

Abstract
To gain insights into the mechanism of action of sclerostin, a protein that regulates bone mass, we performed yeast two-hybrid analyses using human SOST (sclerostin) cDNA cloned into pGBKT7 DNA-binding domain vector as a bait, and a normalized, high-complexity, universal cDNA library in a GAL4 activating domain vector. We identified an interaction between sclerostin and the carboxyl-terminal portion of the receptor tyrosine-protein kinase erbB-3. To determine the biological relevance of this interaction, we treated MC3T3-E1 mouse osteoblast cells transfected with either a SOST expression plasmid or a control vector, with recombinant heregulin/neuregulin. Phospho-p44/42 (Thr202/Tyr204) MAPK was assessed in heregulin/neuregulin treated cells. We observed an increase in phospho-p44/42 (Thr202/Tyr204) MAPK concentrations in SOST transfected cells but not in cells transfected with a control vector, thus demonstrating a modulatory effect of sclerostin on heregulin/neuregulin signaling in osteoblasts. The data demonstrate that sclerostin functions in part, by modulating the activity of erbB-3.
Keywords: Sclerostin, epidermal growth factor receptor, erbB-3, osteoblast, yeast two hybrid
Introduction
Sclerostin is a secreted glycoprotein that plays an important role in regulating bone biology [1; 2]. Human subjects with the sclerosing bone dysplasias, sclerosteosis and van Buchem disease, either lack, or have reduced expression of sclerostin, as a result of stop mutations or splice abnormalities in the SOST gene, or a mutation within an enhancer downstream of the coding region of the SOST gene [2; 3; 4]. Individuals with sclerosteosis have increased bone density throughout the skeleton, but especially within bones at the base of the skull, the latter frequently resulting in narrowing of cranial nerve foramina with attendant cranial nerve palsies, or narrowing of the foramen magnum with increased intracranial pressure [2; 5; 6; 7]. Patients with sclerosteosis frequently die within the second or third decade of life as a result of complications from excessive bone growth. These observations indicate that sclerostin has a profound effect on bone formation in the human skeleton.
Sclerostin expression is observed within osteocytes, osteoblasts, osteoclasts and perichondrial cells [1; 8]. Sclerostin functions by modulating the activity of the bone morphogenic proteins, lipoprotein-receptor related protein 5/6 (LRP 5/6), and the CCN protein, cyr61 [1; 8; 9; 10; 11; 12; 13; 14; 15]. Sclerostin has been shown to directly interact with the bone morphogenic proteins, BMP2, BMP4, BMP6 and BMP7 [8; 15]. In addition, sclerostin binds to the native LRP 5/6 wnt co-receptor but fails to interact with the LRP 5/6 receptor from patients with the high bone mass syndrome, suggesting that it may alter osteoblast function through the LRP 5/6 receptor [11]. We recently demonstrated that sclerostin interacts with the cysteine-rich domain of the CCN protein, cyr61, and observed that sclerostin and cyr61 interact to regulate cell adhesion, growth and migration [15].
We now show that sclerostin interacts with the carboxyl-terminal tail of the epidermal growth factor receptor 3 (erbB-3) and that it modulates the activity of this receptor in osteoblasts.
Materials and methods
Yeast two-hybrid experiments
These were carried out as described previously [15]. A normalized universal Human Mate & Plate Library and the Matchmaker Gold System (Clontech, Mountainview, CA) were used. The following primers specific for human SOST we used to generate a cDNA for cloning in to the pGBKT7 DNA-BD plasmid: 5’ primer: 5’ GAGAGAATTCCAGGGGTGGCAGGCGTTCAAGAATGATGCC 3’, and 3’ primer: 5’ GAGAGGATCCCTAGTAGGCGTTCTCCAGCTCGGCCTGGTTGG 3’.
