

Abstract
Aquaporin-4 (AQP4) is the principle water channel and the primary route for water transport across astrocytic membranes. AQP4 co-localizes with Kir4.1 channels at astrocytic endfeet, and it has been suggested that these channels cooperate in K+ and water homeostasis. In response to injury, two additional aquaporins, AQP1 and AQP9, can be detected in astrocytes, yet neither is found in cultured astrocytes, and therefore their contribution to astrocyte water uptake and biology is poorly investigated. In this study, we used a cortical stab wound assay to demonstrate an upregulation of AQP1 following injury in reactive glia. We were able to mimic such injury in astrocytic cultures and show that AQP1 expression is induced within 16 hr following injury in vitro. This induction could be blocked by inhibition of MEK1/2 using U0126 and suggests that AQP1 is specifically induced in reactive astrocytes via the MAPK signaling pathway.
Keywords: edema, volume regulation, glia, signaling
Introduction
Brain edema presents with marked astrocytic swelling whereby individual cells reach nearly twice their normal cell volume. The associated cellular water uptake is believed to occur via AQP4 water channels, the principle aquaporin expressed in astrocytes (Nagelhus, E.A. et. al., 1999; Papadopoulos, M.C. et. al., 2002). Immuno-gold electron microscopic studies show high levels of AQP4 on astrocytic endfeet associated with blood vessels (Nielsen, S. et. al., 1997; Rash, J.E. et. al., 2004), and AQP4 colocalizes with Kir4.1. It has been suggested that Kir4.1 and AQP4 cooperate in the context of extracellular K+ buffering (Amiry-Moghaddam, M. et. al., 2003; Chen, K.C. and Nicholson, C., 2000; Kofuji, P. and Newman, E.A., 2004). Consistent with this notion, several studies have shown that the loss of AQP4 in astrocytes plays a role in the brain following injury (Manley, G.T. et. al., 2000; Papadopoulos, M.C. et. al., 2004; Zhao, J. et. al., 2005). AQP4 appears to be either beneficial or pathological in the formation of brain edema following injury. Specifically, after acute water intoxication, AQP4 KO mice showed a reduction in cytotoxic edema, suggesting that AQP4 is the mediator of cytotoxic edema (Manley, G.T. et. al., 2000), whereas following vasogenic edema, AQP4 KO mice showed increased edema formation, implicating astrocytic AQP4 in removal of excess fluid from the surrounding parenchyma (Papadopoulos, M.C. et. al., 2004).
AQP4 levels do fluctuate during edema formation, suggesting that there are factors regulating its expression (Badaut, J. et. al., 2007; Ribeiro, M.C. et. al., 2006). Similarly to edemic conditions, hyperosmolarity leads to an increase in AQP4 expression (Arima, H. et. al., 2003), implying a potential role for changes in osmolarity as a mediator of AQP4 protein expression. However, little is known about the role other AQPs play in the formation of edema. As with AQP4, osmotic changes alter the expression levels of both AQP1 and AQP9 (Arima, H. et. al., 2003; Jenq, W. et. al., 1999b; Umenishi, F. and Schrier, R.W., 2002b).
In the CNS, AQP1 expression is highest in the choroid plexus where it functions in the formation of cerebral spinal fluid (Bondy, C. et. al., 1993;Hasegawa, H. et. al., 1994). Recent studies have shown that AQP1 colocalizes with nociceptors in dorsal root ganglia, along the axon and at the synapse (Shields, S.D. et. al., 2007). This suggests that AQP1 potentially regulates the osmotic changes associated with rapid ion fluxes during neuronal activity. Furthermore, chronic osmotic challenges have been shown to upregulate AQP1 expression in kidney cells (Jenq, W. et. al., 1999a; Umenishi, F. and Schrier, R.W., 2002a). The activation of an osmotic response element (ORE) located in the promoter region leads to gene transcription and AQP1 upregulation (Umenishi, F. and Schrier, R.W., 2002a), and these changes were shown to be regulated by mitogen-activated protein kinases (MAPK) (Umenishi, F. and Schrier, R.W., 2003).
MAPKs are serine/threonine specific protein kinases activated by extracellular factors and function in various cellular roles, i.e. gene expression, cell survival, apoptosis, growth factors (Carriere, A. et. al., 2008; Castigli, E. et. al., 2000; Hebert, M.A. and O'Callaghan, J.P., 2000; Katsura, H. et. al., 2008; Santos, S.D. et. al., 2007). During injury, MAPK signaling is upregulated within the first few minutes of insult, spreads to surrounding areas, and is important in regulating the process of reactive gliosis (Lim, J.H. et. al., 2007; Mandell, J.W. et. al., 2001).
In this study, we set out to examine whether AQP1 expression may be upregulated following injury and whether this may invoke MAPK activity. Using a combination of biochemical and immunohistochemical studies, we were able to demonstrate an increase in AQP1 expression in astrocytes following cortical stab injury in rats and were able to mimic this injury using an in vitro scratch injury model. Here, injury caused the re-expression of AQP1 in astrocyte cultures that were lacking AQP1 expression. The re-expression required MAPK activity and was suppressed using MAPK/ERK kinase (MEK) 1/2 inhibitors.
Materials and Methods
Cell Culture
Primary cortical astrocytes were obtained from Sprague-Dawley rats at postnatal day 0-1 (P0-1) by modification of the technique described previously (McCarthy, K.D. and deVellis, J., 1980). The brain tissue was dissected in ice-cold DMEM (Media Tech), supplemented with 20 mM glucose, L-glutamine, and antibiotics and fungicide, and the cortex was removed. The dura mater was then removed, and the tissue was triturated in O2-saturated papain (Worthington, Lakewood, NJ) with 2–5 mg DNase (Worthington) and placed in 37°C for 20 min. The cells were kept at 37°C in a 95% O2/5% CO2 humidified environment atmosphere. Medium was replaced the next day and every third day thereafter. Cells were allowed to grow for 7–10 days. For passaged astrocytes, primary astrocytes were shaken overnight at 37°C at 200 rpm, trypsinized and replated. Secondary astrocyte cultures were used one week after plating. Cells were grown in Dulbecco’s modified Eagle medium (DMEM; Media Tech, University of Alabama at Birmingham Media Preparation Facility) and supplemented with 2 mM glutamine (Media Tech) and 7% heat-inactivated fetal bovine serum (FBS; Hyclone, Logan, UT).
