

Abstract
Introduction
Accumulation of β-amyloid (Aβ) aggregates in the brain is linked to the pathogenesis of Alzheimer’s disease (AD). Imaging probes targeting these Aβ aggregates in the brain may provide a useful tool to facilitate the diagnosis of AD. Recently, [18F]AV-45 ([18F]5) demonstrated high binding to the Aβ aggregates in AD patients. To improve the availability of this agent for widespread clinical application, a rapid, fully automated high yield, cGMP-compliant radiosynthesis was necessary for production of this probe. We report herein an optimal [18F]fluorination, de-protection condition and fully automated radiosynthsis of [18F]AV-45 ([18F]5) on a radiosynthesis module (BNU F-A2).
Methods
The preparation of [18F]AV-45 ([18F]5) was evaluated under different conditions, specifically by employing different precursors (-OTs and -Br as the leaving group), reagents (K222/K2CO3 vs. tributylammonium bicarbonate) and de-protection in different acids. With optimized conditions from these experiments, the automated synthesis of [18F]AV-45 ([18F]5) was accomplished by using a computer-programmed, standard operating procedure, and was purified on an on-line solid-phase cartridge (Oasis HLB).
Results
The optimized reaction conditions were successfully implemented to an automated nucleophilic fluorination module. The radiochemical purity of [18F]AV-45 ([18F]5) was > 95%, and the automated synthesis yield was 33.6 ± 5.2% (no decay corrected, n = 4), 50.1 ± 7.9% (decay corrected) in 50 min at a quantity level of 10~100 mCi (370~3700 MBq). Autoradiography studies of [18F]AV-45 ([18F]5) using postmortem AD brain and Tg mouse brain sections in the presence of different concentration of “cold” AV-136 showed a relatively low inhibition of in vitro binding of [18F]AV-45 ([18F]5) to the Aβ plaques (IC50 = 1-4 μM, a concentration several order of magnitude higher than the expected pseudo carrier concentration in the brain).
Conclusions
Solid phase extraction (SPE) purification and improved labeling conditions were successfully implemented into an automated synthesis module, which is more convenient, highly efficient and simpler in operation than using a semipreparative, high-performance liquid chromatography (HPLC) method. This new, automated procedure for preparation of [18F]AV-45 ([18F]5) is suitable for routine clinical application.
Keywords: Alzheimer’s Disease, automated radiosynthesis, brain imaging, solid-phase extraction, Radiochemistry
1. Introduction
Alzheimer’s disease (AD) is a brain disorder associated with progressive memory loss and decrease in cognitive function. Major neuropathology observations of postmortem AD brains depict the presence of senile plaques (containing β-amyloid (Aβ) aggregates) and neurofibrillary tangles (highly phosphorylated tau proteins). The accumulation of Aβ plaques in the brain is believed to be one of the most significant factors associated with the development of AD [1-5]. Therefore, developing Aβ plaque specific probes for in vivo imaging studies may be important for differential diagnosing and monitoring AD patients [6-11].
There are a number of PET (position emission tomography) and SPECT (single photon emission tomography) tracers that have been reported [12-14] and some of them have been tested clinically, such as 11C-labeled benzothiazole derivative, [11C]PIB [15-17]and a 18F-labeled probe, [18F]FDDNP [18, 19]. They have demonstrated the potential utility of in vivo imaging of Aβ plaque deposition in the brain. However, the short half-life (20 min) of C-11 may limit the usefulness of [11C]PIB for a widespread clinical application. And due to the nonspecific binding and lipophilic nature of [18F]FDDNP, it is relatively difficult to measure, specifically, the signal contributing from the binding of plaques and tangles in the brain.
Currently, many efforts focusing on in vivo imaging agents targeting Aβ plaques have been reported to monitor the amyloid burden following the disease progression and further provide supporting evidence for therapeutic intervention [9, 10, 17, 20-25]. It was reported that (E)-2-(2-(2-(2-[18F]Fluoroethoxy)ethoxy)ethoxy)-5-(4-methylaminostyryl) pyridine ([18F]AV-45) ([18F]5) [22, 23] binds specifically to fibrillar Aβ and has favorable pharmacokinetic properties. Preliminary clinical studies[26, 27] in patients with diagnosed mild AD showed marked retention of [18F]AV-45 ([18F]5) in the cortex, known to contain large amounts of amyloid deposits in AD.
As a new AD imaging agent in phase III clinical trials, [18F]AV-45 ([18F]5) was prepared and purified by using the Synthesizer with pre-HPLC equipment[26]. This method needed HPLC purification, which was costly, cumbersome, laborious to manage in routine preparations, difficult to automate, and took a longer time (more than 70 min) to complete the preparing process. In order to eliminate the HPLC purification system, a one-pot and two-step automated synthesis process with a rapid and simple on-line SPE (solid-phase extraction, Oasis® HLB) purification system was developed. Reported herein is an optimal radiolabeling procedure for [18F]AV-45 ([18F]5) on an automated radiosynthesizer, which would enable and streamline the production, making [18F]AV-45 ([18F]5) readily available for Aβ plaques imaging. Thus, the procedure will supply the agent to a large number of imaging centers and benefit a sizeable population of AD patients.