The underlined sequences are EcoRI and BamHI restriction endonuclease sites. The SOST PCR cDNA construct was treated with EcoRI and BamHI restriction endonucleases and cloned in-frame with the GAL4 DNA-BD of the pGBKT7 DNA-BD vector previously treated with EcoRI and BamHI restriction enzymes. The SOST-pGBKT7 plasmid was used to transform Y2H Gold yeast cells. Appropriate positive and negative control mating, auto-activation and toxicity experiments were performed. One mL of the Mate and Plate Library was combined with 5 mL of the bait strain and 45 mL of 2X YPDA medium (50 µg/mL kanamycin) and incubated for 24 h at 30 °C, after which cells were pelleted, washed with 50 mL of 0.5X YPDA (50 µg/mL kanamycin) and re-suspended in 10 mL of 0.9% NaCl. The culture was plated onto 150-mm SD/-Trp/-Leu/X-αGal/Aureobasidin A plates. The plates were incubated at 30 °C. for 5 days. Positive colonies were re-analyzed on quadruple drop-out (SD/-Leu/-Trp/-His/-Ade) plates containing X-αGal/Aureobasidin A. Aliquots of blue colonies that had been serially selected were used to generate inserts for DNA sequencing. PCR was performed on colonies using Matchmaker Insert Check PCR Mix 2 (Takara Bio/Clontech Laboratories, Mountain View, CA) with flanking primers specific for the pGAD-T7-RecAB plasmid.
Analysis of cells expressing interacting proteins
Yeast cells expressing interacting proteins were lysed in buffer containing 100 mM Tris, pH 7.4, 100 mM NaCl and protease inhibitors by rapid shaking at 4 °C with 0.5 mm glass beads (BioSpec Products, Inc., Bartlesville, OK) on a vortex mixer. The proteins in the clarified lysates were precipitated with HA or c-myc antibodies and protein A beads (40 µL) (Thermo Scientific Pierce, Rockford, IL). The washed, precipitated complexes were separated by SDS–PAGE [16]. Electrophoresed proteins were transferred to PVDF membranes [16]. The presence of the partner protein was determined with a peroxidase-labeled c-myc antibody and chemiluminescence methods [16]. To detect sclerostin in complexes, a monoclonal antibody directed against the N-terminus of sclerostin was used as probe. Peroxidase-labeled anti-mouse IgG was used to detect bound sclerostin with chemiluminescence methods.
Plasmids were rescued from yeast and were used to transform E. coli grown on ampicillin containing plates to isolate the “prey” plasmid as recommended by the manufacturer. Protein interactions were confirmed in yeast cells co-transformed with rescued “prey” and “bait” plasmids by cell lysis, immunoprecipitation with HA or c-myc antibodies and protein A beads and detection as noted above [16]. Proteins was separated by SDS-PAGE and transferred to PVDF membranes. C-myc antibody, or monoclonal antibody to human sclerostin, was used to probe the membranes as described above.
Effects of the SOST expression on heregulin/neuregulin signaling in mouse osteoblasts
MC3T3-E1 mouse osteoblast cells (#CRL-2593, American Type Culture Collection (ATCC), Manassas, VA) were grown in 1X minimum essential medium-α, (MEMα, #A10490), 10% fetal bovine serum (FBS) (#26140079), 10 units/mL penicillin, 10 µg/mL streptomycin (Invitrogen Corp., Carlsbad, CA), at 37 °C, 5% CO2. MC3T3-E1 cells were lifted using 0.05% trypsin-EDTA 1X (#25300, Invitrogen) and plated into 12-well plates (Corning Inc-Costar #3512, Corning, NY). Cells were grown to ~80% confluency. Cells were transfected with lipofectamine (Invitrogen). Cells were rinsed 2X with Opti-Mem I, reduced serum media 1X (#31985, Invitrogen), and then transfected in Opti-Mem I. Cells were transfected with no DNA (control), or 0.8 µg/well pcDNA3.1(+) (Invitrogen) or 0.8 µg/well human sclerostin expression vector SOSTpcDNA3.1(+) (Plasmid 10842, Addgene Inc., Cambridge, MA) for 5 hr. One volume MEMα, 20% FBS was added, and cells were allowed to grow for 48 hours post-transfection. After 48 hours heregulin/neuregulin (recombinant human HRG1β1/NRG1β1 EGF Domain, #396HB/CF, R&D Systems, Inc., Minneapolis, MN) in PBS (Cellgro 21-010-CV) was added to duplicate