RNA Isolation and PCR
Messenger RNA was extracted from the astrocytic cultures following the RNAqueous protocol (Ambion, Austin, TX). Briefly, the cells were lysed, homogenized, and centrifuged. The supernatant was removed and an equal volume of 64% ethanol was added. The mixture was filtered via centrifugation and then washed. The mRNA was eluted from the filter and DNA-free (Ambion) was used to remove contaminating DNA. RNA quality was evaluated by electrophoresis through 1.5% agarose gels.
The cDNA was synthesized and amplified using the OneStep RT-PCR kit (Qiagen, Valencia, CA), per manufacturer’s instructions, by the Eppendorf Mastercycler gradient (Brinkmann Instruments, Westbury, NY). Oligonucleotide primers (Invitrogen, Carlsbad, CA) were designed using the sequences described previously (Wang, W. et. al., 2003). 2 ng of mRNA was loaded per reaction. PCR conditions were as follows; denaturation at 94°C for 5 min followed by 35 cycles at 94°C for 30 s, 56°C for 30 s, 72°C for 2 min, with a final extension at 72°C for 10 min. Amplified products were electrophoresed through 1.5% agarose gels. Kidney cDNA was used for AQP1 and AQP4. Actin was used as loading controls.
Western Blot Analysis
A confluent dish of cells was lysed using RIPA buffer [(50 mM TrisCl, pH 7.5, 150 mM NaCl, 1% Nondet P-40 (NP-40), 0.5% sodium deoxycholate, 1% sodium dodecyl sulfate (SDS)] supplemented with protease inhibitor cocktail (Sigma). Cells were sonicated for 10 s and centrifuged at 14,000 rpm for 10 min, and the supernatant was transferred to a new tube. Protein quantification was performed using a DC protein assay kit (BioRad, Hercules, CA). An equal amount of 6× sample buffer containing 600 mM β-mercaptoethanol was added to the 20–30 μg/ml of cell lysate per lane. Samples were loaded into a 10% precast SDS-PAGE gel (BioRad). Membranes were blocked in blocking buffer (3% nonfat dried milk in TBS plus 0.1% Tween 20). Rabbit antibodies against AQP1 and AQP4 were obtained from Chemicon (Temecula, CA) and used at 1:500. Rabbit antibodies MEK1/2, pMEK1/2, ERK1, ERK2, and p44/p42 (Cell Signaling, Danver, MA) and mouse anti-GFAP (sigma) were used at 1:1000. Membranes were incubated in primary antibody for 1 hr at room temperature and washed 3× for 10 min. The membranes were then incubated in horseradish peroxidase-conjugated secondary antibodies (Sigma) at 1:1000 for 1 hr followed by another round of washing (3× 10 min) and developed using Luminol (Santa Cruz, Santa Cruz, CA). Images were taken on Kodak Imager (Rochester, NY) and analyzed using Kodak Imager software.
Immunocytochemistry
Cells plated on coverslips were fixed in 4% formaldehyde derived from freshly depolymerized paraformaldehyde (Sigma), hence labeled as 4% PFA, for 10 min. Cells were washed 2× 10 min in PBS, blocked and then permeabilized in PBS containing 0.3% Triton X-100 and 10% horse serum (HS) for 30 min. Cells were incubated overnight at 4°C in primary AQP antibodies at 1:500, MAPK 1:1000 and/or mouse anti-GFAP 1:1000. On the following day, astrocytes were washed in PBS (4× 5 min), blocked again and incubated with fluorescein isothiocyanate (FITC)-conjugated goat anti-rabbit secondary antibody (Molecular Probes) diluted at 1:500 and tetramethyl rhodamine iso-thiocyanate (TRITC)-conjugated goat anti-mouse at 1:750 in the dark for 1 hr. Cells were washed in PBS (2× 5 min) and incubated with DAPI, a fluorescent nuclear label, (1:2000, Sigma) for 5 min. Cells were washed 2× with PBS and mounted on slides with GelMount (Biomedia, Foster City, CA). Images were acquired using an inverted Olympus IX-81 spinning disk confocal microscope (Olympus, Center Valley, PA).
Volume Regulation
Cell volume measurements were performed using a Coulter Counter Multisizer 3 (Beckman-Coulter, Miami, FL) as described previously (Parkerson, K.A. and Sontheimer, H., 2003) and modified as described in (McCoy, E. and Sontheimer, H., 2007). Cells were washed in PBS and lifted from the dish using 0.05% trypsin and 0.53 mM EDTA. Trypsin was inactivated with the addition of an equal volume of serum-containing media, and cells were briefly centrifuged to pellet. Cells were resuspended in bath solution [125 mM NaCl, 5.0 mM KCl, 1.2 mM MgSO4, 1.6 mM Na2HPO4, 0.4 mM NaH2PO4, 10.5 mM glucose, 32.5 mM HEPES (acid), 1.0 mM CaCl2, pH 7.4, 300 ± 10 mosmol]. Osmolarity for solutions was measured by a freeze point osmometer (Fiske Micro-Osmometer 210; Fiske-Associates, Norwood, MA). Cells were equilibrated for approximately 5 min, and readings were taken continuously for 3 min. Bath solution was made hypo osmotic with the addition of dH2O. Cells were preincubated for 5 min with 300 μM mercuric chloride (HgCl2, Sigma). Data were collected by Multisizer 3 software, and 5000 pulse listings were exported to EXCEL as the average of 40–50 cells for each 20 ms timepoint. Data were collected as mean diameter and converted to mean cell volume. Mean cell volumes were normalized to baseline values. Data were plotted in Origin 7.0 (MicroCal, Northhampton, MA) ± se. Each time point graphed is an average of the mean cell volume for 40–50 cells per 20 ms.