2. Experimental
2.1 General materials
All reagents used were purchased commercially and were used without further purification, unless otherwise indicated. All reactions were carried out in flame-dried glassware under a nitrogen atmosphere. Flash chromatography (FC) was performed using silica gel 60 (230 - 400 mesh, Sigma-Aldrich). 1H-NMR spectra were obtained at 200MHz in CDCl3 (Bruker DPX 200 Spectrometer). Chemical shifts are reported as δ values (parts per million) relative to residual protons of deuterated solvent. Coupling constants are reported in Hertz. The multiplicity is defined by s (singlet), d (doublet), t (tripled), br (broad), or m (multiplet). Thin layer chromatography (TLC) was run on pre-coated plates of silica gel 60 F254. 18F aqueous solution was purchased from IBA (IBA Molecular North America, Inc.). Solid-phase extraction cartridges were obtained from Waters Corp. (Milford, MA) (Oasis HLB 3cc, QMA light) (Fisher Scientific). Transgenic mice (APPswePSEN1) were purchased from Jackson laboratory. Postmortem human samples were obtained from National Disease Research Interchange (NDRI, Philadelphia, PA).
2.2 Precursors, 1 and 2, and AV-45, 5
The precursors, 1 and 2, and AV-45, 5, were synthesized according to the literatures [24, 28], (Scheme 1).
Scheme 1.

Synthesis of 2, 3, 4, [18F]AV-45 ([18F]5) and AV-136 (6)
Scheme 1. Synthesis of 2, [18F]AV-45-Boc ([18F]3), 4, [18F]AV-45 ([18F]5) and AV-136 (6)
(E)-2-(2-(2-(2-Tosyloxyethoxy)ethoxy)ethoxy)-5-(4-(tertbutoxycarbonyl(methyl)amino)styryl)pyridine (-OTs derivative), 1
1 was synthesized as previously reported [12].
(E)-2-(2-(2-(2-bromoethoxy)ethoxy)ethoxy)-5-(4-(tertbutoxycarbonyl(methyl)amino)styryl)pyridine ((-Br derivative), 2
Lithium bromide (30.3 mg, 0.35 mmol) was added to a solution of 1 (62.6 mg, 0.1 mmol) in dry acetone (3 mL). The reaction mixture was refluxed for 3 h. After cooling to room temperature, the mixture was filtered and evaporated until dry. Pure product, (E)-2-(2-(2-(2-bromoethoxy)ethoxy)ethoxy)-5-(4-(tertbutoxy carbonyl(methyl) amino)styryl)pyridine, was obtained by flash chromatography (Yield: 94%). 1H-NMR (CDCl3, δ ppm) 1.474 (s, 9H); 3.302-3.324 (m, 5H); 3.913-3.697 (m, 8H); 4.495-4.543 (m, 2H); 6.810 (d, 2H, J = 2.6 Hz); 6.919 (s, 2H); 7.236 (d, 2H, J = 8.6 Hz); 7.455 (d, 2H, 8.6 Hz); 7.803 (dd, J1 = 8.8 Hz, J2 = 2.4Hz); 8.196 (d, 1H, 2.2 Hz).
(E)-2-(2-(2-(2-Fluoroethoxy)ethoxy)ethoxy)-5-(4-methylaminostyryl)pyridine, 5, (AV-45)
AV-45 ([19F]5) was synthesized, as previously reported [12].
2.3 Optimization of [18F]fluorination reaction condition for [18F]5 ([18F]AV-45)
Preparation of “dried” [18F]KF/K222 or [18F]TBAF (tetrabutyl ammonium fluoride) complex
An aqueous solution of [18F]F- (3 - 5 mCi, 111-185 MBq), produced by cyclotron using 18O (p, n) 18F reaction, was passed through a Sep-Pak Light QMA cartridge. The cartridge was dried with argon, and the 18F activity was eluted with a 1.0 mL (0.83 mL acetonitrile (ACN)/0.17 mL water) solution containing 11.0 mg Kryptofix222 (K222) and 2.0 mg K2CO3 or a 1.0 mL solution of tetra-butyl ammonium bicarbonate (TBAHCO3, 21.5 mg) in 75% ACN/25% H2O. The volume of eluent was varied according to the number of planned labeling experiments to ensure that each [18F]fluorination vial contained 11.0 mg K222 and 2.0 mg K2CO3 or 21.5 mg of TBAHCO3. The eluent was then evaporated at 110 °C under an argon stream. Furthermore, the residue was azeotropically dried twice with 1.0 mL anhydrous ACN at 110 °C under an argon stream. The residue, containing [18F]KF/K222 or [18F]TBAF complex, was dissolved in solvent (ACN or DMSO), which was dispensed into vials (1.0 mL/vial) for the following [18F]fluorination reactions.
