

Abstract
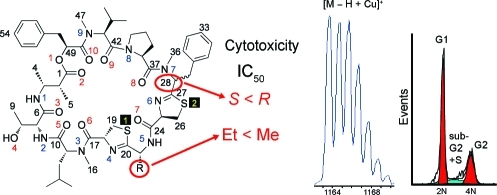
Grassypeptolides A−C (1−3), a group of closely related bis-thiazoline containing cyclic depsipeptides, have been isolated from extracts of the marine cyanobacterium Lyngbya confervoides. Although structural differences between the analogues are minimal, comparison of the in vitro cytotoxicity of the series revealed a structure−activity relationship. When the ethyl substituent of 1 is changed to a methyl substituent in 2, activity is only slightly reduced (3−4-fold), whereas inversion of the Phe unit flanking the bis-thiazoline moiety results in 16−23-fold greater potency. We show that both 1 and 3 cause G1 phase cell cycle arrest at lower concentrations, followed at higher concentrations by G2/M phase arrest, and that these compounds bind Cu2+ and Zn2+. The three-dimensional structure of 2 was determined by MS, NMR, and X-ray crystallography, and the structure of 3 was established by MS, NMR, and chemical degradation. The structure of 3 was explored by in silico molecular modeling, revealing subtle differences in overall conformation between 1 and 3. Attempts to interconvert 1 and 3 with base were unsuccessful, but enzymatic conversion may be possible and could be a novel form of activation for chemical defense.
Introduction
Natural products have historically been a rich source of bioactive compounds and represent a significant portion of marketed drugs, either unchanged or as templates for synthetic modification.1,2 In recent years natural products from marine sources have received increased attention,(3) in part due to the vast biodiversity of the marine environment, a situation that plausibly increases the need for chemical defenses as a result of intense competition and predation. As part of our own search for novel bioactive compounds, we have focused on marine cyanobacteria because of their propensity to produce a variety of structures that are frequently toxic to mammalian cancer cells.(4) Oftentimes, groups of related compounds are produced, sometimes constituting quite large structural families. Such chemical diversity is thought to be an inherent feature of biosynthetic pathways that adapt to changing environments through natural selection(5) and, through differences in activity, exhibit natural structure−activity relationships. An example of this is the lyngbyastatins/micropeptins,6−10 a large group of cyclic depsipeptide enzyme inhibitors that show differing selectivity for elastase, chymotrypsin, and trypsin.
We recently reported the total structure determination of grassypeptolide(11) (1, Figure 1a),(12) a cyclic depsipeptide that contains several interesting features, including a rare β-amino acid (2-methyl-3-aminobutyric acid, Maba), a large number of d-amino acids, and extensive N-methylation. This compound shares some structural features with the cyanobactins, including lissoclinamide 7 (see Figure 1b). Interestingly, although the cyanobactins are now known to be biosynthesized through a novel ribosomal pathway,13−151 is most likely the result of a nonribosomal peptide synthetase (NRPS) system, because of the incorporation of nonproteinogenic units and N-methylation. Although both classes of natural products commonly contain d-amino acids, epimerization is believed to be nonenzymatic in the majority of ribosomal cyanobactins.(16) Like lissoclinamide 7, 1 is cytotoxic to a number of human cancer cell lines. We now report two new analogues of 1, grassypeptolide B (2) and grassypeptolide C (3). While 2 shows cytotoxicity similar to that of 1, compound 3 is 16−23-fold more potent. Additionally, we demonstrate that 1 and 3 can bind to Cu2+ and Zn2+, which may contribute to their activity.
Figure 1.
Structures of grassypeptolide A (1), B (2), and C (3) (a) and of lissoclinamide 7 (b).
Results and Discussion
Isolation and Structure Determination
Samples of Lyngbya confervoides were collected off the Florida Keys as previously described12,17 and in a more recent recollection. Compounds 2 and 3 were obtained as minor components of nonpolar extracts (MeOH−EtOAc, 1:1), following silica chromatography and reversed-phase HPLC.
HRESI/APCIMS and NMR data for 2 suggested a molecular formula of C55H77N9O10S2. Compared to 1, this is a difference of one methylene. The 1H NMR spectrum of 2 in CDCl3 is strikingly similar to 1, except for the presence of a relatively downfield methyl doublet (δH 1.65, see Table 1 and Table S1, Supporting Information), indicative of an alanine. Indeed, examination of the COSY, edited HSQC and HMBC spectra for 2 revealed the presence of the same units found in 1, except that Ala was present in the place of 2-aminobutyric acid (Aba). Overall, both proton and carbon chemical shifts of 1 and 2 were comparable, suggesting that the sequence of units and also the relative configuration of both compounds are the same (see Table 1). The sequence could be confirmed easily by examination of HMBC and ROESY correlations (Table S1, Supporting Information).
Table 1. NMR Spectral Data in CDCl3 for Grassypeptolide A (1) at 500 MHz (1H)/100 MHz (13C) and B (2) at 600 MHz.
