

Abstract
Vaccines based on recombinant proteins require adjuvant systems in order to generate Th1-type immune responses. We have developed a vaccine adjuvant system using a viscous chitosan solution and interleukin (IL)-12, a Th1-inducing cytokine. The chitosan solution is designed to create a depot of antigen and IL-12 at a subcutaneous injection site. We measured the in vivo immune response of a vaccine containing 0.25, 1, or 4 μg murine IL-12 and 75 μg ovalbumin (OVA), formulated in a 1.5% chitosan glutamate solution. The chitosan/IL-12/OVA vaccine, in comparison to chitosan/OVA, IL-12/OVA, or OVA alone, elicited greater antigen-specific CD4+ and CD8+ T-cell responses, as determined by CD4+ splenocyte proliferation, Th1 cytokine release, CD8+ T cell interferon-γ release, and MHC class I peptide pentamer staining. The combination of chitosan and IL-12 also enhanced IgG2a and IgG2b antibody responses to OVA. Co-formulation of chitosan and IL-12 thus promoted the generation of a Th1 immune response to a model protein vaccine.
1. Introduction
There is growing interest in developing adjuvant systems for vaccines based on recombinant proteins [1, 2]. While protein-based vaccines have potential for cancer immunotherapy and treatment of some infectious diseases [3], protein antigens alone are weak stimulators of the immune system and thus require adjuvant systems to achieve robust immune responses [1]. It is hypothesized that effective adjuvant systems for protein antigens will include both a delivery system and an immunopotentiator [2, 4]. Immunostimulatory agents provide the molecular signals needed to stimulate B and T cells or to activate antigen-presenting cells (APCs) for T-cell cross-priming, while optimal delivery systems can extend the residence time of injected antigen and enhance uptake by APCs.
The proinflammatory effects of interleukin (IL)-12 make it a strong candidate as an immunopotentiator in vaccine adjuvant systems. IL-12 induces T cells and natural killer (NK) cells to produce interferon (IFN)-γ, granulocyte-macrophage colony-stimulating factor (GM-CSF), and tumor necrosis factor (TNF)-α, directs CD4+ T cells toward T-helper (Th)1 differentiation, and induces T-cell proliferation [5]. Recombinant IL-12 has been widely studied as a vaccine adjuvant due to its ability to shift protein-based vaccines from a Th2 to a Th1 response [6]. For example, the addition of recombinant IL-12 has been shown to shift the CD4+ T-cell response from Th2 to Th1 in vaccines containing antigens of Leishmania major [7, 8], Schistosoma mansoni [9], and Bordetella pertussis [10]. IL-12 also increased protection against L. major and B. pertussis infection when given with the corresponding antigens [8, 10]. Furthermore, in a pseudorabies virus challenge model, the addition of IL-12 to an inactivated viral vaccine resulted in increased survival and increased IgG2a antibody production [11], an indicator of a Th1-biased immune response [12].
While these models demonstrate the efficacy of IL-12 administered in soluble form, some studies suggest that increasing the residence time of IL-12 may enhance its efficacy. For example, an HIV-1 gp120 vaccine combined with alum-formulated IL-12, but not soluble IL-12, elicited Th1-type immune responses in mice, characterized by serum IFN-γ levels and IgG isotype switching [13]. Similarly, a plasmid DNA encoding IL-12 was used to generate persistent IL-12 expression. As an adjuvant to a whole-cell killed L. major vaccine, IL-12 DNA was more effective than soluble IL-12 or alum-formulated IL-12 in generating long-term protective immunity to L. major infection [14]. Thus, it can be hypothesized that the adjuvanticity of IL-12 may be improved through the use of delivery systems that extend the residence time of IL-12 at the injection site.
A viscous solution of chitosan, a polysaccharide derived from chitin, has considerable potential as a delivery system for soluble proteins. Chitosan is a naturally sourced polymer with a good record of biocompatibility. A review of several studies conducted in various animals concluded that local intranasal, subcutaneous, ocular, or topical administration of chitosan was generally safe and resulted only in mild reactions [15]. Also, subcutaneous or intraperitoneal implantation of chitosan gels resulted in a typical foreign body response and caused no damage to distal organs [16]. Our laboratory has employed viscous chitosan solution as an injectable protein delivery system. Chitosan solution extended the subcutaneous residence time of admixed proteins, including β-galactosidase (β-gal) and GM-CSF, and enhanced the Th1-inducing properties of GM-CSF when coadministered with an inactivated influenza virus [17, 18]. Recently, chitosan has been shown to dramatically enhance the antitumor efficacy of IL-12 via local delivery to subcutaneously implanted tumors and superficial bladder tumors [19, 20].
Based on the delivery potential of chitosan and its compatibility with IL-12 and other proteins, we hypothesized that chitosan and IL-12 could be combined as an adjuvant system for subcutaneously administered protein-based vaccines. In this study, we evaluated the in vivo efficacy of a chitosan/IL-12 adjuvant system using ovalbumin (OVA) as a model protein antigen. The vaccine consisted of a mixture of OVA protein and recombinant murine IL-12 in a 1.5% chitosan glutamate solution. The objective was to determine if combining the immunopotentiating agent IL-12 with a chitosan delivery system could shift the immune response from Th2- to Th1-polarized, as characterized by T-cell and antibody responses following a prime/boost vaccination regimen.