Nucleophilic [18F]fluorination of [18F] 3 ([18F]AV-45-Boc) under different condition (Scheme 1)□
Solvent (ACN or DMSO, 0.5 mL) containing 1 or 2 (5.6 μmol) was added into the vial containing the dried [18F]KF/K222 or [18F]TBAF complex (3 - 5 mCi, 111 - 185 MBq) prepared above in 1.0 mL ACN or DMSO solution. The mixture was thoroughly vortexed (5 seconds) and heated at 100 °C. Aliquots (50 μL) were removed at different time points (0, 3 and 10 min) and the aliquots of the reactions were quenched in ice water and analyzed by TLC and HPLC methods (see below)
De-protection method of [18F] 3 (Scheme 1)
The [18F]3/DMSO solution was diluted with DMSO to make a total volume of 5 mL. Then the [18F]3/DMSO solution was dispensed into vials (1.0 mL/vial) and preheated to 100 °C for the following de-protection reaction. To de-protect the Boc-protection group, 0.25 mL of different acid solutions (3.3 M HCl, 2.0 M H2SO4, 3.0 M H3PO4, and 1.0 M TsOH/DMSO or water) were added, respectively, into the vials. Aliquots (50 μL) were removed at different time points (0, 5 and 10 min) and the aliquots of the reactions were quenched in ice water. The samples were analyzed by TLC and HPLC methods (see below).
Analysis of radiochemical yield, radiochemical purity and stability of the precursors by TLC and HPLC
The radiochemical yields (RCY) were measured by TLC analysis. 1 μL samples were spotted on TLC silica plates and developed with ethyl acetate. The TLC plates were cut into ten sections and the sections were counted in a gamma counter. The radiolabeling yield was calculated by dividing the counts of the measured activity of [18F]3 for initial fluorination) (see above) at the determined Rf value by the total counts on the TLC plates. Similarly, the progress of deprotection (see above) was evaluated by measuring the counts for [18F]5 vs. the total counts on the TLC plates. Rf values were first established by evaluating cold [19F]3 and cold [19F]5 under identical TLC conditions.
The radiochemical purity (RCP) was evaluated by TLC and high pressure liquid chromatography (HPLC) equipped with a gamma ray radio-detector and a UV/Vis detector (Agilent; Gemini C18 (250mm × 4.6 mm). Mobile phase: 1 mL/min with a gradient as follows: from 0 – 2 min isocratic 10 mM ammonium formate buffer (AFB) 100%; from 2 - 5min gradient AFB 100 - 30%, ACN 0 - 70%; from 5 - 10 min gradient AFB 30 - 0%, ACN 70 - 100% and from 10 - 15 min AFB 100%. Additionally, the amount of unchanged 1 or 2 remaining in the final mixture was determined by UV/Vis at 350 nm. Samples (10μL each) were injected into HPLC. The area under the curve for UV peaks of the precursors was compared to the same HPLC peak measured for the initial [18F]fluorination mixture removed at time zero. The difference in the area under the UV/Vis peak, at different time points, was used to determine the stability of the precursors under labeling conditions.
2.5 Automated radiosynthesis and purification of [18F]5 ([18F]AV-45)
Automated synthesis of [18F]5 involves four events: (1) azeotropically drying of [18F]fluoride, (2) fluorination of precursor (displacing the -OTs by [18F]F-), (3) de-protect the Boc-group, and (4) a SPE purification. The reagents for the production of [18F]5 were stored in the reagent vials. (Fig. 1): (A) 1.0 mL (0.83 mL ACN and 0.17 mL H2O) solution containing 11.0 mg K222 and 2.0 mg K2CO3; (B, C) 1.0 mL anhydrous ACN; (D) 1.0 mg 1 dissolved in 1.0 mL anhydrous DMSO; (E) 0.25 mL 3.3 M HCl solution; (F) 3.0 mL 1% sodium hydroxide solution; (G) 3.0 mL deionized water; (H) 1.0 mL EtOH.
Fig. 1.
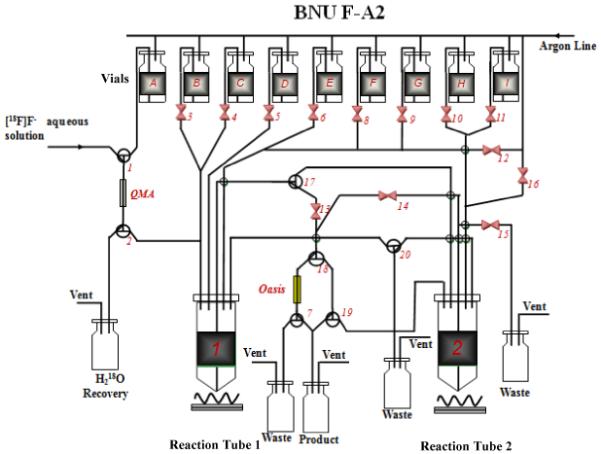
Module diagram of the automated radiosynthesis system (BNU F-A2)
Vials: (A) 1.0 mL of K222 11.0 mg/K2CO3, 2.0 mg in 0.83 mL ACN and 0.17 mL H2O; (B, C) 1.0 mL anhydrous ACN; (D) 1 mg 1 dissolved in 1.0 mL anhydrous DMSO; (E) 0.25 mL 3.3 M HCl; (F) 3 mL 1% sodium hydroxide solution; (G) 3 mL deionized water; (H) 1 mL EtOH.
