

Abstract
There is a strong genetic component for schizophrenia risk, but it is unclear how the illness is maintained in the population given the significantly reduced fertility of those with the disorder. One possibility is that new mutations occur in schizophrenia vulnerability genes. If so, then those with schizophrenia may have older fathers, because advancing paternal age is the major source of new mutations in humans. This review describes several neurodevelopmental disorders that have been associated with de novo mutations in the paternal germ line and reviews data linking increased schizophrenia risk with older fathers. Several genetic mechanisms that could explain this association are proposed, including paternal germ line mutations, trinucleotide repeat expansions, and alterations in genetic imprinting in one or several genes involved in neurodevelopment. Animal models may be useful in exploring these and other explanations for the paternal age effect and they may provide a novel approach for gene identification. Finally, it is proposed that environmental exposures of the father, as well as those of the mother and developing fetus, may be relevant to the etiology of schizophrenia.
Keywords: Development, signaling, schizophrenia, genetics, teratogenesis, neuronal circuits
It was more than a century ago that Kraepelin (1899) first observed the hereditary nature of schizophrenia vulnerability, which has now been confirmed by scores of family, twin, and adoption studies. But now, well into the dawn of the molecular genetic revolution, and despite the overwhelming evidence that schizophrenia is a genetic disorder, the nature of the underlying genetic diathesis remains unclear. Genetic linkage and association studies have produced several interesting leads (Berrettini 2000; Pulver 2000) and scientists have demonstrated that particular gene disruptions can alter neurodevelopment (Impagnatiello et al. 1998; Kao et al. 1998), but there is no consensus that any particular gene plays a meaningful role in the etiology of schizophrenia (Hyman 2000).
Some of the obstacles in genetic research in schizophrenia are those of any complex disorder, and include incomplete penetrance, polygenic interaction (epistasis), diagnostic instability, and variable expressivity. Schizophrenia also does not show a clear Mendelian inheritance pattern, although segregation analyses have variably supported dominant, recessive, additive, sex-linked, and oligogenic inheritance (Book 1953; Slater 1958; Garrone 1962; Elston and Campbell 1970; Slater and Cowie 1971; Karlsson 1972; Stewart et al. 1980; Risch 1990a, 1990b; reviewed by Kendler and Diehl 1993). Furthermore, both nonallelic (Kaufmann et al. 1998) and etiologic heterogeneity (Malaspina et al. 1999, 2000; Tsuang 2000) are likely for schizophrenia, with various exposures and gene-environment interactions presumed to result in a common phenotype.
Another part of the genetic puzzle of schizophrenia is how the disorder is maintained in the population despite the reproductive disadvantage of affected individuals (Fananas et al. 1995; McGrath et al. 1999). One way to explain its persistence would be if schizophrenia genes were constantly being replenished through new mutations.
New Mutations
In addition to sustaining schizophrenia in the population, de novo mutations could account for some of the inconsistencies in segregation and linkage studies. New mutations might also explain why there is a greater recurrence risk for schizophrenia in the children and siblings of schizophrenia probands (∼10%) than in their parents (∼5%) (Gottesman 1991). As early as the 1950s, both Book (1953) and Lewis (1958) suggested a role for new mutations in the etiology of schizophrenia, but the idea was discounted because human mutation rates were considered to be too low to account for the prevalence of schizophrenia (Huxley et al. 1964; Penrose 1968). To the contrary, recent data show that mutations are common in humans; we may acquire 100 mutations per individual per generation, including several deleterious mutations, one or two of which may persist in the gene pool (Crow 1999; Eyre-Walker et al. 1999). The noted geneticist, James F. Crow, has written extensively on the nature and outcome of human mutations and has proposed that a high spontaneous mutation rate could present a genetic risk for the future human population (Crow 1997, 1999).
New Mutations and Human Genetic Disease
Association With Paternal Age
The major source of new mutations in human populations is from advancing paternal age (see Crow 1999). Weinberg (1912) had suggested that aging parental germ cells may be prone to mutation after observing that achondroplasia was more common in last-born siblings. In 1955, Penrose demonstrated that later paternal age, but not maternal age, was predictive of de novo mutations. He proposed that mutations arose by DNA copy errors that accumulate over the many replication cycles that occur in the male germ line. Spermatogonial cells replicate every 16 days, approximating 200 divisions by age 20, and 660 by age 40 (see Drake et al. 1998). By contrast, oocytes undergo only 24 cell divisions, of which all but the last occur before a woman's birth. In addition, as men age, mutations may increase because spermatogenesis occurs in the presence of declining testosterone, lower levels of DNA proofreading and repair enzymes (Tarin et al. 1998), and reduced antioxidant enzyme activity, along with the limitations in vascular supply and reduced cellular efficiency that accompany aging in other tissues. New genetic diseases arising from mutations are likely to minimally affect 1 of every 200 offspring of men older than 40 years (Friedman 1981).
Although a correlation between disease risk and paternal age suggests a role for de novo mutations, a paternal origin of the new mutations can only be confirmed with molecular methods if disease loci are known. The mutations most clearly associated with paternal aging are those involving substitutions at a single base (Crow 1997), whereas small deletions and inversions are more often inherited from the mother (Sapienza 1996) and are independent of maternal age. Other de novo rearrangements of chromosomes, such as translocations, large inversions, or aneuploidies, may be variably related to either maternal or paternal age (Martin et al. 1995). It has long been known that a few autosomal dominant diseases are related to paternal age, but recent findings show that it is related to some complex genetic disorders as well, including prostate cancer (Zhang et al. 1999), nervous system cancer (Hemminiki et al. 1999), and several birth defects (Macintosh et al. 1995).
New Mutations and Neurodevelopment
There has been a recent surge in the discovery of genes that are critical for brain development, many of which have been identified from specific mutations that cause pediatric neurological disorders (Tanaka and Gleeson 2000). De novo mutations arise in a number of these genes in proportion to paternal age, including those that cause several of the craniosynostosis syndromes. These syndromes are among the most common causes of craniofacial anomalies (Hehr and Muenke 1999; Singer et al. 1999), and they may be particularly pertinent to schizophrenia. Waddington et al. (1999), for example, has highlighted in utero cranial facial dysmorphogenesis as an important element in the etiopathology of schizophrenia. Many craniosynostosis syndromes arise from mutations in the fibroblast growth factor receptor gene pathways (Schell et al. 1995; Vajo et al. 2000). These syndromes commonly include midfacial hypoplasia, prognathism, and a high-arched palate as well as developmental delay, mental retardation, and hydrocephalus. These disorders can present a widely variable phenotype, even within families, consistent with epistasis or other modifying in utero exposures. Several of these syndromes are described in table 1.
Table 1. Some craniosynostoses associated with de novo mutations in fibroblast growth factor receptor genes.
| Syndrome | Nervous System Phenotype | Genotype |
|---|---|---|
| Apert Syndrome (Moloney et al. 1996; Tolarova et al.1997; Yu et al. 2000) | Developmental disorder including craniosynostosis, with displacement of the frontal and parietal bone centers, midfacial hypoplasia, facial asymmetry, depressed nasal ridge, and mental retardation. Can include hydrocephalus and hearing difficulties. | 10q25-q26. One of two substitutions, both C→G in the fibroblast growth factor receptor 2 gene (FGFR2) > 98% de novo in association with paternal age. Approximately 5% of craniosynostoses. |
| Pfeiffer Syndrome (Plomp et al. 1998; Robin et al. 1998; Glaser et al. 2000) | Type 1 is an autosomal dominant condition including craniofacial abnormalities, coronal craniostosis, and midface hypoplasia. Type 2 (always de novo) includes a cloverleaf skull, deafness, and midface hypoplasia. Severe cases have significant cognitive impairment. | 8p11.2,10q25-26. Mutation of FGFR2 or FGFR1 genes. Approximately 70% of cases are sporadic. |
| Crouzon Syndrome (Glaser et al. 2000; Yu et al. 2000) | Includes craniosynostosis, hypertelorism, midface hypoplasia, and exophthalmos. Premature closure of some cranial sutures affects brain growth. Includes cognitive difficulties and retardation. | 10q24 heterogeneous FGFR2 mutations. Birth prevalence is 16/million with approximately 30%–60% of cases from de novo events. |
| Thanatophoric Dysplasia (Orioli et al. 1995; Yu et al. 2000) | Characterized by multiple severe bone abnormalities. Includes prominent forehead, hypertelorism, depressed nasal bridge, and a cloverleaf skull. Surviving cases show dramatic growth failure, hydrocephalus, and severe developmental delay. | All cases are de novo and often do not survive. Result from missense mutations in the FGFR3. |
Other sporadic neurodevelopmental disorders arising from de novo mutations include the Klippel-Trenaunay-Weber syndrome (Lorda-Sanchez et al. 1998), which includes hemimegalencephaly or holoprosencephaly (Odent et al. 1998), and results from a developmental defect causing forebrain cleavage failure. Another example is the CHARGE syndrome (Tellier et al. 1998), which includes central nervous system malformations and mental retardation and is attributed to a polytopic developmental field defect of the neural tube and the neural crest cells. Achondroplasia can similarly include craniosynostosis and nonprogressive ventricular dilation consistent with arrested hydrocephalus, as well as corpus callosum hypoplasia (Rousseau et al. 1994; Tolarova et al. 1997; Thompson et al. 1999). Idiopathic torsion dystonia (Fletcher et al. 1990), mental retardation of unknown etiology (Zhang et al. 1992), and Alzheimer's disease (Whalley et al. 1995; Bertram et al. 1998) are among the other conditions related to paternal age and neurodevelopment, as are the conditions described in table 2. Of interest with respect to schizophrenia are some cases of cerebral palsy—previously considered to result from birth asphyxia—that are related to paternal age and gene mutations (Fletcher et al. 1996).