Wound Assay
Astrocytes were grown to a confluent monolayer and scratches were made using a 200 μl plastic pipette tip. Cells were allowed to recover and migrate into the wound site for 24 hrs in the presence and absence of 2 μM U0126. Cells were fixed with 4% PFA and immunostained for GFAP and either AQP1 or AQP4, as stated previously. For Western blotting, lysates were collected from scratched 10 cm3 dishes and probed with rabbit antibodies against MEK1/2, pMEK1/2, ERK1, ERK2, or p44/p42.
Cortical Stab Wound
Six week old male Sprague-Dawley rats were anesthetized using isofluorane, and a 30-gauge syringe was stereotactically inserted 2 mm bregma intracranially. Animals were transcardially perfused 24 hr post injury with 4% PFA. Brains were fixed overnight at 4°C and sectioned coronally at 100 μm using a series 1000 vibratome (Vibratome, St. Louis, MO) and stained as described previously in methods.
Results
AQP1 is upregulated in reactive glia following cortical stab injuries
The primary objective of this study was to examine changes in the expression of AQPs in response to injury, and how the changes may be regulated. Published data suggest that the predominant AQP in astrocytes is AQP4, yet several reports find expression of AQP1 in brain after injury (Badaut, J. et. al., 2003; Simons, K. and Ikonen, E., 1997). As a starting point, we utilized a cortical stab injury in vivo and documented the appearance of AQP1 within 24 hr after injury. Representative images indicate the lesion site at low (Fig. 1A) and high magnification (Fig. 1B). The merged images show expression of AQP1 in GFAP positive astrocytes in the vicinity of the lesion. Staining with secondary antibody only was negative, and AQP1 expression was absent in the contralateral uninjured brain (Supplementary Figure 1). This data is consistent with the reported upregulation of AQP1 that occurs after brain contusion (Suzuki, R. et. al., 2006) or in association with peritumoral edema and subarachnoid hemorrhage (Badaut, J. et. al., 2003).
Figure 1.
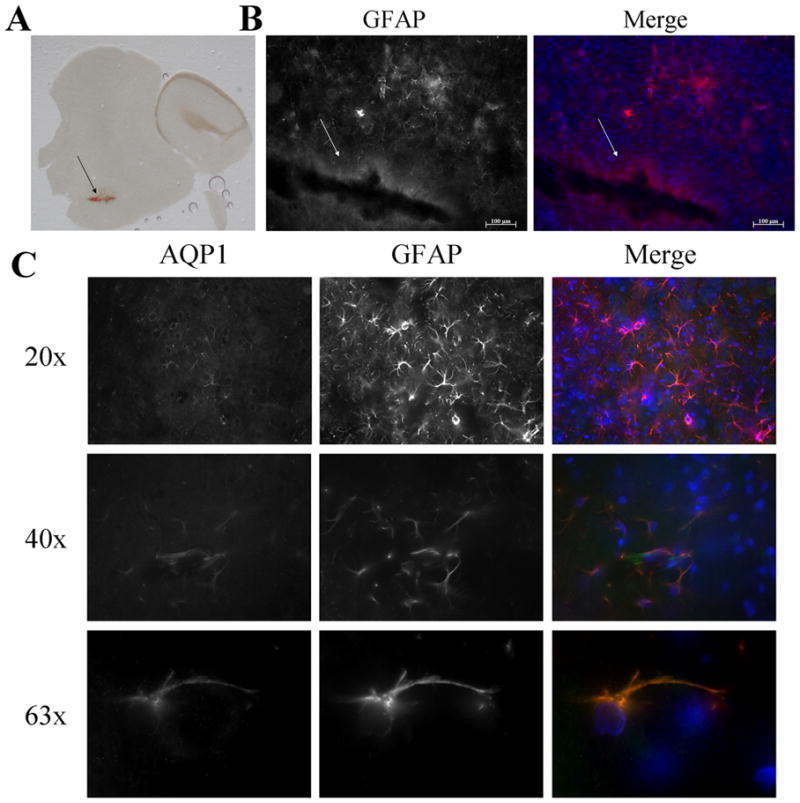
AQP1 expression in astrocytes in a cortical stab wound injury. Representative image of a brain slice indicating the lesion site (A). Fluorescence images show GFAP and DAPI staining at lesion site indicating reactive gliosis (B). Using immunofluorescence, AQP1 was upregulate in cortical astrocytes around the site of injury but not in the contralateral side (C). In addition, no staining was found when cells were stained with secondary antibodies alone. (Arrows indicate site of lesion; Green-AQP1, Red- GFAP, Blue- DAPI)
Primary astrocytes demonstrate AQP1 protein expression
To examine the regulation of AQPs by injury, we next sought to mimic injury conditions using a monolayer of primary cultured astrocytes. As a first step towards examining this preparation, we used PCR, Western blot and immunohistochemistry to assess the complement of AQPs in these cultures. After 7–10 days, primary astrocyte cultures obtained from P0 rats showed prominent expression of AQP1 and AQP4 with all detection methods (Fig. 2). This differs from previous publications that claim an absence of AQP1 in cultured astrocytes (Nakahama, K. et. al., 1999; Nicchia, G.P. et. al., 2000). The latter studies, as most studies in this field, utilized passaged cultures of astrocytes rather than primary cultures. When we passaged astrocytes and once again ran them side-by-side with primary astrocytes on Western blot and immunostainings, it became clear that AQP1 protein was selectively lost in passaged astrocytes (Fig. 2B). However, there were still detectable mRNA levels, suggesting that AQP1 protein is not being translated into protein. To gauge the level of expression, we ran primary astrocyte lysates side-by-side on a Western blot with whole brain and several astrocyte-derived tumor lines and two patient-derived samples (GBM50, GBM62). Highest level of AQP1 expression was observed in whole brain lysates, and one of the patient samples also showed a strong AQP1 band, while all of the highly passaged glioma lines were negative (Fig. 2C). This finding is further supported by immunostaining in Fig. 3 that shows punctate expression of AQP4 in both the primary and passaged astrocytes, whereas AQP1 is only expressed in primary astrocyte cultures. Fig. 3 illustrates staining for morphologically different examples of astrocytes that are typically present in these cultures. No relationship between cell morphology and AQP expression was found. This data suggests that cultured astrocytes lose AQP1 expression within a single passage. Importantly, it gives us an opportunity to examine primary and passaged astrocytes as experimental systems in which either AQP1 is expressed together with AQP4 (primary culture) or AQP4 is expressed alone (passaged cells), the latter mimicking the situation in adult brain.