The synthesis module (Fig. 1) was operated in the following sequence: (1) The [18F]fluoride (10-100 mCi, 370-3700 MBq) was trapped on a Sep-Pak Light QMA cartridge (pre-activated with 10 mL 1 N NaHCO3 followed by 10 mL sterile H2O). The activity was eluted using 1.0 mL eluent (K222 11.0 mg/K2CO3 2.0 mg in 0.83 mL ACN and 0.17 mL H2O), and delivered to Reaction Tube 1. The mixture solution in Tube 1 was evaporated at 110 °C under an argon flow. The residue was dried twice with 1.0 mL anhydrous ACN (Vial B and C) under the same condition. (2) [18F]fluorination of the precursor: After cooling for 60 seconds, 1.0 mg of 1 dissolved in 1.0 mL DMSO (Vial D) was added to the dried [18F]KF/K222 (Tube 1). The reaction occurred at 115 °C for 10 min. (3) De-protection reaction: After cooling for 60 seconds, 0.25 mL of 3.3 M HCl acid solution (Vial E) was added into the tube to deprotect the Boc-group at 100 °C for 10 min; (4) Neutralization and SPE purification by an Oasis HLB cartridge (3 cc, Waters): The reaction mixture in Tube 1 was cooled down to room temperature, then neutralized and diluted with 3 mL of 1% sodium hydroxide solution (Vial F), and loaded onto the Oasis HLB 3cc cartridge (pre-treated with 10 mL EtOH, followed by 10 mL sterile H2O). The cartridge was rinsed with 3 mL of deionized water (Vial G) to eliminate unreacted [18F]fluoride. After drying the cartridge under a flow of argon, the final product was eluted with 1.0 mL EtOH (Vial H) into the product vial containing 9.0 mL of 0.9% saline solution to generate the dose ready for injection. The dose was analyzed by TLC and HPLC as outlined above.
2.6 Autoradiography of [18F]5 ([18F]AV-45) using postmortem AD brain and Tg mouse brain sections
Frozen brain samples from postmortem AD subject and Tg mice ((APPswePSEN1)) were cut into 20 um sections. The sections were incubated with [18F]5 ([18F]AV-45) in 40% ethanol at a concentration of 0.3nM with increasing amounts of “cold” 6, AV-136 (10 different concentrations ranging from 0 to 279 μM). Non-specific binding was defined by adding 280 μM of “cold” 5, (AV-45). The sections were then dipped in saturated Lithium carbonate in 40% ethanol (2 min wash twice) and washed with 40% ethanol (2 min wash once) followed by rinsing with water for 30 seconds. After drying, the sections were exposed to Kodak Biomax MR film for 12-18 hours. After the film was developed, the images were digitized; regions of interest were analyzed and the IC50 values were determined.
3. Results and discussion
3.1 Synthesis of precursors
The 1 (-OTs) was successfully converted to 2 (-Br) by refluxing with lithium bromide in acetone and the reaction proceeded in good yield (94%) (Scheme 1). Both, 1 and 2 were successfully used to label with 18F in nucleophilic fluorination reaction in preparation of [18F]5.
3.2 [18F]fluorination reaction conditions
It is well known that the fluoride ion has very high nucleophilicity, but it is also highly hydrated in aqueous solutions and insoluble in organic solvents. In order to “activate” the fluoride ion and increase its solubility in organic solvent, the surrounding water is stripped, which produces highly “reactive” and “naked” fluoride ions in the organic solution. This is a critical step for a successful fluorination reaction. It can be accomplished by chelating the counter cation, such as K+ or Cs+, using K222 and potassium carbonate or by a phase transfer reagent, such as t-butylammonium fluoride (TBAF). Those reagents act as catalysts for lowering the activation energy of the fluorination reaction, so fluorination of precursor compounds, 1 and 2, under these two popular catalysts conditions can be compared to the preparation of [18F]3.
Table 1 showed that when the reaction was catalyzed by [18F]KF/K222/K2CO3, a commonly used combination catalyst in 18F-radiolabeling, the radiochemical yields of [18F]3 were 79.3 ± 5.3% and 70.0 ± 6.4 for 1 and 2, respectively at 100 °C for 10 min. It showed that the reactivity of 1 (-OTs as a leaving group) and 2 (-Br as a leaving group) was comparable.
Table 1.
Radiochemical yield of [18F]3 and residual precursor amount using different precursors in the presence of different complex agents (n = 4, mean ± SD) *
| Time (min) |
[18F]KF/K222/K2CO3/DMSO | [18F]TBAF/ACN | |||
|---|---|---|---|---|---|
| 1 | 2 | 1 | 2 | ||
| [18F] 3 | 0 | 5.2 ± 2.5 | 4.2 ± 0.9 | 1.4 ± 0.8 | 1.3 ± 0.5 |
| 3 | 70.8 ± 7.5 | 53.0 ± 10.1 | 52.9 ± 6.7 | 35.6 ± 4.0 | |
| Radiochemical | 10 | 79.8 ± 5.3 | 70.0 ± 6.4 | 70.7 ± 4.7 | 54.8 ± 4.8 |
| Yield** (%) | |||||
| Residual | 0 | 100.0 ± 0.0 | 100.0 ± 0.0 | 100.0 ± 0.0 | 100.0 ± 0.0 |
| 3 | 76.9 ± 7.1 | 78.3 ± 4.7 | 59.0 ± 17.5 | 92.3 ± 2.3 | |
| precursor | 10 | 39.6 ± 10.9 | 36.6 ± 3.6 | 24.0 ± 9.7 | 72.4 ± 10.6 |
| amount(%) | |||||
Fluorination condition: 5.6 μmol precursor (either 1 or 2), 11.0 mg K222 and 2.0 mg K2CO3 or 21.5 mg TBAHCO3, 100 °C.