Table 2. Some neurodevelopmental conditions associated with de novo mutations and paternal age.
| Disease | Nervous System Phenotype | Genotype |
|---|---|---|
| Nonsyndromic Holoprosencephaly (Odent et al. 1998) | Developmental failure of forebrain cleavage. Brain malformations are typically associated with facial anomalies ranging from cyclopia to hypotelorism. | 2p21, 7q36,18p11-3, 21q22-3. Sonic hedgehog is the disease-causing gene. Approximately 70% of cases arise de novo. |
| Pelizaeus-Merzbacher Disease (Mimault et al. 1999) | Developmental myelination defect. Includes decreased mature oligodendrocytes, severe hypotonia, and nystagmus. Progressive spastic paraplegia and abnormal movements develop. Impaired early motor development; intellect is generally less affected. | Xq21-q22. Proteolipoprotein (PLP) gene duplication. Affects the major myelin proteins and PLP and its isoform, DM20. De novo duplications arise in the maternal grandfather, but point mutations occur in both parents. |
| Neurofibromatosis type 1 (Evans et al. 1992; North 1993;Bunin et al. 1997; Yin et al. 2000) | Developmental disorder of muscles, bones, skin (café au lait spots) and multiple tumors in the central and peripheral nervous system. Also can include delayed developmental milestones, learning disability, attention deficit disorder, and deficits in visual-motor coordination and language. | 17q11.2. Half of the patients inherit the disease from a parent, but 50% of cases are from a de novo mutation, and more than 90% of these mutations occur in the paternal germ line. |
| Retinoblastoma (Der Kinderen et al. 1990; Dryja et al. 1997) | Retinal cancer from the loss of a pair of tumor-suppressor genes. It can include a variable degree of mental retardation. | 13q14 deletion. Typically de novo, more than 80% arise in the paternal germ line. Approximately 90% penetrance. |
| Cerebral palsy (CP) (Fletcher et al. 1992; McHale et al. 2000) | Aberrant control of movement or posture arising from cortical migration abnormality or hypoxia-ischemia. Intellectual, vision, and speech impairments possible. | 9p12-q12, also X-linked and recessive types possible. |
Paternal Age and Risk for Schizophrenia
If schizophrenia could be caused by de novo mutations, then it should also be associated with paternal age. Indeed there were early reports of advanced paternal age in schizophrenia, including those of Johanson (1958), Gregory (1959), Farina (1963), Schooler (1964), and Bojanovsky and Gerylovova (1967), although Granville-Grossman (1966) found no differences between patients and their siblings. An association with advanced maternal birth age is also reported (Shur 1982; Halmo et al. 1992), but analyses that also included paternal age data found that they accounted for the maternal age effects (Hare and Moran 1979; Kinnell 1983). Bertranpetit and Fananas (1983) critiqued these latter two studies for failing to use appropriate case-control methods, particularly for lack of stratification of the control population. They found no significant parental age differences between patients and controls using (unspecified) sociodemographic variables to select the controls. In general, the data showing later paternal age in schizophrenia received little attention. Although similar contemporaneous findings in other medical conditions implicated a role for de novo mutations in disease etiology, such results in schizophrenia were considered to be artifacts of methodological errors or, if valid, as resulting from delayed marriage and childbearing by psychiatrically vulnerable parents or other confounds.
Late birth order, which could be a proxy for advancing paternal age, has also been described in schizophrenia. This finding prompted speculation about factors that might augment schizophrenia vulnerability, although a genetic explanation for the association was excluded. Schooler (1964) and others presumed that genes affecting schizophrenia risk would have an equal chance of distribution to those in each birth rank. Instead they presumed that the birth order effect resulted from environmental causes, ranging from intrauterine fatigue to economic factors, although a psychological explanation centering on the mother-child relationship unfortunately became the prevailing hypothesis several decades ago (Neill 1990; Hartwell 1996). The basis for this largely nonmedical approach to etiopathology was related to the perception that schizophrenia was more of a functional disorder than an organic illness. More recently, Sham et al. (1993) reported a significantly increased risk of schizophrenia among subjects who had several older siblings. They considered this late birth order effect to be the consequence of maternal viral infections, which were transmitted to the pregnant mother by her older school-aged children.
The major methodological limitation of these studies was the choice of the appropriate comparison groups, considering the many biases inherent in the ascertainment of the patients. The ideal study design for examining parental birth ages and schizophrenia risk is a prospective birth cohort study, in which an entire population of affected and unaffected individuals is identified before illness onset; however, given the relative rareness of schizophrenia in the population and the decades-long duration between birth and disease onset, it is necessary to have a large population size, long study duration, and careful sociodemographic assessments to address this question. A recent study by Malaspina et al. (in press) met these requirements, examining the association of schizophrenia and parental age in an 89,722-member population birth cohort. This analysis revealed a robust association of paternal age and schizophrenia risk, with each decade of the father's age further multiplying the relative risk for schizophrenia by approximately 50 percent in controlled analyses, and no corresponding effect of maternal age (figure 1). An association of an increasing schizophrenia risk with advancing paternal age was subsequently replicated in a second birth cohort by Brown et al. (submitted).
Figure 1. An estimated relationship between paternal age and schizophrenia risk.
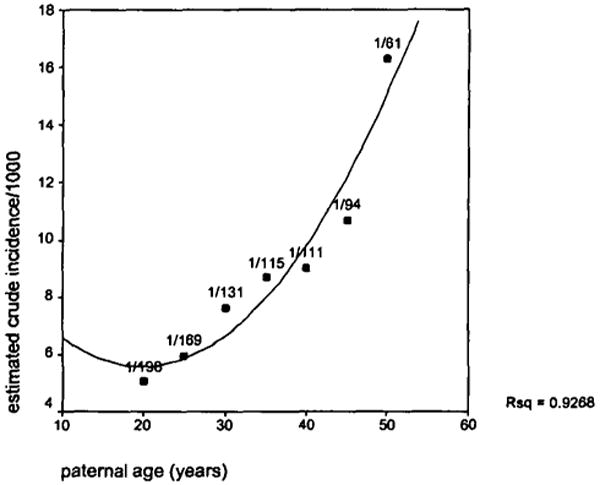
It can be argued that the relationship between paternal age and schizophrenia risk is an artifact of delayed parental childbearing, perhaps because of social inadequacies in psychiatrically vulnerable parents. However, if parental psychiatric vulnerability delays childbearing, then paternal age should be later in familial schizophrenia cases than in sporadic cases. This possibility was examined in a series of schizophrenia research patients (Malaspina et al., submitted) that showed, to the contrary, that paternal age was significantly older for the sporadic cases. Taken together, these findings are consistent with a significant role for new mutations in schizophrenia vulnerability.
Genetic Mechanisms That Could Associate Schizophrenia With Advancing Paternal Age
There are several potential scenarios whereby mutational events might increase schizophrenia vulnerability. These mechanisms include mutations in either a single major schizophrenia gene or in any of several genes involved in neurodevelopment, such as point mutations, trinucleotide repeat expansions, or by impairments in epigenetic modifications in imprinted genes.
A Mutation “Hot Spot” in a Schizophrenia Gene
Because the type of new mutation that most frequently arises in the paternal gametes is a single base pair change, it is possible that a mutation, which enhances schizophrenia risk, occurs in a specific “schizophrenia gene.” This is the case in achondroplastic dwarfism (Tolarova et al. 1997; Wilkin et al. 1998), where more than 90 percent of the cases are sporadic and, in more than 97 percent of people afflicted, the de novo mutation occurs at a “hot spot” in a single codon leading to a missense mutation in the fibroblast growth factor 3 gene (Orioli et al. 1995; Yu et al. 2000).
Such de novo mutations for schizophrenia would need to occur in a dominant or partially dominant gene if inheritance of a single mutated allele confers an increased risk for the disease. Autosomal dominant models for schizophrenia have been supported by some early segregation analyses. Book (1953) was able to account for the observed frequencies of schizophrenia in a geographical isolate with a dominant gene (with gene frequency 0.07) having a homozygous penetrance of 100 percent and a limited heterozygous penetrance (20%). Similarly, Slater (1958) and Slater and Cowie (1971) reanalyzed the earlier family data collected by Kallmann and by Zerbin-Rudin and found fair agreement for a partially dominant gene with frequency 0.015 and a heterozygous penetrance of 26 percent; Gregory (1960) obtained similar results. Overall, dominant gene models were largely discounted because the reduced fertility of schizophrenia patients would select against a dominant gene. But if a dominant gene is being continually introduced into the population through new mutations, then its prevalence will not be dependent on reproductive fitness. Such a major locus may have been undetected in genetic studies if those with the de novo mutation have an even lower fecundity than other schizophrenia probands. Then they would appear to have a sporadic (environmentally induced) disorder. The fathers in whom the mutation arose would be unaffected and the mutated gene would be absent from their somatic cells, further confounding genetic studies that did not allow for this possibility in allele sharing and linkage studies.
Mutations May Occur in Any One of a Number of Genes Involved in Neurodevelopment
Thousands of genes are estimated to play a role in neurodevelopment; therefore, it is possible that mutations in many genes could cause schizophrenia as a final common phenotype. Mutations arising in the male germ line would be amplified in clones of spermatozoa over the repeated cell divisions that occur with paternal aging. Although a full discussion of the agents that augment mutations in spermatogenesis is beyond the scope of this article, it is worth mentioning that they are, by and large, the same ones that induce mutations in somatic cells, including DNA repair enzyme insufficiency, free radicals, oxidative damage, radiation, nutritional deficiency, chemotherapy, and high temperature (Cummins et al. 1994; Robbins 1996; see Cohen 1986). Cigarette smoking has also been linked to genetic damage in spermatozoa, and free radical-induced mutations are theorized to be a common cause of male infertility; both of these exposures have been implicated in the etiology of childhood cancer (Aitken 1999; Shen and Ong 2000; Zenzes 2000).