| grassypeptolide A (1) |
grassypeptolide B (2) |
||||
|---|---|---|---|---|---|
| C/H no. | δH (J in Hz) | δCa | δH (J in Hz) | δCb | |
| Maba | 1 | 172.5, s | 172.7, s | ||
| 2 | 2.51, qd (6.9, 6.2) | 45.5, d | 2.55, qd (7.0, 6.8) | 45.5, d | |
| 3 | 4.18, dqd (6.8, 6.7, 6.2) | 48.6, d | 4.27, m | 48.3, d | |
| 4 | 1.16, d (6.7) | 19.7,c q | 1.22, d (6.7) | 19.5, q | |
| 5 | 1.10, d (6.9) | 14.6, q | 1.15, d (7.0) | 14.5, q | |
| NH | 7.40, br d (6.8) | 7.31, m | |||
| Thr | 6 | 169.8, s | 169.6, s | ||
| 7 | 4.45, dd (7.8, 6.4) | 59.2, d | 4.49, dd (7.9, 6.7) | 58.8, d | |
| 8 | 4.02, dq (6.4, 6.2) | 68.8, d | 4.03, m | 68.5, d | |
| 9 | 1.23, d (6.2) | 19.7,c q | 1.29, d (6.4) | 19.7, q | |
| OH | 3.96, br | 5.02,d br | |||
| NH | 7.12, d (7.8) | 7.13, d (7.9) | |||
| N-Me-Leu | 10 | 170.3, s | 170.3, s | ||
| 11 | 4.92, br | 56.7, d | 5.02, br | 56.1, d | |
| 12a | 1.85, m | 36.9, t | 1.89, m | 36.4, t | |
| 12b | 1.72, ddd (−14, 8.1, 6.2) | 1.76, ddd (−14.3, 7.9, 6.5) | |||
| 13 | 1.55, m | 25.1, d | 1.59, m | 24.8, d | |
| 14 | 0.95, d (6.6) | 23.2, q | 1.02, d (6.6) | 23.0, q | |
| 15 | 0.90, d (6.5) | 22.1, q | 0.96, d (6.4) | 21.9, q | |
| 16 | 3.15, s | 32.3, q | 3.22, s | 31.6, q | |
| Aba-thn-ca/Ala-thn-ca | 17 | 170.4, s | 170.2, s | ||
| 18 | 5.32, ddd (9.5, 9.1, 1.8) | 77.8, d | 5.347, dd (10.4, 10.4) | 77.6, d | |
| 19a | 3.58, dd (−9.9, 9.1) | 33.4, t | 3.71, dd (−10.5, 10.4) | 33.3, t | |
| 19b | 3.27, dd (−9.9, 9.5) | 3.33, dd (−10.5, 10.4) | |||
| 20 | 178.5, s | 178.9, s | |||
| 21 | 4.64, m | 54.4, d | 4.90, dq (7.5, 7.1) | 48.2, d | |
| 22a | 2.18, m | 25.2, t | 1.65, d (7.1) | 18.0, q | |
| 22b | 1.97, m | ||||
| 23 | 0.96, t (7.2) | 11.0, q | |||
| NH | 7.53, d (7.9) | 7.73, d (7.5) | |||
| N-Me-Phe-thn-ca | 24 | 171.0, s | 170.4, s | ||
| 25 | 5.30, m | 79.3, d | 5.351, m | 78.8, d | |
| 26a | 3.70, m (2H) | 37.7, t | 3.77, m (2H) | 37.6, t | |
| 26b | |||||
| 27 | 177.2, s | 177.5, s | |||
| 28 | 3.83, dd (9.0, 3.5) | 69.0, d | 3.91, dd (10.1, 3.2) | 68.9, d | |
| 29a | 3.57, dd (−13.9, 9.0) | 35.3, t | 3.65, dd (−13.4, 10.1) | 34.9, t | |
| 29b | 3.44, dd (−13.9, 3.5) | 3.48, dd (−13.4, 3.2) | |||
| 30 | 138.2, s | 138.2, s | |||
| 31/35 | 7.35, m | 129.8, d | 7.43, m | 129.8, d | |
| 32/34 | 7.34, m | 128.7, d | 7.36, m | 127.1, d | |
| 33 | 7.25, m | 126.7, d | 7.43, m | 128.6, d | |
| 36 | 2.78, s | 39.6, q | 2.81, s | 39.3, q | |
| Pro | 37 | 173.0, s | 172.9, s | ||
| 38 | 4.77, dd (7.4, 5.5) | 57.0, d | 4.81, dd (8.4, 4.2) | 57.0, d | |
| 39a | 2.04, m (2H) | 27.5, t | 2.11, m | 27.4, t | |
| 39b | 2.06, m | ||||
| 40a | 2.12, m | 24.8, t | 2.18, m | 24.6, t | |
| 40b | 1.86, m | 1.93, m | |||
| 41a | 3.69, m | 47.6, t | 3.76, m | 47.4, t | |
| 41b | 3.60, m | 3.67, m | |||
| N-Me-Val | 42 | 167.8, s | 168.1, s | ||
| 43 | 4.93, d (10.9) | 60.3, d | 4.98, d (10.9) | 60.0, d | |
| 44 | 2.42, dqq (10.9, 6.7, 6.4) | 27.3, d | 2.47, dqq (10.9, 6.6, 6.4) | 27.1, d | |
| 45 | 0.97, d (6.4) | 19.5, q | 1.01, d (6.4) | 19.2, q | |
| 46 | 0.87, d (6.7) | 18.2, q | 0.93, d (6.6) | 17.9, q | |
| 47 | 3.11, s | 30.3, q | 3.20, s | 30.1, q | |
| Pla | 48 | 171.1, s | 171.0, s | ||
| 49 | 5.40, dd (9.9, 3.5) | 72.0, d | 5.40, dd (9.9, 3.0) | 72.0, d | |
| 50a | 3.12, dd (−14.5, 9.9) | 37.2, t | 3.17, dd (−14.3, 9.9) | 37.0, t | |
| 50b | 3.00, dd (−14.5, 3.5) | 3.06, dd (−14.3, 2.7) | |||
| 51 | 135.6, s | 135.7, s | |||
| 52/56 | 7.21, m | 129.2, d | 7.27, m | 129.1, d | |
| 53/55 | 7.30, m | 128.6, d | 7.36, m | 128.5, d | |
| 54 | 7.26, m | 127.3, d | 7.32, m | 126.8, d | |
Multiplicity derived from APT and HMQC spectra.
Multiplicity derived from edited HSQC spectrum.
These carbons have the same chemical shift.
OH signal assigned by default.
The obtained yield of 2 was much less than for 1 (1.7 versus 16.4 mg, respectively, from both collections combined). However, by using H2O to produce a saturated solution, a small amount of crystals could be produced that were adequate for X-ray diffraction studies (see Experimental Section). The resulting structure (Figure 2a) is essentially superimposable to that of 1, despite the fact that the crystals were formed under different conditions (Figure 2b). As expected, the relative configuration of all stereocenters was the same as that for 1. Because the Flack x parameter is near zero [0.16 (10)] and the specific rotations shown by 1 and 2 were close in value (+76 versus +109, respectively), the absolute configuration shown is correct.