2. Materials and Methods
2.1 Materials
The vaccine components used in this study were sourced as follows: chitosan, 200-600 kDa, 75% to 90% deacetylated (Protosan UP G213, NovaMatrix; Sandvika, Norway); recombinant murine IL-12 (PeproTech; Rocky Hill, NJ); albumin from chicken egg white, grade VI (OVA, A2512; Sigma-Aldrich; St. Louis, MO). The following reagents were purchased from the suppliers indicated: ß-gal (BG13, Prozyme; Hayward, CA); FITC-, PE-, or PerCP-Cy5.5-labeled antibodies to CD8, CD3 (BD Biosciences; San Jose, CA, and eBioscience; San Diego, CA); PE-labeled MHC class I pentamers with H-2Kb-restricted peptides for OVA (aa 257-264, SIINFEKL) and HSV (aa 498-505, SSIEFARL) (ProImmune; Bradenton, FL); horseradish peroxidase-conjugated goat anti-mouse IgG total, IgG1, IgG2a, and IgG2b antibodies (LifeSpan Biosciences; Seattle, WA); OVA257-264 peptide (SIINFEKL) and VSV-NP52-59 peptide (RGYVYQGL) (CPC Scientific; San Jose, CA). Complete medium was prepared with RPMI-1640 supplemented with 10% FBS, 2 mM L-glutamine, 0.1 mM nonessential amino acids, 1 mM sodium pyruvate, 100 IU/mL penicillin, 100 μg/mL streptomycin, 10 mM HEPES buffer, and 55 μM 2-mercaptoethanol.
2.2 Animals
C57BL/6 mice (8 to 20 weeks old) were obtained from Charles River Laboratories (Wilmington, MA). Mice were housed under pathogen-free conditions, and animal care conformed to the Guide for the Care and Use of Laboratory Animals (National Research Council).
2.3 Vaccinations
Vaccine was prepared by mixing OVA and IL-12 with chitosan solution (1.5% (w/v) final concentration) in DPBS. The dosage was 75 μg OVA and either 0.25, 1, or 4 μg IL-12 per vaccination. Controls included OVA alone, OVA + IL-12, and OVA + chitosan. Mice were anesthetized with ketamine/xylazine prior to vaccination and primed on day 0 with a subcutaneous injection of 100 μL vaccine on one side of the lumbar region. On day 14 mice were given a boost with the same volume of vaccine on the contralateral side of the lumbar region. In each case the vaccine components were coinjected using a single syringe. On day 21, blood was collected and centrifuged to generate serum samples, which were stored at −20°C. Harvested spleens were dispersed by filtering through a 70-μm nylon mesh strainer (BD Biosciences), then pooled and treated with ACK red blood cell lysis buffer (Lonza; Allendale, NJ).
2.4 Serum Antibody ELISA
OVA-specific serum antibodies were measured by ELISA [17]. Briefly, 96-well plates were sensitized overnight at 4°C with 100 ng/well of antigen (OVA) or control (β-gal) protein in 50 μL of DPBS. Plates were aspirated and blocked with 100 μL DPBS plus 5% BSA (Mediatech; Herndon, VA) for 1 h at 37°C, washed once with DPBS plus 1% BSA, then incubated with 50 μL of serially diluted serum samples (1:20 to 1:1,562,500 in DPBS containing 1% BSA) for 1.5 h. After washing 3 times, plates were incubated with 50 μL of a 1:4000 dilution of HRP-conjugated antimouse IgG antibodies (IgG total, IgG1, IgG2a, and IgG2b) for 1 h. Plates were washed 3 times, and HRP was quantified by adding 100 μL of TMB Substrate (Pierce Protein Research Products; Rockford, IL). After 20 min, the reaction was stopped with 100 μL of 1 M HCl, and the optical density was measured at 450 nm.
2.5 Splenic CD4+ Proliferation Assay
CD4+ cells were isolated from splenocyte suspensions using a Dynal negative isolation kit per the manufacturer’s instructions (Invitrogen; Carlsbad, CA). CD4+ splenocytes (200,000 cells/well) from vaccinated mice were incubated with irradiated (20 Gy) splenocytes (500,000 cells/well) from naive syngeneic mice in a volume of 200 μL/well in a 96-well plate. The cultures were pulsed with 6.25–100 μg/mL OVA protein for 5 days. As a positive control, cells were pulsed with 0.06–2 μg/mL concanavalin A for 3 days. In the last 18 to 24 h of each culture, plates were treated with 1 μCi/well 3H-thymidine (Perkin-Elmer; Waltham, MA). Following the culture, plates were harvested onto glass fiber filtermats using a Tomtec Harvester 96 (Hamden, CT), and incorporated radioactivity was measured by liquid scintillation counting on a Wallac 1450 MicroBeta (Perkin-Elmer). Results from triplicate wells were combined to yield a mean +/− standard error of the mean (SEM) for each animal or pooled treatment group.
2.6 Splenic CD4+ Cell Cytokine Release
Mice were vaccinated on days 0 and 14, and spleens were harvested on day 21. CD4+ splenocytes were cultured under the same conditions as the proliferation assay, at antigen protein (OVA) and control protein (β-gal) concentrations of 100 μg/mL. Supernatants were sampled after 4 days of incubation and analyzed for IFN-γ and IL-4 by ELISA per the manufacturer’s protocol (Pierce Protein Research Products). Data are presented as the mean of duplicate wells.
2.7 MHC Class I Pentamer Staining
Prior to pentamer staining, splenocytes were depleted of B cells using a Dynabeads Mouse Pan B (B220) isolation kit (Invitrogen) in order to reduce background pentamer signal. B cell-depleted splenocytes were stained with MHC class I pentamers according to the manufacturer’s protocol. Briefly, 1 × 106 cells were transferred to a 12 × 75-mm polystyrene tube, washed, and resuspended in the residual liquid. Cells were stained for 30 min with 5 μL PE-labeled pentamer and 1.5 μL FITC-labeled anti-CD8 antibody. All samples were stained with antigen-specific OVA257-264 (SIINFEKL) or control HSV498-505 (SSIEFARL) H-2Kb pentamers. Cells were washed twice, fixed with BD Cytofix (BD Biosciences), and stored overnight before reading on an LSR-II Cell Analyzer (BD Biosciences). Cells were gated by forward/side scatter and by FITC-CD8. Results are expressed as the percentage of CD8+ cells that are positive for the MHC-I/peptide pentamer.