Figure 2.
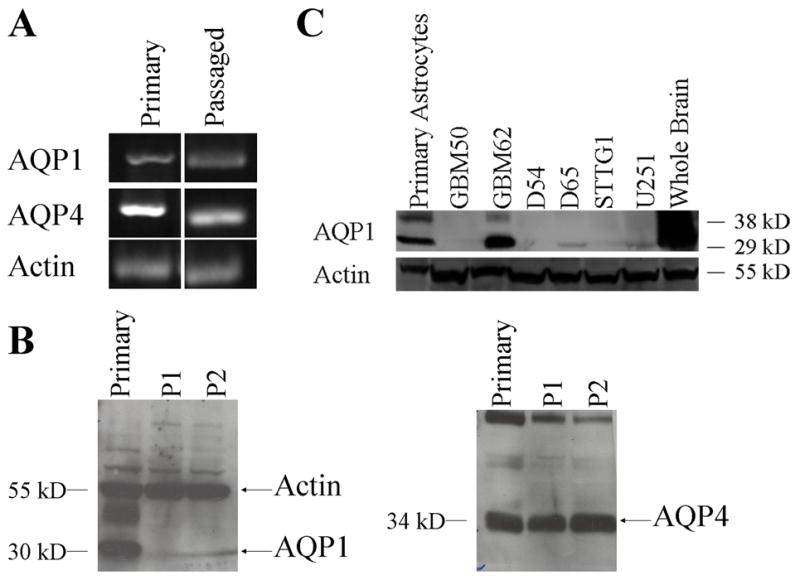
AQP expression in astrocyte cultures. Aquaporin mRNA expression in astrocytes (A). Western blot analysis was used to examine AQP1 and AQP4 expression levels in primary and passaged astrocyte cultures (B). AQP1 expression was compared to whole brain lysates and several glioblastoma cell lines (D54, G65, STTG1, U251) and patient derived samples (GBM50, GBM62) to gauge the level of expression (C). Highest level of AQP1 expression was observed in whole brain lysates, primary astrocytes and one of the patient samples also showed a strong AQP1 band, while all of the highly passaged glioma lines were negative.
Figure 3.

Immunoflourescence demonstrate expression of AQP1 in primary astrocyte cultures. AQP1 protein was found only in primary astrocytes, while AQP4 was located in both primary and secondary astrocyte cultures. No staining was found in cells stained with secondary antibodies alone. (Green- AQP1, Red- GFAP, Blue- DAPI)
Primary astrocytes exhibit mercury-sensitive cell swelling following a hypo osmotic challenge
An important question is the degree to which each of these AQPs contributes to homeostasis in astrocytes, i.e. indicative of functional AQPs. To determine if there were changes to the water permeability in those passaged astrocytes that express only AQP4 as compared to primary cultures expressing both AQP1 and AQP4, we examined water transport directly by measuring cell swelling in response to a hypo osmotic challenge with a Coulter-Counter Cell Sizer. Cell size was measured continuously, with 20 msec time resolution over a 3 min period. A 60 s baseline volume was followed by the addition of a 50% hypo osmotic challenge. Recordings were taken for an additional 2 min (see Methods for details). The average cell volume +/− S.E.M. for 40–50 cells was plotted every 20 msec. Figure 4 shows volume changes caused by water movement in primary and passaged astrocyte cultures (Fig. 4A,B respectively). Both cell types showed a rapid influx of water following the hypo osmotic challenge, indicative of the expression of functional aquaporins. Only in primary astrocytes did addition of 300 μM HgCl2, a known inhibitor of AQP1 (Jung, J.S. et. al., 1994) show a markedly reduced rate of cell swelling (Fig. 4A), whereas passaged astrocytes lacking AQP1 expression did not respond to HgCl2 at all (Fig 4B). To demonstrate the selectivity of HgCl2 for AQP1, we repeated the same experiments in two astrocyte-derived cell lines that selectively express either AQP1 or AQP4; Supplementary Figure 2). Here too, only AQP1 expressing cells responded to HgCl2. These data indicate that primary astrocytes utilize AQP1 as the predominant aquaporin for water movement. These studies also show that the degree and time-course of swelling was identical in primary and passaged cells, suggesting that AQP4 alone can confer sufficient water permeability in astrocytes. These results suggest that when AQP1 is co-expressed in astrocytes with AQP4, AQP4 function must be restricted. AQP4 function could be reduced due to a decrease in its plasma membrane expression, or it may have reduced water permeability, which may occur through phosphorylation by PKC (unpublished data).
Figure 4.

Functional expression of AQP1 and AQP4 in astrocytes. Using a Coulter-Counter, mean cell volume was measured for 3 min in primary (n=6) and passaged astrocyte cultures (n= 6) following a 50% hypo osmotic challenge. Primary astrocytes (A) preincubated with 300 μM HgCl2 show mercury sensitive water movement that is not found in the passaged astrocytes (B).