Radiochemical yield of [18F]3 was determined by TLC.
When using [18F]TBAF as the phase transfer catalyst, the nucleophilic fluorination reactivity between [18F]F− and the –OTs leaving group was as high as the fluorination reactivity for [18F]KF/K222/K2CO3 (radiochemical yield at 100 °C for 10 min: 70.7 ± 4.7% for [18F]TBAF and 79.8 ± 5.3 for [18F]KF/K222/K2CO3). On the contrary, the use of –Br as the leaving group resulted in lower fluorination yield for [18F]TBAF (radiochemical yield at 100 °C for 10 min: 54.8 ± 4.8% for [18F]TBAF and 70.0 ± 6.4 for [18F]KF/K222/K2CO3).
The stability and reactivity of the precursors were also investigated by measuring the residual amount of the 1 and 2 remaining in the final mixture after the [18F]fluorination. The main chemical impurity was the hydroxide byproduct, 4 after hydrolysis of the precursor. The hydrolysis of the precursors paralleled to the reactivity of nucleophilic F-displacement. The radiochemical yield and the amount of residual precursor in the reaction mixture was compared in Table 1.
When fluorination of 1 was carried out in the presence of [18F]TBAF at 100 °C for 10 min, the radiochemical yield of [18F]3 was 70.7 ± 4.7% (n = 10), and the amount of 1 noticeably decreased to 24.0 ± 9.7%, while the radiochemical yield of [18F]3 was 54.8 ± 4.8% for 2 in the same conditions. Nearly 70% of 2 remained, which suggested that 2 was more stable than 1 under this reaction condition.
When fluorinated in [18F]KF/K222/K2CO3/DMSO, 1 and 2 displayed a similar stability, and 39.6 ± 10.9% and 36.6 ± 3.6% of the precursors remained, respectively at 10 min post reaction.
The radiochemical yield was also dependent on the reaction time. The radiochemical yield with 2 was increased from 53.0 ± 10.1% at the first 3 min to 70.0 ± 6.4% at 10 min post reaction in K222/K2CO3/DMSO. And the radiochemical yield ascended dramatically after a few minutes of initial fluorination, and the reaction headed a balance as time extended.
3.3 De-protection
A series of N-Boc deprotection reactions of [18F]3 were performed at 100 °C using different acids, such as HCl, H2SO4, H3PO4 and TsOH. The hydrogen ion concentrations of inorganic acids were 2-3.3 mol/L, and organic acid was 1 mol/L for the consideration of convenience for preparation of injection solutions. The results are shown in Table 2. The cleavage of N-Boc did not complete at 5 min under the four strong acidic conditions. By prolonging the reaction time from 5 min to 10 min, the removal of N-Boc was completed with use of the HCl acid solution. The percent of product [18F]5 for H2SO4/H2O, TsOH/H2O and TsOH/DMSO was 68%, 75% and 54%, respectively. The N-Boc deprotection was not observed at all under H3PO4 condition (100% of [18F]3 remained). These results indicated that the removal of N-Boc in the presence of HCl achieved optimal results.
Table 2.
Deprotecting reaction of [18F]3 under acidic conditions at 100 °C***
| Acid | 5 min | 10 min | ||
|---|---|---|---|---|
| [18F]3* | [18F]5** | [18F]3* | [18F]5** | |
| 3.3 M HCl/H2O | 11% | 88% | 0% | 100% |
| 2.0 M H2SO4/H2O | 61% | 27% | 31% | 68% |
| 3.0 M H3PO4/H2O | 100% | 0% | 100% | 0% |
| 1.0 M TsOH/DMSO | 62% | 22% | 45% | 54% |
| 1.0 M TsOH/H2O | 58% | 31% | 31% | 75% |
Residual [18F]3 amount (%) after deprotection reaction.
Product [18F]5 amount (%) after deprotection reaction.
single determinations except for the use of 3.3 M HCl/H2O with 10 minutes reaction time (n = 10).
3.4 Automated radiosynthesis and purification
The automated synthesis yield of the [18F]AV-45 ([18F]5) was 33.6 ± 5.2% (no decay corrected, n = 4), 50.1 ± 7.9% (decay corrected) in the quantity of 10-100 mCi (370-3700 MBq) with a RCP > 95% (Fig. 2), after a total time of about 50 min. Purification by solid-phase cartridge (Oasis HLB) removed other impurities, but did not eliminate non-polar chemical impurities such as hydroxide derivative, AV-136 (6)(Scheme 1). The effective specific activity was 43 ± 36 Ci/mmole (1.59 GBq/μM). In the future when the preparation is scaled up for routine production at ten to twenty times higher activity, the specific activity will likely be improved.
Fig. 2.