Figure 2.

Grassypeptolide crystal structures: (a) displacement ellipsoids (50% probability) for the X-ray crystal structure of grassypeptolide B (2); (b) overlay of X-ray crystal structures of grassypeptolides A (1, magenta) and B (2, cyan).
An additional analogue, compound 3, was obtained. The HRESI/APCIMS spectrum of 3 suggested a molecular formula of C56H79N9O10S2, the same as that for 1. Examination of the 1H NMR, COSY, edited HSQC, HMBC, and ROESY spectra for 3 revealed the presence of the same units present in 1, namely, 2-methyl-3-aminobutyric acid (Maba), Thr, N-Me-Leu, N-Me-Phe-derived thiazoline carboxylic acid (N-Me-Phe-thn-ca), Pro, N-Me-Val, and phenyllactic acid (Pla). The presence of a 2-aminobutyric acid derived thiazoline carboxylic acid unit was proposed by default, as there were no HMBC or ROESY correlations between the thiazoline ring and the aminobutyric acid portion. Importantly, although the proton and carbon chemical shifts were very similar to those of 1 and 2, those at position 28 were distinct (δH/δC was 3.83/69.0 and 5.48/59.2 for 1 and 3, respectively). Second, the methyl signal at position 36 also showed substantially different chemical shifts (δH/δC was 2.78/39.6 for 1 and 3.169/30.7 for 3). This suggested that 3 was the epimer of 1 at C-28. Examination of correlations in the HMBC and ROESY spectra easily allowed the construction of the sequences of Maba−Thr−N-Me-Leu and Aba−N-Me-Phe-thn-ca−Pro−N-Me-Val−Pla. A ROESY correlation was present between H-28 and H-38 (N-Me-Phe and Pro α-protons, respectively), suggesting that the intervening amide bond was cis, in contrast to the corresponding bond in the crystal structures of 1 and 2. To confirm the complete sequence of 3, ESIMS/MS studies were carried out using 1 and 2 for comparison (see Figure 3). Analysis of the fragmentation patterns of 1 and 2, as well as MS/MS/MS of the prominent ions 859.3 and 845.3 (see Figure 3) allowed the assignments shown. We found a dominant continuous series of mainly b ions, resulting from ring-opening at the Pro−N-Me-Val amide bond. This is consistent with previous studies of cyclic peptide fragmentation, where only b ions were observed,(18) and it was noted that in some proline-containing molecules, ring opening predominantly occurs at this residue.(19) None of the other possible b ion series were observed. However, the major series resulted from loss of Pla−N-Me-Val to give a pseudo c ion (859.3/845.3, see Figure 3), followed by loss of Thr−Maba to give a b ion (641.2/627.2), Pro−N-Me-Phe (411.0/397.1), and finally Aba/Ala-thn (241.2/241.1). Compound 3 showed a fragmentation pattern identical to that of both 1 and 2, and therefore it shares the same sequence.
Figure 3.

MS/MS fragmentation data for grassypeptolides A−C (1−3).
Attempts to crystallize 3 failed, perhaps because the isolated yield (0.6 mg) was insufficient. Compound 3 was hydrolyzed with 6 N HCl (110 °C, 18 h), and the hydrolyzate was subjected to chiral HPLC-MS to reveal the presence of d-allo-Thr, N-Me-l-Val, l-Pro, N-Me-d-Leu, and l-Pla. Compound 3 was treated with ozone at −78 °C followed by oxidative workup and acid hydrolysis, and then the product was analyzed by chiral HPLC-MS to detect the presence of d-Aba. Under these conditions two peaks were observed corresponding to N-Me-l-Phe and N-Me-d-Phe in the ratio 1.28:1, respectively. More stringent ozonolysis conditions were then used to treat both 1 and 3 (30 min, rt) in order to convert N-Me-Phe to N-Me-Asp. Analysis of the reaction product of 3 by chiral HPLC-MS again showed the presence of both enantiomers of N-Me-Asp in the ratio of 1.22:1 l to d. Two peaks in the ratio of 1:1.85 corresponding to N-Me-l-Asp and N-Me-d-Asp, respectively, were also apparent in the ozonolysis product of 1 under the same conditions. In an attempt to liberate N-Me-Phe under milder conditions, leading to less epimerization, we treated both 1 and 3 with RuCl3/NaIO4, which in the past has been used both to cleave double bonds and to oxidize thioethers.(20) After hydrolysis of the reaction product with 6 N HCl (110 °C, 16 h), chiral HPLC-MS analysis revealed the presence of N-Me-l-Phe and N-Me-d-Phe in the ratios 1:4.88 for 1 and 3.35:1 for 3, clearly indicating l-configuration in 3 and d-configuration in 1. The difference in configuration at that position is consistent with NMR chemical shift evidence, as pointed out above. Additionally, compounds 1 and 3 have very different specific rotations ([α]20D of +78 and +18°, respectively), supporting a difference in configuration. Consistent with previous experience with 1, both l-cysteic acid and d-cysteic acid (Cya) were detected in the ozonolysis product of 3. Since both enantiomers were observed in a similar ratio when derived from 1 and 3 (∼2:1), and also because the 1H and 13C NMR chemical shifts of the thiazoline moiety signals are both much the same as those of 1, the configuration of both are likely R in 3 as well as 1. Consequently, we conclude that 1 and 3 differ indeed only in the configuration at C-28.