Despite the high fidelity of DNA replication, a small number of new mutations are generated at every cell division and are fixed by repair enzymes. The amount of new mutations tolerated by an organism is a trade-off between the cellular energy expenditures by DNA proofreading and repair and the loss of offspring viability from mutations. Because most mutations decrease fitness, evolution will sustain genomes with efficient DNA repair enzymes. However, there has likely been little evolutionary pressure to maintain high DNA replication accuracy in older men (Drake et al. 1998). Of note, the offspring of very young fathers also have an elevated risk for de novo genetic disorders, which may result from the immaturity of spermatids or from low activity of DNA repair or antioxidant enzymes. This leads to a “U-shaped curve” for paternal age and the offspring's risk for several genetic conditions, including type 1 diabetes mellitus (Tai et al. 1998), cardiac ventricular and atrial septal deficits (Olshan et al. 1994), and neural tube defects (Mclntosh et al. 1995).
Trinucleotide Repeat Expansions
Increased schizophrenia risk could also be a consequence of progressive DNA trinucleotide expansions in one or several genes that participate in neurodevelopment. This effect would be more marked as paternal age advances because the possibility for further trinucleotide expansion occurs at each DNA replication. This molecular mechanism, discovered by Richards and Sutherland in 1992, underlies the clinical phenomenon of anticipation, which is the greater risk of illness or increasing disease severity in subsequent generations. Repeat expansions have been demonstrated in a number of neuropsychiatric disorders, including myotonic dystrophy, fragile X syndrome, spinocerebellar ataxias (Takano et al. 1996), and Huntington's disease (Orr 1994). The sex of the transmitting parent is frequently a major factor influencing anticipation (Telenius et al. 1993), with many disorders showing greater trinucleotide repeat expansion with paternal inheritance (Wellington et al. 1997). For example, early-onset Huntington's disease is usually inherited paternally and offspring have a significantly younger age of illness onset than their father (Ridley et al. 1988). The DNA in individual spermatozoa of an affected individual shows high variability in Huntington trinucleotide repeat lengths (MacDonald 1993; Leeflang 1999), suggesting that repeat expansions could be related to chance molecular events during the many cell divisions that occur during spermatogenesis.
For schizophrenia, too, the onset appears to be earlier in successive generations of multiply affected pedigrees, consistent with anticipation (Gorwood et al. 1995; Petronis and Kennedy 1995; Heiden et al. 1999) or possibly resulting from biases in methodology and ascertainment. Clinically, anticipation has reported to be stronger for paternal than for maternal transmission of schizophrenia in some (Gorwood et al. 1997; Johnson et al. 1997; Husted et al. 1998), but not all, studies (Imamura et al. 1998). Furthermore, Mclnnis et al. (1999) reported the presence of anticipation for aunt:niece/nephew pairs, but not for uncle:niece/nephew pairs. Parent of origin effects are consistent with both anticipation and with genetic imprinting (see following).
Only some studies have found longer trinucleotide repeats in schizophrenia patients (Morris et al. 1995; O'Donovan et al. 1996; Bowen et al. 2000). It is further unclear which genes have these expansions and whether such genes play a meaningful role in the pathogenesis of schizophrenia (O'Donovan and Owen 1999; Vincent et al. 2000).
Impairments in Imprinting
Imprinting is a form of gene regulation in which gene expression depends on whether the allele was inherited from the male or female parent in the prior generation. Imprinted genes that are only expressed if paternally inherited are reciprocally silenced at the maternal allele, and the contrary is true for maternally expressed genes. Genes are silenced by DNA methylation—which may preclude transcription factor binding—and by alterations in chromatin structure. The inherited methylation pattern is maintained in somatic cell divisions, but is erased in the primordial germ cells and reestablished late in gametogenesis. The monoallelic pattern of gene expression is maintained in offspring of the same sex and is reversed when genes are transmitted through individuals of the opposite sex. Imprinted genes do not conform to Mendelian principles because only one allele from the prior generation is expressed, even though an equivalent amount of genetic material, other than sex chromosomes, is inherited from both parents.
Paternal age–related factors might have a detrimental effect on genetic imprinting because paternal genes are imprinted during spermatogenesis. Environmental exposures, in addition to genetic mechanisms, can cause epigenetic changes in imprinted genes (Jirtle et al. 2000), and spermatozoan DNA may be especially susceptible to noxious exposures because of the ongoing replication during spermatogenesis and the unstable nature of genetic imprinting. Many of the mutations associated with advancing paternal age are at CpG nucleotides, which are methylation sites (Sapienza 1994) consistent with imprinted gene involvement.
Although our understanding of genetic imprinting is nascent, there are several characteristics of imprinted genes that make them reasonable candidates for schizophrenia vulnerability. First, imprinted genes play a key role in brain development, leading to lasting changes in cognition and behavior (Keverne et al. 1996; Isles and Wilkinson 2000). Paternal and maternal genes both are necessary for embryo-genesis (Surani et al. 1990), playing greater parts, respectively, in placental and embryo development (Kato et al. 1999). The influence of paternal genes in the placenta may represent a mechanism for the father to ensure that his offspring derive adequate resources from the maternal in utero environment, even if it may be in the best interest of the mother to limit these resources (Iwasa 1998).
Second, there appears to be a neuroanatomic localization pattern for the expression of certain imprinted genes in mice that the paternal or maternal allele expression patterns correspond, respectively, to limbic and neocortical regions (Allen et al. 1995; Keverne et al. 1996). The conceptualization of schizophrenia symptoms as deriving from an imbalance or modulatory disturbance between these regions (Weinberger et al. 1992) might be pertinent to these expression differences. In addition, genes for several neurotransmitters implicated in schizophrenia may be imprinted, including those for the serotonin 2A receptor, the dopamine 3 receptor, and several GABA A receptors (Meguro et al. 1997; Petronis 2000).
Third, imprinted genes and schizophrenia are both associated with language development and social functioning. A role for imprinted genes in determining language and social capacity was shown in an elegant set of studies by Skuse et al. (1997) in patients with Turner syndrome, a sporadic female disorder in which all or part of either the maternal or paternal X chromosome is missing. Turner syndrome females who inherited their father's X chromosome had relatively better social adjustment, superior verbal skills, and higher executive functioning than those who had inherited the maternal X chromosome, who, in turn, had relatively better spatial skills. Bishop et al. (2000) suggests that imprinted X chromosome genes influence the neurodevelopment of the brain lateralization observable for different neuropsychological tasks. The sexual dimorphism in the expression of schizophrenia, with males showing an earlier onset, more severe course, and a greater disability, could also involve imprinting of the X chromosome. Human sexual dimorphisms may be partially determined by imprinted genes on the X chromosome, according to Skuse (1999). Only normal female offspring receive a paternal X chromosome and normal male offspring only receive a maternal X chromosome. Because the paternal X chromosome genes are associated with superior social communication skills, their absence in male offspring may account for their increased vulnerability for developmental disorders involving social behavior and language, such as schizophrenia.
Finally, parent of origin effects have been linked with several other neuropsychiatric disorders, including autism (Cook et al. 1997; Schroer et al. 1998), bipolar affective disorder (McMahon et al. 1995), epilepsy (Gurrieri et al. 1999), and Tourette syndrome (Lichter et al. 1995; Eapen et al. 1997). In schizophrenia, parent of origin effects include greater negative symptoms, a worse course, and greater sibling concordance in those who have inherited schizophrenia paternally (Crow 1989; Asherson et al. 1994; Ohara et al. 1997). Schizophrenia has also shown genetic linkage to chromosome 15ql3-ql4 (Freedman 1997), a region near many imprinted genes, including those for Angelman and Prader-Willi syndromes (Mann and Bautolomei 1999). Moreover, Prader-Willi syndrome, in which the paternal gene is deleted, is frequently characterized by a schizophrenia-like psychosis (Clarke 1998).
Male Germline Mutations May Participate in Other Risk Pathways for Schizophrenia
It is anticipated that vulnerability genes may be inherited from either parent, but little consideration has been given to the influence of paternal health on schizophrenia risk, other than for a few postnatal psychosocial variables. Studies of deleterious exposures have centered on the mother (both before and during the pregnancy) and the in utero environment, as well as on the developing fetus and neonate.
Paternal Occupation
Recent data suggest that environmental exposures of the male parent could also be pertinent to schizophrenia risk. Paternal exposures have already been related to several congenital conditions, typically by using occupation to index probable environmental factors. Olshan et al. (1991) found that paternal occupations in forestry and logging, printing, and janitorial service were related to birth defects, the latter two occupations being associated with central nervous system anomalies. An elevated risk of Wilms tumor has been demonstrated in offspring whose fathers were auto mechanics, automobile body repairmen, and welders in the preconception period (Olshan et al. 1990). Occupational hydrocarbon exposure is elevated among fathers of Prader-Willi syndrome patients (Cassidy et al. 1989) and Blatter (1991) showed that spina bifida was related to paternal exposures to welding fumes, UV radiation, cleaning agents, and high levels of pesticides, but not with organic solvent exposures. Whalley et al. (1995) found that both paternal age and occupation as a coal miner made significant contributions to the risk for Alzheimer's disease.
Season of Birth Effects
There is a well-documented excess of winter and early spring births for those with schizophrenia—on the order of 8 to 10 percent (Torrey et al. 1977; Bradbury and Miller 1985)—that may preferentially comprise sporadic patients (O'Callaghan et al. 1991). These data suggest that some seasonally varying factor bearing a relationship to the time of birth may influence the subsequent development of schizophrenia. Infections are often seasonably variable, and there are good data that maternal infections are related to schizophrenia vulnerability (Mednick et al. 1988; Adams et al. 1993; McGrath and Castle 1995; Takei et al. 1996).
However other environmental factors are also seasonally variable, including ambient temperature in many climates. Because heat increases exposure of spermatozoa to mutagenic metabolites (Setchell 1998), a physiological adaptation to high temperatures is necessary. The scrotal location of the testes and the countercurrent heat exchange between the spermatic artery and vein keep the testicular temperature 4 to 7 degrees cooler than the rest of the body. Heat exposure in the period before conception could theoretically increase de novo mutations or alter the epigenetic modification of imprinted genes, particularly because late spermatids and mature spermatozoa do not have repair enzymes (Tarin et al. 1998). The effects of elevated external temperature on sperm may persist for 3 months after exposure (Rachootin and Olsen 1983). Somewhat akin to this idea, T. Crow (1987) previously theorized that heat-related mutagenesis during the warmer summer months could affect “an integrated virogene” in spermatozoa that might account for the winter-spring birth excess in schizophrenia.