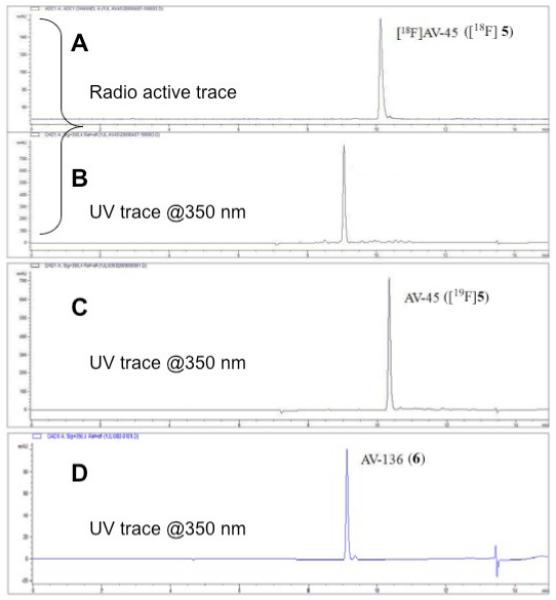

HPLC profile of a [18F]AV-45 ([18F]5) sample purified by SPE method (trace A: radio trace of sample; trace B: UV trace of sample @ 350 nm). Trace C is an UV trace of non - radioactive reference compound AV-45 (@350 nm). The bottom trace (D) is an UV trace of AV-136 (6) (@350 nm).
HPLC profile of [18F]AV-45 ([18F]5) sample purified by SPE method (HPLC conditions: Agilent HPLC 1100 ; Gemini C18 column (250mm × 4.6 mm); mobile phase: 1 mL/min with a gradient as follows: from 0 – 2 min isocratic 10 mM ammonium formate buffer (AFB) 100%; from 2 - 5min gradient AFB 100 - 30%, ACN 0 - 70%; from 5 - 10 min gradient AFB 30 - 0%, ACN 70 - 100% and from 10 - 15 min AFB 100%).
A: radioactive trace of sample
B: UV trace of sample @ 350 nm
C: UV trace of cold AV-45 ([19F]5) under identical HPLC conditions as above.
D: UV trace of synthesized AV-136 @ 350 nm under identical HPLC conditions as above. The “identical” retention times of AV-136 compared to the UV material of the labeled sample suggest that the byproduct in the labeling is AV-136 (6).
3.5 Autoradiography of [18F]5 ([18F]AV-45) using postmortem AD brain and Tg mouse brain sections
Autoradiography of [18F]5 ([18F]AV-45) using postmortem AD brain and Tg mouse brain sections with or without the presence of increasing concentration of “cold” pseudo-carrier, 6, AV-136, were evaluated. We are particularly interested in measuring the competition due to the presence of the pseudo-carrier, 6, AV-136, because previously it was reported that The autoradiography of the “HPLC” purified [18F]5 ([18F]AV-45) showed excellent labeling of the Aβ plaques in the brain sections (Fig. 3). The images are consistent with those previously reported preclinical data [26]. Based on the optical density of the digitized brain images (n = 3 for each data point for two different brain regions), the changes of the signal vs the concentration of “cold” pseudo carrier - AV-136 were plotted (Fig. 3). The autoradiography images showed that human AD brain and Tg mouse brain sections displayed an IC50 of 1.68 and 3.47 μM, respectively. The preparation of [18F]5 ([18F]AV-45) for human studies usually started with the N-Boc, -OTs precursor (1, 5 mg, 8.17 μmole). Using a very conservative assumption, 50% of the pseudo-carrier, 6, AV-136, remains after the preparation, all of this pseudo-carrier is inject into a human subject and 5% of this pseudo carrier is localized in the brain, then the maximum concentration in the human brain (1.5 L volume), the concentration will be 8.17 μmole × 0.5 × 0.05/1.5L = 0.14 μM. For the actual clinical preparation, there will be multiple doses prepared in one run and each subject will likely receive a small fraction of the pseudo-carrier, 6, AV-136, as calculated above. It will be highly un-likely that the concentration of the pseudo-carrier, 6, AV-136, will be sufficient to compete and saturate the Aβ plaque-binding sites in the brain. We, therefore, conclude that the [18F]5 ([18F]AV-45) prepared by the SPE preparation (without HPLC purification) will likely be suitable for human use.
Fig. 3.
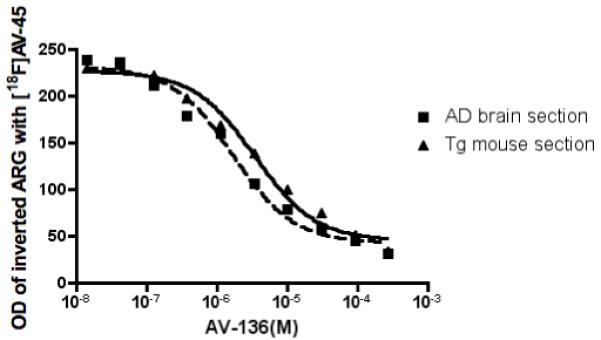
Autoradiography (ARG) images of [18F]AV-45 ([18F]5, HPLC purified samples) using postmortem human brain and transgenic (tg) mice (APPswePSEN1) brain sections. Increasing concentration of pseudo-carrier, 6,AV-136, were added (10 different concentrations ranging from 0 to 279 μM) and the optical density (OD) of brain sections were analyzed and the inhibition by the presence of pseudo-carrier, 6, AV-136 was plotted. The postmortem human brain and transgenic (tg) mice (APPswePSEN1) brain sections displayed comparable values, IC50 = 1.68 and 3.47 μM, respectively. Non-specific binding (NSB) was defined by using 280 μM of “cold” 5, (AV-45).