Considering that we isolated two epimers, 1 and 3, from cyanobacterial extracts, a relevant question is whether the intact molecules can interconvert. This has some precedence, for example, a synthetic analogue of trunkamide A containing l-Phe adjacent to the thiazoline in place of d-Phe showed spontaneous epimerization to the natural product.(21) Likewise, the synthetic 31S isomer of lissoclinamide 7 showed conversion to lissoclinamide 7 (see Figure 1b) in the presence of pyridine and CDCl3 at 60 °C, although the reverse conversion was not observed.(22) Using these same conditions we were not able to convert intact 3 to 1 or vice versa, perhaps suggesting that neither are isolation artifacts. A reduced tendency for base-induced interconversion may reflect less overall strain in the macrocycle; compounds 1 and 3 are 31-membered, whereas lissoclinamide 7 and trunkamide A are both 21-membered. The fact that the 28S isomer, compound 3, was isolated in much reduced yield compared with 1 suggests that 3 could be a minor side-product of the biosynthesis of 1 or else is an activated form (vide infra).(23)
Molecular Modeling of Grassypeptolide C (3)
To further explore the potential conformational differences between 1, 2, and 3, we carried out molecular modeling of 3. Apart from the differences in chemical shifts already noted, the NMR spectra of 1 and 3 are similar, which therefore suggested a fairly similar conformation for both compounds. In particular, the two 3JNH-αH values that could be measured in 3 (indicative of Φ angles)(24) had almost the same values in 1. Therefore, the same ROESY distances and angle constraints previously used for modeling of 1(12) were used. Most of the ROESY correlations observed for 1 were also seen for 3 (see Table 2, common correlations shown in bold), but four extra interresidue correlations were added for modeling purposes (see Table 2, shown in red). In addition, the N-Me-Phe−Pro amide bond was set to cis as previously noted. As with modeling for 1, 10 random structures were subjected to distance geometry followed by simulated annealing and then constrained molecular dynamics (see Experimental Section). This resulted in two conformational families; however, one clearly had higher calculated energies and a larger number of constraint violations (3 structures, average 61.2 kcal/mol and 8 violations; see Table S4−S14 and Figure S1, Supporting Information). The other family was all within 1 Å rmsd of the lowest energy conformation and showed closely related backbone conformations (7 structures, average 35.1 kcal/mol and 2.3 constraint violations; see Figure 4a and Table S4). Interestingly, this family contained two orientations of the N-Me-Phe side chain, with H-28 either pointing into the center of the macrocycle or away from it. The interconversion of these two forms would have resulted from a rotation of the C27−C28 bond (see Figure 4b). The backbone conformation for the lower energy conformational family is also like that of the crystal structure of 1 (see Figure 4c).
Table 2. NMR Spectral Data for Grassypeptolide C (3) in CDCl3 at 600 MHz.
 |
Multiplicity derived from edited HSQC spectrum.
Correlations to NH protons in the same unit unless otherwise indicated.
Protons showing long-range correlation with indicated carbon.
Correlations also observed in the ROESY spectrum of grassypeptolide A (1) in CDCl3 are shown in bold, and correlations that were used as extra constraints in molecular modeling are shown in red.
(w) indicates a weak correlation.
This correlation was previously observed in the ROESY spectrum of grassypeptolide A (1) in DMSO-d6 but not the ROESY spectrum in CDCl3.
Figure 4.

Molecular modeling of grassypeptolide C (3): (a) lower-energy conformational family of models of grassypeptolide C (3); (b) select models showing two possible orientations of the N-Me-Phe side chain (lowest-energy examples of each); (c) comparison of lowest energy conformation of 3 (green) and the crystal structure of 1 (purple), showing N-Me-Phe α-protons to illustrate configuration at this center and the amide bond that is trans and cis in 1 and 3, respectively.
Antiproliferative Activity
Previously, we found that 1 was cytotoxic against a number of cancer cell lines with IC50s in the low micromolar range.(12) Compounds 2 and 3 were tested alongside 1 in two cell lines, HT29 (colorectal adenocarcinoma) and HeLa (cervical carcinoma; see Table 3). Surprisingly, 3 was 16−23 times more potent than 1 and 65-fold more potent than 2. Presumably this is due to differences in conformation and suggests that the region around the N-Me-Phe is crucial to cytotoxicity. Interestingly, the synthetic 31S-isomer of lissoclinamide 7 (Figure 1b) was also found to be more potent than the isolated natural compound in some cell lines.(22) To test if the antiproliferative effects were mediated by stage specific cell cycle arrest, we investigated the effect of both 1 and 3 on cell cycle in HT29 cells (see Figure 5) using FACS-based DNA content analysis. Both compounds exhibited G1 arrest at lower concentrations, followed by G2/M arrest at higher concentrations, with 3 showing these effects at lower concentrations than 1 (see Figure 5). An increase in apoptotic sub-G1 or “sub-G2” populations is also seen. It is interesting that 3, the most active of the series, was recovered as the most minor component from the cyanobacterial extract. This could be an indication that 1 requires activation by epimerization, possibly as a strategy for self-resistance by the producing organism.
Table 3. IC50 Values for Cytotoxicity Exhibited by Grassypeptolides A−C (1−3) against Two Cancer Cell Lines.
| cell line | grassypeptolide A (1) | grassypeptolide B (2) | grassypeptolide C (3) |
|---|---|---|---|
| HT29 | 1.22 μM | 4.97 μM | 76.7 nM |
| HeLa | 1.01 μM | 2.93 μM | 44.6 nM |
Figure 5.

Cell cycle analysis of HT29 cells treated with grassypeptolides A (1) and C (3) for 24 h. Taxol served as a positive control for G2 arrest.
Metal Binding
Cyclic peptides that have tandem thiazole/oxazole or thiazoline/oxazoline rings, for example, patellamides A and C and ascidiacyclamide (see Figure 6), have been shown to bind to metals such as Cu2+ and Zn2+.25−27 In the crystal structure of ascidiacyclamide,(27) the molecule is complexed with two Cu2+ ions bridged by a carbonate anion. Each Cu2+ is coordinated to one thiazole, one oxazoline, and the intervening deprotonated amide (see Figure 6), which has been termed a “TAO” domain, and is also thought to bind Cu2+ in the patellamides.28,29 Additionally, a nonspecific cytotoxic metal chelator (tachpyridine) has been shown to cause G2/M phase cell-cycle arrest in HeLa cells.(30) Since compounds 1−3 also promoted G2/M cell cycle arrest and contained a motif comprising two thiazolines and an intervening amide (similar to TAO), we investigated the binding of 1 to Cu2+ and Zn2+.