Astrocytes upregulate AQP1 following injury
Based on the above studies, secondary astrocyte cultures, expressing exclusively AQP4, should be a suitable model system to question whether injury may lead to an induction of AQP1 expression in vitro as has been observed in vivo. We employed an artificial wound assay in which a 200 μl pipette tip was used to produce multiple scratches through a confluent monolayer of astrocytes. Cells were then allowed to recover for 10 min, 30 min, 60 min, 16 hr and 24 hr prior to harvesting cell lysates for Western blot analysis. As shown in Fig. 5A, AQP1 became detectable 16h after injury and was prominently expressed at 24 hr. AQP1 expression could also be identified by immunostaining of astrocytes at the lesion 24 hr post-injury (Fig. 5C), yet no AQP1 positive cells were detectable 30 minutes after lesioning. Cell nuclei are visible by DAPI staining.
Figure 5.

AQP1 expression increases in passaged astrocytes following an in vitro wound assay. A confluent monolayer of astrocytes was scratched using a 200 μl pipette tip and allowed to recover for 10 min, 30 min, 60 min, 16 hr, and 24 hr and protein expression was examined using Western blotting (n=6). AQP1 expression was detectable at 16 hr and was maintained at 24 hr in controls (B) but expression was abolished by the addition of U0126 (B). Immunofluorescence confirms the Western blot results showing a lack of AQP1 expression following treatment with U0126 at 24 hr (C).
Gliosis, which occurs with mechanical injury, is also a hallmark of neurological diseases, including Alzheimer’s disease (Unger, J.W., 1998), Creutzfeldt-Jakob (Rodriguez, A. et. al., 2006), ALS, Parkinson’s disease and multiple sclerosis (Drew, P.D. et. al., 2006). Gliosis has also been shown to activate multiple signaling cascades in astrocytes, including the MAPK signaling pathway, which had been suggested as a possible regulator of AQPs (Arima, H. et. al., 2003;Umenishi, F. and Schrier, R.W., 2003). To examine the possibility that MAPK activation may underlie the observed induction of AQP1 after injury, we repeated the above experiments in the presence of 2 μM U0126, a MEK1/2 inhibitor. As illustrated in Fig. 5B & C, inhibition of MAPK signaling by preincubation with U0126 for 30 min completely inhibited the induction of AQP1 expression following injury, and no AQP1 positive cells could be identified at the lesion by immunohistochemistry. However, western blotting revealed an increase in pMEK1/2 and p44/p42 at 10 min, 30 min and 60 min (Fig. 6), and later time points showed no increase in phosphorylation of MEK1/2 and ERK1/2. Further, increased activity was inhibited by addition of U0126 (Fig. 6). This data is further supported by immunostaining, which reveals an increase in pMEK1/2 and p44/p42 at 30 min at the lesion site, but was absent by 24 hr (Fig. 7). This occurred without changes in the overall MEK1/2 and ERK1/2 levels. Taken together this data suggest that the MAPK signaling pathway is involved in the upregulation of AQP1 expression following injury.
Figure 6.
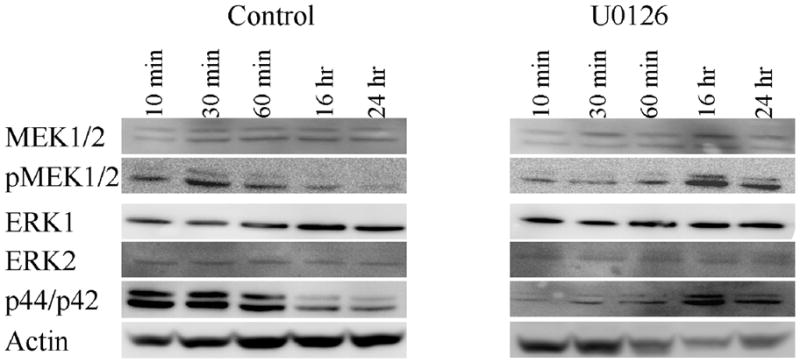
MAPK expression is upregulated immediately following injury. Time course for MEK1/2 and ERK1/2 activity was examined at 10 min, 30 min, 60 min, 16 hr and 24 hr (n=4). Western blot analysis showed increased expression of pMEK1/2 and p44/p42 within 60 min following injury, which can be inhibited by U0126.
Figure 7.

Expression of MAPK signaling molecules over 24 hr time course in a wound assay using passaged astrocytes. Examining the time course discussed in Figure 6, MEK1/2 and ERK1/2 expression at both 30 min and 24 hr time points in migrating astrocytes remained unchanged using immunofluorescence. However, their phosphorylated forms were only detected at the 30 min time point.
Discussion
In this study, we demonstrated an upregulation of AQP1 in astrocytes following a cortical stab wound. This process could be mimicked in an in vitro injury model, where it was possible to show a requirement for MAPK signaling in this induction. This finding is in agreement with previous work in the kidney, where chronic hypertonic challenges increased AQP1 protein expression in a MAPK-dependent fashion (Umenishi, F. and Schrier, R.W., 2003) and with a similar time-dependence as observed here. Following injury, we detected an increase in phosphorylation of MEK1/2, ERK1 and ERK2 by 30-60 min, and at 16 hr, only baseline levels were detectable. Highest expression of AQP1 was present at 16 hr and 24 hr. Pretreating the astrocytes with the MEK1/2 inhibitor U0126 could inhibit these effects.
We were surprised to find increased expression of pMEK1/2 and pERK1/2 at the 16 hr time point in the cells treated with U0126. It is possible that the incubation time was longer than the half-life of the inhibitor. Once the inhibitory effect was lost, the cells would increase the phosphorylation of the signaling molecules, as with the earlier time points, leading to an increase in activation. Also, in our immunostaining, we did not see this increase in the phosphorylated forms, which may have been due to the fact that we were only examining the site of the lesion. It has been demonstrated that MAPK signaling following injury spreads to the surrounding astrocytes (Mandell et al. 2001). Therefore, by Western blot one would detect expression in all astrocytes rather than only cells localized to the lesion.