wells transfected with SOSTpcDNA3.1(+) or pcDNA3.1(+) for 10 min or 1 hr. At the appropriate time cells were rinsed 2X with PBS, and harvested by scraping cells in lysis buffer (2% SDS, 150 mM NaCl, 50 mM Tris pH 8.2 with complete protease inhibitor cocktail (#11697498001, Roche Diagnostics, Indianapolis, IN)). Protein was assayed in the lysate using the BCA reagent with bovine serum albumin (BSA) standard (Thermo Fisher Scientific, Waltham, MA). Cell lysates were combined with 4X SDS sample buffer (25 mM Tris base, 20% beta-mercaptoethanol, 40% glycerol 8% sodium dodecyl sulfate, 0.04% bromophenol blue), heated to 100 °C for 5 min. Five µg protein/well or pre-stained low molecular weight standard, were loaded to 15% acrylamide gels (15% Ready Gels, Bio-Rad Hercules, CA). Gels were soaked in transfer buffer (25 mM Tris, 192 mM glycine, 20% methanol) for 10 min proteins were transferred electrophoretically to polyvinylidene diflouride (PVDF) membrane (Bio-Rad). Before immunoblotting, membranes were blocked with 1% blocking reagent (#11500694001, Roche Diagnostics) for 1 hr at room temperature. Membranes were probed with 1:1000 primary antibody to phospho-p44/42 (Thr202/Tyr204) MAPK (#9101S, Cell Signaling Technology, Inc., Danvers, MA) overnight in 5% BSA in TBST (50 mM Tris, 150 mM NaCl, pH 7.5 with 0.1% Tween 20). Membranes were then washed with TBST, and treated with 1:2000 secondary antibody, goat anti-rabbit IgG-horseradish peroxidase, 0.5 % Roche block (P0448;Dako, Carpinteria, CA), and developed using chemiluminescent substrate (Roche Diagnostics). After development blots were washed 2 X 10 min with stripping buffer (0.2 M glycine, 0.1% SDS, 1% tween 20, pH 2.2), 2 X 10 min with PBS and 2 X 10 min with then with TBST. Membranes were re-blocked and probed using mouse anti-β-actin monoclonal antibody (#A2228, Sigma-Aldrich, St. Louis, MO) and anti-mouse IgG gamma-chain specific horseradish peroxidase (#A3673, Sigma-Aldrich) as loading control.
Results
Sclerostin interacts with the erbB-3
Yeast two-hybrid experiments demonstrated that sclerostin bound to the carboxyl-terminal tail of the erbB3 receptor. The SOST cDNA was cloned in-frame into the pGBKT7 DNA-BD plasmid and the chimeric “bait” plasmid was used to transform Y2H Gold MATa Saccharomyces cerevisiae cells. Following mating with a normalized universal Human Mate & Plate Library (Y187 MATα Saccharomyces cerevisiae cells) transformed with pGADT7-RecAB “prey” plasmids and selection on appropriate media, blue colonies capable of growing on quadruple dropout medium were identified. Interactions between sclerostin and the protein encoded by the “prey plasmid” were confirmed by co-immunoprecipitation methods (Figure 1). In lane A, proteins were precipitated with a c-myc antibody and in lane B an HA antibody was used for immunoprecipitation. In lane C no antibody was used for immunoprecipitation. The immunoblot in lanes A–C was probed with an anti-c-myc antibody that is expressed as an amino-terminal fusion with sclerostin from the pBKT7 plasmid. As anticipated, in lane A the c-myc immunoprecipitating antibody precipitated the c-myc-sclerostin fusion that was subsequently detected by the c-myc antibody. In lane B, the anti-HA antibody precipitated the "prey" protein to which the c-myc-sclerostin fusion was bound. Subsequent probing with the c-myc antibody detected the bound c-myc- sclerostin fusion. In lane C, no antibody was used for immunoprecipitation and no signal was noted. In lane D, a c-myc antibody was used for immunoprecipitation and an anti-sclerostin monoclonal antibody was used as a probe. As expected, the c-myc-sclerostin fusion was immunoprecipitated by the c-myc antibody and the protein was subsequently detected by the anti-sclerostin monoclonal antibody. In lane E, anti-HA antibody was used for immunoprecipitation. The antibody immunoprecipitated the HA-prey fusion protein to which sclerostin was bound. Sclerostin was subsequently detected by the monoclonal antibody against sclerostin.
Figure 1.

