

SUMMARY
Caenorhabditis elegans exhibit avoidance behavior when presented with diverse bacterial pathogens. We hypothesized that exposure to pathogens might not only cause worms to move away but also simultaneously activate pathways that promote resistance to the pathogen. We show that brief exposure to the virulent or avirulent strains of the bacterial pathogen enteropathogenic E. coli (EPEC) “conditions” or “immunizes” C. elegans to survive a subsequent exposure that would otherwise prove lethal. Conditioning requires dopaminergic neurons. Conditioning also requires the p38 MAP Kinase pathway, which regulates innate immunity, and the insulin/IGFR pathway, which regulates lifespan. Our findings suggest that the molecular pathways that regulate innate immunity and lifespan and provide protection may, in nature, be regulated or “conditioned” by exposure to pathogens, and perhaps allow survival in noxious environments.
Keywords: EPEC, C. elegans, pathogenesis, aging, model systems
INTRODUCTION
In its natural habitat, the nematode Caenorhabditis elegans encounters diverse microorganisms. It utilizes odors or other chemical cues to detect bacteria on which it then feeds (Bargmann, 2006). However, C. elegans is susceptible to natural bacterial pathogens such as Microbacterium nematophilum (Hodgkin et al., 2000), as well as to a wide variety of gram-positive and gram-negative bacteria and even fungi that are pathogenic in humans (e.g. Pseudomonas aeruginosa (Darby et al., 1999; Tan, 1999b)). Studies of C. elegans-pathogen interactions indicate that some bacteria, such as Salmonella typhimurium, colonize and persistently infect C. elegans causing animals to succumb within days (Aballay et al., 2000), whereas others, such as P. aeruginosa and enteropathogenic E. coli (EPEC)), secrete toxins that kill within hours (Anyanful et al., 2005; Mahajan-Miklos et al., 1999). Significantly, many virulence factors or toxins responsible for killing C. elegans also contribute to disease in plant and mammalian systems (Hendrickson et al., 2001; Tan, 1999a), suggesting conservation of virulence mechanisms among diverse eukaryotic hosts.
Studies of C. elegans-pathogen interactions also indicate that nematodes have evolved behavioral mechanisms that facilitate survival. C. elegans can distinguish virulent from avirulent strains and thereby undergo avoidance behavior (Zhang et al., 2005). For example, Serratia marcescens produce a cyclic lipodepsipentapeptide, serrawettin W2 that is detected by AWB chemosensory neurons in C. elegans (Pradel et al., 2007) and causes animals to move away from bacteria. Moreover, Bargmann and colleagues found that C. elegans modifies its olfactory preferences after exposure to pathogenic bacteria, so as to avoid noxious strains and prefer nonpathogenic ones (Zhang et al., 2005). This change in preference is mediated by serotonin in the ADF sensory neurons, and by serotonin-gated chloride channels in sensory interneurons (Zhang et al., 2005).
Besides aversive behavior, C. elegans have also evolved protective mechanisms against pathogens. These include the p38 Mitogen-Activated Protein (MAP) kinase (Kim et al., 2002) and the insulin/IGF-1 receptor (IGFR) signaling systems (Garsin et al., 2003). The p38 MAP kinase pathway, which includes NSY-1, SEK-1 and PMK-1 kinases, has been proposed to mediate pathogen-specific responses by regulating expression of ~86 protective immune response genes (Troemel et al., 2006). The insulin/IGF-1 signaling pathway controls longevity and dauer formation in C. elegans (Kenyon et al., 1993). Activation of the IGFR homologue DAF-2 initiates a signaling cascade that negatively regulates the FOX-O family transcription factor DAF-16 (Ogg et al., 1997). DAF-16 up-regulates expression of ~263 genes that promote longevity, and down-regulates expression of ~251 life-shortening genes (Hamilton et al., 2005; Murphy et al., 2003). Genes up- or down-regulated by DAF-16 include those likely to metabolize toxins (e.g. cytochrome p450s) or to destroy bacteria (e.g. lysozyme, saposins). It has been proposed that DAF-16 controls basal innate immune responses in C. elegans, whereas MAP kinase signaling regulates pathogen-induced responses (Troemel et al., 2006).
Using both C. elegans and mammalian systems, we have been studying EPEC (O127:H6 (Levine et al., 1985)) and the related pathogen enterohemmorhagic E. coli (EHEC O157:H7; (Frankel et al., 1998)). EPEC is a human gastrointestinal pathogen that is transmitted via contaminated food and water and causes severe diarrhea in humans, leading to high mortality particularly among infants in developing nations (Clarke et al., 2002). We have shown previously that EPEC and EHEC paralyze and kill C. elegans via a secreted toxin (Anyanful et al., 2005). Both activities depend on the presence of tryptophan in the growth media and on the bacterial tryptophanase gene (tnaA) as well as on genes encoding virulence factors (e.g. espF). Importantly, we have also identified the MAP kinase and aging pathways as mediators of a protective response to EPEC in C. elegans.
In contrast to the wealth of information on aversive behavioral responses to pathogens in C. elegans and on innate immune and aging genes that provide protection, information on how neuronal sensing of pathogens or behavior itself might regulate expression of protective genes, is much more limited. Kenyon and colleagues have shown that specific neurons within the chemosensory organs in the front of the animal, called amphids, can perceive and mediate environmental cues that regulate lifespan (Alcedo and Kenyon, 2004; Apfeld and Kenyon, 1999). These data raise the possibility that detection of pathogens by olfactory or other sensory cues might induce protracted changes in the expression of protective genes.
We report here that, as expected, the brief exposure to EPEC induces aversive behavior. The important advance in this work is that brief exposure to either EPEC or to avirulent EPEC strains conditions worms to survive a subsequent exposure to EPEC that would otherwise prove lethal. Conditioning requires dopaminergic neurons as well as genes in the innate immunity and insulin/IGFR1 signaling pathways. Together, our findings suggest that C. elegans uses sensory mechanisms both to trigger aversive behavior and to induce expression of protective genes.
RESULTS
C. elegans avoid contact with EPEC
Exposure of C. elegans to EPEC grown in a lawn causes paralysis within 30 minutes and subsequently death, scored as lack of movement and pharyngeal muscle activity (pumping) in worms 24 hours after transfer to plates containing the nonpathogenic E. coli strain OP50, the laboratory food source (Anyanful et al., 2005). Paralysis and killing by EPEC depend on the presence of tryptophan in the media, and on the activity of bacterial tryptophanase (TnaA) and EPEC virulence factors (e.g. EspF) (Anyanful et al., 2005). The rate of killing depends in part on formulation of the agar media: EPEC grown on Luria-Bertani (LB) agar containing tryptophan (LBT) kills ~90% of wildtype worms (N2) within 3 hours (Fig. 1a), whereas EPEC grown on ECD media kills with the same efficacy but within ~8 hours (Anyanful et al., 2005). Killing upon exposure to EPEC/LBT was evident with similar kinetics using N2 strains from three different laboratories (not shown) and with rol-6(su1006) roller worms (Supp. Fig. 1a).
Figure 1. Conditioning of C. elegans by pre-exposure to virulent or avirulent EPEC strains.

For A, and B, mean values +/- SEM are presented. For D, E, G, and H, statistically significant differences, calculated by ANOVA, of mean survival values are evident as a lack of overlap of 95% confidence intervals. A. Time course of killing of C. elegans by constitutive exposure to an EPEC lawn. N2 worms were exposed to EPEC grown on LBT agar for the times indicated and then removed to OP50 grown on NGM agar. After 24 hours, survival was scored. Conditioning and challenge times used were 0.5 and 3 hrs respectively. The linear trend was highly significant (p<0.0001). B. Time course of C. elegans movement away from an 8mm diameter spot of EPEC or EPECΔtnaA. The percentage of worms outside the spot was scored at each time point. C. Conditioning scheme. Conditioned exposure of C. elegans to EPEC lawns on LBT plates for 30 minutes (conditioning period), followed by exposure to OP50 on NGM plates for 3 hours (waiting period), and then exposure to EPEC on LBT plates for 3 hours (challenge). D. Conditioning of C. elegans using the scheme in C increases survival. E. A waiting period of 3 hrs induces optimal survival, and by 48 hours no conditioning was evident. F. Alternative conditioning scheme with avirulent strains of EPEC (EPECΔtnaA, EPECΔespF) or with EPEC grown in lawns on LB plates, which renders the bacteria unable to kill. An exposure time of 3 hours induces optimal survival. G. Exposure of C. elegans to avirulent EPEC strains or to EPEC grown in lawns on LB, to Pseudomonas aeruginosa strain PAK1, or to C. rodentium using the conditioning scheme in F. Survival of C. elegans conditioned on EPEC as in C is shown for comparison. H. EPEC or EPECΔtnaA were grown on filters on top of LBT agar overnight, the filters discarded, and the animals pre-exposed on the plates for 40 minutes for EPEC or 3 hours for EPECΔtnaA. Animals were then exposed to OP50 for 3 hours (waiting), and then to EPEC for 3 hours (challenge).
When wildtype C. elegans (N2) were placed within a spot of EPEC (8mm in diameter), approximately 50% of the animals exit the spot within thirty minutes, and those remaining became paralyzed and later died (Fig. 1b). By contrast, such avoidance behavior was not evident with EPECΔtnaA (Fig. 1b), a strain that does not cause paralysis or death (Anyanful et al., 2005). Thus, toxins produced by EPEC induce behavioral avoidance.
Conditioning facilitates survival of C. elegans upon lethal exposure to EPEC
We next determined whether brief exposures to EPEC might activate protective responses within C. elegans and thus increase their capacity to survive a subsequent exposure that would otherwise prove lethal. To do this, we developed a “conditioning” protocol (Fig. 1c) so as to more precisely control the time and degree of exposure to EPEC. N2 worms were incubated with EPEC grown in a lawn on LBT agar for thirty minutes (“pre-exposure” period; Fig. 1c), and then moved to NGM plates containing OP50 for three hours (waiting period). Worms were then re-exposed to an EPEC lawn on LBT plates for an additional three hours (challenge) and the number of survivors scored. The EPEC lawn covered the 6cm plate so that worms could not exit during the pre-exposure or challenge period. The duration of the pre-exposure period was chosen so as to maximize survival, and the duration of the challenge period to maximize lethality and minimize variance (Fig. 1a). Pre-exposure of N2 worms to EPEC/LBT induced survival of ~40% of animals, compared to ~9.9% of animals without previous exposure, a ~4 fold increase in survival (Fig. 1d). Similarly, pre-exposure of rol-6(su1006) worms to EPEC induced a statistically significant increase in survival of ~4.9 fold (34% survival with pre-exposure compared to 7% without; Supp. Fig. 1b). Pre-exposure of N2 worms to LBT plates lacking bacteria was without effect (data not shown). Pre-exposure to EPEC for periods greater or less than thirty minutes proved less effective in promoting survival (Supp. Fig. 1c). Likewise, waiting periods of less than three hours or more than four hours proved less effective (Fig. 1e), and though increased survival was still evident after 24 hours, none was evident after 48 hours (Fig. 1e). These data suggest that pre-exposure to EPEC coupled with a waiting period promotes increased survival of C. elegans, and that the effect lasts for extended periods.