Figure 6.
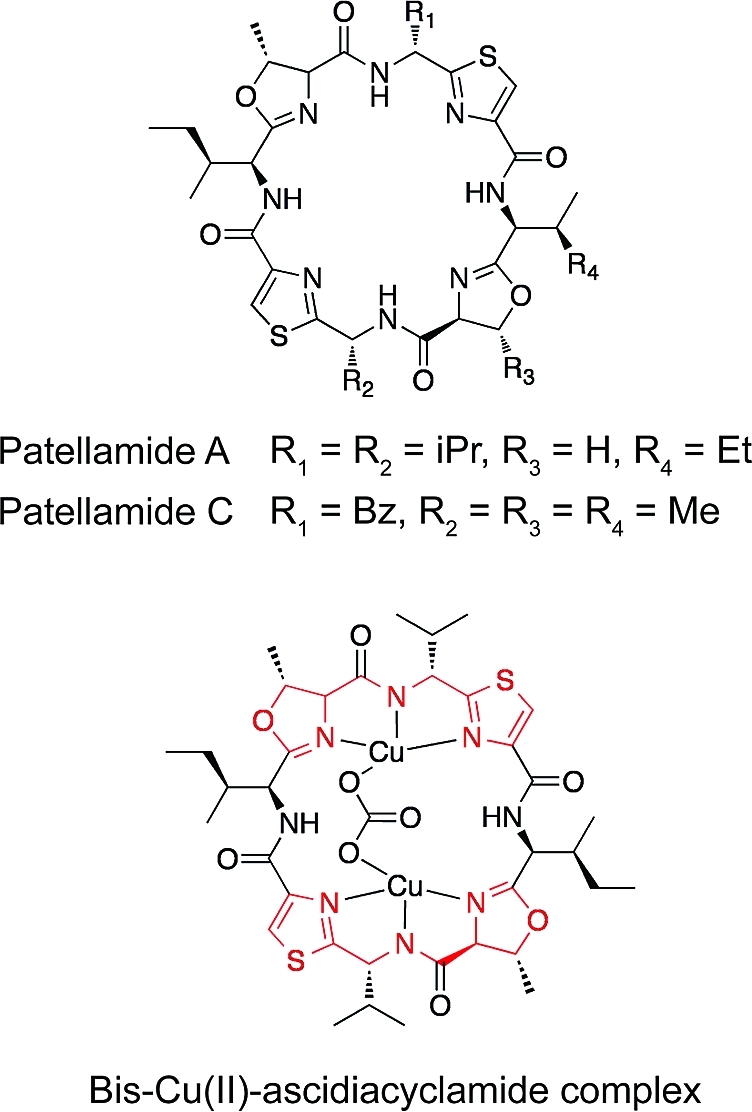
Structures of patellamides A and C and the structure of the bis-Cu(II)-ascidiacyclamide complex, with TAO domains in red.
The circular dichroism spectrum of 1 revealed positive maxima at 218, 230, and 253 nm (see Figure 7). Previously the CD spectra of patellamides A and C, lissoclinamides 9 and 10, and ulithiacyclamide had been investigated.26,31 The near-UV CD maximum at 253 nm could be due to the aromatic N-Me-Phe and Pla residues, while the far-UV maximum at 218 nm is similar to that observed in the random coil conformation of poly(Lys).(32) The remaining maximum at 230 nm could be attributed to the tight turn at N-Me-Phe-thn-ca observed in the solid state and solution structures of 1. The CD spectra of β-turns are fairly variable,(33) but some examples exist with positive maxima near 230 nm. Upon addition of one equivalent of Cu2+, the shape of the CD spectrum of 1 changed (see Figure 7b), indicating that the binding of Cu2+ to 1 occurs, and that it is accompanied by a slight change in conformation. A second equivalent of Cu2+ did not cause a further change, indicating a 1:1 stoichiometry. Addition of 1 equiv of Zn2+ to 1 also brought about a slight change in the CD spectrum (see Figure 7c and d). While Cu2+ caused both positive and negative changes in the CD spectrum of 1 (see Figure 7b), Zn2+ generated only positive changes (see Figure 7d), perhaps indicating different conformations for the resulting complexes. These results are qualitatively reminiscent of the changes in CD spectrum seen when Cu2+ and Zn2+ are added to lissoclinamide 10, a 21-membered cyclic peptide that contains two thiazolines, an oxazoline and a proline.(31) In this compound, Cu2+ causes both positive and negative changes in the CD spectrum with two isobesic points, whereas Zn2+ causes only a negative change. Unfortunately, there were insufficient amounts of 3 to obtain a CD spectrum; however, we were able to obtain evidence of both Cu2+ and Zn2+ adducts by MS for both 1 and 3 (see Figure 8). Both compounds exhibited [M − H + Cu]+ ions with the expected isotope distribution, as well as several Zn2+ adducts ([M + Zn]2+, [M − H + Zn]+, and [M + ZnCl]+).
Figure 7.

Circular dichroism spectra of grassypeptolide A (1) in the presence and absence of Cu2+ and Zn2+: (a) CD spectra of 1 alone and after addition of 1 and 2 equiv of Cu2+ to 1; (b) differences induced in the CD spectrum of 1 by addition of Cu2+; (c) CD spectra of 1 alone and after addition of 1 or 2 equiv of Zn2+; (d) differences induced in the CD spectrum of 1 by addition of Zn2+.
Figure 8.

(a) Cu2+ and Zn2+ adducts observed for 1; (b) Cu2+ and Zn2+ adducts observed for 3; (c) calculated isotope patterns for the observed metal adducts.