Yeast cells containing the c-myc-sclerostin and HA-tagged "prey" protein expression are plasmids were grown for three days at 30 °C. Cells were lysed and immuno-precipitation was performed using c-myc (lanes A and D) or HA (lanes B and E) antibodies or no antibodies (lane C). The antibody complexes were captured by protein A beads, the protein precipitates were separated by SDS-PAGE and transferred onto PVDF membranes. The membranes were probed with c-myc or sclerostin monoclonal antibodies.
The cDNA insert in the pGADT7-RecAB “prey” vector was sequenced. The insert, coding for the protein interacting with the human sclerostin bait protein is shown in Figure 2. The cDNA encodes nucleotides 3493–4029 (through the stop codon) of the ErbB-3 receptor (GenBank NM_001982.2 with G 3807 to A (no coding change) nucleotide difference from the GenBank entry)). These nucleotides code for the last 178 amino acids, His 1165 to Thr 1342 (last amino acid), in the human erbB-3 receptor intracellular domain.
Figure 2.
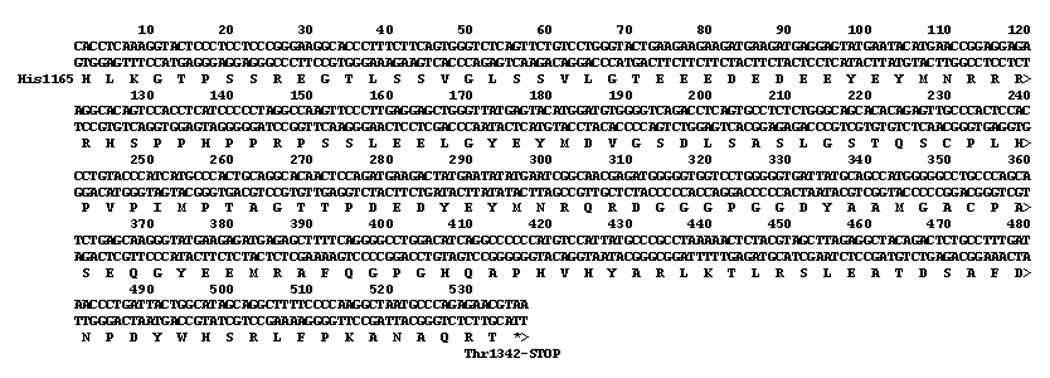
Insert of pBKT7-AD plasmid that interacted with sclerostin expressing pGBK7-DB plasmid. The sequence corresponds to nucleotides 3493–4029 (through stop codon) in GenBank NM_001982.2, human ErbB-3.
Interactions were confirmed by isolating and purifying each plasmid encoding sclerostin or the erbB-3 receptor intracellular domain. The purified plasmids were used to co-transform yeast cells. Co-immunoprecipitation experiments with appropriate antibodies confirmed the interaction between sclerostin and erbB-3 (Figure 3). Immunoprecipitation was performed as described in figure 1. In lane A, the immunoprecipitated antibody was a c-myc antibody which immunoprecipitated the amino-terminal c-myc-sclerostin fusion protein. This was readily detected by the c-myc antibody used as a probe. In lane B, an HA antibody was used for immunoprecipitation which precipitated the HA-erbB-3 fusion to which the c-myc-sclerostin fusion protein was bound. The latter protein was then detected by the c-myc antibody. In lane C, the immunoprecipitated antibody was a c-myc antibody that immunoprecipitated the c-myc-sclerostin fusion that was subsequently detected by the sclerostin monoclonal antibody. In lane D, the immunoprecipitated antibody was an HA antibody that immunoprecipitated the HA-erbB-3 fusion to which sclerostin was bound. Sclerostin was subsequently detected by the sclerostin monoclonal antibody. This data confirms the interaction between sclerostin and erbB-3 carboxyl-terminal domain.
Figure 3.

Transformed yeast cells containing the c-myc-sclerostin and HA-tagged erbB-3 carboxyl-terminal domain expression plasmids were grown for three days at 30 °C. Cells were lysed and immuno-precipitation was performed using c-myc (lanes A and D) or HA (lanes B and E) antibodies or no antibodies (lane C). The antibody complexes were captured by protein A beads, the protein precipitates were separated by SDS-PAGE and transferred onto PVDF membranes. The membranes were probed with c-myc or sclerostin monoclonal antibodies.