We next determined whether survival of C. elegans induced by pre-exposure required virulence factors expressed by EPEC. To do this, we pre-exposed C. elegans to EPEC grown on LB without added tryptophan, or to the EPEC strains EPECΔespF or EPECΔtnaA grown on LBT. Growth of EPEC on LB renders the bacteria avirulent, as no killing was observed even with extended exposure (up to 96 hrs, the longest time tested; not shown). Likewise, EPECΔespF and EPECΔtnaA contain isogenic mutations in espF and tnaA respectively, and are avirulent when grown on LBT even with extended exposures (up to 48 hrs for EPECΔespF or 96 hrs for EPECΔtnaA, the longest times tested; not shown). The capacity to kill C. elegans can be restored to EPECΔespF and EPECΔtnaA by complementation with plasmid- or chromosomally-encoded espF or tnaA, respectively (S. Bhatt and D.K. unpublished, and (Anyanful et al., 2005)). Neither pre-exposure to EPEC grown under avirulent conditions, nor to the avirulent strains EPECΔespF and EPECΔtnaA for thirty minutes (the protocol shown in Fig. 1c), induced a statistically significant increase in survival (Fig. 1g). Together, these data suggest that the transfer protocol itself did not induce enhanced survival, and that virulence factors or toxins produced by EPEC facilitate survival induced by pre-exposure.
Avirulent EPEC strains and other pathogens induce conditioning upon extended exposure
Pre-exposure to EPEC grown on LB, or to either EPECΔespF or EPECΔtnaA grown on LBT for an extended period (three hours; Figs. 1f,g) induce survival to the same extent as pre-exposure to EPEC grown on LBT for 30 minutes (4.3 fold for N2 and 5.4 fold for rol-6(su1006); Fig. 1d & Supp. Fig. 1b). Pre-exposure to EPEC grown on LBT for thirty minutes, and then to EPECΔespF, EPECΔtnaA, or to EPEC/LB during the waiting period, did not result in a significant difference in survival compared to exposure to OP50/NGM during the waiting period (not shown). Thus, survival can be enhanced by pre-exposure to EPEC under both virulent and avirulent conditions, as well as by avirulent EPEC strains. However, for all the conditioning protocols tested, no more than 40-50% of the animals survived.
We next grew EPEC or EPECΔtnaA overnight atop 0.2 μm nitrocellulose filters. After removal of the filters, the plates were used to pre-expose the animals for forty minutes to EPEC, or for three hours to EPECΔtnaA. After a waiting period of three hours, animals were challenged for three hours with EPEC. We observed a significant increase in survival on plates pre-exposed to EPEC using this protocol, though the level was lower than that observed with direct contact. By contrast no such effects were evident on plates pre-exposed to EPECΔtnaA on filters. These data suggest that survival induced by pre-exposure to EPEC did not require direct contact whereas that induced by EPECΔtnaA did (Fig. 1h). The contact-dependence of pre-exposure and the difference in pre-exposure times required for EPEC grown under virulent or avirulent conditions or for avirulent strains, suggests that EPEC produces two “conditioning” factors; one is secreted, acts quickly, and depends on the toxin, whereas the other acts slowly, requires direct contact, and acts independently of the toxin.
We next assessed whether pre-exposure to other pathogenic bacteria could enhance survival upon exposure to EPEC. Some Pseudomonas aeruginosa strains (e.g. PA01, PA14; (Darby et al., 1999; Tan, 1999b) and PAK1 (Laws et al., 2006)) kill C. elegans. PAK1 produced no detectable deleterious effects in C. elegans, even over extended periods on LBT plates (96 hrs, the longest time tested; not shown). However, pre-exposure to PAK1 for 3 hours followed by exposure to EPEC for 3 hours induced a 2.7-fold increase in survival compared to animals exposed to OP50/NGM for 3 hours (Fig. 1g). By contrast, the rodent pathogen Citrobacter rodentium, which also does not kill C. elegans, did not induce a significant increase in survival to EPEC (Fig. 1g). These data suggest that some bacterial species can induce responses in C. elegans that are protective against other species.
Conditioning depends on the insulin/IGFR-1 pathway
The insulin/IGFR-1 signaling pathway, which regulates lifespan (Kenyon, 2001; Kenyon et al., 1993; Lin et al., 1997; Ogg et al., 1997), also protects C. elegans from a variety of pathogens (Garsin et al., 2003), including EPEC (Anyanful et al., 2005). Worms with mutations in daf-2 (e.g. daf-2(e1370)), which have extended lifespan (Kenyon et al., 1993), also are more resistant to EPEC, and mutations in daf-16 abrogate such effects ((e.g. daf-16(m26);daf-2(e1370), (Anyanful et al., 2005)). Thus, DAF-2 negatively regulates DAF-16. daf-16 mutants also succumb more quickly than N2 upon constitutive exposure to EPEC (Fig. 2a).
Figure 2. Genes that promote longevity and innate immunity mediate conditioning.

A. daf-16 and hsf-1 mutants succumb more quickly that N2 upon constitutive exposure to EPEC, suggesting a protective role for these genes. B. hsf-1(sy441), but not daf-16 mutants, are as conditionable as N2 by EPECΔtnaA. daf-16 mutants carrying an extrachromosomal array are partially rescued for conditioning. Mean survival +/- SEM are presented, and fold differences relative to the unconditioned control are indicated above the bar. For statistical analysis see C and D. C. Death rate during constitutive exposure to EPEC is not correlated with fold increase in conditioned survival. Death rates and are plotted as a function of fold-increase in survival with conditioning. Fold conditioning was calculated as a quotient (percent survival with conditioning divided by the percent survival without), and death rates were estimated by regression for 30 mutants described in this study. Regression analysis for only N2, rol-6(su1006) and daf-16, and innate immunity mutants (small black dots) showed a strong negative correlation. Regression analysis for all mutants (small and large black dots) showed no correlation. D. Adjusted mean survival with conditioning for mutants shown in B using an ANCOVA model (see Methods). For this panel and G below, nonoverlap of 95% confidence intervals indicates statistical significance. No significant differences exist between control strains N2 or rol-6(su1006), and hsf-1(sy441). However, statistically significant differences are evident between N2 and all daf-16 mutants, between the daf-16 parental strains and strains rescued with the integrated array (I.A.) and the extrachromosomal array (Ex.A.). There was no difference between daf-16 mutants and the baseline unconditioned control, with the exception of daf-16(mu86), which exhibited slight conditioning. E. Representative images showing nuclear translocation of DAF-16∷GFP in N2 animals upon exposure to EPEC for 30 minutes or EPECΔtnaA for 3 hours, but not in unexposed animals. In some experiments, C. elegans were co-stained with DAPI to visualize nuclei. Note that GFP-fluorescence colocalizes with DAPI staining in images of C. elegans exposed to EPECΔtnaA. All images were acquired and fluorescence adjusted in an identical manner. Scale bar, 30 μm. F. Time course of survival of sek-1(km4) and pmk-1(km25) animals following constitutive exposure to EPEC. G. Adjusted mean survival with conditioning upon pre-exposure to EPECΔtnaA for N2, sek-1(km4) and pmk-1(km25) animals .
We next assessed whether daf-16 mutants exhibited increased survival upon pre-exposure to EPEC or EPECΔtnaA. In contrast to wild-type C. elegans (N2), neither daf-16(m26), daf-16(mgDf50), daf-16(mu86), nor daf-16(mgDf47) animals exhibited survival upon pre-exposure to EPECΔtnaA or to EPEC (1.6 to 1.8 fold; Fig. 2b with EPECΔtnaA; Supp. Fig. 2a with EPEC) to the same extent as N2 (e.g. 4 to 4.3 fold for N2). Increased survival upon pre-exposure to other avirulent strains (EPECΔespF) or by EPEC grown under nonpathogenic conditions (LB medium) also required daf-16 (not shown). Because results with pre-exposure to EPEC and EPECΔtnaA were nearly identical in our assays, subsequent experiments presented below show only results with EPECΔtnaA. Data with EPEC are presented in Supplementary Information or not shown. Enhanced survival upon pre-exposure was evident in transgenic daf-16(mu86) or daf-16(mgDf47) animals expressing DAF-16 under its own promoter either as an integrated array (I.A.; e.g. 3.5 to 4.8 fold for the rescued strains compared to 1.7 fold for the parental strains) or as an extrachromosomal array (Ex. A.; Fig. 2b; Supp. Fig. 2a). Notably, the daf-16 mutants and their integrated complemented strains still succumbed faster than N2 (Fig. 2a), though daf-16(mu86) animals expressing DAF-16 under its own promoter as an extrachromosomal array had a slightly slower death rate (Fig. 2b).
To determine whether the effect of lifespan genes on conditioning was specific, we assessed whether enhanced survival upon pre-exposure required other genes that mediate protective responses to pathogens. HSF-1 activates a different subset of genes than DAF-16 (Singh and Aballay, 2006), and hsf-1(RNAi) worms have a shorter lifespan than N2 (Hsu et al., 2003). Moreover, hsf-1 is required for innate immunity to a variety of bacterial pathogens (Singh and Aballay, 2006). hsf-1(sy441) suffer a severe egg laying defect (Hadju-ronin et al., 2004) and die from internal hatching of young. To minimize contributions from such defects in our assays, we only used young adults, and our experiments were completed within 7 hours. We did not observe the “bag of worms” phenotype associated with internal hatching suggesting that egg laying defects play little if any role in phenotypes we have studied. Both hsf-1 RNAi (not shown) and hsf-1(sy441) succumbed faster than N2 when exposed to EPEC, indicating a protective role (Fig. 2a). However, hsf-1(sy441) animals exhibited 5.2 fold more survival upon pre-exposure to EPECΔtnaA, compared to unconditioned animals, an increase comparable to that seen with N2 animals (Fig. 2b; see also Supp. Fig. 2a for EPEC).
These data suggest that lack of conditioning is not due to increased susceptibility, and that conditioning is a specific response independent of other protective genes. Nevertheless, the observation that daf-16 mutations cause animals to succumb more quickly raised the possibility that the apparent lack of conditioning of daf-16 mutations results from an increased sensitivity to EPEC. To establish quantitatively whether a correlation existed between the rate of killing and the degree of conditioning, we first estimated the death rate of each mutant using regression techniques. We then regressed this rate against the fold conditioning, calculated as the fold increase in survival with pre-exposure to EPECΔtnaA/LBT (as per protocol in Fig. 1f) or EPEC (as per protocol in Fig. 1c) compared with pre-exposure only to OP50/NGM. When we considered only N2 and rol-6(su1006) together with strains that showed little conditioning (e.g. daf-16 mutations, mutations in innate immunity genes, and dopamine signaling mutants; see below) we found a statistically significant negative correlation between death rate and conditioning (small black dots in Fig. 2c). However, when we considered these mutations, together with mutations in hsf-1, serotonin signaling, and other genes (see below), we found a regression line with slope ~0 (small and large black dots in Fig. 2c), indicating that no correlation existed between the rate of killing and the degree of conditioning. Thus, from a statistical standpoint, the apparent lack of conditioning in several mutant strains was not simply a reflection of a faster death rate.