4. Conclusion
The preparation of [18F]AV-45 ([18F]5) was evaluated using -OTs and -Br as leaving groups with different catalysts. There is no marked difference in achieving high radiochemical yields between 1 (-OTs) and 2 (-Br) in [18F]KF/K222/K2CO3, but the radiochemical yield of 1 (-OTs) is slightly higher than that of 2 (-Br) in [18F]TBAF. The N-Boc deprotection of [18F]3 was best achieved with a 3.3 M HCl/H2O acid solution at 100 °C for 10 min. The automated radiosynthesis of [18F]AV-45 ([18F]5) was successfully complemented by a simple and fast SPE purification, avoiding a more complex and time-consuming HPLC purification. [18F]AV-45 ([18F]5) was prepared in a high radiochemical purity (> 95%, within 50 min), which would be suitable for PET imaging studies in a clinical setting. We have performed evaluations comparing the preparations, with or without HPLC purification, using in vitro autoradiography binding assay which suggest that the concentration of the pseudo-carrier, 6, AV-136, will not be sufficient to compete for the Aβ plaque-binding sites in the brain. The improved [18F]AV-45 ([18F]5) preparation may streamline the preparation; thus, making the agent widely available.
Acknowledgemen
The authors are grateful for the financial support from the Chinese National 973 Program (2006CB500705 to L.Z.), the Chinese National 863 Program (2006AA02A408 to L.Z.), NIH grant (AG-022559 to H.F.K.) and Avid Radiopharmaceuticals Inc. (to H.F.K.). Authors also thank Dr. Michael Kilbourn, University of Michigan for his helpful discussion. Authors thank Mrs. Alex Paine for helpful editorial assistance.
Abbreviations
- AD
Alzheimer’s Disease
- Aβ
β-amyloid
- AV-45
((E)-2-(2-(2-(2-[18F]Fluoroethoxy)ethoxy)ethoxy)-5-(4- methylaminostyryl)pyridine)
- K222
Kryptofix®222
- SPE
solid phase extraction
- HPLC
high-performance liquid chromatography
- TLC
thin layer chromatography
- PET
positron emission tomography
- SPECT
single photon emission tomography
- FC
flash chromatography
- TLC
thin layer chromatography
- TBAHCO3
tetra-butyl ammonium bicarbonate
- AFB
ammonium formate buffer
- TBAF
t-butylammonium fluoride
- RCY
radiochemical yields
- DMSO
dimethyl sulfoxide
- ACN
CH3CN or acetonitrile
- tg mouse
transgenic muse
- ARG
autoradiography
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference
- [1].Selkoe DJ. Imaging Alzheimer’s amyloid. Nat Biotechnol. 2000;18:823–24. doi: 10.1038/78422. [DOI] [PubMed] [Google Scholar]
- [2].Goedert M, Spillantini MG. A century of Alzheimer’s disease. Science. 2006;314:777–81. doi: 10.1126/science.1132814. [DOI] [PubMed] [Google Scholar]
- [3].Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–56. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- [4].Hardy J. Is amyloid plaque imaging the key to monitoring brain pathology of Alzheimer’s disease in vivo? Eur J Nucl Med Mol Imaging. 2004;31:1539–40. doi: 10.1007/s00259-004-1676-9. [DOI] [PubMed] [Google Scholar]
- [5].Querfurth HW, LaFerla FM. Alzheimer’s disease. N Engl J Med. 2010;362:329–44. doi: 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- [6].Drzezga A. Amyloid-plaque imaging in early and differential diagnosis of dementia. Ann Nucl Med. 2010 doi: 10.1007/s12149-009-0330-9. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- [7].Wolk DA, Price JC, Saxton JA, Snitz BE, James JA, Lopez OL, et al. Amyloid imaging in mild cognitive impairment subtypes. Ann Neurol. 2009;65:557–68. doi: 10.1002/ana.21598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Cairns NJ, Ikonomovic MD, Benzinger T, Storandt M, Fagan AM, Shah A, et al. Absence of PIttsburgh Compound B Detection of CerebralAmyloid Beta in a Patient With Clinical, Cognitive, and Cerebrospinal FluidMarkers of Alzheimer Disease. Arch Neurol. 2009;66:1557–62. doi: 10.1001/archneurol.2009.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Rinne JO, Brooks DJ, Rossor MN, Fox NC, Bullock R, Klunk WE, et al. 11C-PiB PET assessment of change in fibrillar amyloid-beta load in patients with Alzheimer’s disease treated with bapineuzumab: a phase 2, double-blind, placebo-controlled, ascending-dose study. Lancet Neurol. 2010;9:363–72. doi: 10.1016/S1474-4422(10)70043-0. [DOI] [PubMed] [Google Scholar]
- [10].Nordberg A, Rinne JO, Kadir A, Langstrom B. The use of PET in Alzheimer disease. Nat Rev Neurol. 2010;6:78–87. doi: 10.1038/nrneurol.2009.217. [DOI] [PubMed] [Google Scholar]