2.8 In Vitro Cytokine Release (IFN-γ)
Splenocytes were pulsed for 6 days with 1 μg/mL SIINFEKL peptide in 10 mL of complete medium in upright T-25 flasks. Following in vitro stimulation, cells were washed with complete medium and incubated overnight at 6 × 106 cells per well in 6-well plates without peptide. Cultured splenocytes (0.5 × 106 cells/well) were then incubated with 5 × 106 irradiated (20 Gy) splenocytes from naive syngeneic mice, along with 1 μg/mL antigen peptide (OVA257-264, SIINFEKL) or control peptide (VSV-NP52-59, RGYVYQGL), in a 24-well plate with a volume of 2 mL/well. Supernatants were collected at 24 h, and IFN-γ concentration was measured by ELISA per the manufacturer’s protocol (Pierce Protein Research Products).
2.9 Statistical Analysis
CD4+ splenoctye proliferation data are presented as the mean ± SEM. Differences in means between treatment groups were analyzed using a 2-tailed Welch’s t-test assuming unequal variances (Prism; GraphPad Software, Inc; La Jolla, CA). Differences in means were accepted as significant if P was less than 0.05.
3. Results
3.1 Serum Antibody ELISA
Antigen-specific serum antibody responses (total IgG and subtypes G1, G2a, and G2b) were measured 7 days after the boost vaccination (Fig. 1). Chitosan/OVA/IL-12 vaccine generated significant levels of IgG1, IgG2a, and IgG2b antibodies. The presence of IgG2a and IgG2b antibodies is indicative of a Th1-polarized immune response. In contrast, the chitosan/OVA vaccine generated a high level of IgG1 and a moderate level of IgG2a and IgG2b antibodies, indicating a mixed Th1/Th2 response with a bias toward Th2 [12, 21]. OVA/IL12 vaccine, however, elicited low levels of IgG antibodies. These data demonstrate that the combination of chitosan and IL-12 is required to induce Th1-biased IgG isotype switching.
Figure 1.
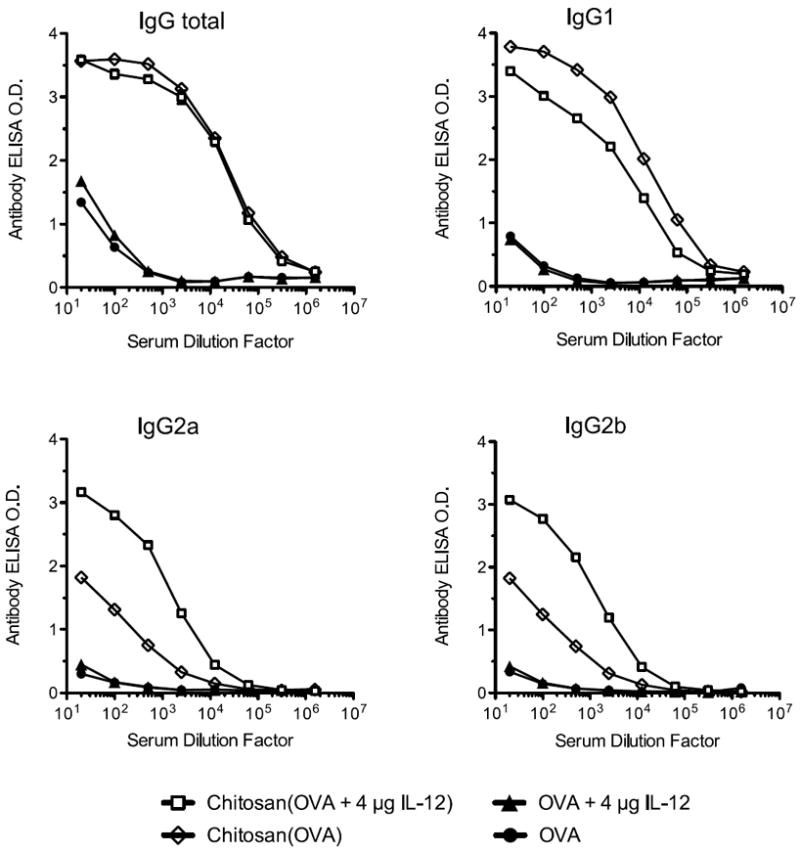
Chitosan/OVA/4 μg IL-12 vaccine elicits IgG1, IgG2a, and IgG2b antibody response. Mice were vaccinated on days 0 and 14 with OVA ± chitosan ± 4 μg IL-12 and sacrificed on day 21. Blood serum was pooled (n = 5), and OVA-specific IgG antibody responses were measured by ELISA. Data are plotted as mean of triplicate wells. Results are representative of 3 independent experiments with similar trends.
3.2 Splenic CD4+ Proliferation Assay
The recall response of T-helper cells was assayed 7 days following the boost vaccination (Fig. 2). Chitosan/OVA/IL-12 vaccine induced a strong antigen-specific proliferation of CD4+ splenocytes in response to OVA protein stimulation. The maximum CD4+ cell proliferation for chitosan/OVA/4 μg IL-12 was 2-fold higher (P < 0.05) than for OVA/4 μg IL-12 and 3.2-fold higher (P < 0.05) than for chitosan/OVA. These data indicate that coformulation of chitosan and IL-12 significantly enhances the CD4+ helper T-cell response in comparison to either adjuvant alone.