The variability in rates of susceptibility to EPEC across a wide variety of strains raised questions of whether background mutations contribute to fold conditioning, complicating comparison of conditioning in different strains. To address this issue, we developed an analysis of covariance (ANCOVA) model to assess the contribution to conditioning of baseline survival without pre-exposure. Using ANCOVA, we quantitatively defined the contribution of extraneous variability that derives from pre-existing individual strain differences to the mean conditioned survival, insofar as those differences are reflected in the baseline survival. We then adjusted the mean conditioned survival to compensate for the fact that different mutants have different levels of survival without pre-exposure. The ANCOVA analysis accounts for procedural and strain differences because the baseline used for comparison of different mutants is the adjusted mean survival for all mutant animals considered. Notably, because the rate of death and degree of conditioning appeared highly uncorrelated, the ANCOVA correction was in most cases minimal (e.g. compare Figs. 2b and 2d), though differences between the corrected levels and fold conditioning were evident with particular mutants that showed very low basal levels of survival with pre-exposure (see Fig. 4e and Supp. Fig. 4i below). Adjusted mean survival with pre-exposure, together with 95% confidence intervals, are shown for daf-16 mutants, the rescued strains, and hsf-1(sy441) in Fig. 2d, and below for other mutants. For ease of comparing different mutants, we hereafter refer to the adjusted mean survival with pre-exposure as “conditioned survival,” and present non-normalized data with and without pre-exposure to EPEC or EPECΔtnaA in Supplementary Information.
Figure 4. Dopaminergic neuron signaling mediates conditioned survival from EPEC.

A. Adjusted mean survival with conditioning for N2, rol-6(su1006), or daf-16 (mu86) animals expressing DAF-16∷GFP in neurons (unc-119 promoter) or intestinal cells (ges-1 promoter). B. Adjusted mean survival with conditioning of cat-1(e1111). C. Dopaminergic signaling mutants or N2 animals treated with 6-OH-DA, which kills dopaminergic neurons, eliminates conditioned survival, except minimal conditioning of cat-2(e1112) was evident. D. Rescue of conditioned survival in dop-3(vs106) animals expressing aqp-1 or spp-1. E. Serotonergic signaling mutants exhibit conditioned survival. ser-1(ok345) exhibited far less conditioning than other mutants, but baseline survival of this mutant were particularly low (see Supp Fig. 4I).
The lack of overlap of confidence intervals indicates that all the mutations in daf-16 tested cause a statistically significant decrease in conditioned survival compared to N2. All but one of these mutants, daf-16(mu86), did not show significant conditioned survival compared to the baseline adjusted mean survival without conditioning (“no conditioning”; Fig 2d). All the daf-16 strains rescued with overexpressed DAF-16 show a statistically significant increase compared to the baseline. Conditioned survival of daf-16(mu86) rescued with DAF-16 in an integrated array was not significantly different from the parental strain, daf-16(mu86). However, conditioned survival of daf-16(mu86) rescued with an extrachromosomal array or daf-16(mgDf47) rescued with an integrated array were statistically different from their respective parental strains. These data suggest that the rescue of both mutants was incomplete, in accordance with the observation that the rate of death for both mutants was not significantly different from their parental strains, and different from N2 (Fig. 2A). It is not without precedent that rescue by integrated or extrachromosomal arrays is not always complete. Transgenic arrays contain many copies of the wild type gene and thus there is overexpression, and the complete up- or downstream regulatory sequences may not be included in the rescue construct, thus gene regulation may not be “native” (Mello, 1995). Nevertheless, the observations that all four daf-16 mutants tested exhibited significantly lower conditioning levels compared to N2, and that conditioned survival of two of the rescued daf-16 mutant strains was significantly higher than baseline levels or levels seen with the parental strains, suggest a role for daf-16 in mediating conditioning.
DAF-16 protein translocates into the nucleus upon activation by environmental stimuli to promote longevity (Henderson and Johnson, 2001). To determine whether exposure to EPEC likewise induces DAF-16 translocation, we assessed nuclear localization of DAF-16 in N2 worms carrying an integrated DAF-16∷GFP transgene (N2;zls356[ExDAF-16∷DAF-16-GFP,rol-6(su1006]) that were exposed to EPEC for thirty minutes, or EPECΔtnaA for three hours, or left unexposed. Without exposure, GFP fluorescence was predominantly cytoplasmic. DAF-16∷GFP translocation into the nucleus, identified by DAPI staining, was evident within 30 minutes following exposure to EPEC and within 3 hrs to EPECΔtnaA (Fig. 2e). Together, these data suggest that DAF-16 translocates into the nucleus upon pre-exposure to bacteria.
Conditioning also depends on the MAP kinase pathway
Whereas genes activated by DAF-2/DAF-16 pathway have been proposed to confer basal resistance to pathogens, the SEK-1/PMK-1 pathway mediates expression of several immune response genes upon exposure to pathogens and regulate both innate immunity and longevity in C. elegans (Kim et al., 2002; Troemel et al., 2006). sek-1 and pmk-1 encode MAP kinase kinase and MAP kinase orthologs respectively. Both sek-1(km4) and pmk-1(km25) succumbed faster than N2 when exposed to EPEC indicating that these genes mediate protection against the pathogen (Fig. 2f and (Anyanful et al., 2005)). Moreover, neither sek-1(km4) nor pmk-1(km25) animals exhibited as much conditioned survival as N2 upon pre-exposure with EPECΔtnaA (Fig. 2g; Supp. Fig. 2b) or EPEC (Supp. Fig. 2c), and levels were similar those seen with daf-16 mutations (Fig. 2d). Together, these experiments indicate that daf-16, sek-1 and pmk-1 mediate not only protection against EPEC but also conditioning.
The DAF-16-regulated genes spp-1 and aqp-1 genes mediate conditioning
DAF-16 regulates genes that promote longevity, including factors that have antibacterial activity (e.g. lysozyme), inactivate toxins (e.g. cytochrome p450s), and facilitate responses to stress (Hamilton et al., 2005; Murphy et al., 2003). Murphy et al. (2003) found that DAF-16 upregulates ~263 genes that promote longevity and downregulates ~251 life-shortening genes. We reasoned that a subset of these genes might regulate susceptibility to EPEC by providing protection when activated by daf-16 during pre-exposure. We first determined which of the upregulated genes are responsible for increasing resistance to EPEC. To do this, we devised a screen in which E. coli HT115 bacteria expressing dsRNA for 85 selected genes of the ~263 upregulated genes described by Murphy et al. (2003) were fed to daf-2(e1370) worms. daf-2(e1370) mutants are not paralyzed by EPEC after 30 minutes and greater than 60% survive after 3 hours (Anyanful et al., 2005). Moreover, these animals appear to exhibit maximal conditioning constitutively, likely due to nuclear translocation of DAF-16. Thus, upon pre-exposure to EPEC or EPECΔtnaA, we could detect no differences in survival compared to daf-2(e1370) worms pre-exposed to OP50 (not shown). To identify genes up-regulated by DAF-16 that mediate survival in response to EPEC, we challenged daf-2(e1370) worms fed with various dsRNAs with EPEC, and scored for animals that readily succumbed.
We identified two genes, spp-1 and aqp-1, which, when inactivated, suppressed the capacity of daf-2(e1370) worms to survive a four hour exposure to EPEC (Fig. 3a). When inactivated, several other genes partially abrogated daf-2-mediated survival but not to the same extent as spp-1 and aqp-1, and were not pursued further (not shown). Inactivation of other DAF-16-inducible genes, such as lys-8, was without effect (Fig. 3a).
Figure 3. Genes regulated by DAF-16 and PMK-1 mediate protective responses to EPEC.

A. Inactivation of spp-1 and aqp-1 but not lys-8 by RNAi suppress survival of daf-2(e1370) upon constitutive exposure to EPEC for 4 hrs. The differences between the control and spp-1 (p<.001) or aqp-1 (p=.006), but not lys-8 (p=.50) are statistically significant. B. Time course of survival upon exposure to EPEC of N2 animals, or N2 animals in which spp-1, aqp-1, or lys-8 were inactivated by RNAi. C. Adjusted mean survival with conditioning for N2 animals, or N2 animals in which spp-1, aqp-1, or lys-8 were inactivated by RNAi. D. Time course of survival of N2, rol-6(su1006), aqp-1(tm2309), spp-1(ok2703) and rescued strains following exposure to EPEC. E. Adjusted mean survival with conditioning by pre-exposure to EPECΔtnaA of N2, rol-6(su1006), aqp-1(tm2309), spp-1(ok2703) and rescued strains. F. Rescue of conditioned survival in daf-16(mu86) or pmk-1(km25) by extrachromosomal arrays of aqp-1 or spp-1. G. Quantitative RTPCR of aqp-1 or spp-1 mRNA in N2, dop-3(vs106), daf-16(mu86), or pmk-1(km25) worms before and after conditioning, For each worm strain, the mRNA levels of aqp-1 and spp-1 before and after conditioning were normalized to those of act-1. Fold differences in the mRNA levels of aqp-1 and spp-1 were calculated relative to pre-exposure levels, which were arbitrarily set to a value of 100 for each (black bars).
We next determined whether spp-1 or aqp-1 mediated conditioned survival in N2 animals. To do this we fed N2 animals E. coli HT115 containing dsRNA for spp-1, aqp-1 or, as controls, lys-8, or the empty vector. As shown in Fig. 3b, inactivation of aqp-1 or spp-1 in N2 animals increased the sensitivity to EPEC. Moreover, knockdown of either spp-1, or aqp-1 resulted in no conditioned survival (Fig. 3c; see also Supp. Fig. 3a). In accordance with the RNAi results, the aquaporin mutant aqp-1(tm2309) and the saposin mutant spp-1(ok2703) succumbed faster than N2 (Fig. 3d), indicating a protective role, and spp-1(ok2703) was not conditionable whereas aqp-1(tm2309) was only slightly conditionable relative to non conditioned controls (Fig. 3e).
We next set out to rescue aqp-1(tm2309) or spp-1(ok2703) with the wildtype gene under its own promoter using an extrachromosomal array. Rescue of aqp-1(tm2309) with aqp-1, restored conditioned survival to levels comparable to those seen in N2 and rol-6(su1006), and partial restoration of conditioning was achieved upon rescue of spp-1(ok2703) with spp-1 (Fig. 3e). Expression of aqp-1 or spp-1 in both daf-16(mu86) and pmk-1(km25) animals also significantly increased conditioned survival, suggesting that each is sufficient (Fig 3f; Supp Fig 3c,d). However, expression of aqp-1 or spp-1 in N2 did not result in a significant increase in conditioned survival lending support to the idea that an upper limit to conditioning exists using our protocols (not shown).