MAPK signaling is important for regulating gene transcription, differentiation and cellular response to growth factors. Several AQPs have been shown to be regulated by MAPK. Specifically, AQP1 and AQP5 are upregulated following chronic hypertonicity in kidney cells, and this is due to activation of MAPK (Umenishi, F. and Schrier, R.W., 2003). Similarly, AQP4 and AQP9 are increased under hyperosmolar conditions (Arima, H. et. al., 2003). Previous reports have shown an increased expression of AQP1 in reactive astrocytes following contusion injury (Simons, K. and Ikonen, E., 1997), and we extended this finding to a cortical stab wound in vivo. Following subarachnoid hemorrhage, AQP1 expression is also increased around blood vessels due to an increased activation of JNK signaling (Yatsushige, H. et. al., 2007). AQP1 upregulation is thought to mediate the increase in vascular permeability found in SAH, which is consistent with this study where AQP1 became the predominant water channel for mediating water permeability in primary cortical astrocyte cultures.
MAPK signaling has been implicated in regulating the expression of numerous proteins following brain injury. For example, the upregulation in expression of GFAP is a hallmark of reactive gliosis dependent on MAPK signaling (O'Callaghan, J.P. et. al., 1998), and studies suggest that the overall process of reactive gliosis is dependent on MAPK (Mandell, J.W. and VandenBerg, S.R., 1999). In the retina, acute injury induces Muller glial proliferation that is directly regulated by MAPK via ERK1/2 activation (Fischer, A.J. et. al., 2008). Of course, gliosis is often associated with a remodeling of injured brain tissue and may well involve the migration of surrounding cells to the injury site. In light of this, AQP1 expression may signify a more migratory phenotype for astrocytes as AQP1 expression has been shown to promote cell migration in epithelial cells (Hara-Chikuma, M. and Verkman, A.S., 2006) and glial tumors (Papadopoulos, M.C. et. al., 2008). Alternatively, AQP1 expression in reactive astrocytes may contribute to edema associated with the injury as has been illustrated for a number of brain injuries (Papadopoulos, M.C. et. al., 2002). Expression of AQP1 is indicative of a subset of cortical astrocytes with the ability to differentially regulate water transport following cerebral edema.
Supplementary Material
Specificity of AQP1 staining in brain slices. To illustrate the specificity of the staining with AQP1 antibodies in tissue, brain sections from injured animals were stained with AQP1 antibodies or secondary only (green on merge), as well as GFAP (red) and nuclei counterstained with DAPI (blue). Only in the injured ipsilateral brain are GFAP positive AQP1 positive cells visible, and neither the contralateral brain nor the secondary control shows any AQP1 positive cells. This is more readily visible in cultured astrocytes at 40x magnification (bottom). (Arrows indicate site of the lesion).
HgCl2 specifically inhibits AQP1 water permeability. Astrocyte derived tumor cell lines which stably express either AQP1 or AQP4 were challenged with 50% water, and cell volume recorded in the presence and absence of HgCl2, or the Cl− channel inhibitors Cd2+ and NPPB. Only AQP1 expressing cells responded to HgCl2, and this response was not enhanced by Cd2+ or NPPB; AQP4 expressing cells were unaffected by either drug. Hence, HgCl2 is a specific AQP1 inhibitor.
Acknowledgments
This work was supported by NIH 5R01NS031234 and NIH 2R01NS036692.
References
- Amiry-Moghaddam M, Williamson A, Palomba M, Eid T, de Lanerolle NC, Nagelhus EA, Adams ME, Froehner SC, Agre P, Ottersen OP. Delayed K+ clearance associated with aquaporin-4 mislocalization: phenotypic defects in brains of alpha-syntrophin-null mice. Proc Natl Acad Sci U S A. 2003;100:13615–13620. doi: 10.1073/pnas.2336064100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arima H, Yamamoto N, Sobue K, Umenishi F, Tada T, Katsuya H, Asai K. Hyperosmolar mannitol simulates expression of aquaporins 4 and 9 through a p38 mitogen-activated protein kinase-dependent pathway in rat astrocytes. J Biol Chem. 2003;278:44525–44534. doi: 10.1074/jbc.M304368200. [DOI] [PubMed] [Google Scholar]
- Badaut J, Ashwal S, Tone B, Regli L, Tian HR, Obenaus A. Temporal and regional evolution of aquaporin-4 expression and magnetic resonance imaging in a rat pup model of neonatal stroke. Pediatr Res. 2007;62:248–254. doi: 10.1203/PDR.0b013e3180db291b. [DOI] [PubMed] [Google Scholar]
- Badaut J, Brunet JF, Grollimund L, Hamou MF, Magistretti PJ, Villemure JG, Regli L. Aquaporin 1 and aquaporin 4 expression in human brain after subarachnoid hemorrhage and in peritumoral tissue. Acta Neurochir Suppl. 2003;86:495–498. doi: 10.1007/978-3-7091-0651-8_101. [DOI] [PubMed] [Google Scholar]
- Bondy C, Chin E, Smith BL, Preston GM, Agre P. Developmental gene expression and tissue distribution of the CHIP28 water-channel protein. Proc Natl Acad Sci U S A. 1993;90:4500–4504. doi: 10.1073/pnas.90.10.4500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carriere A, Ray H, Blenis J, Roux PP. The RSK factors of activating the Ras/MAPK signaling cascade. Front Biosci. 2008;13:4258–4275. doi: 10.2741/3003. [DOI] [PubMed] [Google Scholar]