Conclusions
We have described two new cytotoxic cyclic depsipeptides, grassypeptolides B (2) and C (3). Compounds 1−3 show a natural structure−activity relationship. While the replacement of d-Aba (1) with d-Ala (2) only slightly reduced potency, inversion of N-Me-d-Phe (1) to N-Me-l-Phe (3) lowered the IC50 by 16−23-fold (see Table 3). Like the thiazole- and oxazoline-containing cyclic peptides patellamides A and C and ulithiacyclamide, compounds 1 and 3 can bind to metals. This activity may be a potential mechanism for cytotoxicity in mammalian cells, as many classes of enzymes are known to rely on metal cofactors. For example, ribonucleotide reductase (RR) requires iron for catalysis of the conversion of ribonucleotides to deoxyribonucleotides, the precursors needed for the synthesis of DNA during S phase of the cell cycle. Iron depletion by exogenous chelators therefore causes cell cycle arrest at the G1/S phase,(34) through inhibition of RR and other mechanisms.(35) However, metal chelators have been shown to induce G2/M cell cycle arrest under some conditions, such as desferrioxamine(36) and tachpyridine.(30) The latter was shown to mediate this effect through activation of CHK1 and CHK2, potentially through ataxia-telangiectasia-mutated and Rad3-related kinase (ATR). Another potential target for metal chelators is Cu/Zn superoxide dismutase (SOD1),37,38 which protects cells from superoxide radicals produced during normal metabolism and especially at times of oxidative stress. Inhibition of SOD1 can lead to buildup of reactive oxygen species (ROS), which can result in activation of both G1 and G2 checkpoint functions through the ataxia telangiectasia gene product (ATM).(39) Our cell cycle data for 1 and 3, where a degree of both G1 and G2/M phase arrest is seen, is consistent with these compounds’ abilities to bind metals. Compound 1 has a low micromolar IC50 in a number of cancer cell lines, which is a potency similar to that of desferrioxamine(40) and tachpyridine.(30) Compound 3 was more potent, and this could be due to an increased affinity for metals. However, it should be noted that the direct involvement of metals in the cytotoxicity of 1−3 was not demonstrated, and metal-independent major pleiotropic mechanisms of action for these compounds may exist. In light of previous examples of intricate protection/activation strategies in natural products,(23) a potential hypothesis is that compound 3 may represent an activated form of 1, and this process may constitute a self-resistance strategy by the producing organism. We were unable to convert 1 to 3 under basic conditions, but this does not exclude conversion by a specific enzyme. Compound 3 could also be a minor byproduct of the biosynthesis of 1, or it could be the result of a modified pathway in a minority of cells in the collected material.
Experimental Section
General Experimental Methods
NMR spectra for 2 and 3 were acquired on an NMR spectrometer using a 1-mm triple-resonance high-temperature superconducting cryogenic probe.(41) Spectra were referenced to the residual solvent signals of CDCl3 [δH/C 7.26/77.0]. Edited HSQC experiments were optimized for 145 Hz, and HMBC experiments were optimized for 7 Hz.
Extraction and Isolation
Samples of L. confervoides were collected off Grassy Key, Florida, on May 26, 2004, and fractionated as previously described.(12) Samples of the same species were collected off Key Largo, Florida, on May 8, 2003, and fractionated as previously described.(17) Silica gel fractions from both extracts eluting with i-PrOH and MeOH were subjected to preparative HPLC [column, Luna 10 μm C18(2) 100A AXI, 100 mm × 21.2 mm, 10.0 mL/min, UV detection at 220 and 254 nm] using a MeOH−H2O linear gradient (60−100% MeOH over 30 min, then 100% MeOH for 5 min). From each fraction, impure 2 eluted at tR = 21.5 min and 1 eluted at tR = 22.1 (14.5 mg). The collected peaks appeared the same by 1H NMR and so were combined before being subjected to another round of HPLC purification [column,YMC-Pack ODS-AQ, 250 mm × 10 mm, 2.0 mL/min; PDA detection, 190−500 nm] using a linear MeOH−H2O gradient (60−97.5% MeOH over 28 min, then 97.5% MeOH for 20 min) to give 2 (1.7 mg) at tR = 32.7 min.
The 20:80 i-PrOH/CH2Cl2 fraction from the silica column of the extract of the Grassy Key material was subjected to reversed-phase HPLC [column, YMC-Pack ODS-AQ, 250 mm × 10 mm, 2.0 mL/min; PDA detection, 190−500 nm], using a linear MeOH−H2O gradient (60−100% MeOH over 30 min, then 100% MeOH for 20 min), to furnish 3 (0.6 mg) and 1 (1.9 mg) at tR 31.4 and 33.3 min, respectively.
Reisolation of Grassypeptolides
Samples of L. confervoides collected off Big Pine Shoals, Florida, on May 30, 2009, were extracted as previously described.(12) The nonpolar extract was subjected to solvent partitioning to give hexanes-, butanol-, and water-soluble fractions. The butanol-soluble portion was directly fractionated over silica gel eluting with increasing amounts of i-PrOH in CH2Cl2. Fractions eluted with 20% and 50% i-PrOH/CH2Cl2 (15.8 mg and 32.2 mg, respectively) consisted of mixtures of grassypeptolides A (1), B (2), and C (3). Each fraction was subjected to repetitive semipreparative HPLC [column, Synergi Hydro-RP 4 μm 80A, 250 mm × 10.0 mm or Luna Phenyl-Hexyl 5 μm 100A, 250 mm × 10.0 mm, 2.0 mL/min, UV detection at 220 and 240 nm] using a MeOH−H2O linear gradient (60−100% MeOH over 30 min, then 100% MeOH for 20 min or 80−100% MeOH over 20 min, then 100% MeOH for 10 min) to give grassypeptolides A (1, 3.8 mg), B (2, 0.4 mg), and C (3, 0.6 mg), confirmed by 1H NMR and MS analysis.
Grassypeptolide B (2)
Colorless amorphous solid; [α]20D +109; HRESI/APCIMS m/z [M + Na]+ 1110.5115 (calcd for C55H77N9O10S2Na, 1110.5133), [M + H]+ 1088.5298 (calcd for C55H78N9O10S2, 1088.5313); UV (CH2Cl2) λmax (log ε) 224 (3.20), 250 (sh, 2.80), 280 (sh, 2.58); IR (film) νmax 3306 (br), 3027 (w), 2921, 2851, 2361 (w), 1731, 1641, 1531, 1453, 1266, 1225, 1160, 1122, 1085, 1026, 967, 913 cm−1; 1H NMR, COSY, edited HSQC, HMBC and ROESY, see Table 1 and Table S1, Supporting Information.