To determine whether pre-exposure induced changes in aqp-1 or spp-1 gene expression, we next assessed RNA levels. We found that in N2 animals, but not in daf-16(mu86) or pmk-1(km25) animals, mRNA levels of aqp-1 or spp-1 increased by ~1.5 fold upon pre-exposure to EPEC for thirty minutes followed by three hour waiting period on OP50/NGM (Fig. 3g). Notably, the levels of aqp-1 and spp-1 were lower in conditioned compared to unconditioned pmk-1(km25) animals (Fig. 3g), raising the possibility that PMK-1 regulates basal levels of expression and DAF-16 regulates induced levels. In daf-2(e1370) animals, in which DAF-16-regulated genes are constitutively activated, basal mRNA levels of aqp-1 or spp-1 were significantly higher than those seen in N2 (Supp. Fig. 3e). Together these data suggest that both aqp-1 and spp-1 mediate conditioned responses to EPEC upon activation of DAF-16 and PMK-1 signaling.
A neuronal circuit mediates conditioning
We next set out to determine how C. elegans detects bacterial factors that initiate conditioning. Previous reports indicate that pathogenic bacteria induce changes in olfactory neurons that cause aversion of C. elegans to pathogenic strains, and increase the preference for nonpathogenic ones (Zhang et al., 2005). Moreover, specific chemosensory neurons within the amphids perceive and integrate environmental cues that regulate lifespan (Alcedo and Kenyon, 2004; Apfeld and Kenyon, 1999). Finally, daf-16 expression in neurons and intestines has been shown to regulate lifespan (Libina et al., 2003). Therefore, we assessed whether daf-16 expression in neurons or intestines was sufficient for conditioning. We used daf-16(mu86) worms containing DAF-16∷GFP expressed in either neurons (using the unc-119 promoter) or intestines (using the ges-1 promoter) (Libina et al., 2003). Expression of daf-16 in either cell type was sufficient to permit enhanced conditioned survival in animals pre-exposed to EPECΔtnaA (Fig. 4a; see also Supp. Fig. 4a). Similar results were obtained in daf-16(mgDf47) (data not shown). The sufficiency of daf-16 expression in neurons and intestines suggests the involvement of both neurons and intestinal cells in conditioned survival. Kenyon and colleagues (Libina et al., 2003) have reported that DAF-16 activity in neurons can up-regulate DAF-16 in specific responding tissues such as intestinal cells via insulin-like peptides. These data raised the possibility that a neuronal circuit might sense bacterial conditioning factors and communicate the signal to other tissues such as the intestines, where expression of aqp-1 and spp-1 are prominent. In this paper, we have focused on the role of neuronal signaling in regulating conditioned survival.
To determine whether specific sets of neurons mediate conditioning, we assessed conditioned survival in several worm strains carrying mutations in the molecules that generate or regulate effects of serotonin or dopamine, neurotransmitters implicated in responses to bacteria (Chase et al., 2004; Suo et al., 2004; Suo et al., 2003; Zhang et al., 2005). We first tested mutations in cat-1, which encodes a vesicular monoamine transporter that loads dopamine and serotonin into presynaptic vesicles (Duerr et al., 1999). cat-1(e1111) animals also have defects in associative learning (Zhang et al., 2005) and detection of bacteria on lawns (Sawin et al., 2000). cat-1(e1111) animals succumb at a faster rate than N2 upon constitutive exposure (Supp. Fig. 4b), but do not exhibit conditioning (Fig. 4b; Supp. Fig. 4c). These data raise the possibility that dopamine or serotonin or both might mediate conditioning.
Dopaminergic neurons, but not serotonergic neurons mediate conditioning
To distinguish between serotonergic and dopaminergic mechanisms, we first assessed conditioning in animals with defects in cat-2, which encodes tyrosine hydroxylase (Lints and Emmons, 1999), the rate limiting enzyme in dopamine biosynthesis, and in dop-1, dop-2, and dop-3, which encode dopamine receptors (Chase et al., 2004; Sugiura et al., 2005; Suo et al., 2004; Suo et al., 2002; Suo et al., 2003). We also assessed conditioning in animals with mutations in goa-1, which encodes an ortholog of a heterotrimeric G protein α-subunit, dgk-1, which encodes an ortholog of diacylglycerol kinase, and dat-1, which encodes a dopamine reuptake transporter (Nass et al., 2005). Both goa-1 and dgk-1 mediate signaling distal to DOP-3 receptor (Chase et al., 2004). Notably, like cat-1(e1111), dop-3(vs106), and to a lesser extent dop-1(vs100), and dop-2(vs105), have been shown to be defective in detecting bacteria on lawns (Chase et al., 2004; Suo et al., 2004; Suo et al., 2003).
We found that cat-2(e1112), dop-3(vs106), goa-1(sa734), dat-1(ok157) and dgk-1(sy428) all succumbed faster than N2 upon constitutive exposure to EPEC (Supp. Fig. 4d). Notably, conditioned survival by EPEC or by EPECΔtna was significantly lower in dop-3(vs106) mutants compared to N2, and none was evident in any of the other dopamine signaling mutants (Fig. 4c; Supp. Fig. 4e). Additionally, we found that dop-1(vs100) and dop-2(vs105) were only partially conditionable (not shown). Expression of aqp-1 or spp-1 in dop-3(vs106) under control of their own promoters restored conditioned survival to levels similar to those seen in N2 animals (Fig. 4d; Supp. Fig. 4f,g). To further verify a role for dopaminergic signaling in conditioning, we treated C. elegans with 6-hydroxy-dopamine (6-OH-DA), which selectively kills dopaminergic neurons (Nass et al., 2002). C. elegans previously exposed to 6-OH-DA were not conditionable (Fig. 4c). Finally, we found that mRNA for aqp-1 and spp-1 were not induced in dop-3(vs106) following exposure to EPEC (Fig. 3g). Together, these data provide evidence that dopaminergic signaling in neurons mediates conditioning upon exposure to EPEC.
We next assessed conditioned survival in animals with mutations in tph-1, which encodes tryptophan hydroxylase, the rate limiting enzyme in serotonin biosynthesis (Sze et al., 2000), in mod-1, which encodes a serotonin-gated chloride channel (Ranganathan et al., 2000), and in three serotonin receptors (ser-1(ok345), ser-4(ok512), and ser-7(tm1325)). mod-1(ok103) and tph-1(mg280) have defects in associative learning in response to Pseudomonas aeruginosa PA14 (Zhang et al., 2005). Similar to hsf-1(sy441), all serotonin mutants succumb more quickly than N2 in response to EPEC indicating that serotonergic signaling is protective upon constitutive exposure to EPEC (Supp. Fig. 4h). However, all exhibited conditioned survival levels significantly greater than unconditioned animals, with ser-4(ok512) having levels comparable to those seen in N2, and ser-7(tm1325), tph-1(mg280), and mod-1(ok103) somewhat less than N2. Only ser-1(ok345) exhibited conditioned survival that was only slightly above baseline levels (Fig. 4e). Notably, all the serotonin mutants had significantly lower baseline levels of conditioning, with ser-1(ok345) having the lowest. Moreover, fold increases over the baseline were either greater than or comparable to those seen with N2 (Supp. Fig. 4I). Thus, although ser-1(ok345) appeared to have little conditioning relative to all the mutants, the fold conditioning was nonetheless comparable to levels seen with N2. We therefore conclude that serotonergic signaling is not required for conditioning (Supp. Fig. 4i). These data suggest that some genes mediate only protective responses, whereas others mediate both protection and conditioning.
Dopaminergic signaling, aqp-1 and spp-1 are required for conditioning by EPEC but not by stressors
We next determined whether conditioned survival in EPEC is a specific response that utilizes a particular signaling pathway or a general stress response. To do this, we exposed N2 or various mutants to stressors such as starvation (3 hours), heat shock (32°C, 2 hours), heavy metals (CuSO4, 2 hours), or oxidative stress (H2O2; 2 hours). After a three hour waiting period on OP50, animals were then exposed to EPEC for three hours. Heat shock and, to a lesser extent, copper and peroxide, induced conditioned survival in a manner that depended on daf-16, aqp-1, spp-1, and pmk-1 whereas starvation did not (Fig. 5a). These data are in accordance with previous reports suggesting that these stressors activate daf-16 signaling (Liang et al., 2006). As shown above, dop-3(vs106) and cat-2(e1112) succumb more rapidly than N2 when exposed to EPEC (Supp. Fig. 4d) and pre-exposure to EPEC or EPECΔtnaA does not significantly increase conditioned survival (e.g. Fig. 4c). However, pre-exposure to heat shock significantly increases survival of dop-3(vs106) and to a lesser extent cat-2(e1112) (Fig 5a). Heavy metal and peroxide induced more modest levels of conditioned survival in dop-3(vs106). The effect of these stressors was not evident in cat-2(e1112), perhaps because these animals had higher levels of basal conditioning compared to dop-3(vs106). Together these data indicate that survival induced by pre-exposure to bacteria specifically depends on dopaminergic signaling. Moreover, aqp-1(tm2309), spp-1(ok2703), and dop-3(vs106) were as sensitive as N2 to H2O2, and as resistant as N2 to heavy metals, at least under the conditions tested (Fig. 5b), suggesting that the effects of aqp-1, spp-1, and dop-3 were specific to EPEC, and that these genes do not nonspecifically mediate responses to all stressors. Thus, whereas many stressors can activate daf-16 and downstream effectors (e.g. aqp-1 and spp-1) that protect against EPEC, bacteria activate these effectors via dopaminergic signaling pathways.
Figure 5. Stressors condition C. elegans against EPEC by bypassing neuronal signaling.

A. CuSO4, 1 mM H2O2 (peroxide), and heat shock, but not starvation induce conditioned survival against EPEC in N2, dop-3(vs106), and cat-2(e1112) animals, but not pmk-1(km25), daf-16(mu86), aqp-1(tm2309), or spp-1(ok2703) animals. Conditioning schema is shown to the left. Bars in A indicate 95% confidence intervals derived from ANOVA analysis. B. aqp-1(tm2309), spp-1(ok2703), and dop-3(vs106) were as sensitive as N2 to 5 mM H2O2, and as resistant as N2 to heavy metals (e.g. 5 mM CuSO4). Bars in B represent standard errors of the mean.
Discussion
Our observations suggest that C. elegans detect EPEC directly or indirectly via dopaminergic neurons. Once secreted, dopamine may act postsynaptically on metabotropic DOP-3 receptors to induce sustained increases in the levels of a subset of DAF-16 dependent genes that control longevity (e.g. aqp-1) and allow C. elegans to survive subsequent lethal exposure. Protection develops over a period of hours and lasts for approximately 24 hours, and involves a different set of genes than those mediating protection upon acute exposure. Thus, some genes mediate only protection (e.g. ser-1, hsf-1), whereas others mediate both protection and conditioning (e.g. dop-3, daf-16, pmk-1). Conditioning requires master regulators of longevity (e.g. daf-16) and innate immunity (e.g. pmk-1), which respond to many environmental challenges, including pathogens (Garsin et al., 2003; Troemel et al., 2006). Sensory inputs into these pathways activate a specific subset of responses that can protect against some environmental insults and not others. Thus, C. elegans appear to utilize sensory receptors and neuronal signaling networks in conjunction with innate immunity and aging pathways to generate a protective immune response that is specific, adaptive, and persistent. These observations are summarized in Fig. 6.