Sclerostin interacts with the erbB-3 receptor to modulate erbB-3 signaling in osteoblast-like cells
To determine if the sclerostin-erbB-3 receptor interaction he is a biological significance, we transfected MC3T3 osteoblast-like cells with a sclerostin expression plasmid or a blank vector. Cells were then treated with neuregulin/heregulin and erbB-3 receptor signaling was assessed by determining phospho p44/42 amounts in the presence or absence of expressed sclerostin (Figure 4). The amount of phospho p44/42 was increased in cells expressing sclerostin compared with cells not expressing sclerostin (compare lanes 3A versus 4A and lanes 3B versus 4B). This data suggests that in the presence of sclerostin expression, neuregulin/heregulin increase erbB-3 receptor signaling in osteoblast cells.
Figure 4.

MC3T3 cells were transfected with a sclerostin expression plasmid (SostpcDNA3.1 (+)) or an empty vector (pcDNA3.1 (+)). The transfected cells were then treated with neuregulin/heregulin (HRGβ1) for 10 minutes or 1 hour. Phospho p44/42 was assessed by immunoblotting.
Discussion and Conclusion
Sclerostin is a secreted glycoprotein that influences osteoblast function [1; 8]. When sclerostin expression is absent or greatly reduced, sclerosteosis or van Buchem disease develop [2; 3; 4]. Sclerostin functions by interacting with the bone morphogenic proteins 2, 4, 6, and 7, with the LRP 5/6 Wnt co-receptor and with the CCN protein, cyr61 [1; 8; 9; 10; 11; 12; 13; 14; 15]. Although the increase in bone mass observed in patients with sclerosteosis and van Buchem syndrome is similar to that observed in patients with the high bone mass syndrome due to mutations in the LRP 5/6 Wnt co-receptor, other phenotypic features (such as syndactyly, and the predominant basilar skull bone increase observed in sclerosteosis) suggest that sclerostin may function by additional pathways [2; 7; 17; 18; 19].
We previously observed that sclerostin interacted with, and modulated the function of cyr61, a member of the CCN family of proteins important for chondrocyte, osteoblast and vascular function [15]. We now demonstrate that sclerostin also interacts with the carboxyl-terminal portion of the erbB-3 receptor and modulates the activity of the receptor in osteoblast-like cells. Epidermal growth factor (EGF) and EGF-ligands and the erb tyrosine-kinase receptors play an important role in the cartilage and bone formation [20; 21; 22; 23; 24; 25; 26; 27]. Mice with EGF overexpression have excessive chondrocyte hypertrophy and altered bone formation [28]. Based on our data, it is conceivable that sclerostin expression within osteoblast and chondrocyte cell lineages could alter epidermal growth factor-ligand activity.
In summary, we have demonstrated that sclerostin binds to the carboxyl-terminal domain of erbB-3 and could function by modulating the activity of epidermal growth factor pathways in bone and cartilage.
Acknowledgments
Supported by NIH grant DK 76829 and a grant from the Dr. Ralph and Marian C. Falk Medical Research Trust (RK).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Winkler DG, Sutherland MK, Geoghegan JC, Yu C, Hayes T, Skonier JE, Shpektor D, Jonas M, Kovacevich BR, Staehling-Hampton K, Appleby M, Brunkow ME, Latham JA. Osteocyte control of bone formation via sclerostin, a novel BMP antagonist. Embo J. 2003;22:6267–6276. doi: 10.1093/emboj/cdg599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beighton PH, Hanmersma H, Brunkow ME. SOST-Related Sclerosing Bone Dysplasias. GeneReviews NCBI Bookshelf; 2010. [Google Scholar]
