nitric oxide and large conductance calcium-activated potassium channels. Br J Pharmacol. 2010 doi: 10.1111/j.1476-5381.2009.00622.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prusakiewicz JJ, Kingsley PJ, Kozak KR, Marnett LJ. Selective oxygenation of N-arachidonylglycine by cyclooxygenase-2. Biochem Biophys Res Commun. 2002;296:612–617. doi: 10.1016/s0006-291x(02)00915-4. [DOI] [PubMed] [Google Scholar]
- Ross HR, Gilmore AJ, Connor M. Inhibition of human recombinant T-type calcium channels by the endocannabinoid N-arachidonoyl dopamine. Br J Pharmacol. 2009;156:740–750. doi: 10.1111/j.1476-5381.2008.00072.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseau F, Aubrey KR, Supplisson S. The glycine transporter GlyT2 controls the dynamics of synaptic vesicle refilling in inhibitory spinal cord neurons. J Neurosci. 2008;28:9755–9768. doi: 10.1523/JNEUROSCI.0509-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux MJ, Supplisson S. Neuronal and glial glycine transporters have different stoichiometries. Neuron. 2000;25:373–383. doi: 10.1016/s0896-6273(00)80901-0. [DOI] [PubMed] [Google Scholar]
- Santos SF, Rebelo S, Derkach VA, Safronov BV. Excitatory interneurons dominate sensory processing in the spinal substantia gelatinosa of rat. J Physiol. 2007;581:241–254. doi: 10.1113/jphysiol.2006.126912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheskin T, Hanus L, Slager J, Vogel Z, Mechoulam R. Structural requirements for binding of anandamide-type compounds to the brain cannabinoid receptor. J Med Chem. 1997;40:659–667. doi: 10.1021/jm960752x. [DOI] [PubMed] [Google Scholar]
- Singer JH, Talley EM, Bayliss DA, Berger AJ. Development of glycinergic synaptic transmission to rat brain stem motoneurons. J Neurophysiol. 1998;80:2608–2620. doi: 10.1152/jn.1998.80.5.2608. [DOI] [PubMed] [Google Scholar]
- Spike RC, Watt C, Zafra F, Todd AJ. An ultrastructural study of the glycine transporter GLYT2 and its association with glycine in the superficial laminae of the rat spinal dorsal horn. Neuroscience. 1997;77:543–551. doi: 10.1016/s0306-4522(96)00501-5. [DOI] [PubMed] [Google Scholar]
- Succar R, Mitchell VA, Vaughan CW. Actions of N-arachidonyl-glycine in a rat inflammatory pain model. Mol Pain. 2007;3:24. doi: 10.1186/1744-8069-3-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe M, Takasu K, Yamaguchi S, Kodama D, Ono H. Glycine transporter inhibitors as a potential therapeutic strategy for chronic pain with memory impairment. Anesthesiology. 2008;108:929–937. doi: 10.1097/ALN.0b013e31816c9044. [DOI] [PubMed] [Google Scholar]
- Turecek R, Trussell LO. Presynaptic glycine receptors enhance transmitter release at a mammalian central synapse. Nature. 2001;411:587–590. doi: 10.1038/35079084. [DOI] [PubMed] [Google Scholar]
- Vuong LAQ, Mitchell VA, Vaughan CW. Actions of N-arachidonyl-glycine in a rat neuropathic pain model. Neuropharmacology. 2008;54:189–193. doi: 10.1016/j.neuropharm.2007.05.004. [DOI] [PubMed] [Google Scholar]
- Wiles AL, Pearlman RJ, Rosvall M, Aubrey KR, Vandenberg RJ. N-Arachidonyl-glycine inhibits the glycine transporter, GLYT2a. J Neurochem. 2006;99:781–786. doi: 10.1111/j.1471-4159.2006.04107.x. [DOI] [PubMed] [Google Scholar]
- Williams JR, Khandoga AL, Goyal P, Fells JI, Perygin DH, Siess W, et al. Unique ligand selectivity of the GPR92/LPA(5) lysophosphatidate receptor indicates role in human platelet activation. J Biol Chem. 2009;284:17304–17319. doi: 10.1074/jbc.M109.003194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson RI, Nicoll RA. Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nature. 2001;410:588–592. doi: 10.1038/35069076. [DOI] [PubMed] [Google Scholar]
- Xu TX, Gong N, Xu TL. Inhibitors of GlyT1 and GlyT2 differentially modulate inhibitory transmission. Neuroreport. 2005;16:1227–1231. doi: 10.1097/00001756-200508010-00019. [DOI] [PubMed] [Google Scholar]
- Yang Z, Aubrey KR, Alroy I, Harvey RJ, Vandenberg RJ, Lynch JW. Subunit-specific modulation of glycine receptors by cannabinoids and N-arachidonyl-glycine. Biochem Pharmacol. 2008;76:1014–1023. doi: 10.1016/j.bcp.2008.07.037. [DOI] [PubMed] [Google Scholar]
- Yin H, Chu A, Li W, Wang B, Shelton F, Otero F, et al. Lipid G protein-coupled receptor ligand identification using beta-arrestin pathhunter (TM) assay. J Biol Chem. 2009;284:12328–12338. doi: 10.1074/jbc.M806516200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zafra F, Aragon C, Olivares L, Danbolt NC, Gimenez C, Storm-Mathisen J. Glycine transporters are differentially expressed among CNS cells. J Neurosci. 1995;15:3952–3969. doi: 10.1523/JNEUROSCI.15-05-03952.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeilhofer HU. The glycinergic control of spinal pain processing. Cell Mol Life Sci. 2005;62:2027–2035. doi: 10.1007/s00018-005-5107-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeilhofer HU, Studler B, Arabadzisz D, Schweizer C, Ahmadi S, Layh B, et al. Glycinergic neurons expressing enhanced green fluorescent protein in bacterial artificial chromosome transgenic mice. J Comp Neurol. 2005;482:123–141. doi: 10.1002/cne.20349. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.