Figure 6. Model of C. elegans conditioning by virulent or avirulent EPEC strains.
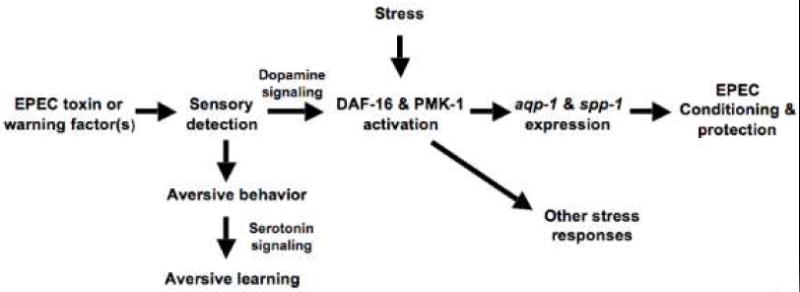
Conditioning initiated by toxin or by factors on avirulent EPEC strains or other pathogens triggers a neuronal circuit mediated by dopaminergic neurons that activates DAF-16 and PMK-1 signaling. These mediators of longevity and innate immunity in turn activate aqp-1 and spp-1, which protect C. elegans. Note that stressors such as heavy metals, oxidative stress, or heat shock, but not starvation, can also activate DAF-16 and PMK-1 in a manner that does not depend on neuronal signaling, and which protect C. elegans against both EPEC and other stressors. Likewise, although exposure to EPEC induces genes such as aqp-1 and spp-1 that are specifically required for protection against subsequent lethal exposure to EPEC, the response elicited is protective against a variety of stressors, including other pathogens.
Several lines of evidence argue against the idea that mutants may not appear conditionable simply because they are more sensitive to EPEC. First, heat shock and other stressors markedly increase conditioned survival of dop-3(vs106) and cat-2(e1112) (Fig. 5a). Thus, despite the sensitivity of these mutants, protective mechanisms can be activated by bypassing dopamine signaling. Second, no correlation was evident between susceptibility and fold conditioning (Fig. 2c). As an example, hsf-1 mutants die as fast as daf-16 mutants, but whereas hsf-1 mutants are as conditionable as N2, daf-16 mutants are not (Figs. 2d). A similar result was obtained with the serotonin mutants (Supp. Fig. 4h, Fig. 4e).
At least two factors from EPEC induce conditioning: one is secreted and diffusible, whereas the other requires direct contact with the worm. That some bacterial strains (e.g. EPEC, EPECΔtnaA, PAK1), virulent or not, can induce protection against EPEC, but others cannot (C. rodentium, OP50), suggests that conditioning may be a general strategy to discriminate classes of bacteria as beneficial or deleterious food sources. Noxious stimuli from bacterial pathogens induce olfactory learning that changes olfactory preferences and trains C. elegans to avoid the pathogen (Zhang et al., 2005). Our data suggest that in addition to behavioral avoidance, such stimuli may also upregulate protective responses that allow the animal to survive subsequent exposure to toxins, and perhaps even establish a feeding niche in an otherwise noxious environment. Thus, aversive behavior may provide a window for protective responses to develop. Notably, whereas olfactory learning is mediated by serotonergic circuits (Zhang et al., 2005), our data suggest that dopaminergic neurons mediate induction of long term protective responses. Our current efforts are focused on defining EPEC factors that stimulate conditioning, and the sensory detectors of such factors in C. elegans. Moreover, because the signaling pathways regulating longevity and innate immunity are highly conserved from C. elegans to humans, we hypothesize that such detection mechanisms are coupled to protective responses mediated by analogous pathways in mammalian systems.
Previous work based on identification of C. elegans mutants that are resistant to various pathogens or toxins raised the possibility that the degree of host susceptibility was based on basal expression of protective genes, mediated by IGF-1R/DAF-16 signaling, and induced expression of protective genes, mediated by the MAP kinase signaling (Troemel et al., 2006). Our data provide a refinement of this idea and raise the possibility that when encountering pathogens, C. elegans use a neuronal circuit to regulate both aversive behavior and induction of protective responses via aging and innate immune signaling pathways. As noted above, different circuits appear to mediate particular functions. Thus, whereas serotonergic signaling is required for aversive behavioral responses, dopaminergic signaling is required for long-term protective responses. Moreover, both circuits are plastic and modifiable by previous exposure or experience. Therefore, aversive behavior and neuronal-mediated induction of protective genes evident in ancient organisms such as C. elegans serve analogous functions to the innate and adaptive immune immune systems found in vertebrates.
The question arises as to whether acquired resistance to EPEC is likely to represent a specific response to bacteria versus a generalized stress response. Our data suggest that the response comprises elements of both. As noted above, the capacity to discriminate pathogens from food sources is in part achieved by coupling of neuronal detection and signaling systems to either behavioral responses or to master stress regulators such as PMK-1 and DAF-16. Effectors that regulate responses to aging or stress include factors that inactivate toxins or have antimicrobial activity. However, effectors are not restricted to antibacterial functions, likely because the stimuli that activate these pathways are so diverse and enhanced longevity requires survival from challenges other than just infection. It is interesting to note that the contribution of daf-16-regulated genes as a group to longevity has been proposed to be cumulative. Thus, loss of function of particular life-shortening or life-extending genes only has an incremental effect on lifespan (Kenyon, 2005). Our data suggest that two genes, aqp-1 and spp-1, are major contributors to protection against EPEC induced by not only EPEC toxins and warning factors but also by other pathogens (e.g. Pseudomonas) and stressors (Figs. 1, 3 and 5). These genes are specific for protection against bacteria; aqp-1 and spp-1 are not important for survival from heavy metals (Fig. 5b). Together these data suggest that neuronal activation of aging and innate immunity pathways likely results in activation of a broad range of protective genes, including aqp-1 and spp-1. Should the dopaminergic signaling pathway we have identified transduce signals from diverse sensory receptors, then we expect that the conditioning paradigm identified here may be activated in response to a wide variety of pathogens.
Using a different exposure protocol than ours, Evans et al. (Evans et al., 2008) report that infection of C. elegans with P. aeruginosa activates DAF-2 signaling, leading to translocation of DAF -16 protein from nuclei to cytosol of intestinal cells, and down-regulation of some DAF-16 targets (thn-2, spp-1 and lys-7), but not others (abf-2, lys-2 and F08G5.6). These changes depended upon the bacterial two-component regulator gacA and the quorum-sensing regulators lasR and rhlR. In contrast, exposure to S. typhimurium or E. faecalis induced all six genes. Evans et al. (Evans et al., 2008) conclude that the down-regulation is characteristic of P. aeruginosa infections and is not applicable to S. typhimurium and E. faecalis. Our data likewise suggest that EPEC induces different responses than P. aeruginosa. Accordingly, we knocked down lys-7 and thn-2 by RNAi and found no increase sensitivity of daf-2(e1370) RNAized worms upon EPEC exposure and no effect of these genes on conditioning. Together, these results suggest that particular bacterial factor, together with the timing or dynamics of its presentation to the worm may have significant effect on the nature or extent of the protective response engendered.
aqp-1 encodes an aquaglyceroporin (Huang et al., 2007), which facilitates transport of water and glycerol across cell membranes and in addition regulates lifespan (Murphy et al., 2003). We surmise that such channels may prove important in uptake of EPEC toxins or warning factors. spp-1 encodes a peptide similar to amoebaphores produced by Entoamoeba histolytica and is thought to function by forming pores in the membranes of bacterial target cells, and limits Salmonella infection (Alegado and Tan, 2008; Banyai and Patthy, 1998). spp-1 may confer protection by a novel means because EPEC-mediated killing does not involve direct contact. Data presented here suggests that aqp-1 and spp-1 mediate conditioning, but understanding the roles of these genes at the molecular level will require that a definition of the cellular basis for conditioning.
Materials and Methods
Bacterial and nematode strains
Experiments were mainly carried out with enteropathogenic E. coli serotype 0127:H6 strain E2348/69 (Levine et al., 1985) and E. coli OP50 (Brenner, 1974). EPECΔtnaA, EPECΔespF, Pseudomonas aeruginosa strain PAK1 and Citrobacter rodentium have been described previously (Anyanful et al., 2005; McNamara and Donnenberg, 1998). The following C. elegans mutants were obtained from the Caenorhabditis Genetics Center: daf-16(m26), daf-16(mgDf50), daf-16(mu86), daf-2(e1370), hsf-1(sy441), sek-1(km4), pmk-1(km25), spp-1(ok2703), cat-1(e1111), cat-2(e1112), dop-1(vs100), dop-1(vs101), dop-2(vs105), dop3(vs106), goa-1(sa734), dgk-1(sy428), dat-1(ok157), ser-1(ok345), ser-4(ok512), ser-7(tm1325), tph-1(mg280), mod-1(ok103), daf-16(mgDf47);xrls87[daf-16a∷gfp∷DAF-16B, rol-6(su1006)] (strain GR1352), daf-16(mu86);muIs61[daf-16∷GFP, rol-6(su1006)] (strain CF1139), N2;zls356[Ex DAF-16-GFP, rol-6(su1006)] (strain TJ356), rol-6(su1006) and wild-type Bristol strain N2. aqp-1(tm2309) and daf-16(mgDf47) and additional N2 worms were generously provided by Shohei Mitani, Siu Silvia Lee, Shoichiro Ono and Guy Benian respectively. All C. elegans strains were maintained on Nematode Growth Medium (NGM) under standard culturing conditions with E. coli OP50 as food source (Sulston, 1988). All assays were performed at 25° C.
Worm killing assays
Worm killing assays were carried out essentially as described previously (Anyanful et al., 2005), though times of exposure and the media were altered. Briefly, EPEC, EPECΔtnaA, EPECΔespF, PAK1 or C. rodentium were cultured in Luria-Bertani (LB) broth overnight to an OD600 of 0.8-1.0 and 170μL spread on 6 cm LB agar (Fisher) plates containing 2mg/mL tryptophan (LBT plates). After incubation for 20 hours at 37°C, the plates were cooled in 25°C incubators for an hour. Young adult worms maintained at 20°C were transferred to each plate, and at least 180 worms/strain were tested for each experiment. For killing assays, worms were exposed to EPEC for 3 hours at 25°C before being transferred to OP50 on NGM plates. After 24 hours worms were gently prodded with a platinum wire and considered dead if they failed to respond to touch and showed no indication of pharyngeal pumping.
Worm conditioning assays
For EPEC conditioning assays, young adult worms were exposed to LBT/EPEC for 30 minutes, transferred to OP50 on NGM plates for 3 hours and then again to fresh LBT/EPEC for 3 hours. Worms were then transferred to OP50 on NGM plates and scored after 24 hours. For non-pathogenic conditioning assays, worms were exposed for 3 hours to either EPECΔtnaA, EPECΔespF, Pseudomonas aeruginosa (PAK1) or Citrobacter rodentium on LBT plates, or EPEC on LB plates before being transferred to LBT/EPEC plates for 3 hours and subsequent recovery on NGM/OP50 for 24 hours. For most of the experiments we used EPECΔtnaA/LBTfor the conditioning. For experiments with 0.2 μm nitrocellulose filters (Fig. 1h), we confirmed that bacteria did not penetrate the filters by culturing sample plates at 37° for 48 hours following removal of filters. We never observed bacterial growth on plates overlaid with filters.