Grassypeptolide C (3)
Colorless amorphous solid; [α]20D +18; HRESI/APCIMS m/z [M + Na]+ 1124.5299 (calcd for C56H79N9O10S2Na, 1124.5289), [M + H]+ 1102.5428 (calcd for C56H80N9O10S2, 1102.5470); UV (CH2Cl2) λmax (log ε) 224 (3.82), 260 (3.41); IR (film) νmax 3419 (br), 3056, 2961, 2920, 2851, 2306, 2125, 1750, 1640, 1515, 1456, 1265, 1030, 895 cm−1; 1H NMR, COSY, edited HSQC, HMBC and ROESY, see Table 2 and Supporting Information.
X-ray Crystallography of Grassypeptolide B (2)
The entire isolated yield of 2 was dissolved in a small amount of filtered CH2Cl2, and then MeOH was added, followed by H2O until the saturation point was reached. The solution was left to slowly evaporate at rt for 10 days to yield needle-shaped crystals. Data were collected at 173 K on an X-ray diffractometer equipped with A CCD area detector and a graphite monochromator utilizing Mo Kα radiation (λ = 0.71073 Å). Cell parameters were refined using up to 8192 reflections. A full sphere of data (1850 frames) was collected using the ω-scan method (0.3° frame width). The first 50 frames were remeasured at the end of data collection to monitor instrument and crystal stability (maximum correction on I was <1%). Absorption corrections by integration were applied on the basis of measured indexed crystal faces.
The structure was solved by the Direct Methods in SHELXTL6 and refined using full-matrix least-squares. The non-H atoms were treated anisotropically, whereas the hydrogen atoms were calculated in ideal positions and were riding on their respective carbon atoms. In addition to the molecule, there is a methanol and water molecule in the asymmetric unit. They are both disordered and were refined in two parts with their site occupation factors dependently refined. A total of 730 parameters were refined in the final cycle of refinement using 3804 reflections with I > 2σ(I) to yield R1 and wR2 of 5.71% and 8.84%, respectively. Refinement was performed using F2.
Ozonolysis and Acid Hydrolysis of Grassypeptolide C (3)
A portion (50 μg) of 3 was subjected to acid hydrolysis (6 N HCl, 110 °C, 18 h) and then evaporated to dryness. The sample was reconstituted in 100 μL of H2O and subjected to chiral HPLC analysis [column, Chirobiotic TAG (4.6 mm × 250 mm), Supelco; solvent, MeOH−10 mM NH4OAc (40:60 pH 5.39); flow rate, 0.5 mL/min; detection by ESIMS in positive ion mode (MRM scan)]. d-allo-Thr, N-Me-l-Val, l-Pro, and N-Me-d-Leu eluted at tR = 12.6, 13.1, 14.8, and 112 min, respectively. The retention times (tR, min; MRM ion pair, parent → product) of the authentic amino acids were as follows: l-Thr (8.0; 120 → 74), l-allo-Thr (8.6), d-Thr (9.5), d-allo-Thr (12.6), N-Me-l-Val (13.1; 132 → 86), N-Me-d-Val (40.4), l-Pro (14.8; 116 → 70), d-Pro (41.9), N-Me-l-Leu (16.2, 146 → 100), N-Me-d-Leu (112). Compound-dependent MS parameters were as follows: Thr, DP 28, EP 9, CEP 5, CE 14, CXP 3.5; N-Me-Val, DP 37, EP 6, CEP 12, CE 16, CXP 3; Pro, DP 36, EP 10, CEP 8, CE 22, CXP 2.3; N-Me-Leu, DP 35, EP 6, CEP 10, CE 17, CXP 3. Source-dependent MS parameters were as follows: CUR 50, CAD medium, IS 5500, TEM 750, GS1 70, GS2 70. l-Pla was detected with different conditions [column, Chirobiotic TAG (4.6 mm × 250 mm), Supelco; solvent, MeOH−H2O (both with 10 mM NH4OAc, pH 5.43); flow rate, 0.5 mL/min; detection by ESIMS in negative ion mode (MRM scan)], at tR = 10.6. The retention times (tR, min; MRM ion pair, parent → product) of the authentic amino acids were as follows: l-Pla (10.6, 165 → 147), d-Pla (11.5). The assignment of l-Pla was confirmed by coinjection with both authentic standards. Compound-dependent MS parameters were as follows: DP −37, EP −3, CEP −9, CE −17, CXP −2.8. Source-dependent MS parameters were as follows: CUR 50, CAD high, IS −4500, TEM 750, GS1 70, GS2 70.
Another portion of 3 (50 μg) in 3 mL of CH2Cl2 was cooled to −78 °C. Ozone was bubbled through the solution for 30 min, which was then allowed to reach room temperature and evaporated to dryness. The residue was treated with H2O2/HCOOH (15% w/v and 50% v/v in H2O, respetively, 70 °C, 20 min), evaporated to dryness, and reconstituted in 100 μL of H2O. Analysis by chiral HPLC [column, Chirobiotic TAG (4.6 × 250 mm), Supelco; solvent, MeOH−10 mM NH4OAc (40:60, pH 5.39); flow rate, 0.5 mL/min; detection by ESIMS in positive ion mode (MRM scan)], revealed the presence of d-Aba at tR = 15.4, but both N-Me-l-Phe and N-Me-d-Phe were detected at tR = 25.3 and 47.3 min, respectively, in the ratio 1.28:1. The retention times (tR, min; MRM ion pair, parent → product) of the authentic amino acids were as follows: l-Aba (9.1, 104 → 58), d-Aba (15.4), N-Me-l-Phe (25.3, 180 → 134), N-Me-d-Phe (47.3). Compound-dependent MS parameters were as follows: Aba, DP 41, EP 2, CEP 8, CE 14, CXP 2.3; N-Me-Phe, DP 30, EP 10, CEP 13, CE 20, CXP 3. Source-dependent MS parameters were as follows: CUR 50, CAD medium, IS 5500, TEM 750, GS1 70, GS2 70.