Birth Events
Adverse birth events and hypoxia are considered to contribute to schizophrenia risk (McNeil 1988; Parnas et al. 1982; Magrath et al. 1995), although Jones et al. (1998) reported that the characteristics of the offspring (low birth weight and early birth), not of the delivery, were related to later schizophrenia. Because genetic imprinting by paternal genes contributes to the development and maintenance of the placenta, placental insufficiency and hypoxia could be a proximal cause of schizophrenia from mutations or epigenetic events arising in paternal genes. For example, the paternal insulin-like growth factor II receptor allele facilitates the use of maternal resources by the fetus whereas the maternal gene acts to limit these resources (Haig et al. 1997). An unopposed maternal allele could limit the growth of the fetus, leading to low birth weight. Studies in mice show that imprinted genes may even influence the future maternal behavior of female offspring onto their young (Lefebvre 1998; Li et al. 1999).
Implications for Translational Studies
Schizophrenia Phenotype
A phenotype for the subtype of schizophrenia that is related to paternal age is suggested from the results of birth-order studies. Farina (1963) reported that schizophrenia patients with many older siblings were less likely to recover than were other patients, and Schooler (1961) found that schizophrenia patients who were last-born siblings were more likely to have catatonia and were more socially isolated. Similarly, last-born female patients were found to have lower social competence, more bizarre and self-destructive behavior, less education, and, if they worked, lower-status jobs and were less likely to marry than first-born female patients, despite the fact that social class of origin did not differ between the birth rank groups (Schooler 1964).
Animal Models
Animal models can afford us a bridge to understand the complex genetic systems that are involved in the regulation of behavior. It is thus of interest that paternal age is associated with learning capacity in rodent models. The offspring of older rats are comparatively impaired in several tests of cognition and exploration, despite having no noticeable physical anomalies (Auroux 1983). In mice, the offspring of either young postpubescent fathers or older fathers have less spontaneous activity and worse learning capacity than offspring of mature fathers, leading to a U-shaped curve for learning and paternal age (Auroux et al. 1998; Auroux et al. 1999). Although spontaneous mutation rates in mice and humans are not comparable (Sankaranarayanan et al. 1998) and the genes that are involved in rodent cognition may be dissimilar to any human genes involved in schizophrenia risk, the rodent data minimally suggest that animal models of the advancing paternal age might be useful in identifying genes involved in learning and, optimally, might be useful in identifying schizophrenia vulnerability genes.
Conclusion
This article has reviewed data that support an association between schizophrenia risk and advancing paternal age. As such, schizophrenia may be one of a number of neurodevelopmental disorders caused by de novo mutations in the male germ line. This mutation mechanism may contribute a significant proportion of schizophrenia risk within a population in proportion to the demographics of paternal childbearing age. Furthermore, this risk may have implications for public health and for the primary prevention of schizophrenia.
Gene environment interaction in schizophrenia is most often considered to result from interplay between fetal genes and the in utero environment. This perspective of this review suggests that we should expand our epidemiological viewpoint to include the effects of environmental exposures on spermatozoan DNA. Animal studies offer an opportunity to test more complex models and, optimally, to generate unexpected candidate genes for schizophrenia or other human cognitive disorders. Our understanding of de novo mutations and schizophrenia risk may provide a new paradigm for identifying a genetic basis for some forms of schizophrenia.
Acknowledgments
This work is supported by RO1 MH59114, MH01699 K24 and the G. Harold and Leila Y. Mathers Charitable Foundation. The author would like to thank J. Harkavy-Friedman, S. Yale, and C. Corcoran for their assistance with this manuscript and acknowledge her collaborators in the Jerusalem Perinatal Cohort Schizophrenia Study and the Schizophrenia Research Unit Case Series Study: S. Harlap, E. Susser, S. Fennig, M. Davidson, J. Rabinowitz, A. Brown, D. Goetz, A. Berman, C. Fahim, R. Goetz, X. Amador, and J. Gorman.
References
- Adams W, Kendell RE, Hare EH, Munk-Jorgensen P. Epidemiological evidence that maternal influenza contributes to the aetiology of schizophrenia: An analysis of Scottish, English, and Danish data. British Journal of Psychiatry. 1993;163:522–534. doi: 10.1192/bjp.163.4.522. [DOI] [PubMed] [Google Scholar]
- Asherson P, Walsh C, Williams J, Sargeant M, Taylor C, Clements A, Gill M, Owen M, McGuffin P. Imprinting and anticipation: Are they relevant to genetic studies of schizophrenia? British Journal of Psychiatry. 1994;164:619–624. doi: 10.1192/bjp.164.5.619. [DOI] [PubMed] [Google Scholar]
- Auroux M. Decrease of learning capacity in offspring with increasing paternal age in the rat. Teratology. 1983;27:141–148. doi: 10.1002/tera.1420270202. [DOI] [PubMed] [Google Scholar]
- Auroux M, Nawar NN, Naguib M, Baud M, Lapaquellerie N. Post-pubescent to mature fathers: increase in progeny quality? Human Reproduction. 1998;13:55–59. doi: 10.1093/humrep/13.1.55. [DOI] [PubMed] [Google Scholar]
- Auroux MR, Mayaux MJ, Guihard-Moscato ML, Fromantin M, Barthe J, Schwartz D. Paternal age and mental functions of progeny in man. Human Reproduction. 1989;4:794–797. doi: 10.1093/oxfordjournals.humrep.a136988. [DOI] [PubMed] [Google Scholar]
- Berrettini WH. Genetics of psychiatric disease. Annual Review of Medicine. 2000;51:465–79. doi: 10.1146/annurev.med.51.1.465. [DOI] [PubMed] [Google Scholar]
- Bertram L, Busch R, Spiegl M, Lautenschlager NT, Muller U, Kurz A. Paternal age is a risk factor for Alzheimer disease in the absence of a major gene. Neurogenetics. 1998;1:277–280. doi: 10.1007/s100480050041. [DOI] [PubMed] [Google Scholar]
- Bertranpetit J, Fananas L. Parental age in schizophrenia in a case-controlled study [letter] British Journal of Psychiatry. 1993;162:574. doi: 10.1192/bjp.162.4.574. [DOI] [PubMed] [Google Scholar]
- Bishop DV, Canning E, Elgar K, Morris E, Jacobs PA, Skuse DH. Distinctive patterns of memory function in subgroups of females with Turner syndrome: Evidence for imprinted loci on the X-chromosome affecting neurodevelopment. Neuropsychologia. 2000;38:712–721. doi: 10.1016/s0028-3932(99)00118-9. [DOI] [PubMed] [Google Scholar]
- Blatter BM, Hermens R, Bakker M, Roeleveld N, Verbeek AL, Zielhuis GA. Paternal occupational exposure around conception and spina bifida in offspring. American Journal of Industrial Medicine. 1997;32:283–291. doi: 10.1002/(sici)1097-0274(199709)32:3<283::aid-ajim15>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- Bojanovsky J, Gerylovova A. Schizophrenia the parental age of patients. Preliminary report. Ceskka Psychiatrie. 1967;63:85–87. [PubMed] [Google Scholar]
- Book JA. Schizophrenia as a gene mutation. Acta Geneticae. 1953;4:133–139. [PubMed] [Google Scholar]
- Bowen T, Guy CA, Cardno AG, Vincent JB, Kennedy JL, Jones LA, Gray M, Sanders RD, McCarthy G, Murphy KC, Owen MJ, O'Donovan MC. Repeat sizes at CAG/CTG loci CTG18.1, ERDA1 and TGC13–7a in schizophrenia. Psychiatric Genetics. 2000;10:33–37. doi: 10.1097/00041444-200010010-00006. [DOI] [PubMed] [Google Scholar]
- Bradbury TN, Miller GA. Season of birth in schizophrenia: A review of evidence, methodology, and etiology. Psychology Bulletin. 1985;98:569–594. [PubMed] [Google Scholar]
- Brown A, Schaefer CA, Wyatt RJ, Bresnahan M, Goetz R, Harkavy-Friedman J, Gorman J, Malaspina D, Susser ES. Paternal age and risk of schizophrenia in adult offspring. doi: 10.1176/appi.ajp.159.9.1528. Submitted for publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunin GR, Needle M, Riccardi VM. Paternal age and sporadic neurofibromatosis: I. A case-control study and consideration of the methodological issues. Genetic Epidemiology. 1997;14:507–516. doi: 10.1002/(SICI)1098-2272(1997)14:5<507::AID-GEPI5>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Cassidy SB, Gainey AJ, Butler MG. Occupational hydrocarbon exposure among fathers of Prader-Willi syndrome patients with and without deletions of 15q. American Journal of Human Genetics. 1989;44:806–810. [PMC free article] [PubMed] [Google Scholar]