DAF-16∷GFP nuclear localization assay
Young adult N2;zls356 [daf-16∷DAF-16∷GFP, rol-6(su1006)] (strain TJ356) worms were left unexposed or exposed to EPEC for 30 minutes or EPECΔtnaA for 3 hours. GFP fluorescence was monitored using a Zeiss 200M inverted microscope with a 63x N.A.1.4 lens. For some experiments, worms were stained with DAPI to visualize nuclei.
Real time PCR assays
Synchronized young adults of N2, dop-3(vs106), pmk-1(km25), daf-16(mu86) and daf-2(e1370) were exposed to EPEC for 30 minutes and then transferred to OP50 on NGM for three hours. Worms were washed free of bacteria with M9 buffer and frozen at -80°C. Unexposed worms grown on NGM/OP50 for control were collected at the young adult stage and frozen. Total RNA from a ~250μL worm pellet was extracted using Trizol (Life Technologies) following manufacturer’s protocol and quantitated by UV absorbance. cDNAs were synthesized with Oligo (dT) Primers using a RETROscript kit (Ambion). Real time PCR was performed in 50μL volume using SYBR-green detection in a 96 well plate on an iCycler machine (Biorad). Duplicates for each sample were included for each reaction and three independent reactions were performed. Primer pairs and product lengths for act-1 and sod-3 have been described (Li et al., 2007). Primers for spp-1 and aqp-1 are shown below. The PCR reaction was initiated at 95°C for 3 minutes followed by 15s at 95°C, 60s at 60°C and 30s at 68°C for 40 cycles. The real time PCR experiments were repeated three times using independently derived RNA preparations. The mRNA levels of the aqp-1 and spp-1 from N2, dop-3(vs106), daf-16(mu86) and pmk-1(km25) were expressed relative to that of actin by calculation using the cycle threshold (Ct). The following equation described by Maeda et al., (2006) was used: mRNA expression level (%) = 2 (Ct post-exposure) − (Ct pre-exposure) × 100. Ct post-exposure = [Ct values of aqp-1 and spp-1 in N2, dop-3, daf-16 and pmk-1 post-exposure - Ct values of act-1 in N2, dop-3, daf-16 and pmk-1 post-exposure] and Ct pre-exposure = [Ct values of aqp-1 and spp-1 in N2, dop-3, daf-16 and pmk-1 pre-exposure - Ct values of act-1 in N2, dop-3, daf-16 and pmk-1 pre-exposure].
spp-1
5’-GATGATCTCGATGCATGGCTTGATG-3’ (forward)
5’-CCTTGCACGCCTTGTCTGGAGAATCC-3’ (reverse).
aqp-1
5’- GGAGGTAATCGCACAATCCTCGGAGC-3’ (forward)
5’-GTATGCGAGAACTCCGGTACCGACGC-3’ (reverse)
RNA interference (RNAi) assays
We performed RNAi using the E. coli strain HT115(DE3) carrying the RNAi vector L4440 or L4440-derived plasmids engineered to express double stranded RNA (dsRNA) targeting genes of interest, obtained from the C. elegans RNAi library (Kamath et al., 2003). Briefly, specific HT115(DE3) bacteria from the library were cultured overnight at 37°C on LB plates containing 100μg/mL ampicillin and 15μg/mL tetracycline. Single colonies were then cultured overnight at 37°C in LB broth containing 50μg/mL ampicillin and spotted onto 10 cm plates containing NGM agar supplemented with 5mM IPTG and 25μg/ml carbenicillin. The plates were incubated overnight at room temperature to induce the dsRNA expression. 15 N2 or daf-2(e1370) worms at the L3 stage were transferred onto the plates and cultured at 20°C to purge any eggs. After 48 hours, 12 worms were then transferred to fresh plates and cultured at 20°C. After removing the parents 48 hours later, F1 worms grown to the young adult stage were subject to EPEC killing and conditioning assays.
6-hydroxydopamine experiments
Synchronized L4 to young adult worms were collected and washed with M9 buffer and added to a mixture of 1mL 10mM ascorbic acid and 50mM 6-hydroxydopamine (Nass et al., 2002) for 1 hour at 24°C, mixing gently every 15 minutes. Worms were washed thoroughly with M9 buffer and transferred to NGM/OP50 plates. After 15 hours, worms were subjected to killing and conditioning assays.
Construction of transgenic worms
To generate AQP-1∷GFP, a 6036bp genomic fragment including 3635bp of putative promoter sequence and 2401bp of total coding sequence was amplified using platinum taq DNA polymerase (Invitrogen) and primers AQP-1F and AQP-1R (see below). The fragment was cloned in-frame into the BamHI site of the expression vector pPD95.75 (a gift from Andy Fire, Stanford University). To generate SPP-1∷GFP, a 2466bp genomic fragment including 2094bp promoter sequence and 373bp of total coding sequence was amplified similarly with primers SPP-1F and SPP-1R. The fragment was cloned into the BamHI site of pPD95.75. For rescue experiments, these constructs were microinjected into aqp-1(tm2309) and spp-1(ok2703) respectively, using 20μg/mL of vector construct and 80μg/mL of pRF4 as marker. Transgenic F2 progeny were selected by their roller phenotype and expression of GFP in the specific tissues confirmed. For over-expression experiments, the constructs were injected into daf-16(mu86), pmk-1(km25) and dop-3(vs106) and transgenic worms selected as above. In all cases two independent lines were used for analysis. To generate daf-16 tissue-specific promoter fusions, we used a PCR fusion-based approach as described (Hobert, 2002). We first generated the gfp coding sequence plus the 3’ untranslated region from vector pPD95.75 using primers C and D. We then generated DAF-16 from N2 cDNA using primers A and B. Primer B had a 24bp overlap with primer C. Finally, we used primers UNC-119F & UNC-119R and GES-1F & GES-1R to generate the promoter regions of unc-119 and ges-1 respectively from genomic DNA. Primers UNC-119R and GES-1R have a 33bp overlap with DAF-16 cDNA. To perform fusion PCR, we used 10ng of each fragment and Nested D primer as reverse and Nested UNC-119F and Nested GES-1F primers as forward. We used Clontech polymerase for all fusion PCR reactions. After sequence confirmation, we injected and selected transgenics as above. Using the same procedure, we generated Pdaf-16∷DAF-16∷GFP to confirm the rescue of the daf-16 mutation with our construct.
| AQP-1F | CTTCGGATCCGGAAAAGCGGCCGCCGCACATTTCG |
| AQP-1R | CAACGGATCCCAGCTTGAAGCAATTTTTGTTGCTCTAC |
| SPP-1F | CAACGGATCCGCCCTGAAGCGGAATCTGCTCCGC |
| SPP-1R | CAACGGATCCCGCACAAATCAACATCCTTGCACGC |
| Primer D | AAGGGCCCGTACGGCCGACTAGTAGG |
| Nested D | GGAAACAGTTATGTTTGGTATATTGGG |
| Primer C | AGCTTGCATGCCTGCAGGTCGACT |
| Primer B | AGTCGACCTGCAGGCATGCAAGCTCAAATCAAAATGAATATGCTG CCCTCC |
| Primer A | ATG AAC GAC TCA ATA GAC GAC G |
| UNC-119R1 | CGGCGGAAAATCGTCGTCTATTGAGTCGTTCATATATGCTGTT GT AGCTGAAAATTTTGG |
| UNC-119F | CTAGGCCATCGGTGACGTCATTACTC |
| Nested UNC-119F | CATGGCATTGCCTATGCCATTTTCAC |
| GES-1R1 | CGGCGGAAAATCGTCGTCTATTGAGTCGTTCATCTGAATTCAA AG ATAAGATATGTAATAG |
| GES-1F | CTACTGGAATCCGCCAAATTGTCGACAAC |
| Nested GES-1F | CTTGAGTGTTAGCGGCGTCTTCACC |
| DAF-16-R1 | CGGCGGAAAATCGTCGTCTATTGAGTCGTTCATTCTGGAAGCT GTGCTCCTCCGAAGGGG |
| DAF-16-FO | CAACGGATCCCAGGGATAAGGG AGATTCGAACAG |
| Nested DAF-16-FO | CAACGGATCCCTAGTCATAAGTAGTCAGGCAGGC |
Stressor conditioning and killing assays
For conditioning assays, worms were subjected to heat shock by incubating them at 32°C for 2 hours, or exposed to 200μM CuSO4 for 2 hours, or exposed to 1mM H2O2 or starvation conditions for two hours. Afterwards, the worms were transferred to NGM/OP50 for 3 hours before being exposed to EPEC/LBT for 3 hours. For starvation experiments, no incubation with NGM/OP50 was done. Worms were transferred to NGM/OP50 after EPEC exposure and % survival determined 24 hours later. For killing assays, worms were exposed to either 5mM H2O2 or 5mM CuSO4 for two hours. They were then transferred to NGM/OP50 and % survival determined after 24 hours.
Statistical analysis
ANOVA analysis and test for trend
In Figure 1a, we used an ANOVA and a test for linear trend in survival over time. For this experiment, a linear trend test over the seven time points was significant (p < 0.0001). . For Figures 1d, 1e, 1g, and 1h, and for Figure 5, survival was compared using ANOVA. The comparisons were highly significant (p<0.0001). 95% confidence intervals are shown for each.
Calculation of fold survival with conditioning and survival rates
Fold increases in survival with conditioning were calculated as the quotient of the percent of animals surviving following procedure in Fig. 1c (EPEC/LBT pre-exposure) or Fig. 1f (EPECΔtnaA/LB pre-exposure) divided by the percent surviving following procedure in Fig. 1c (OP50/LB pre-exposure) or Fig. 1f (OP50/LB pre-exposure). For Figure 2d, the survival rate for each mutant was estimated by linear regression using the percentage of animals surviving following exposure to EPEC for 0, 1, 2, or 3 hours. The estimated rates of survival rate were then regressed against fold increase in survival with conditioning (Fig. 2d). Linear regression of survival rate (the dependent variable) on fold change (the independent variable) for all mutants yielded the following relationship: Survival Rate = -.146 − (.00444*Fold Change); Intercept = -.146 (standard error = .01729); slope = -.00444(standard error = .00316); When comparing the estimated slope to zero using a t-test, a value of p=.17 was obtained indicating the difference was not statistically significant. Linear regression of survival rate on fold change for only daf-16 mutants, N2 and rol-6(su1006) yielded: Survival Rate = -.0573 − (.04686*Fold Change); Intercept = -.0573 (standard error = .0207); slope = -.04686 (standard error = .0089); p < .0001 when comparing the estimated slope to zero using a t-test; SD of the regression = .04476, mean square error = 0.002. Note that a significant negative correlation was evident when considering only a subset of mutants, but not with all mutants
ANCOVA
We used ANCOVA to remove the effects of pre-existing mutant differences, and ensure that mutants are starting out approximately equal, on average, with respect to all factors that might be pertinent to how well they are likely to respond to the conditioning paradigms. Such a correction is useful because individual differences in conditioning displayed by a particular mutant could potentially be correlated with survival rate. The mean survival and 95% confidence intervals for all 39 mutants without conditioning was 0.05335 or 5.3% +/- 0.243% (mean +/- sem), and the slope estimate was 0.05585. For N2, the adjusted mean survival with conditioning, Y, was 0.42595 + .05585*mean survival without conditioning: Y = .42595 + (.05585*.05335) = .429 or 42.9%.