- 3.Staehling-Hampton K, Proll S, Paeper BW, Zhao L, Charmley P, Brown A, Gardner JC, Galas D, Schatzman RC, Beighton P, Papapoulos S, Hamersma H, Brunkow ME. A 52-kb deletion in the SOST-MEOX1 intergenic region on 17q12-q21 is associated with van Buchem disease in the Dutch population. Am J Med Genet. 2002;110:144–152. doi: 10.1002/ajmg.10401. [DOI] [PubMed] [Google Scholar]
- 4.Brunkow ME, Gardner JC, Van Ness J, Paeper BW, Kovacevich BR, Proll S, Skonier JE, Zhao L, Sabo PJ, Fu Y, Alisch RS, Gillett L, Colbert T, Tacconi P, Galas D, Hamersma H, Beighton P, Mulligan J. Bone dysplasia sclerosteosis results from loss of the SOST gene product, a novel cystine knot-containing protein. Am J Hum Genet. 2001;68:577–589. doi: 10.1086/318811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beighton P, Horan F, Hamersma H. A review of the osteopetroses. Postgrad Med J. 1977;53:507–516. doi: 10.1136/pgmj.53.622.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beighton P, Davidson J, Durr L, Hamersma H. Sclerosteosis - an autosomal recessive disorder. Clin Genet. 1977;11:1–7. doi: 10.1111/j.1399-0004.1977.tb01269.x. [DOI] [PubMed] [Google Scholar]
- 7.Beighton P, Durr L, Hamersma H. The clinical features of sclerosteosis. A review of the manifestations in twenty-five affected individuals. Ann Intern Med. 1976;84:393–397. doi: 10.7326/0003-4819-84-4-393. [DOI] [PubMed] [Google Scholar]
- 8.Kusu N, Laurikkala J, Imanishi M, Usui H, Konishi M, Miyake A, Thesleff I, Itoh N. Sclerostin is a novel secreted osteoclast-derived bone morphogenetic protein antagonist with unique ligand specificity. J Biol Chem. 2003;278:24113–24117. doi: 10.1074/jbc.M301716200. [DOI] [PubMed] [Google Scholar]
- 9.Lowik CW, van Bezooijen RL. Wnt signaling is involved in the inhibitory action of sclerostin on BMP-stimulated bone formation. J Musculoskelet Neuronal Interact. 2006;6:357. [PubMed] [Google Scholar]
- 10.van Bezooijen RL, Svensson JP, Eefting D, Visser A, van der Horst G, Karperien M, Quax PH, Vrieling H, Papapoulos SE, ten Dijke P, Lowik CW. Wnt but not BMP signaling is involved in the inhibitory action of sclerostin on BMP-stimulated bone formation. J Bone Miner Res. 2007;22:19–28. doi: 10.1359/jbmr.061002. [DOI] [PubMed] [Google Scholar]
- 11.Ellies DL, Viviano B, McCarthy J, Rey JP, Itasaki N, Saunders S, Krumlauf R. Bone density ligand, Sclerostin, directly interacts with LRP5 but not LRP5G171V to modulate Wnt activity. J Bone Miner Res. 2006;21:1738–1749. doi: 10.1359/jbmr.060810. [DOI] [PubMed] [Google Scholar]
- 12.Winkler DG, Sutherland MS, Ojala E, Turcott E, Geoghegan JC, Shpektor D, Skonier JE, Yu C, Latham JA. Sclerostin inhibition of Wnt-3a-induced C3H10T1/2 cell differentiation is indirect and mediated by bone morphogenetic proteins. J Biol Chem. 2005;280:2498–2502. doi: 10.1074/jbc.M400524200. [DOI] [PubMed] [Google Scholar]
- 13.Winkler DG, Yu C, Geoghegan JC, Ojala EW, Skonier JE, Shpektor D, Sutherland MK, Latham JA. Noggin and sclerostin bone morphogenetic protein antagonists form a mutually inhibitory complex. J Biol Chem. 2004;279:36293–36298. doi: 10.1074/jbc.M400521200. [DOI] [PubMed] [Google Scholar]
- 14.van Bezooijen RL, Roelen BA, Visser A, van der Wee-Pals L, de Wilt E, Karperien M, Hamersma H, Papapoulos SE, ten Dijke P, Lowik CW. Sclerostin is an osteocyte-expressed negative regulator of bone formation, but not a classical BMP antagonist. J Exp Med. 2004;199:805–814. doi: 10.1084/jem.20031454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Craig TA, Bhattacharya R, Mukhopadhyay D, Kumar R. Sclerostin binds and regulates the activity of cysteine-rich protein 61. Biochem Biophys Res Commun. 2010;392:36–40. doi: 10.1016/j.bbrc.2009.12.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K. Current Protocols in Molecular Biology. John Wiley and Sons; 2007. [Google Scholar]