- Clarke D. Prader-Willi syndrome and psychotic symptoms: II. A preliminary study of prevalence using the Psychopathology Assessment Schedule for Adults with Developmental Disability checklist. Journal of Intellectual and Disability Research. 1998;42(Pt 6):451–454. doi: 10.1046/j.1365-2788.1998.4260451.x. [DOI] [PubMed] [Google Scholar]
- Cohen FL. Paternal contributions to birth defects. Nursing Clinics of North America. 1986;21:49–64. [PubMed] [Google Scholar]
- Cook EH, Jr, Lindgren V, Leventhal BL, Courchesne R, Lincoln A, Shulman C, Lord C, Courchesne E. Autism or atypical autism in maternally but not paternally derived proximal 15q duplication. American Journal of Human Genetics. 1997;60:928–934. [PMC free article] [PubMed] [Google Scholar]
- Crow JF. The high spontaneous mutation rate: Is it a health risk? Proceedings of the National Academy of Sciences of the United States of America. 1997;94:8380–8386. doi: 10.1073/pnas.94.16.8380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow JF. Spontaneous mutation in man. Mutation Research. 1999;437:5–9. doi: 10.1016/s1383-5742(99)00063-0. [DOI] [PubMed] [Google Scholar]
- Crow TJ. Mutation and psychosis: A suggested explanation of seasonality of birth. Psychological Medicine. 1987;17:821–828. doi: 10.1017/s0033291700000611. [DOI] [PubMed] [Google Scholar]
- Crow TJ, DeLisi LE, Johnstone EC. Concordance by sex in sibling pairs with schizophrenia is paternally inherited: Evidence for a pseudoautosomal locus. British Journal of Psychiatry. 1989;155:92–97. doi: 10.1192/bjp.155.1.92. [DOI] [PubMed] [Google Scholar]
- Cummins JM, Jequier AM, Kan R. Molecular biology of human male infertility: Links with aging, mito-chondrial genetics, and oxidative stress? Molecular Reproduction and Development. 1994;37:345–362. doi: 10.1002/mrd.1080370314. [DOI] [PubMed] [Google Scholar]
- DerKinderen DJ, Koten JW, Tan KE, Beemer FA, Van Romunde LK, Den Otter W. Parental age in sporadic hereditary retinoblastoma. American Journal of Ophthalmology. 1990;110:605–609. doi: 10.1016/s0002-9394(14)77056-4. [DOI] [PubMed] [Google Scholar]
- Drake JW, Charlesworth B, Charlesworth D, Crow JF. Rates of spontaneous mutation. Genetics. 1998;148:1667–1686. doi: 10.1093/genetics/148.4.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dryja TP, Morrow JF, Rapaport JM. Quantification of the paternal allele bias for new germline mutations in the retinoblastoma gene. Human Genetics. 1997;100:446–449. doi: 10.1007/s004390050531. [DOI] [PubMed] [Google Scholar]
- Eapen V, O'Neill J, Gurling HM, Robertson MM. Sex of parent transmission effect in Tourette's syndrome: Evidence for earlier age at onset in maternally transmitted cases suggests a genomic imprinting effect. Neurology. 1997;48:934–937. doi: 10.1212/wnl.48.4.934. [DOI] [PubMed] [Google Scholar]
- Elston RC, Campbell MA. Schizophrenia: Evidence for the major gene hypothesis. Behavioral Genetics. 1970;1:3–10. doi: 10.1007/BF01067366. [DOI] [PubMed] [Google Scholar]
- Evans DG, Huson SM, Donnai D, Neary W, Blair V, Newton V, Harris R. A clinical study of type 2 neurofibromatosis. Quarterly Journal of Medicine. 1992;84:603–618. [PubMed] [Google Scholar]
- Evans DG, Huson SM, Donnai D, Neary W, Blair V, Newton V, Strachan T, Harris R. A genetic study of type 2 neurofibromatosis in the United Kingdom: II. Guidelines for genetic counselling. Journal of Medical Genetics. 1992;29:847–852. doi: 10.1136/jmg.29.12.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans DG, Huson SM, Donnai D, Neary W, Blair V, Teare D, Newton V, Strachan T, Ramsden R, Harris R. A genetic study of type 2 neurofibromatosis in the United Kingdom: I. Prevalence, mutation rate, fitness, and confirmation of maternal transmission effect on severity. Journal of Medical Genetics. 1992;29:841–846. doi: 10.1136/jmg.29.12.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyre-Walker A, Keightley PD. High genomic deleterious mutation rates in hominids. Nature. 1999;397:344–347. doi: 10.1038/16915. [DOI] [PubMed] [Google Scholar]
- Fananas L, Bertranpetit J. Reproductive rates in families of schizophrenic patients in a case-control study. Acta Psychiatrica Scandinavica. 1995;91:202–204. doi: 10.1111/j.1600-0447.1995.tb09767.x. [DOI] [PubMed] [Google Scholar]
- Farina A, Storrs C, Barry H, III, Garmezy N. Birth order of recovered and nonrecovered schizophrenics. Archives of General Psychiatry. 1963;9:224–228. doi: 10.1001/archpsyc.1963.01720150034005. [DOI] [PubMed] [Google Scholar]
- Fletcher NA, Harding AE, Marsden CD. A genetic study of idiopathic torsion dystonia in the United Kingdom. Brain. 1990;113(Pt 2):379–395. doi: 10.1093/brain/113.2.379. [DOI] [PubMed] [Google Scholar]
- Fletcher NA, Marsden CD. Dyskinetic cerebral palsy: A clinical and genetic study. Developmental Medicine and Child Neurology. 1996;38:873–880. doi: 10.1111/j.1469-8749.1996.tb15043.x. [DOI] [PubMed] [Google Scholar]
- Freedman R, Coon H, Myles-Worsley M, Orr-Urtreger A, Olincy A, Davis A, Polymeropoulos M, Holik J, Hopkins J, Hoff M, Rosenthal J, Waldo MC, Reimherr F, Wender P, Yaw J, Young DA, Breese CR, Adams C, Patterson D, Adler LE, Kruglyak L, Leonard S, Byerley W. Linkage of a neurophysiological deficit in schizophrenia to a chromosome 15 locus. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:587–592. doi: 10.1073/pnas.94.2.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman JM. Genetic disease in the offspring of older fathers. Obstetrics and Gynecology. 1981;57:745–749. [PubMed] [Google Scholar]
- Glaser RL, Jiang W, Boyadjiev SA, Tran AK, Zachary AA, Van Maldergem L, Johnson D, Walsh S, Oldridge M, Wall SA, Wilkie AO, Jabs EW. Paternal origin of FGFR2 mutations in sporadic cases of Crouzon syndrome and Pfeiffer syndrome. American Journal of Human Genetics. 2000;66:768–777. doi: 10.1086/302831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorwood P, Leboyer M, Falissard B, Rouillon F, Jay M, Feingold J. Further epidemiological evidence for anticipation in schizophrenia. Biomedical Pharmacotherapy. 1997;51:376–380. doi: 10.1016/s0753-3322(97)89429-2. [DOI] [PubMed] [Google Scholar]
- Gottesman I. Schizophrenia Genesis: The Origins of Madness. New York: W.H. Freeman and Company; 1991. [Google Scholar]
- Granville-Grossman KL. Parental age and schizophrenia. British Journal of Psychiatry. 1966;112:899–905. doi: 10.1192/bjp.112.490.899. [DOI] [PubMed] [Google Scholar]
- Gregory I. An analysis of family data on 1000 patients admitted to a Canadian mental hospital. Acta Genetica et Statistica Medica. 1959;9:54–96. [PubMed] [Google Scholar]
- Gurrieri F, Battaglia A, Torrisi L, Tancredi R, Cavallaro C, Sangiorgi E, Neri G. Pervasive developmental disorder and epilepsy due to maternally derived duplication of 15q11-q13. Neurology. 1999;52:1694–1697. doi: 10.1212/wnl.52.8.1694. [DOI] [PubMed] [Google Scholar]
- Haig D, Graham C. Genomic imprinting and the strange case of the insulin-like growth factor II receptor. Cell. 1991;64:1045–1046. doi: 10.1016/0092-8674(91)90256-x. [DOI] [PubMed] [Google Scholar]
- Halmo M, Pogady J, Letko E. Maternal age in the etiology of schizophrenia. Bratislavske Lekarske Library. 1991;92:512–514. [PubMed] [Google Scholar]
- Hare EH, Moran PA. Raised parental age in psychiatric patients: Evidence for the constitutional hypothesis. British Journal of Psychiatry. 1979;134:169–177. doi: 10.1192/bjp.134.2.169. [DOI] [PubMed] [Google Scholar]
- Hartwell CE. The schizophrenogenic mother concept in American psychiatry. Psychiatry. 1996;59:274–297. doi: 10.1080/00332747.1996.11024768. [DOI] [PubMed] [Google Scholar]
- Hehr U, Muenke M. Craniosynostosis syndromes: From genes to premature fusion of skull bones. Molecular Genetics and Metabolism. 1999;68:139–151. doi: 10.1006/mgme.1999.2915. [DOI] [PubMed] [Google Scholar]
- Heiden A, Willinger U, Scharfetter J, Meszaros K, Kasper S, Aschauer HN. Anticipation in schizophrenia. Schizophrenia Research. 1999;35:25–32. doi: 10.1016/s0920-9964(98)00112-1. [DOI] [PubMed] [Google Scholar]
- Hemminki K, Kyyronen P. Parental age and risk of sporadic and familial cancer in offspring: Implications for germ cell mutagenesis. Epidemiology. 1999;10:747–751. [PubMed] [Google Scholar]
- Husted J, Scutt LE, Bassett AS. Paternal transmission and anticipation in schizophrenia. American Journal of Medical Genetics. 1998;81:156–162. [PMC free article] [PubMed] [Google Scholar]