- Castigli E, Arcuri C, Giovagnoli L, Luciani R, Giovagnoli L, Secca T, Gianfranceschi GL, Bocchini V. Interleukin-1beta induces apoptosis in GL15 glioblastoma-derived human cell line. Am J Physiol Cell Physiol. 2000;279:C2043–C2049. doi: 10.1152/ajpcell.2000.279.6.C2043. [DOI] [PubMed] [Google Scholar]
- Chen KC, Nicholson C. Spatial buffering of potassium ions in brain extracellular space. Biophys J. 2000;78:2776–2797. doi: 10.1016/S0006-3495(00)76822-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drew PD, Xu J, Storer PD, Chavis JA, Racke MK. Peroxisome proliferator-activated receptor agonist regulation of glial activation: relevance to CNS inflammatory disorders. Neurochem Int. 2006;49:183–189. doi: 10.1016/j.neuint.2006.04.003. [DOI] [PubMed] [Google Scholar]
- Fischer AJ, Scott MA, Tuten W. Mitogen-activated protein kinase-signaling stimulates Muller glia to proliferate in acutely damaged chicken retina. Glia. 2008 doi: 10.1002/glia.20743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara-Chikuma M, Verkman AS. Aquaporin-1 facilitates epithelial cell migration in kidney proximal tubule. J Am Soc Nephrol. 2006;17:39–45. doi: 10.1681/ASN.2005080846. [DOI] [PubMed] [Google Scholar]
- Hasegawa H, Lian SC, Finkbeiner WE, Verkman AS. Extrarenal tissue distribution of CHIP28 water channels by in situ hybridization and antibody staining. Am J Physiol. 1994;266:t-903. doi: 10.1152/ajpcell.1994.266.4.C893. [DOI] [PubMed] [Google Scholar]
- Hebert MA, O'Callaghan JP. Protein phosphorylation cascades associated with methamphetamine-induced glial activation. Ann N Y Acad Sci. 2000;914:238–262. doi: 10.1111/j.1749-6632.2000.tb05200.x. [DOI] [PubMed] [Google Scholar]
- Jenq W, Cooper DR, Bittle P, Ramirez G. Aquaporin-1 expression in proximal tubule epithelial cells of human kidney is regulated by hyperosmolarity and contrast agents. Biochem Biophys Res Commun. 1999a;256:240–248. doi: 10.1006/bbrc.1999.0306. [DOI] [PubMed] [Google Scholar]
- Jenq W, Cooper DR, Bittle P, Ramirez G. Aquaporin-1 expression in proximal tubule epithelial cells of human kidney is regulated by hyperosmolarity and contrast agents. Biochem Biophys Res Commun. 1999b;256:240–248. doi: 10.1006/bbrc.1999.0306. [DOI] [PubMed] [Google Scholar]
- Jung JS, Preston GM, Smith BL, Guggino WB, Agre P. Molecular structure of the water channel through aquaporin CHIP. The hourglass model. J Biol Chem. 1994;269:14648–14654. [PubMed] [Google Scholar]
- Katsura H, Obata K, Miyoshi K, Kondo T, Yamanaka H, Kobayashi K, Dai Y, Fukuoka T, Sakagami M, Noguchi K. Transforming growth factor-activated kinase 1 induced in spinal astrocytes contributes to mechanical hypersensitivity after nerve injury. Glia. 2008;56:723–733. doi: 10.1002/glia.20648. [DOI] [PubMed] [Google Scholar]
- Kofuji P, Newman EA. Potassium buffering in the central nervous system. Neurosci. 2004;129:1043–1054. doi: 10.1016/j.neuroscience.2004.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim JH, Gibbons HM, O'Carroll SJ, Narayan PJ, Faull RL, Dragunow M. Extracellular signal-regulated kinase involvement in human astrocyte migration. Brain Res %20. 2007;1164:1–13. doi: 10.1016/j.brainres.2007.06.020. [DOI] [PubMed] [Google Scholar]
- Mandell JW, Gocan NC, VandenBerg SR. Mechanical trauma induces rapid astroglial activation of ERK/MAP kinase: Evidence for a paracrine signal. Glia. 2001;34:283–295. doi: 10.1002/glia.1062. [DOI] [PubMed] [Google Scholar]
- Mandell JW, VandenBerg SR. ERK/MAP kinase is chronically activated in human reactive astrocytes. Neuroreport. 1999;10:3567–3572. doi: 10.1097/00001756-199911260-00019. [DOI] [PubMed] [Google Scholar]
- Manley GT, Fujimura M, Ma T, Noshita N, Filiz F, Bollen AW, Chan P, Verkman AS. Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat Med. 2000;6:159–163. doi: 10.1038/72256. [DOI] [PubMed] [Google Scholar]
- McCarthy KD, deVellis J. Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J Physiol (London) 1980;85:890–902. doi: 10.1083/jcb.85.3.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy E, Sontheimer H. Expression and function of water channels (aquaporins) in migrating malignant astrocytes. Glia. 2007;55:1034–1043. doi: 10.1002/glia.20524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagelhus EA, Horio Y, Inanobe A, Fujita A, Haug FM, Nielsen S, Kurachi Y, Ottersen OP. Immunogold evidence suggests that coupling of K+ siphoning and water transport in rat retinal Muller cells is mediated by a coenrichment of Kir4.1 and AQP4 in specific membrane domains. Glia. 1999;26:47–54. doi: 10.1002/(sici)1098-1136(199903)26:1<47::aid-glia5>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- Nakahama K, Nagano M, Fujioka A, Shinoda K, Sasaki H. Effect of TPA on aquaporin 4 mRNA expression in cultured rat astrocytes. Glia. 1999;25:240–246. [PubMed] [Google Scholar]
- Nicchia GP, Frigeri A, Liuzzi GM, Santacroce MP, Nico B, Procino G, Quondamatteo F, Herken R, Roncali L, Svelto M. Aquaporin-4-containing astrocytes sustain a temperature- and mercury-insensitive swelling in vitro. Glia. 2000;31:29–38. doi: 10.1002/(sici)1098-1136(200007)31:1<29::aid-glia30>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Nielsen S, Nagelhus EA, Amiry-Moghaddam M, Bourque C, Agre P, Ottersen OP. Specialized membrane domains for water transport in glial cells: high-resolution immunogold cytochemistry of aquaporin-4 in rat brain. J Neurosci. 1997;17:171–180. doi: 10.1523/JNEUROSCI.17-01-00171.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Callaghan JP, Martin PM, Mass MJ. The MAP kinase cascade is activated prior to the induction of gliosis in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) model of dopaminergic neurotoxicity. Ann N Y Acad Sci. 1998;844:40–49. [PubMed] [Google Scholar]