Supplementary Material
Supplementary Figure 1. A. Time course of killing of rol-6(su1006) mutant worms by constitutive exposure to an EPEC lawn. rol-6(su1006) worms were exposed to EPEC/LBT for the times indicated, removed to OP50/NGM and survival scored after 24 hours. B. Conditioned exposure of rol-6(1006) to EPEC/LBT plates for 30 minutes (conditioning period), followed by exposure to OP50/NGM plates for 3 hours (waiting period), and then exposure to EPEC/LBT plates for 3 hours (challenge). Also shown is conditioned exposure to EPECΔtnaA using the protocol shown in Fig. 1f. 95% confidence intervals are indicated by bars. C. A conditioning time of 30 minutes induces optimal survival of N2.
Supplementary Figure 2. A. hsf-1(sy441) is conditionable by EPEC, but daf-16 mutants are not. daf-16 mutants carrying an extrachromosomal array of wild type daf-16 are partially rescued. These data mirror those shown in Fig. 2b with pre-exposure to EPECΔtnaA. B, C. sek-1(km4) and pmk-1(km25) animals are not conditionable as N2 upon pre-exposure to EPECΔtnaA (B) or EPEC (C).
Supplementary Figure 3. A. Inactivation of spp-1 and aqp-1 by RNAi suppress conditioned survival of N2 upon pre-exposure to EPECΔtnaA for 3 hrs. Similar results were obtained with EPEC (not shown). B. Rescued strains, N2, and rol-6(su1006), but not aqp-1(tm2309) and spp-1(ok2703) are conditionable by EPECΔtnaA. C. Time course of survival of N2, rol-6(su1006), daf-16(mu86), pmk-1(km25), or mutants overexpressing aqp-1 or spp-1. D. Conditioning of mutants in C. E. Quantitative RTPCR of aqp-1 or spp-1 mRNA in daf-2(e1370) animals upon pre-exposure to EPEC for 30 minutes, followed by three hour waiting period on OP50. The mRNA levels of aqp-1 and spp-1 before and after conditioning were normalized to those of act-1. Note that basal levels of these genes are significantly higher in daf-2(e1370) animals, which is consistent with constitutive activation of DAF-16. Note also that no effect of conditioning was evident in these animals.
Supplementary Figure 4. Survival data used to calculate adjusted percent survival with conditioning (Fig 4). A.Conditioning for N2, rol-6(su1006), or daf-16 (mu86) animals expressing DAF-16∷GFP in neurons (unc-119 promoter) or intestinal cells (ges-1 promoter). B.Time course of survival of cat-1(e1111) upon constitutive exposure to EPEC. C. Conditioning of cat-1(e1111) with EPECΔtnaA. D. Time course of survival of dopaminergic signaling mutants. E. Conditioning of dopaminergic signaling mutants. F. Time course of survival of dop-3(vs106), or of dop-3(vs106) expressing aqp-1 or spp-1. G. Conditioning of mutants in F. H. Time course of survival of serotonergic signaling mutants. I. Conditioning of mutants in H.
Acknowledgments
The authors would like to thank Hang Lu, Vladimir Brezina, David Kalman and Paul O’Lague for comments on the manuscript; Alyson Swimm for preparation of figures; Christa McKenzie for technical assistance; Melanie Sherman, and colleagues in the Kalman Lab for insightful discussion. Most worm strains used in this study were provided by the Caenorhabditis Genetics Center, which is supported by the National Center for Research Resources of the NIH. This work was supported by grants from NIDDK (R01DK074731-01A2) and NIAID (R01AI056067) to D.K.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature Cited
- Aballay A, Yorgey P, Ausubel FM. Salmonella typhimurium proliferates and establishes a persistent infection in the intestine of Caenorhabditis elegans. Curr Biol. 2000;10:1539–1542. doi: 10.1016/s0960-9822(00)00830-7. [DOI] [PubMed] [Google Scholar]
- Alcedo J, Kenyon C. Regulation of C. elegans longevity by specific gustatory and olfactory neurons. Neuron. 2004;41:45–55. doi: 10.1016/s0896-6273(03)00816-x. [DOI] [PubMed] [Google Scholar]
- Alegado RA, Tan MW. Resistance to antimicrobial peptides contributes to persistence of Salmonella typhimurium in the C. elegans intestine. Cell Microbiol. 2008;10:1259–1273. doi: 10.1111/j.1462-5822.2008.01124.x. [DOI] [PubMed] [Google Scholar]
- Anyanful A, Dolan-Livengood J, Lewis T, Sheth S, DeZalia MN, Sherman M, Kalman LV, Benian GM, Kalman D. Paralysis and killing of C. elegans by enteropathogenic E. coli requires the bacterial tryptophanase gene. Molecular Microbiology. 2005;57:988–1007. doi: 10.1111/j.1365-2958.2005.04739.x. [DOI] [PubMed] [Google Scholar]
- Apfeld J, Kenyon C. Regulation of lifespan by sensory perception in Caenorhabditis elegans. Nature. 1999;402:804–809. doi: 10.1038/45544. [DOI] [PubMed] [Google Scholar]
- Banyai L, Patthy L. Amoebapore homologs of Caenorhabditis elegans. Biochim Biophys Acta. 1998;1429:259–264. doi: 10.1016/s0167-4838(98)00237-4. [DOI] [PubMed] [Google Scholar]
- Bargmann CI. Chemosensation in C elegans. WormBook. 2006:1–29. doi: 10.1895/wormbook.1.123.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chase DL, Pepper JS, Koelle MR. Mechanism of extrasynaptic dopamine signaling in Caenorhabditis elegans. Nat Neurosci. 2004;7:1096–1103. doi: 10.1038/nn1316. [DOI] [PubMed] [Google Scholar]
- Clarke SC, Haigh RD, Freestone PP, Williams PH. Enteropathogenic Escherichia coli infection: history and clinical aspects. Br J Biomed Sci. 2002;59:123–127. doi: 10.1080/09674845.2002.11783647. [DOI] [PubMed] [Google Scholar]
- Darby C, Cosma CL, Thomas JH, Manoil C. Lethal paralysis of Caenorhabditis elegans by Pseudomonas aeruginosa. Proc Natl Acad Sci U S A. 1999;96:15202–15207. doi: 10.1073/pnas.96.26.15202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duerr JS, Frisby DL, Gaskin J, Duke A, Asermely K, Huddleston D, Eiden LE, Rand JB. The cat-1 gene of Caenorhabditis elegans encodes a vesicular monoamine transporter required for specific monoamine-dependent behaviors. J Neurosci. 1999;19:72–84. doi: 10.1523/JNEUROSCI.19-01-00072.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans EA, Kawli T, Tan MW. Pseudomonas aeruginosa suppresses host immunity by activating the DAF-2 insulin-like signaling pathway in Caenorhabditis elegans. PLoS Pathog. 2008;4:e1000175. doi: 10.1371/journal.ppat.1000175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankel G, Phillips AD, Rosenshine I, Dougan G, Kaper JB, Knutton S. Enteropathogenic and enterohaemorrhagic Escherichia coli: more subversive elements. Mol Microbiol. 1998;30:911–921. doi: 10.1046/j.1365-2958.1998.01144.x. [DOI] [PubMed] [Google Scholar]
- Garsin DA, Villanueva JM, Begun J, Kim DH, Sifri CD, Calderwood SB, Ruvkun G, Ausubel FM. Long-lived C. elegans daf-2 mutants are resistant to bacterial pathogens. Science. 2003;300:1921. doi: 10.1126/science.1080147. [DOI] [PubMed] [Google Scholar]
- Hamilton B, Dong Y, Shindo M, Liu W, Odell I, Ruvkun G, Lee SS. A systematic RNAi screen for longevity genes in C. elegans. Genes Dev. 2005;19:1544–1555. doi: 10.1101/gad.1308205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson ST, Johnson TE. daf-16 integrates developmental and environmental inputs to mediate aging in the nematode Caenorhabditis elegans. Curr Biol. 2001;11:1975–1980. doi: 10.1016/s0960-9822(01)00594-2. [DOI] [PubMed] [Google Scholar]
- Hendrickson EL, Plotnikova J, Mahajan-Miklos S, Rahme LG, Ausubel FM. Differential roles of the Pseudomonas aeruginosa PA14 rpoN gene in pathogenicity in plants, nematodes, insects, and mice. J Bacteriol. 2001;183:7126–7134. doi: 10.1128/JB.183.24.7126-7134.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobert O. PCR fusion-based approach to create reporter gene constructs for expression analysis in transgenic C. elegans. Biotechniques. 2002;32:728–730. doi: 10.2144/02324bm01. [DOI] [PubMed] [Google Scholar]
- Hodgkin J, Kuwabara PE, Corneliussen B. A novel bacterial pathogen, Microbacterium nematophilum, induces morphological change in the nematode C. elegans. Curr Biol. 2000;10:1615–1618. doi: 10.1016/s0960-9822(00)00867-8. [DOI] [PubMed] [Google Scholar]
- Hsu AL, Murphy CT, Kenyon C. Regulation of aging and age-related disease by DAF-16 and heat-shock factor. Science. 2003;300:1142–1145. doi: 10.1126/science.1083701. [DOI] [PubMed] [Google Scholar]
- Huang CG, Lamitina T, Agre P, Strange K. Functional analysis of the aquaporin gene family in Caenorhabditis elegans. Am J Physiol Cell Physiol. 2007;292:C1867–1873. doi: 10.1152/ajpcell.00514.2006. [DOI] [PubMed] [Google Scholar]
- Kamath RS, Fraser AG, Dong Y, Poulin G, Durbin R, Gotta M, Kanapin A, Le Bot N, Moreno S, Sohrmann M, et al. Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature. 2003;421:231–237. doi: 10.1038/nature01278. [DOI] [PubMed] [Google Scholar]
- Kenyon C. A conserved regulatory system for aging. Cell. 2001;105:165–168. doi: 10.1016/s0092-8674(01)00306-3. [DOI] [PubMed] [Google Scholar]
- Kenyon C. The plasticity of aging: insights from long-lived mutants. Cell. 2005;120:449–460. doi: 10.1016/j.cell.2005.02.002. [DOI] [PubMed] [Google Scholar]
- Kenyon C, Chang J, Gensch E, Rudner A, Tabtiang R. A C. elegans mutant that lives twice as long as wild type. Nature. 1993;366:461–464. doi: 10.1038/366461a0. [DOI] [PubMed] [Google Scholar]
- Kim DH, Feinbaum R, Alloing G, Emerson FE, Garsin DA, Inoue H, Tanaka-Hino M, Hisamoto N, Matsumoto K, Tan MW, Ausubel FM. A conserved p38 MAP kinase pathway in Caenorhabditis elegans innate immunity. Science. 2002;297:623–626. doi: 10.1126/science.1073759. [DOI] [PubMed] [Google Scholar]