- Huxley J, Mayr E, Osmond H. Schizophrenia [letter] Nature. 1965;206:1112. doi: 10.1038/2061112a0. [DOI] [PubMed] [Google Scholar]
- Hyman SE. The genetics of mental illness: Implications for practice. Bulletin of the World Health Organization. 2000;78:455–63. [PMC free article] [PubMed] [Google Scholar]
- Imamura A, Honda S, Nakane Y, Okazaki Y. Anticipation in Japanese families with schizophrenia. Journal of Human Genetics. 1998;43:217–223. doi: 10.1007/s100380050076. [DOI] [PubMed] [Google Scholar]
- Impagnatiello F, Guidotti AR, Pesold C, Dwivedi Y, Caruncho H, Pisu MG, Uzunov DP, Smalheiser NR, Davis JM, Pandey GN, Pappas GD, Tueting P, Sharma RP, Costa E. A decrease of reelin expression as a putative vulnerability factor in schizophrenia. Proceedings of the National Academy of Sciences USA. 1998;95:15718–15723. doi: 10.1073/pnas.95.26.15718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isles AR, Wilkinson LS. Imprinted genes, cognition and behaviour. Trends in Cognitive Science. 2000;4:309–318. doi: 10.1016/s1364-6613(00)01504-7. [DOI] [PubMed] [Google Scholar]
- Iwasa Y. The conflict theory of genomic imprinting: How much can be explained? Current Topics in Developmental Biology. 1998;40:255–293. doi: 10.1016/s0070-2153(08)60369-5. [DOI] [PubMed] [Google Scholar]
- Jirtle RL, Sander M, Barrett JC. Genomic imprinting and environmental disease susceptibility. Environmental Health Perspectives. 2000;108:271–278. doi: 10.1289/ehp.00108271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johanson E. A study of schizophrenia in the male. Acta Psychiatrica Scandiavica. 1958;33(suppl. 125):7–107. [PubMed] [Google Scholar]
- Johnson JE, Cleary J, Ahsan H, Harkavy FJ, Malaspina D, Cloninger CR, Faraone SV, Tsuang MT, Kaufmann CA. Anticipation in schizophrenia: Biology or bias? American Journal of Medical Genetics. 1997;74:275–280. doi: 10.1002/(sici)1096-8628(19970531)74:3<275::aid-ajmg7>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- Jones PB, Rantakallio P, Hartikainen AL, Isohanni M, Sipila P. Schizophrenia as a long-term outcome of pregnancy, delivery, and perinatal complications: A 28-year follow-up of the 1966 north Finland general population birth cohort. American Journal of Psychiatry. 1998;155:355–364. doi: 10.1176/ajp.155.3.355. [DOI] [PubMed] [Google Scholar]
- Kao HT, Porton B, Czernik AJ, Feng J, Yiu G, Haring M, Benfenati F, Greengard P. A third member of the synapsin gene family. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:4667–4672. doi: 10.1073/pnas.95.8.4667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato Y, Rideout WM, III, Hilton K, Barton SC, Tsunoda Y, Surani MA. Developmental potential of mouse primordial germ cells. Development. 1999;126:1823–1832. doi: 10.1242/dev.126.9.1823. [DOI] [PubMed] [Google Scholar]
- Karlsson JL. A two-locus hypothesis for inheritance of schizophrenia. In: Kaplan AR, editor. Genetic Factors in Schizophrenia. Springfield IL: Charles C. Thomas; 1972. [Google Scholar]
- Kaufmann CA, Suarez B, Malaspina D, Pepple J, Svrakic D, Markel PD, Meyer J, Zambuto CT, Schmitt K, Matise TC, Harkavy Friedman JM, Hampe C, Lee H, Shore D, Wynne D, Faraone SV, Tsuang MT, Cloninger CR. NIMH Genetics Initiative Millenium Schizophrenia Consortium: Linkage analysis of African-American pedigrees. American Journal of Medical Genetics. 1998;81:282–289. [PubMed] [Google Scholar]
- Keverne EB, Martel FL, Nevison CM. Primate brain evolution: Genetic and functional considerations. Proceedings of the Royal Society of London, Series B: Biological Series. 1996;263:689–696. doi: 10.1098/rspb.1996.0103. [DOI] [PubMed] [Google Scholar]
- Kinnell HG. Parental age in schizophrenia [letter] British Journal of Psychiatry. 1983;142:204. doi: 10.1192/bjp.142.2.204a. [DOI] [PubMed] [Google Scholar]
- Kraepelin E. Ein Lehrbuch fur Studirende und Aerzte: Vol II. Leipzig, Germany: Verlagvon, Barth; 1899. [Google Scholar]
- Leeflang EP, Tavare S, Marjoram P, Neal CO, Srinidhi J, MacFarlane H, MacDonald ME, Gusella JF, de Young M, Wexler NS, Arnheim N. Analysis of germline mutation spectra at the Huntington's disease locus supports a mitotic mutation mechanism. Human Molecular Genetics. 1999;8:173–183. doi: 10.1093/hmg/8.2.173. [DOI] [PubMed] [Google Scholar]
- Lefebvre L, Viville S, Barton SC, Ishino F, Keverne EB, Surani MA. Abnormal maternal behaviour and growth retardation associated with loss of the imprinted gene Mest. Nature Genetics. 1998;20:163–169. doi: 10.1038/2464. [DOI] [PubMed] [Google Scholar]
- Lewis A. Fertility and mental illness. Eugenics Review. 1958;50:91–106. [PMC free article] [PubMed] [Google Scholar]
- Li L, Keverne EB, Aparicio SA, Ishino F, Barton SC, Surani MA. Regulation of maternal behavior and offspring growth by paternally expressed Peg3. Science. 1999;284:330–333. doi: 10.1126/science.284.5412.330. [DOI] [PubMed] [Google Scholar]
- Lichter DG, Jackson LA, Schachter M. Clinical evidence of genomic imprinting in Tourette's syndrome. Neurology. 1995;45:924–928. doi: 10.1212/wnl.45.5.924. [DOI] [PubMed] [Google Scholar]
- Lorda-Sanchez I, Prieto L, Rodriguez-Pinilla E, Martinez-Frias ML. Increased parental age and number of pregnancies in Klippel-Trenaunay-Weber syndrome. Annals of Human Genetics. 1998;62(Pt 3):235–239. doi: 10.1046/j.1469-1809.1998.6230235.x. [DOI] [PubMed] [Google Scholar]
- MacDonald ME, Barnes G, Srinidhi J, Duyao MP, Ambrose CM, Myers RH, Gray J, Conneally PM, Young A, Penney J. Gametic but not somatic instability of CAG repeat length in Huntington's disease. Journal of Medical Genetics. 1993;30:982–986. doi: 10.1136/jmg.30.12.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malaspina D, Goetz RR, Yale S, Berman A, Friedman JH, Tremeau F, Printz D, Amador X, Johnson J, Brown A, Gorman JM. Relation of familial schizophrenia to negative symptoms but not to the deficit syndrome. American Journal of Psychiatry. 2000;157:994–1003. doi: 10.1176/appi.ajp.157.6.994. [DOI] [PubMed] [Google Scholar]
- Malaspina D, Harlap S, Fennig S, Heiman D, Nahon D, Feldman D, Susser E. Advancing paternal age and the risk of schizophrenia. Archives of General Psychiatry. 2001;58:361–367. doi: 10.1001/archpsyc.58.4.361. [DOI] [PubMed] [Google Scholar]
- Malaspina D, Harlap S, Susser E, Berman A, Harkavy Friedman J, Brown A, Fahim C, Gorman J. Further data supporting new mutations in the etiology of sporadic schizophrenia [Abstract] Schizophrenia Research. under review. [Google Scholar]
- Malaspina D, Sohler NL, Susser E. Interaction of genes and prenatal exposures in schizophrenia. In: Susser E, Brown AS, Gorman JM, editors. Prenatal Exposures in Schizophrenia. Washington, DC: American Psychiatric Press; 1999. pp. 35–61. [Google Scholar]
- Mann MR, Bartolomei MS. Toward a molecular understanding of Prader-Willi and Angelman syndromes. Human Molecular Genetics. 1999;8:1867–1873. doi: 10.1093/hmg/8.10.1867. [DOI] [PubMed] [Google Scholar]
- Martin RH, Spriggs E, Ko E, Rademaker AW. The relationship between paternal age, sex ratios, and aneuploidy frequencies in human sperm, as assessed by multicolor FISH. American Journal of Human Genetics. 1995;57:1395–1399. [PMC free article] [PubMed] [Google Scholar]
- McGrath J, Castle D. Does influenza cause schizophrenia? A five year review. Australian and New Zealand Journal of Psychiatry. 1995;29:23–31. doi: 10.3109/00048679509075888. [DOI] [PubMed] [Google Scholar]
- McGrath JJ, Hearle J, Jenner L, Plant K, Drummond A, Barkla JM. The fertility and fecundity of patients with psychoses. Acta Psychiatrica Scandinavica. 1999;99:441–446. doi: 10.1111/j.1600-0447.1999.tb00990.x. [DOI] [PubMed] [Google Scholar]
- McHale DP, Jackson AP, Campbell DA, Levene MI, Corry P, Woods CG, Lench NJ, Mueller RF, Markham AF. A gene for ataxic cerebral palsy maps to chromosome 9pl2-ql2. European Journal of Human Genetics. 2000;8:267–272. doi: 10.1038/sj.ejhg.5200445. [DOI] [PubMed] [Google Scholar]
- Mclnnis MG, McMahon FJ, Crow T, Ross CA, DeLisi LE. Anticipation in schizophrenia: A review and reconsideration. American Journal of Medical Genetics. 1999;88:686–693. doi: 10.1002/(sici)1096-8628(19991215)88:6<686::aid-ajmg19>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- Mclntosh GC, Olshan AF, Baird PA. Paternal age and the risk of birth defects in offspring. Epidemiology. 1995;6:282–288. doi: 10.1097/00001648-199505000-00016. [DOI] [PubMed] [Google Scholar]
- McMahon FJ, Stine OC, Meyers DA, Simpson SG, DePaulo JR. Patterns of maternal transmission in bipolar affective disorder [see comments] American Journal of Medical Genetics. 1995;56:1277–1286. [PMC free article] [PubMed] [Google Scholar]