Figure 2.
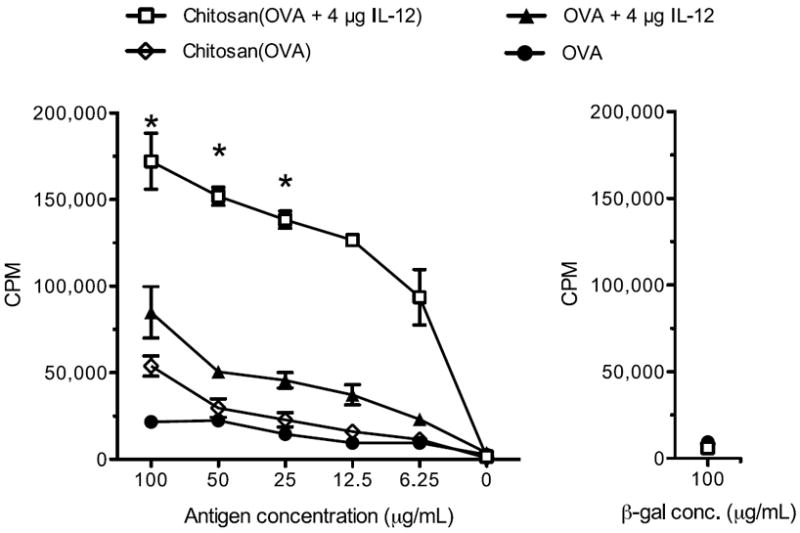
Chitosan/OVA/IL-12 vaccine elicits antigen-specific CD4+ T-cell responses. Mice (n = 5) were vaccinated on days 0 and 14 with OVA ± chitosan ± 4 μg IL-12, and splenocytes were harvested 7 days after the boost injection. Proliferation of OVA protein-pulsed CD4+-sorted splenocytes was measured by 3H-thymidine incorporation. Cells incubated with β-gal protein were a negative control. Data are plotted as mean ± SEM of triplicate wells. Results are representative of 3 independent experiments with similar trends. *P < 0.05 compared to OVA + 4 μg IL-12.
3.3 Splenic CD4+ Cell Cytokine Release
CD4+ splenocytes from mice vaccinated with chitosan/OVA/4 μg IL-12 produced 565,000 pg/mL of IFN-γ (a Th1 cytokine) after 4 days of incubation with OVA protein (Fig. 3). This represents a 9-fold increase over the next highest group, OVA/4 μg IL-12 (64,300 pg/mL). The response was antigen-specific, as cells stimulated with β-gal control protein produced less than 600 pg/mL of IFN-γ. Production of IL-4 (a Th2 cytokine) was 78 pg/mL for chitosan/OVA/4 μg IL-12 and 107 pg/mL for OVA/4 μg IL-12. The ratio of IFN-γ to IL-4 production was 7200 for chitosan/OVA/4 μg IL-12 and 600 for OVA/4 μg IL-12, indicating that the addition of chitosan to OVA/IL-12 enhances Th1 polarization.
Figure 3.
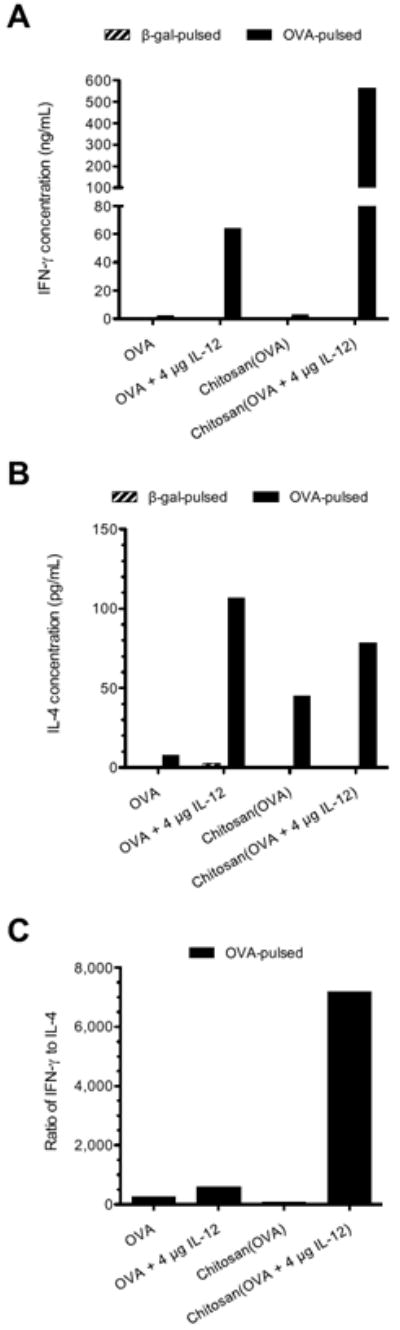
Chitosan/OVA/IL-12 vaccine elicits Th1 polarization in splenocytes. Mice (n = 5) were vaccinated on days 0 and 14 with OVA ± chitosan ± 4 μg IL-12, and splenocytes were harvested 7 days after the boost injection. Pooled splenocytes were cultured with irradiated APCs and 100 μg/mL antigen protein (OVA) or control protein (β-gal) for 4 days, and released (A) IFN-γ and (B) IL-4 were measured by ELISA. Results are plotted as the mean of duplicate wells. (C) Calculated ratio of IFN-γ to IL-4 concentrations for OVA-pulsed cells.