- Papadopoulos MC, Krishna S, Verkman AS. Aquaporin water channels and brain edema. Mt Sinai J Med. 2002;69:242–248. [PubMed] [Google Scholar]
- Papadopoulos MC, Manley GT, Krishna S, Verkman AS. Aquaporin-4 facilitates reabsorption of excess fluid in vasogenic brain edema. FASEB J. 2004;18:1291–1293. doi: 10.1096/fj.04-1723fje. [DOI] [PubMed] [Google Scholar]
- Papadopoulos MC, Saadoun S, Verkman AS. Aquaporins and cell migration. Pflugers Arch. 2008;456:693–700. doi: 10.1007/s00424-007-0357-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkerson KA, Sontheimer H. Contribution of chloride channels to volume regulation of cortical astrocytes. Am J Physiol Cell Physiol. 2003;284:C1460–C1467. doi: 10.1152/ajpcell.00603.2002. [DOI] [PubMed] [Google Scholar]
- Rash JE, Davidson KG, Yasumura T, Furman CS. Freeze-fracture and immunogold analysis of aquaporin-4 (AQP4) square arrays, with models of AQP4 lattice assembly. Neurosci. 2004;129:915–934. doi: 10.1016/j.neuroscience.2004.06.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro MC, Hirt L, Bogousslavsky J, Regli L, Badaut J. Time course of aquaporin expression after transient focal cerebral ischemia in mice. J Neurosci Res. 2006;83:1231–1240. doi: 10.1002/jnr.20819. [DOI] [PubMed] [Google Scholar]
- Rodriguez A, Perez-Gracia E, Espinosa JC, Pumarola M, Torres JM, Ferrer I. Increased expression of water channel aquaporin 1 and aquaporin 4 in Creutzfeldt-Jakob disease and in bovine spongiform encephalopathy-infected bovine-PrP transgenic mice. Acta Neuropathol (Berl) 2006;112:573–585. doi: 10.1007/s00401-006-0117-1. [DOI] [PubMed] [Google Scholar]
- Santos SD, Verveer PJ, Bastiaens PI. Growth factor-induced MAPK network topology shapes Erk response determining PC-12 cell fate. Nat Cell Biol. 2007;9:324–330. doi: 10.1038/ncb1543. [DOI] [PubMed] [Google Scholar]
- Shields SD, Mazario J, Skinner K, Basbaum AI. Anatomical and functional analysis of aquaporin 1, a water channel in primary afferent neurons. Pain. 2007;131:8–20. doi: 10.1016/j.pain.2006.11.018. [DOI] [PubMed] [Google Scholar]
- Simons K, Ikonen E. Functional rafts in cell membranes. Nature. 1997;387:569–572. doi: 10.1038/42408. [DOI] [PubMed] [Google Scholar]
- Umenishi F, Schrier RW. Identification and characterization of a novel hypertonicity-responsive element in the human aquaporin-1 gene. Biochem Biophys Res Commun. 2002a;292:771–775. doi: 10.1006/bbrc.2002.6709. [DOI] [PubMed] [Google Scholar]
- Umenishi F, Schrier RW. Identification and characterization of a novel hypertonicity-responsive element in the human aquaporin-1 gene. Biochem Biophys Res Commun. 2002b;292:771–775. doi: 10.1006/bbrc.2002.6709. [DOI] [PubMed] [Google Scholar]
- Umenishi F, Schrier RW. Hypertonicity-induced aquaporin-1 (AQP1) expression is mediated by the activation of MAPK pathways and hypertonicity-responsive element in the AQP1 gene. J Biol Chem. 2003;278:15765–15770. doi: 10.1074/jbc.M209980200. [DOI] [PubMed] [Google Scholar]
- Unger JW. Glial reaction in aging and Alzheimer's disease. Microsc Res Tech. 1998;43:24–28. doi: 10.1002/(SICI)1097-0029(19981001)43:1<24::AID-JEMT4>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Wang W, Hart PS, Piesco NP, Lu X, Gorry MC, Hart TC. Aquaporin expression in developing human teeth and selected orofacial tissues. Calcif Tissue Int. 2003;72:222–227. doi: 10.1007/s00223-002-1014-9. [DOI] [PubMed] [Google Scholar]
- Yatsushige H, Ostrowski RP, Tsubokawa T, Colohan A, Zhang JH. Role of c-Jun N-terminal kinase in early brain injury after subarachnoid hemorrhage. J Neurosci Res. 2007;85:1436–1448. doi: 10.1002/jnr.21281. [DOI] [PubMed] [Google Scholar]
- Zhao J, Moore AN, Clifton GL, Dash PK. Sulforaphane enhances aquaporin-4 expression and decreases cerebral edema following traumatic brain injury. J Neurosci Res. 2005;82:499–506. doi: 10.1002/jnr.20649. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Specificity of AQP1 staining in brain slices. To illustrate the specificity of the staining with AQP1 antibodies in tissue, brain sections from injured animals were stained with AQP1 antibodies or secondary only (green on merge), as well as GFAP (red) and nuclei counterstained with DAPI (blue). Only in the injured ipsilateral brain are GFAP positive AQP1 positive cells visible, and neither the contralateral brain nor the secondary control shows any AQP1 positive cells. This is more readily visible in cultured astrocytes at 40x magnification (bottom). (Arrows indicate site of the lesion).
HgCl2 specifically inhibits AQP1 water permeability. Astrocyte derived tumor cell lines which stably express either AQP1 or AQP4 were challenged with 50% water, and cell volume recorded in the presence and absence of HgCl2, or the Cl− channel inhibitors Cd2+ and NPPB. Only AQP1 expressing cells responded to HgCl2, and this response was not enhanced by Cd2+ or NPPB; AQP4 expressing cells were unaffected by either drug. Hence, HgCl2 is a specific AQP1 inhibitor.