- McNeil TF. Obstetric factors and perinatal injuries. In: Tsuang MT, Simpson JC, editors. Nosology, Epidemiology and Genetics of Schizophrenia. New York: Elsevier; 1988. pp. 319–344. [Google Scholar]
- Mednick SA, Machon RA, Huttunen MO, Bonett D. Adult schizophrenia following prenatal exposure to an influenza epidemic. Archives of Genetic Psychiatry. 1988;45:189–192. doi: 10.1001/archpsyc.1988.01800260109013. [DOI] [PubMed] [Google Scholar]
- Meguro M, Mitsuya K, Sui H, Shigenami K, Kugoh H, Nakao M, Oshimura M. Evidence for uni-parental, paternal expression of the human GABAA receptor subunit genes, using microcell-mediated chromosome transfer. Human Molecular Genetics. 1997;6:2127–2133. doi: 10.1093/hmg/6.12.2127. [DOI] [PubMed] [Google Scholar]
- Mimault C, Giraud G, Courtois V, Cailloux F, Boire JY, Dastugue B, Boespflug-Tanguy O. Proteolipoprotein gene analysis in 82 patients with sporadic Pelizaeus-Merzbacher Disease: Duplications, the major cause of the disease, originate more frequently in male germ cells, but point mutations do not. The Clinical European Network on Brain Dysmyelinating Disease. American Journal of Human Genetics. 1999;65:360–369. doi: 10.1086/302483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moloney DM, Slaney SF, Oldridge M, Wall SA, Sahlin P, Stenman G, Wilkie AO. Exclusive paternal origin of new mutations in Apert syndrome. Nature Genetics. 1996;13:48–53. doi: 10.1038/ng0596-48. [DOI] [PubMed] [Google Scholar]
- Morris AG, Gaitonde E, McKenna PJ, Mollon JD, Hunt DM. CAG repeat expansions and schizophrenia: Association with disease in females and with early age-at-onset. Human Molecular Genetics. 1995;4:1957–1961. doi: 10.1093/hmg/4.10.1957. [DOI] [PubMed] [Google Scholar]
- Neill J. Whatever became of the schizophrenogenic mother? American Journal of Psychotherapy. 1990;44:499–505. doi: 10.1176/appi.psychotherapy.1990.44.4.499. [DOI] [PubMed] [Google Scholar]
- North K. Neurofibromatosis type 1: Review of the first 200 patients in an Australian clinic. Journal of Child Neurology. 1993;8:395–402. doi: 10.1177/088307389300800421. [DOI] [PubMed] [Google Scholar]
- O'Callaghan E, Gibson T, Colohan HA, Walshe D, Buckley P, Larkin C, Waddington JL. Season of birth in schizophrenia: Evidence for confinement of an excess of winter births to patients without a family history of mental disorder. British Journal of Psychiatry. 1991;158:764–769. doi: 10.1192/bjp.158.6.764. [DOI] [PubMed] [Google Scholar]
- Odent S, Le Marec B, Munnich A, Le Merrer M, Bonaiti-Pellie C. Segregation analysis in nonsyndromic holoprosencephaly. American Journal of Medical Genetics. 1998;77:139–143. [PubMed] [Google Scholar]
- O'Donovan MC, Guy C, Craddock N, Bowen T, McKeon P, Macedo A, Maier W, Wildenauer D, Aschauer HN, Sorbi S, Feldman E, Mynett-Johnson L, Claffey E, Nacmias B, Valente J, Dourado A, Grassi E, Lenzinger E, Heiden AM, Moorhead S, Harrison D, Williams J, McGuffin P, Owen MJ. Confirmation of association between expanded CAG/CTG repeats and both schizophrenia and bipolar disorder. Psychological Medicine. 1996;26:1145–1153. doi: 10.1017/s0033291700035868. [DOI] [PubMed] [Google Scholar]
- Ohara K, Xu HD, Mori N, Suzuki Y, Xu DS, Ohara K, Wang ZC. Anticipation and imprinting in schizophrenia. Biological Psychiatry. 1997;42:760–766. doi: 10.1016/s0006-3223(97)00022-x. [DOI] [PubMed] [Google Scholar]
- Olshan AF, Breslow NE, Daling JR, Falletta JM, Grufferman S, Robison LL, Waskerwitz M, Hammond GD. Wilms' tumor and paternal occupation. Cancer Research. 1990;50:3212–3217. [PubMed] [Google Scholar]
- Olshan AF, Teschke K, Baird PA. Paternal occupation and congenital anomalies in offspring. American Journal of Industrial Medicine. 1991;20:447–475. doi: 10.1002/ajim.4700200403. [DOI] [PubMed] [Google Scholar]
- Orioli IM, Castilla EE, Scarano G, Mastroiacovo P. Effect of paternal age in achondroplasia, thanatophoric dysplasia, and osteogenesis imperfecta. American Journal of Medical Genetics. 1995;59:209–217. doi: 10.1002/ajmg.1320590218. [DOI] [PubMed] [Google Scholar]
- Orr HT. Unstable trinucleotide repeats and the diagnosis of neurodegenerative disease. Human Pathology. 1994;25:598–601. doi: 10.1016/0046-8177(94)90226-7. [DOI] [PubMed] [Google Scholar]
- Parnas J, Schulsinger F, Teasdale TW, Schulsinger H, Feldman PM, Mednick SA. Perinatal complications and clinical outcome within the schizophrenia spectrum. British Journal of Psychiatry. 1982;140:416–420. doi: 10.1192/bjp.140.4.416. [DOI] [PubMed] [Google Scholar]
- Penrose LS. Parental age and mutation. Lancet. 1955;2:312. doi: 10.1016/s0140-6736(55)92305-9. [DOI] [PubMed] [Google Scholar]
- Penrose LS. Critical survey of schizophrenia genetics. In: Howell FG, editor. Modern Perspectives in World Psychiatry. Edinburgh, United Kingdom: Oliver and Boyd; 1968. pp. 3–19. [Google Scholar]
- Petronis A, Kennedy JL. Unstable genes—unstable mind? American Journal of Psychiatry. 1995;152:164–172. doi: 10.1176/ajp.152.2.164. [DOI] [PubMed] [Google Scholar]
- Petronis A. The genes for major psychosis: Aberrant sequence or regulation? Neuropsychopharmacology. 2000;23:1–12. doi: 10.1016/S0893-133X(00)00127-5. [DOI] [PubMed] [Google Scholar]
- Plomp AS, Hamel BC, Cobben JM, Verloes A, Offermans JP, Lajeunie E, Fryns JP, Die-Smulders CE. Pfeiffer syndrome type 2: Further delineation and review of the literature. American Journal of Medical Genetics. 1998;75:245–251. [PubMed] [Google Scholar]
- Pulver AE. Search for schizophrenia susceptibility genes. Biological Psychiatry. 2000;47:221–230. doi: 10.1016/s0006-3223(99)00281-4. [DOI] [PubMed] [Google Scholar]
- Rachootin P, Olsen J. The risk of infertility and delayed conception associated with exposures in the Danish workplace. Journal of Occupational Medicine. 1983;25:394–02. [PubMed] [Google Scholar]
- Richards RI, Sutherland GR. Heritable unstable DNA sequences. Nature Genetics. 1992;1:7–9. doi: 10.1038/ng0492-7. [DOI] [PubMed] [Google Scholar]
- Risch N. Linkage strategies for genetically complex traits: I. Multilocus models. American Journal of Human Genetics. 1990a;46:222–228. [PMC free article] [PubMed] [Google Scholar]
- Risch N. Genetic linkage and complex diseases, with special reference to psychiatric disorders. Genetic Epidemiology. 1990b;7:3–16. doi: 10.1002/gepi.1370070103. [DOI] [PubMed] [Google Scholar]
- Robbins WA. Cytogenetic damage measured in human sperm following cancer chemotherapy. Mutation Research. 1996;355:235–252. doi: 10.1016/0027-5107(96)00030-9. [DOI] [PubMed] [Google Scholar]
- Robin NH, Scott JA, Arnold JE, Goldstein JA, Shilling BB, Marion RW, Cohen MM., Jr Favorable prognosis for children with Pfeiffer syndrome types 2 and 3: Implications for classification. American Journal of Medical Genetics. 1998;75:240–244. [PubMed] [Google Scholar]
- Rousseau F, Bonaventure J, Legeai-Mallet L, Pelet A, Rozet JM, Maroteaux P, Le Merrer M, Munnich A. Mutations in the gene encoding fibroblast growth factor receptor–3 in achondroplasia. Nature. 1994;371:252–254. doi: 10.1038/371252a0. [DOI] [PubMed] [Google Scholar]
- Roy MA, Flaum MA, Gupta S, Jaramillo L, Andreasen NC. Epidemiological and clinical correlates of familial and sporadic schizophrenia. Acta Psychiatria Scandinavica. 1994;89:324–328. doi: 10.1111/j.1600-0447.1994.tb01523.x. [DOI] [PubMed] [Google Scholar]
- Sankaranarayanan K. Ionizing radiation and genetic risks IX. Estimates of the frequencies of mendelian diseases and spontaneous mutation rates in human populations: A 1998 perspective. Mutation Research. 1998;411:129–178. doi: 10.1016/s1383-5742(98)00012-x. [DOI] [PubMed] [Google Scholar]
- Sapienza C. Parental origin effects, genome imprinting, and sex-ratio distortion: Double or nothing? American Journal of Human Genetics. 1994;55:1073–1075. [PMC free article] [PubMed] [Google Scholar]
- Sapienza C. A paternal wash in Apert syndrome. Nature Genetics. 1996;13:9–10. doi: 10.1038/ng0596-9. [DOI] [PubMed] [Google Scholar]
- Schell U, Hehr A, Feldman GJ, Robin NH, Zackai EH, Die-Smulders C, Viskochil DH, Stewart JM, Wolff G, Ohashi H. Mutations in FGFR1 and FGFR2 cause familial and sporadic Pfeiffer syndrome. Human Molecular Genetics. 1995;4:323–328. doi: 10.1093/hmg/4.3.323. [DOI] [PubMed] [Google Scholar]
- Schooler C. Birth order and schizophrenia. Archives of General Psychiatry. 1961;4:91–97. doi: 10.1001/archpsyc.1961.01710070093013. [DOI] [PubMed] [Google Scholar]
- Schooler C. Birth order and hospitalization for schizophrenia. Journal of Abnormal and Social Psychology. 1964;69:574–579. doi: 10.1037/h0048880. [DOI] [PubMed] [Google Scholar]