3.4 MHC Class I Pentamer Staining
B cell-depleted splenocytes from mice treated with chitosan/OVA/IL-12 were stained with MHC class I OVA257-264 (SIINFEKL) antigen and HSV498-505 (SSIEFARL) control pentamers to measure antigen-specific CD8+ T-cell responses. Figure 4 shows pentamer staining for varying doses of IL-12 administered with chitosan and OVA, measured 7 days after the boost vaccination. In mice treated with chitosan/OVA/4 μg IL-12, 1.8% of CD8+ splenocytes stained positive for OVA257-264 (SIINFEKL) pentamer, compared to 0.8% for chitosan/OVA/1 μg IL-12 and less than 0.3% for chitosan/OVA/0.25 μg IL-12. OVA/4 μg IL-12 vaccine administered in PBS resulted in background levels of OVA257-264 pentamer staining. Thus, the chitosan delivery system was required for IL-12 to induce CD8+ T cells to recognize the MHC class I OVA257-264 peptide.
Figure 4.
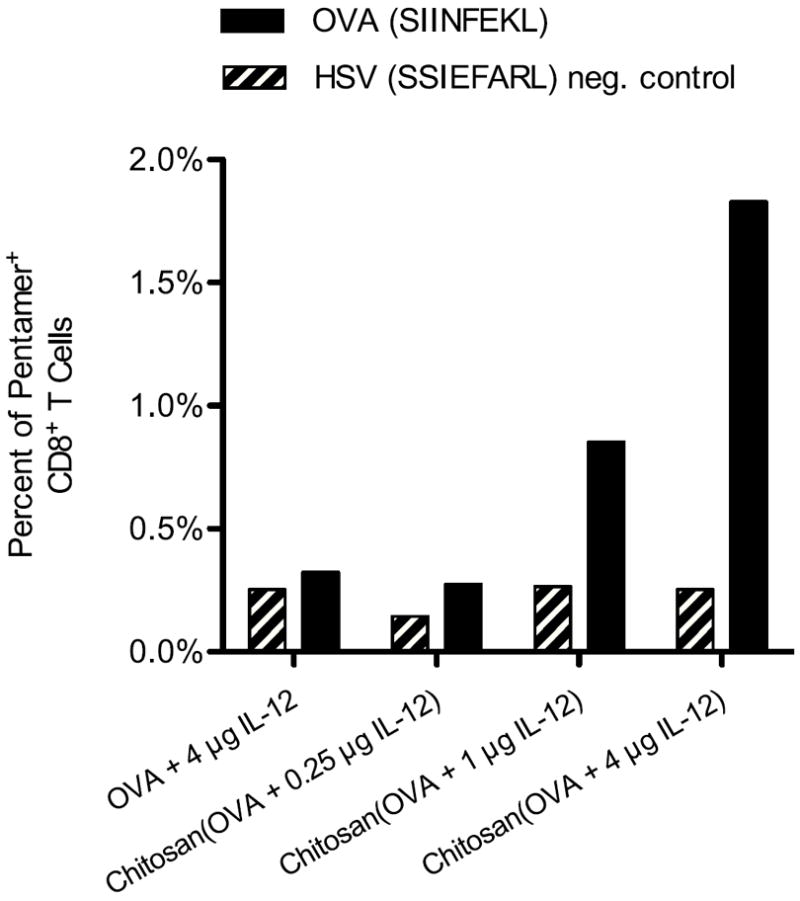
Chitosan/OVA/IL-12 vaccine induces production of CD8+ T cells that recognize MHC class I-restricted antigen peptide. Mice (n = 5) were vaccinated on days 0 and 14 with OVA ± chitosan + IL-12 at doses of 0.25, 1, or 4 μg IL-12, and splenocytes were harvested on day 21. B cell-depleted splenocytes were pooled and stained with FITC-CD8 and either PE-labeled OVA257-264 (SIINFEKL) or PE-labeled HSV498-505 (SSIEFARL) MHC class I pentamer. Data are expressed as the percentage of CD8+ cells that were positive for MHC class I pentamer. Results are representative of 2 independent experiments with similar trends.
3.5 In Vitro Cytokine Release (IFN-γ)
Splenocytes isolated from vaccinated mice were stimulated with OVA257-264 (SIINFEKL) peptide for 6 days, rested overnight in peptide-free medium, and then restimulated with peptide and fresh syngeneic irradiated splenocytes (APCs). IFN-γ levels in the supernatant at 24 h indicated CD8+ T-cell activation in response to the antigenic MHC class I peptide. Restimulated cells from mice treated with chitosan/OVA/4 μg IL-12 (Fig. 5) produced high levels of IFN-γ (1480 pg/mL for 500,000 cells in 2 mL), which was 10-fold higher than the next highest group, which was mice treated with chitosan/OVA/1 μg IL-12. Cells restimulated with control peptide (VSV-NP) produced low levels of IFN-γ, indicating that the CD8+ T-cell response to the chitosan/OVA/4 μg IL-12 vaccine is specific to the OVA antigen. The combination of chitosan and IL-12 was required to achieve the CD8+ T-cell response, as the OVA/4 μg IL-12 group (without chitosan) showed negligible IFN-γ production.
Figure 5.