- Laws TR, Atkins HS, Atkins TP, Titball RW. The pathogen Pseudomonas aeruginosa negatively affects the attraction response of the nematode Caenorhabditis elegans to bacteria. Microb Pathog. 2006;40:293–297. doi: 10.1016/j.micpath.2006.03.001. [DOI] [PubMed] [Google Scholar]
- Levine MM, Nataro JP, Karch H, Baldini MM, Kaper JB, Black RE, Clements ML, O’Brien AD. The diarrheal response of humans to some classic serotypes of enteropathogenic Escherichia coli is dependent on a plasmid encoding an enteroadhesiveness factor. J Infect Dis. 1985;152:550–559. doi: 10.1093/infdis/152.3.550. [DOI] [PubMed] [Google Scholar]
- Li J, Tewari M, Vidal M, Lee SS. The-14-33 protein FTT-2 regulates DAF-16 in Caenorhabditis elegans. Dev Biol. 2007;301:82–91. doi: 10.1016/j.ydbio.2006.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang B, Moussaif M, Kuan CJ, Gargus JJ, Sze JY. Serotonin targets the DAF-16/FOXO signaling pathway to modulate stress responses. Cell Metab. 2006;4:429–440. doi: 10.1016/j.cmet.2006.11.004. [DOI] [PubMed] [Google Scholar]
- Libina N, Berman JR, Kenyon C. Tissue-specific activities of C. elegans DAF-16 in the regulation of lifespan. Cell. 2003;115:489–502. doi: 10.1016/s0092-8674(03)00889-4. [DOI] [PubMed] [Google Scholar]
- Lin K, Dorman JB, Rodan A, Kenyon C. daf-16: An HNF-3/forkhead family member that can function to double the life-span of Caenorhabditis elegans. Science. 1997;278:1319–1322. doi: 10.1126/science.278.5341.1319. [DOI] [PubMed] [Google Scholar]
- Lints R, Emmons SW. Patterning of dopaminergic neurotransmitter identity among Caenorhabditis elegans ray sensory neurons by a TGFbeta family signaling pathway and a Hox gene. Development. 1999;126:5819–5831. doi: 10.1242/dev.126.24.5819. [DOI] [PubMed] [Google Scholar]
- Mahajan-Miklos S, Tan MW, Rahme LG, Ausubel FM. Molecular mechanisms of bacterial virulence elucidated using a Pseudomonas aeruginosa-Caenorhabditis elegans pathogenesis model. Cell. 1999;96:47–56. doi: 10.1016/s0092-8674(00)80958-7. [DOI] [PubMed] [Google Scholar]
- McNamara BP, Donnenberg MS. A novel proline-rich protein, EspF, is secreted from enteropathogenic Escherichia coli via the type III export pathway. FEMS Microbiol Lett. 1998;166:71–78. doi: 10.1111/j.1574-6968.1998.tb13185.x. [DOI] [PubMed] [Google Scholar]
- Mello C, F A. DNA Transformation. Vol. 48. Academic Press; 1995. [Google Scholar]
- Murphy CT, McCarroll SA, Bargmann CI, Fraser A, Kamath RS, Ahringer J, Li H, Kenyon C. Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans. Nature. 2003;424:277–283. doi: 10.1038/nature01789. [DOI] [PubMed] [Google Scholar]
- Nass R, Hahn MK, Jessen T, McDonald PW, Carvelli L, Blakely RD. A genetic screen in Caenorhabditis elegans for dopamine neuron insensitivity to 6-hydroxydopamine identifies dopamine transporter mutants impacting transporter biosynthesis and trafficking. J Neurochem. 2005;94:774–785. doi: 10.1111/j.1471-4159.2005.03205.x. [DOI] [PubMed] [Google Scholar]
- Nass R, Hall DH, Miller DM, 3rd, Blakely RD. Neurotoxin-induced degeneration of dopamine neurons in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2002;99:3264–3269. doi: 10.1073/pnas.042497999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogg S, Paradis S, Gottlieb S, Patterson GI, Lee L, Tissenbaum HA, Ruvkun G. The Fork head transcription factor DAF-16 transduces insulin-like metabolic and longevity signals in C. elegans. Nature. 1997;389:994–999. doi: 10.1038/40194. [DOI] [PubMed] [Google Scholar]
- Pradel E, Zhang Y, Pujol N, Matsuyama T, Bargmann CI, Ewbank JJ. Detection and avoidance of a natural product from the pathogenic bacterium Serratia marcescens by Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2007;104:2295–2300. doi: 10.1073/pnas.0610281104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranganathan R, Cannon SC, Horvitz HR. MOD-1 is a serotonin-gated chloride channel that modulates locomotory behaviour in C. elegans. Nature. 2000;408:470–475. doi: 10.1038/35044083. [DOI] [PubMed] [Google Scholar]
- Sawin ER, Ranganathan R, Horvitz HR. C. elegans locomotory rate is modulated by the environment through a dopaminergic pathway and by experience through a serotonergic pathway. Neuron. 2000;26:619–631. doi: 10.1016/s0896-6273(00)81199-x. [DOI] [PubMed] [Google Scholar]
- Singh V, Aballay A. Heat-shock transcription factor (HSF)-1 pathway required for Caenorhabditis elegans immunity. Proc Natl Acad Sci U S A. 2006;103:13092–13097. doi: 10.1073/pnas.0604050103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiura M, Fuke S, Suo S, Sasagawa N, Van Tol HH, Ishiura S. Characterization of a novel D2-like dopamine receptor with a truncated splice variant and a D1-like dopamine receptor unique to invertebrates from Caenorhabditis elegans. J Neurochem. 2005;94:1146–1157. doi: 10.1111/j.1471-4159.2005.03268.x. [DOI] [PubMed] [Google Scholar]
- Sulston J, H J. In: Methods, In The Nematode Caenorhabditis elegans. Wood WB, editor. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1988. pp. 587–606. [Google Scholar]
- Suo S, Ishiura S, Van Tol HH. Dopamine receptors in C. elegans. Eur J Pharmacol. 2004;500:159–166. doi: 10.1016/j.ejphar.2004.07.021. [DOI] [PubMed] [Google Scholar]
- Suo S, Sasagawa N, Ishiura S. Identification of a dopamine receptor from Caenorhabditis elegans. Neurosci Lett. 2002;319:13–16. doi: 10.1016/s0304-3940(01)02477-6. [DOI] [PubMed] [Google Scholar]
- Suo S, Sasagawa N, Ishiura S. Cloning and characterization of a Caenorhabditis elegans D2-like dopamine receptor. J Neurochem. 2003;86:869–878. doi: 10.1046/j.1471-4159.2003.01896.x. [DOI] [PubMed] [Google Scholar]
- Sze JY, Victor M, Loer C, Shi Y, Ruvkun G. Food and metabolic signalling defects in a Caenorhabditis elegans serotonin-synthesis mutant. Nature. 2000;403:560–564. doi: 10.1038/35000609. [DOI] [PubMed] [Google Scholar]
- Tan M-W, Rahme Laurence G, Sternberg Jeffrey A, Tompkins Ronald G, Ausubel Frederick M. Pseudomonas aeruginosa killing of Caenorhabditis elegans used to identify P. aeruginosa virulence factors. Proc Natl Acad Sci USA. 1999a;96:2408–2413. doi: 10.1073/pnas.96.5.2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan M-W, Mahajan-Miklos Shalina, Ausubel Frederick M. Killing of Caenorhabditis elegans by Pseudomonas aeruginosa used to model mammalian bactreial pathogenesis. Proc Natl Acad Sci USA. 1999b;96:715–720. doi: 10.1073/pnas.96.2.715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troemel ER, Chu SW, Reinke V, Lee SS, Ausubel FM, Kim DH. p38 MAPK regulates expression of immune response genes and contributes to longevity in C. elegans. PLoS Genet. 2006;2:e183. doi: 10.1371/journal.pgen.0020183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Lu H, Bargmann CI. Pathogenic bacteria induce aversive olfactory learning in Caenorhabditis elegans. Nature. 2005;438:179–184. doi: 10.1038/nature04216. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. A. Time course of killing of rol-6(su1006) mutant worms by constitutive exposure to an EPEC lawn. rol-6(su1006) worms were exposed to EPEC/LBT for the times indicated, removed to OP50/NGM and survival scored after 24 hours. B. Conditioned exposure of rol-6(1006) to EPEC/LBT plates for 30 minutes (conditioning period), followed by exposure to OP50/NGM plates for 3 hours (waiting period), and then exposure to EPEC/LBT plates for 3 hours (challenge). Also shown is conditioned exposure to EPECΔtnaA using the protocol shown in Fig. 1f. 95% confidence intervals are indicated by bars. C. A conditioning time of 30 minutes induces optimal survival of N2.
Supplementary Figure 2. A. hsf-1(sy441) is conditionable by EPEC, but daf-16 mutants are not. daf-16 mutants carrying an extrachromosomal array of wild type daf-16 are partially rescued. These data mirror those shown in Fig. 2b with pre-exposure to EPECΔtnaA. B, C. sek-1(km4) and pmk-1(km25) animals are not conditionable as N2 upon pre-exposure to EPECΔtnaA (B) or EPEC (C).
Supplementary Figure 3. A. Inactivation of spp-1 and aqp-1 by RNAi suppress conditioned survival of N2 upon pre-exposure to EPECΔtnaA for 3 hrs. Similar results were obtained with EPEC (not shown). B. Rescued strains, N2, and rol-6(su1006), but not aqp-1(tm2309) and spp-1(ok2703) are conditionable by EPECΔtnaA. C. Time course of survival of N2, rol-6(su1006), daf-16(mu86), pmk-1(km25), or mutants overexpressing aqp-1 or spp-1. D. Conditioning of mutants in C. E. Quantitative RTPCR of aqp-1 or spp-1 mRNA in daf-2(e1370) animals upon pre-exposure to EPEC for 30 minutes, followed by three hour waiting period on OP50. The mRNA levels of aqp-1 and spp-1 before and after conditioning were normalized to those of act-1. Note that basal levels of these genes are significantly higher in daf-2(e1370) animals, which is consistent with constitutive activation of DAF-16. Note also that no effect of conditioning was evident in these animals.
Supplementary Figure 4. Survival data used to calculate adjusted percent survival with conditioning (Fig 4). A.Conditioning for N2, rol-6(su1006), or daf-16 (mu86) animals expressing DAF-16∷GFP in neurons (unc-119 promoter) or intestinal cells (ges-1 promoter). B.Time course of survival of cat-1(e1111) upon constitutive exposure to EPEC. C. Conditioning of cat-1(e1111) with EPECΔtnaA. D. Time course of survival of dopaminergic signaling mutants. E. Conditioning of dopaminergic signaling mutants. F. Time course of survival of dop-3(vs106), or of dop-3(vs106) expressing aqp-1 or spp-1. G. Conditioning of mutants in F. H. Time course of survival of serotonergic signaling mutants. I. Conditioning of mutants in H.