- Setchell BP. The Parkes Lecture: Heat and the testes. Journal of Reproduciton and Fertility. 1998;114:179–194. doi: 10.1530/jrf.0.1140179. [DOI] [PubMed] [Google Scholar]
- Sham PC, MacLean CJ, Kendler KS. Risk of schizophrenia and age difference with older siblings: Evidence for a maternal viral infection hypothesis? British Journal of Psychiatry. 1993;163:627–633. doi: 10.1192/bjp.163.5.627. [DOI] [PubMed] [Google Scholar]
- Shen H, Ong C. Detection of oxidative DNA damage in human sperm and its association with sperm function and male infertility. Free Radical Biology and Medicine. 2000;28:529–536. doi: 10.1016/s0891-5849(99)00234-8. [DOI] [PubMed] [Google Scholar]
- Shur E. Family history and schizophrenia: Characteristics of groups with and without positive family histories. Psychological Medicine. 1982;12:591–594. doi: 10.1017/s0033291700055690. [DOI] [PubMed] [Google Scholar]
- Singer S, Bower C, Southall P, Goldblatt J. Craniosynostosis in Western Australia, 1980–1994: A population-based study. American Journal of Medical Genetics. 1999;83:382–387. doi: 10.1002/(sici)1096-8628(19990423)83:5<382::aid-ajmg8>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- Skuse DH. Genomic imprinting of the X chromosome: A novel mechanism for the evolution of sexual dimorphism. Journal of Laboratory and Clinical Medicine. 1999;133:23–32. doi: 10.1053/lc.1999.v133.a94575. [DOI] [PubMed] [Google Scholar]
- Skuse DH, James RS, Bishop DV, Coppin B, Dalton P, Aamodt-Leeper G, Bacarese-Hamilton M, Creswell C, McGurk R, Jacobs PA. Evidence from Turner's syndrome of an imprinted X-linked locus affecting cognitive function. Nature. 1997;387:705–708. doi: 10.1038/42706. [DOI] [PubMed] [Google Scholar]
- Slater E. The monogenic theory of schizophrenia. Acta Genetica Statistica Medica. 1958;8:50–56. [PubMed] [Google Scholar]
- Slater E, Cowie V. The Genetics of Mental Disorders London. United Kingdom: Oxford University Press; 1971. [Google Scholar]
- Stewart J, Debray Q, Caillard V. Schizophrenia: The testing of genetic models by pedigree analysis. American Journal of Human Genetics. 1980;32:55–63. [PMC free article] [PubMed] [Google Scholar]
- Surani MA, Kothary R, Allen ND, Singh PB, Fundele R, Ferguson-Smith AC, Barton SC. Genome imprinting and development in the mouse. Development. 1990;(Suppl):89–98. [PubMed] [Google Scholar]
- Tai TY, Wang CY, Lin LL, Lee LT, Tsai ST, Chen CJ. A case-control study on risk factors for type 1 diabetes in Taipei City. Diabetes Research and Clinical Practices. 1998;42:197–203. doi: 10.1016/s0168-8227(98)00105-3. [DOI] [PubMed] [Google Scholar]
- Takano H, Onodera O, Takahashi H, Igarashi S, Yamada M, Oyake M, Ikeuchi T, Koide R, Tanaka H, Iwabuchi K, Tsuji S. Somatic mosaicism of expanded CAG repeats in brains of patients with dentatorubral-pallidoluysian atrophy: Cellular population-dependent dynamics of mitotic instability. American Journal of Human Genetics. 1996;58:1212–1222. [PMC free article] [PubMed] [Google Scholar]
- Takei N, Mortensen PB, Klaening U, Murray RM, Sham PC, O'Callaghan E, Munk-Jorgensen P. Relationship between in utero exposure to influenza epidemics and risk of schizophrenia in Denmark. Biological Psychiatry. 1996;40:817–824. doi: 10.1016/0006-3223(95)00592-7. [DOI] [PubMed] [Google Scholar]
- Tarin JJ, Brines J, Cano A. Long-term effects of delayed parenthood. Human Reproduction. 1998;13:2371–2376. doi: 10.1093/humrep/13.9.2371. [DOI] [PubMed] [Google Scholar]
- Telenius H, Kremer HP, Theilmann J, Andrew SE, Almqvist E, Anvret M, Greenberg C, Greenberg J, Lucotte G, Squitieri F. Molecular analysis of juvenile Huntington disease: The major influence on (CAG)n repeat length is the sex of the affected parent. Human Molecular Genetics. 1993;2:1535–1540. doi: 10.1093/hmg/2.10.1535. [DOI] [PubMed] [Google Scholar]
- Tellier AL, Cormier-Daire V, Abadie V, Amiel J, Sigaudy S, Bonnet D, Lonlay-Debeney P, Morrisseau-Durand MP, Hubert P, Michel JL, Jan D, Dollfus H, Baumann C, Labrune P, Lacombe D, Philip N, LeMerrer M, Briard ML, Munnich A, Lyonnet S. CHARGE syndrome: Report of 47 cases and review. American Journal of Medical Genetics. 1998;76:402–409. doi: 10.1002/(sici)1096-8628(19980413)76:5<402::aid-ajmg7>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- Thompson NM, Hecht JT, Bohan TP, Kramer LA, Davidson K, Brandt ME, Fletcher JM. Neuroanatomic and neuropsychological outcome in school-age children with achondroplasia. American Journal of Medical Genetics. 1999;88:145–153. doi: 10.1002/(sici)1096-8628(19990416)88:2<145::aid-ajmg10>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- Tolarova MM, Harris JA, Ordway DE, Vargervik K. Birth prevalence, mutation rate, sex ratio, parents' age, and ethnicity in Apert syndrome. American Journal of Medical Genetics. 1997;72:394–398. doi: 10.1002/(sici)1096-8628(19971112)72:4<394::aid-ajmg4>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- Torrey EF, Miller J, Rawlings R, Yolken RH. Seasonality of births in schizophrenia and bipolar disorder: A review of the literature. Schizophrenia Research. 1997;28:1–38. doi: 10.1016/s0920-9964(97)00092-3. [DOI] [PubMed] [Google Scholar]
- Tsuang M. Schizophrenia: Genes and environment. Biological Psychiatry. 2000;47:210–220. doi: 10.1016/s0006-3223(99)00289-9. [DOI] [PubMed] [Google Scholar]
- Vajo Z, Francomano CA, Wilkin DJ. The molecular and genetic basis of fibroblast growth factor receptor 3 disorders: The achondroplasia family of skeletal dysplasias, Muenke craniosynostosis, and Crouzon syndrome with acanthosis nigricans. Endocrine Reviews. 2000;21:23–39. doi: 10.1210/edrv.21.1.0387. [DOI] [PubMed] [Google Scholar]
- Vincent JB, Paterson AD, Strong E, Petronis A, Kennedy JL. The unstable trinucleotide repeat story of major psychosis. American Journal of Medical Genetics. 2000;97:77–97. doi: 10.1002/(sici)1096-8628(200021)97:1<77::aid-ajmg11>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Waddington JL, Lane A, Larkin C, O'Callaghan E. The neurodevelopmental basis of schizophrenia: Clinical clues from cerebro-craniofacial dysmorphogenesis, and the roots of a lifetime trajectory of disease. Biological Psychiatry. 1999;46:31–39. doi: 10.1016/s0006-3223(99)00055-4. [DOI] [PubMed] [Google Scholar]
- Weinberg W. Zur Verebung des Zwergwuchses. Archiv Fuer Rassen und Gesellschafts-Biologie. 1912;9:710. [Google Scholar]
- Weinberger DR, Berman KF, Suddath R, Torrey EF. Evidence of dysfunction of a prefrontal-limbic network in schizophrenia: A magnetic resonance imaging and regional cerebral blood flow study of discordant monozygotic twins. American Journal of Psychiatry. 1992;149:890–897. doi: 10.1176/ajp.149.7.890. [DOI] [PubMed] [Google Scholar]
- Wellington CL, Brinkman RR, O'Kusky JR, Hayden MR. Toward understanding the molecular pathology of Huntington's disease. Brain Pathology. 1997;7:979–1002. doi: 10.1111/j.1750-3639.1997.tb00897.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whalley LJ, Thomas BM, Starr JM. Epidemiology of presenile Alzheimer's disease in Scotland (1974–88) II. Exposures to possible risk factors. British Journal of Psychiatry. 1995;167:732–738. doi: 10.1192/bjp.167.6.732. [DOI] [PubMed] [Google Scholar]
- Wilkin DJ, Szabo JK, Cameron R, Henderson S, Bellus GA, Mack ML, Kaitila I, Loughlin J, Munnich A, Sykes B, Bonaventure J, Francomano CA. Mutations in fibroblast growth-factor receptor 3 in sporadic cases of achondroplasia occur exclusively on the paternally derived chromosome. American Journal of Human Genetics. 1998;63:711–716. doi: 10.1086/302000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu K, Herr AB, Waksman G, Ornitz DM. Loss of fibroblast growth factor receptor 2 ligand-binding specificity in apert syndrome. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:14536–14541. doi: 10.1073/pnas.97.26.14536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenzes MT. Smoking and reproduction: Gene damage to human gametes and embryos. Human Reproduction Update. 2000;6:122–131. doi: 10.1093/humupd/6.2.122. [DOI] [PubMed] [Google Scholar]
- Zhang SL. A study on effects of parents' age, birth order and mental retardation of unknown etiology. Zhonghua Sheng Jing Jing Shen Ke Za Xhi. 1992;25:303–318. [PubMed] [Google Scholar]
- Zhang Y, Kreger BE, Dorgan JF, Cupples LA, Myers RH, Splansky GL, Schatzkin A, Ellison RC. Parental age at child's birth and son's risk of prostate cancer: The Framingham Study. American Journal of Epidemiology. 1999;150:1208–1212. doi: 10.1093/oxfordjournals.aje.a009947. [DOI] [PubMed] [Google Scholar]