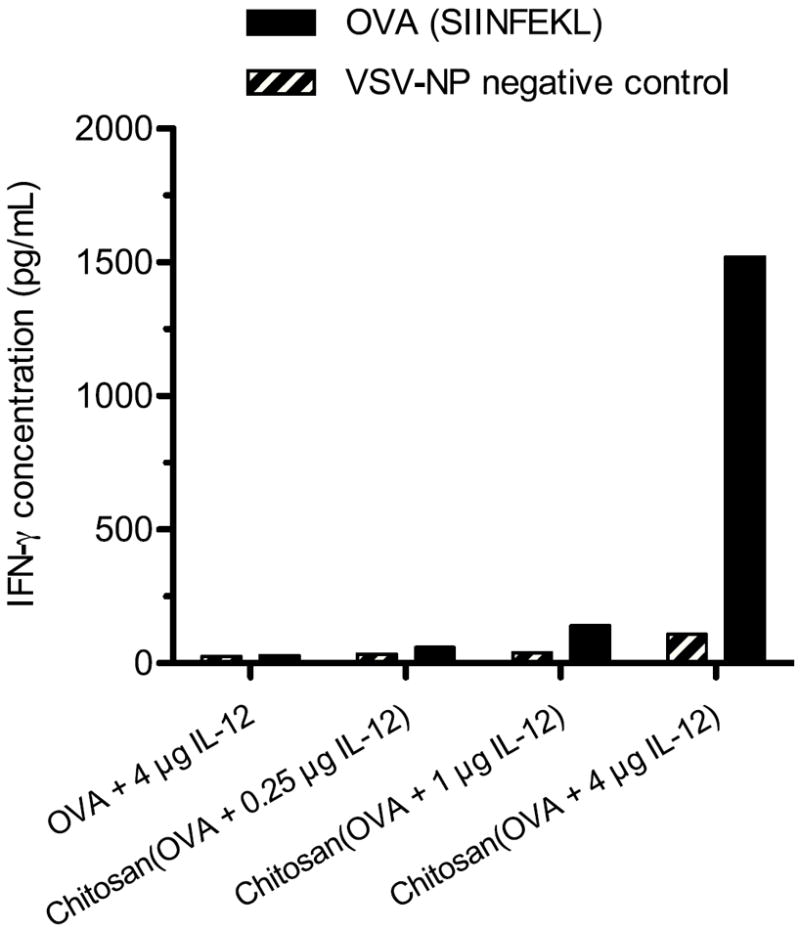
Chitosan/OVA/IL-12 stimulates IFN-γ-producing CD8+ T cells. Mice (n = 5) were vaccinated on days 0 and 14 and sacrificed on day 21. Splenocytes were pooled and cultured for 6 days with 1 μg/mL OVA257-264 (SIINFEKL) MHC class I peptide, then rested overnight in plain medium. Splenocytes (500,000 cells in 2 mL) were subsequently cultured with irradiated APCs and 1 μg/mL OVA257-264 (SIINFEKL) or control (VSV-NP) peptide for 24 h, and released IFN-γ was measured by ELISA. Results are plotted as the mean of duplicate wells.
4. Discussion
In this study, we demonstrated that the chitosan/IL-12 adjuvant system elicits a strong Th1-polarized cellular and humoral immune response to a protein antigen. The chitosan/IL-12 adjuvant system generated a strong antigen-specific CD4+ helper T-cell response (Fig. 2). Th1 polarization was demonstrated by CD4+ (helper) T-cell cytokine release profiles, which indicated a high level of IFN-γ produced in response to antigen restimulation (Fig. 3). The chitosan/IL-12 adjuvanted vaccine produced an antigen-specific CD8+ T-cell response (cellular immune response), which is associated with Th1 polarization. The CD8+ T-cell response was characterized by pentamer staining and in vitro IFN-γ production (Figs. 4 and 5). The humoral response generated by the chitosan/IL-12 adjuvant system was also consistent with a Th1-polarized response, as demonstrated by high levels of OVA-specific IgG2a and IgG2b antibodies in addition to IgG1 (Fig. 1). Collectively, these results demonstrate that the chitosan/IL-12 adjuvant system is capable of inducing a strong Th1 immune response to a protein-based vaccine.
An important finding is that both the delivery vehicle (chitosan) and the immunopotentiating agent (IL-12) are required to achieve a robust Th1 immune response. Chitosan/OVA and OVA/IL-12 elicited moderate levels of CD4+ T-cell proliferation; however, these were significantly less than the CD4+ T-cell response achieved by chitosan/OVA/IL-12. Similarly, chitosan/OVA and OVA/IL-12 each generated negligible CD8+ T-cell responses, which demonstrates that the combination of chitosan and IL-12 is needed to stimulate a cellular immune response. Chitosan adjuvant alone was sufficient to generate high levels of IgG (total) and IgG1 antibodies to OVA, similar to that generated by chitosan/IL-12. However, chitosan generated lower levels of IgG2a and IgG2b compared to chitosan/IL-12, demonstrating that the combination of chitosan and IL-12 was needed for immunoglobulin isotype switching. Vaccination with IL-12 adjuvant alone resulted in substantial levels of antigen-specific CD4+ T-cell IFN-γ release; however, the IFN-γ levels were 9-fold less than with chitosan/IL-12 combined. Thus, neither component alone is sufficient to generate a Th1 immune response, but rather the combination of chitosan and IL-12 is required to achieve a Th1 immune response to protein antigen.
The efficacy of the Th1-inducing cytokine IL-12 was greatly enhanced by formulation with the chitosan delivery vehicle. This may be explained by examining the various pathways by which IL-12 may stimulate the immune system. For example, IL-12 may stimulate NK cells at the injection site to produce IFN-γ [5], which may in turn stimulate dendritic cells (DCs) [22] that are taking up antigen. This would result in increased activation of DCs and enhanced cross-priming of T cells by DCs after they have migrated to the draining lymph node. By formulating IL-12 in chitosan, its effects on IFN-γ-producing cells are potentially prolonged, resulting in greater activation of DCs taking up protein antigen. This is in addition to the antigen depot effect of chitosan [17, 18], which prolongs exposure of antigen to DCs. A second possible pathway for IL-12 stimulation of the immune system is that IL-12 drains to the lymph node, where it directly stimulates T cells that are being cross-primed by DCs. This process would mimic the natural secretion of IL-12 by activated DCs. The chitosan depot effect would enhance this mechanism because IL-12 would drain to the lymph node over a prolonged period of time, such that the timing of T-cell stimulation at the lymph node would potentially coincide with antigen cross-presentation by migrating DCs. A third possible pathway is the diffusion of IL-12 into the circulation, whereby IL-12 acts systemically on NK or T cells to stimulate IFN-γ production or clonal expansion of T cells. While further studies are needed to elucidate the relative contributions of these pathways, the present data demonstrate that chitosan plays an important role in enhancing the immunostimulatory properties of IL-12.