- 17.Little RD, Carulli JP, Del Mastro RG, Dupuis J, Osborne M, Folz C, Manning SP, Swain PM, Zhao SC, Eustace B, Lappe MM, Spitzer L, Zweier S, Braunschweiger K, Benchekroun Y, Hu X, Adair R, Chee L, FitzGerald MG, Tulig C, Caruso A, Tzellas N, Bawa A, Franklin B, McGuire S, Nogues X, Gong G, Allen KM, Anisowicz A, Morales AJ, Lomedico PT, Recker SM, Van Eerdewegh P, Recker RR, Johnson ML. A mutation in the LDL receptor-related protein 5 gene results in the autosomal dominant high-bone-mass trait. Am J Hum Genet. 2002;70:11–19. doi: 10.1086/338450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Little RD, Recker RR, Johnson ML. High bone density due to a mutation in LDL-receptor-related protein 5. N Engl J Med. 2002;347:943–944. doi: 10.1056/NEJM200209193471216. author reply 943-4. [DOI] [PubMed] [Google Scholar]
- 19.Boyden LM, Mao J, Belsky J, Mitzner L, Farhi A, Mitnick MA, Wu D, Insogna K, Lifton RP. High bone density due to a mutation in LDL-receptor-related protein 5. N Engl J Med. 2002;346:1513–1521. doi: 10.1056/NEJMoa013444. [DOI] [PubMed] [Google Scholar]
- 20.Centrella M, Canalis E. Isolation of EGF-dependent transforming growth factor (TGF beta-like) activity from culture medium conditioned by fetal rat calvariae. J Bone Miner Res. 1987;2:29–36. doi: 10.1002/jbmr.5650020106. [DOI] [PubMed] [Google Scholar]
- 21.Coffin-Collins PA, Hall BK. Chondrogenesis of mandibular mesenchyme from the embryonic chick is inhibited by mandibular epithelium and by epidermal growth factor. Int J Dev Biol. 1989;33:297–311. [PubMed] [Google Scholar]
- 22.Dealy CN, Scranton V, Cheng HC. Roles of transforming growth factor-alpha and epidermal growth factor in chick limb development. Dev Biol. 1998;202:43–55. doi: 10.1006/dbio.1998.8988. [DOI] [PubMed] [Google Scholar]
- 23.Ishizeki K, Takahashi N, Nawa T. Formation of the sphenomandibular ligament by Meckel's cartilage in the mouse: possible involvement of epidermal growth factor as revealed by studies in vivo and in vitro. Cell Tissue Res. 2001;304:67–80. doi: 10.1007/s004410100354. [DOI] [PubMed] [Google Scholar]
- 24.Miettinen PJ, Chin JR, Shum L, Slavkin HC, Shuler CF, Derynck R, Werb Z. Epidermal growth factor receptor function is necessary for normal craniofacial development and palate closure. Nat Genet. 1999;22:69–73. doi: 10.1038/8773. [DOI] [PubMed] [Google Scholar]
- 25.Nonaka K, Shum L, Takahashi I, Takahashi K, Ikura T, Dashner R, Nuckolls GH, Slavkin HC. Convergence of the BMP and EGF signaling pathways on Smad1 in the regulation of chondrogenesis. Int J Dev Biol. 1999;43:795–807. [PubMed] [Google Scholar]
- 26.Wong RW. Transgenic and knock-out mice for deciphering the roles of EGFR ligands. Cell Mol Life Sci. 2003;60:113–118. doi: 10.1007/s000180300007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yoon YM, Oh CD, Kim DY, Lee YS, Park JW, Huh TL, Kang SS, Chun JS. Epidermal growth factor negatively regulates chondrogenesis of mesenchymal cells by modulating the protein kinase C-alpha, Erk-1, and p38 MAPK signaling pathways. J Biol Chem. 2000;275:12353–12359. doi: 10.1074/jbc.275.16.12353. [DOI] [PubMed] [Google Scholar]
- 28.Chan SY, Wong RW. Expression of epidermal growth factor in transgenic mice causes growth retardation. J Biol Chem. 2000;275:38693–38698. doi: 10.1074/jbc.M004189200. [DOI] [PubMed] [Google Scholar]