- [11].Berti V, Osorio RS, Mosconi L, Li Y, De Santi S, de Leon MJ. Early detection of Alzheimer’s disease with PET imaging. Neurodegener Dis. 7:131–5. doi: 10.1159/000289222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Henriksen G, Yousefi BH, Drzezga A, Wester HJ. Development and evaluation of compounds for imaging of beta-amyloid plaque by means of positron emission tomography. Eur J Nucl Med Mol Imaging. 2008;35:S75–81. doi: 10.1007/s00259-007-0705-x. [DOI] [PubMed] [Google Scholar]
- [13].Ono M. Development of positron-emission tomography/single-photon emission computed tomography imaging probes for in vivo detection of beta-amyloid plaques in Alzheimer’s brains. Chem Pharm Bull (Tokyo) 2009;57:1029–39. doi: 10.1248/cpb.57.1029. [DOI] [PubMed] [Google Scholar]
- [14].Cai L, Innis RB, Pike VW. Radioligand Development for PET Imaging of beta-Amyloid (Abeta)-Current Status. Curr Med Chem. 2007;14:19–52. doi: 10.2174/092986707779313471. [DOI] [PubMed] [Google Scholar]
- [15].Mathis CA, Wang Y, Holt DP, Huang G-F, Debnath ML, Klunk WE. Synthesis and Evaluation of 11C-Labeled 6-Substituted 2-Arylbenzothiazoles as Amyloid Imaging Agents. J Med Chem. 2003;46:2740–54. doi: 10.1021/jm030026b. [DOI] [PubMed] [Google Scholar]
- [16].Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, et al. Imaging Brain Amyloid in Alzheimer’s Disease with Pittsburgh Compound-B. Ann Neurol. 2004;55:306–19. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- [17].Mathis CA, Lopresti BJ, Klunk WE. Impact of amyloid imaging on drug development in Alzheimer’s disease. Nucl Med Biol. 2007;34:809–22. doi: 10.1016/j.nucmedbio.2007.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Small GW, Kepe V, Ercoli LM, Siddarth P, Bookheimer SY, Miller KJ, et al. PET of brain amyloid and tau in mild cognitive impairment. N Engl J Med. 2006;355:2652–63. doi: 10.1056/NEJMoa054625. [DOI] [PubMed] [Google Scholar]
- [19].Agdeppa ED, Kepe V, Liu J, Flores-Torres S, Satyamurthy N, Petric A, et al. Binding characteristics of radiofluorinated 6-dialkylamino-2-naphthylethylidene derivatives as positron emission tomography imaging probes for β-amyloid plaques in Alzheimer’s disease. J Neurosci. 2001;21:RC189. doi: 10.1523/JNEUROSCI.21-24-j0004.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kung HF, Choi SR, Qu W, Zhang W, Skovronsky D. (18)F Stilbenes and Styrylpyridines for PET Imaging of Abeta Plaques in Alzheimer’s Disease: A Miniperspective. J Med Chem. 2009;53:933–41. doi: 10.1021/jm901039z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Zhang W, Oya S, Kung MP, Hou C, Maier DL, Kung HF. F-18 Stilbenes as PET Imaging Agents for Detecting beta-Amyloid Plaques in the Brain. J Med Chem. 2005;48:5980–8. doi: 10.1021/jm050166g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zhang W, Oya S, Kung MP, Hou C, Maier DL, Kung HF. F-18 PEG Stilbenes as PET Imaging Agents Targeting Aβ Aggregates in the Brain. Nucl Med Biol. 2005;32:799–809. doi: 10.1016/j.nucmedbio.2005.06.001. [DOI] [PubMed] [Google Scholar]
- [23].Zhang W, Kung MP, Oya S, Hou C, Kung HF. (18)F-labeled styrylpyridines as PET agents for amyloid plaque imaging. Nucl Med Biol. 2007;34:89–97. doi: 10.1016/j.nucmedbio.2006.10.003. [DOI] [PubMed] [Google Scholar]
- [24].Qu W, Kung MP, Hou C, Oya S, Kung HF. Quick Assembly of 1,4-Diphenyltriazoles as Probes Targeting beta-Amyloid Aggregates in Alzheimer’s Disease. J Med Chem. 2007;50:3380–7. doi: 10.1021/jm070467l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Qu W, Kung MP, Hou C, Jin LW, Kung HF. Radioiodinated aza-diphenylacetylenes as potential SPECT imaging agents for beta-amyloid plaque detection. Bioorg Med Chem Lett. 2007 doi: 10.1016/j.bmcl.2007.04.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Choi SR, Golding G, Zhuang Z, Zhang W, Lim N, Hefti F, et al. Preclinical Properties of 18F-AV-45: A PET Agent for A{beta} Plaques in the Brain. J Nucl Med. 2009;50:1887–94. doi: 10.2967/jnumed.109.065284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kung HF, Choi SR, Qu W, Zhang W, Skovronsky D. 18F stilbenes and styrylpyridines for PET imaging of A beta plaques in Alzheimer’s disease: a miniperspective. J Med Chem. 2010;53:933–41. doi: 10.1021/jm901039z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Goswami R, Ponde DE, Kung MP, Hou C, Kilbourn MR, Kung HF. Fluoroalkyl derivatives of dihydrotetrabenazine as positron emission tomography imaging agents targeting vesicular monoamine transporters. Nucl Med Biol. 2006;33:685–94. doi: 10.1016/j.nucmedbio.2006.05.006. [DOI] [PubMed] [Google Scholar]