We have demonstrated the in vivo efficacy of chitosan/IL-12 as an adjuvant system for a protein-based vaccine. In the field of vaccine research, there is expected to be an increased use of recombinant proteins as vaccine components [2]. A primary reason is that vaccines using a protein subunit are potentially safer and easier to produce than traditional vaccines based on attenuated or inactivated whole pathogens. This is particularly important in treating infectious diseases such as HIV, where whole-virus vaccines arouse safety concerns [3]. Protein-based vaccines also have a potential role in cancer immunotherapy, where the objective is to mount an immune response against tumor-associated antigens. The success of protein-based vaccines, however, has been limited by the weak immunogenicity of purified protein antigens [1, 2]. To overcome this limitation, various immunopotentiating agents, such as cytokines and Toll-like receptor agonists, have been explored as adjuvants to protein antigens. In addition, many types of delivery systems have been developed that increase the residence time of injected antigens and adjuvants. By increasing exposure of the vaccine to APCs, delivery systems such as chitosan can potentially reduce the dose of antigen and adjuvant required, and thus lower costs and minimize systemic side effects.
Chitosan has several advantages as a biomaterial for vaccine delivery. An important characteristic of a successful vaccine delivery system is that it be readily translated to clinical applications. Critical components of an effective translational pathway are sourcing of clinical-grade materials and minimal formulation required in the clinical setting. The chitosan delivery system meets both of these requirements. Chitosan is currently being evaluated as an intranasal vaccine excipient [23], and there are plans to employ chitosan in a clinical trial at the NCI for treating bladder cancer. The chitosan/IL-12 adjuvant system can be formulated in a clinical setting with minimal steps (heating and mixing under sterile conditions). In addition, chitosan has been well tolerated, with only mild adverse events, in clinical trials of intranasal and intramuscular vaccine delivery systems [24, 25]. These characteristics suggest that the chitosan delivery system is well suited for translation to clinical applications in cancer or infectious diseases.
5. Conclusions
We have developed a vaccine adjuvant system composed of a viscous chitosan solution and IL-12. Chitosan, a polysaccharide derived from natural sources, is designed to provide an injection-site depot for protein antigen and the immunostimulatory cytokine IL-12. A vaccine containing chitosan, IL-12, and OVA elicited Th1-polarized immune responses characterized by antigen-specific CD4+ and CD8+ T-cell responses, Th1 cytokine production, and IgG2a/IgG2b antibody responses. OVA with chitosan alone generated primarily an IgG1 antibody response, and OVA with IL-12 alone generated only moderate levels of CD4+ T-cell proliferation and IFN-γ secretion. The collective results demonstrate that both the delivery vehicle (chitosan) and immunopotentiating agent (IL-12) are required to achieve the desired Th1 immune response to protein antigen. The chitosan/IL-12 adjuvant system may be combined with whole-protein vaccine antigens, such as pathogen-derived proteins or tumor-associated proteins, to develop a clinically relevant vaccine for treating cancer or infectious diseases.
Acknowledgments
The authors acknowledge Garland Davis, Bertina Gibbs, and Curtis Randolph for their excellent technical support in performing vaccine injections and immunological assays, and Bonnie L. Casey for editorial assistance in the preparation of this manuscript. This work was funded by the Intramural Research Program of the Center for Cancer Research at the National Cancer Institute, NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Reed SG, Bertholet S, Coler RN, Friede M. New horizons in adjuvants for vaccine development. Trends Immunol. 2009;30(1):23–32. doi: 10.1016/j.it.2008.09.006. [DOI] [PubMed] [Google Scholar]
- 2.O’Hagan DT, De Gregorio E. The path to a successful vaccine adjuvant--‘the long and winding road’. Drug Discov Today. 2009;14(11-12):541–51. doi: 10.1016/j.drudis.2009.02.009. [DOI] [PubMed] [Google Scholar]
- 3.Lore K, Karlsson Hedestam GB. Novel adjuvants for B cell immune responses. Curr Opin HIV AIDS. 2009;4(5):441–6. doi: 10.1097/COH.0b013e32832da082. [DOI] [PubMed] [Google Scholar]
- 4.Pashine A, Valiante NM, Ulmer JB. Targeting the innate immune response with improved vaccine adjuvants. Nat Med. 2005;11(4 Suppl):S63–8. doi: 10.1038/nm1210. [DOI] [PubMed] [Google Scholar]
- 5.Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol. 2003;3(2):133–46. doi: 10.1038/nri1001. [DOI] [PubMed] [Google Scholar]
- 6.Scott P, Trinchieri G. IL-12 as an adjuvant for cell-mediated immunity. Semin Immunol. 1997;9(5):285–91. doi: 10.1006/smim.1997.0084. [DOI] [PubMed] [Google Scholar]
- 7.Afonso LC, Scharton TM, Vieira LQ, Wysocka M, Trinchieri G, Scott P. The adjuvant effect of interleukin-12 in a vaccine against Leishmania major. Science. 1994;263(5144):235–7. doi: 10.1126/science.7904381. [DOI] [PubMed] [Google Scholar]
- 8.Mougneau E, Altare F, Wakil AE, Zheng S, Coppola T, Wang ZE, et al. Expression cloning of a protective Leishmania antigen. Science. 1995;268(5210):563–6. doi: 10.1126/science.7725103. [DOI] [PubMed] [Google Scholar]
- 9.Mountford AP, Anderson S, Wilson RA. Induction of Th1 cell-mediated protective immunity to Schistosoma mansoni by co-administration of larval antigens and IL-12 as an adjuvant. J Immunol. 1996;156(12):4739–45. [PubMed] [Google Scholar]
- 10.Mahon BP, Ryan MS, Griffin F, Mills KH. Interleukin-12 is produced by macrophages in response to live or killed Bordetella pertussis and enhances the efficacy of an acellular pertussis vaccine by promoting induction of Th1 cells. Infect Immun. 1996;64(12):5295–301. doi: 10.1128/iai.64.12.5295-5301.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schijns VE, Haagmans BL, Horzinek MC. IL-12 stimulates an antiviral type 1 cytokine response but lacks adjuvant activity in IFN-gamma-receptor-deficient mice. J Immunol. 1995;155(5):2525–32. [PubMed] [Google Scholar]
- 12.Mosmann TR, Coffman RL. TH1 and TH2 cells: different patterns of lymphokine secretion lead to different functional properties. Annu Rev Immunol. 1989;7:145–73. doi: 10.1146/annurev.iy.07.040189.001045. [DOI] [PubMed] [Google Scholar]
- 13.Jankovic D, Caspar P, Zweig M, Garcia-Moll M, Showalter SD, Vogel FR, et al. Adsorption to aluminum hydroxide promotes the activity of IL-12 as an adjuvant for antibody as well as type 1 cytokine responses to HIV-1 gp120. J Immunol. 1997;159(5):2409–17. [PubMed] [Google Scholar]
- 14.Gurunathan S, Prussin C, Sacks DL, Seder RA. Vaccine requirements for sustained cellular immunity to an intracellular parasitic infection. Nat Med. 1998;4(12):1409–15. doi: 10.1038/4000. [DOI] [PubMed] [Google Scholar]
- 15.Baldrick P. The safety of chitosan as a pharmaceutical excipient. Regul Toxicol Pharmacol. 2010;56(3):290–9. doi: 10.1016/j.yrtph.2009.09.015. [DOI] [PubMed] [Google Scholar]
- 16.Azab AK, Doviner V, Orkin B, Kleinstern J, Srebnik M, Nissan A, et al. Biocompatibility evaluation of crosslinked chitosan hydrogels after subcutaneous and intraperitoneal implantation in the rat. J Biomed Mater Res A. 2007;83(2):414–22. doi: 10.1002/jbm.a.31256. [DOI] [PubMed] [Google Scholar]
- 17.Zaharoff DA, Rogers CJ, Hance KW, Schlom J, Greiner JW. Chitosan solution enhances both humoral and cell-mediated immune responses to subcutaneous vaccination. Vaccine. 2007;25(11):2085–94. doi: 10.1016/j.vaccine.2006.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zaharoff DA, Rogers CJ, Hance KW, Schlom J, Greiner JW. Chitosan solution enhances the immunoadjuvant properties of GM-CSF. Vaccine. 2007;25(52):8673–86. doi: 10.1016/j.vaccine.2007.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zaharoff DA, Hoffman BS, Hooper HB, Benjamin CJ, Jr, Khurana KK, Hance KW, et al. Intravesical immunotherapy of superficial bladder cancer with chitosan/interleukin-12. Cancer Res. 2009;69(15):6192–9. doi: 10.1158/0008-5472.CAN-09-1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zaharoff D, Hance K, Rogers C, Schlom J, Greiner J. Intratumoral immunotherapy of established solid tumors with chitosan/IL-12. J Immunother. 2010;33(7):697–705. doi: 10.1097/CJI.0b013e3181eb826d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Klinman DM, Klaschik S, Sato T, Tross D. CpG oligonucleotides as adjuvants for vaccines targeting infectious diseases. Adv Drug Deliv Rev. 2009;61(3):248–55. doi: 10.1016/j.addr.2008.12.012. [DOI] [PubMed] [Google Scholar]
- 22.Mocikat R, Braumuller H, Gumy A, Egeter O, Ziegler H, Reusch U, et al. Natural killer cells activated by MHC class I(low) targets prime dendritic cells to induce protective CD8 T cell responses. Immunity. 2003;19(4):561–9. doi: 10.1016/s1074-7613(03)00264-4. [DOI] [PubMed] [Google Scholar]
- 23.Phase I Norwalk Vaccine Study. [cited 3 August 2010]; Available from: http://www.clinicaltrials.gov/ct2/show/NCT00806962?term=NCT00806962&rank=1.
- 24.McNeela EA, Jabbal-Gill I, Illum L, Pizza M, Rappuoli R, Podda A, et al. Intranasal immunization with genetically detoxified diphtheria toxin induces T cell responses in humans: enhancement of Th2 responses and toxin-neutralizing antibodies by formulation with chitosan. Vaccine. 2004;22(8):909–14. doi: 10.1016/j.vaccine.2003.09.012. [DOI] [PubMed] [Google Scholar]
- 25.Read RC, Naylor SC, Potter CW, Bond J, Jabbal-Gill I, Fisher A, et al. Effective nasal influenza vaccine delivery using chitosan. Vaccine. 2005;23(35):4367–74. doi: 10.1016/j.vaccine.2005.04.021. [DOI] [PubMed] [Google Scholar]