Samples of both 1 and 3 (50 μg each) were subjected to ozonolysis at room temperature for 30 min followed by oxidative workup and then subjected to chiral HPLC analysis [column, Chirobiotic TAG (4.6 mm × 250 mm), Supelco; solvent, MeOH−10 mM NH4OAc (30:70, pH 5.14); flow rate, 0.5 mL/min; detection by ESIMS in negative ion mode (MRM scan)]. For the ozonolysis product of 3, both N-Me-l-Asp and N-Me-d-Asp were detected at tR = 6.6 and 10.6 min, respectively, in the ratio of 1.22:1. For the ozonolysis product of 1, the ratio was 1:1.85. The retention times (tR, min; MRM ion pair, parent → product) of the authentic amino acids were as follows: N-Me-l-Asp (6.6, 146 → 102), N-Me-d-Asp (10.6). Compound-dependent MS parameters were as follows: DP −33, EP −4.5, CEP −14, CE −18, CXP −5. Source-dependent MS conditions were as follows: CUR 50, CAD high, IS −4500, TEM 750, GS1 70, GS2 70. The ozonolysis product of 3 was also examined under different chiral HPLC conditions [column, Chirex phase 3126 (D) (4.6 mm × 250 mm), Phenomenex; solvent, 2 mM CuSO4−MeCN (97.5:2.5); flow rate, 0.8 mL/min; UV detection at 254 nm], to confirm the presence of d-allo-Thr, N-Me-l-Val, l-Pro and d-Aba at tR = 14.5, 16.3, 17.4, and 21.0, respectively. In addition, both l- and d-Cya were detected at tR = 19.8 and 23.7, respectively, consistent with previous results for 1 (ratio ∼2:1).(12) The retention times (tR, min) of the authentic standards were as follows: l-Thr (10.3), d-Thr (11.6), l-allo-Thr (13.9), d-allo-Thr (14.5), l-Aba (15.0), d-Aba (21.0), N-Me-l-Val (16.3), N-Me-d-Val (22.2), l-Pro (17.4), d-Pro (33.6), l-Cya (19.8), d-Cya (23.7).
Thiazoline Cleavage and Oxidation
Both 1 and 3 were treated with RuCl3/NaIO4 using the procedure of Jayaramann et al.,(42) with minor differences. Portions of both compounds (100 μg of 1 and 200 μg of 3) were dissolved in CH3CN/CCl4/H2O (2:2:3, 150 μL total). To this was added powdered NaIO4 (3 equiv) followed by RuCl3 (0.7 mg), and the reactions were stirred at rt for 1 h. The reaction mixtures were then diluted with water and extracted with CH2Cl2. The combined CH2Cl2 fractions were dried under N2 and then treated with 6 N HCl at 110 °C for 16 h and evaporated to dryness. The resulting hydrolysates were subjected to chiral HPLC analysis [column, Chirobiotic TAG (4.6 mm × 250 mm), Supelco; solvent, MeOH−10 mM NH4OAc (40:60, pH 5.39); flow rate, 0.5 mL/min; detection by ESIMS in positive ion mode (MRM scan)]. For 1, both N-Me-l-Phe and N-Me-d-Phe were detected at tR = 25.3 and 47.3 min, respectively, in the ratio 1:4.88. For 3, the ratio was reversed, with N-Me-l-Phe and N-Me-d-Phe detected in the ratio 3.35:1. The retention times (tR, min; MRM ion pair, parent → product) of the authentic amino acids were as follows: N-Me-l-Phe (25.3, 180 → 134), N-Me-d-Phe (47.3). Compound-dependent MS parameters were as follows: DP 30, EP 10, CEP 13, CE 20, CXP 3. Source-dependent MS parameters were as follows: CUR 50, CAD medium, IS 5500, TEM 750, GS1 70, GS2 70.
Advanced Marfey’s Analysis
The N-benzoyl O-methyl esters of (2R,3R)- and (2R,3S)-2-methyl-3-aminobutyric acid (Maba) were treated with 6 N HCl at 110 °C for 22 h. The products of each reaction were dried down and made up to 50 mM solutions in water. Then, 10 μL of 1 M NaHCO3 and 50 μL of 1-fluoro-2,4-dinitrophenyl-5-l-leucinamide (l-FDLA, 1% w/v in acetone) were added to 25 μL of the Maba stock solutions. The mixtures were heated to 35 °C for 1 h with frequent mixing, then neutralized with 5 μL of 2 N HCl, concentrated to dryness, and then reconstituted with 250 μL of MeCN−H2O (1:1). The FDLA derivative of the ozonolysis product of 3 was prepared in a similar way using ∼45 μg of starting material. Derivatives were analyzed by reversed phase HPLC [column, Kinetex 2.6 μm C18 100A (4.6 mm × 100 mm), Phenomenex; flow rate, 0.5 mL/min; ESIMS detection in negative ion mode], using a MeOH−H2O (both with 0.1% HCOOH) linear gradient (40−100% MeOH over 50 min). Under these conditions two peaks were detected in the l-FDLA derivative of the ozonolysate of 3, corresponding to (2R,3R)- and (2S,3R)-Maba-l-FDLA, respectively, in the ratio 1.51:1. This is consistent with the extent of epimerization at the 2-position observed after treatment of the standards with acid,(12) allowing the assignment of (2R,3R)-Maba. The retention times (tR, min) of the authentic standards were as follows: (2R,3S)-Maba-l-FDLA (25.6), (2R,3R)-Maba-d-FDLA(43) (25.6), (2R,3R)-Maba-l-FDLA (27.1), (2R,3S)-Maba-d-FDLA(44) (27.5).
Attempted Interconversion of Grassypeptolides A (1) and C (3)
Compounds 1 and 3 were dissolved in CDCl3 (150 μL) and transferred to a 2.5 mm NMR tube. Pyridine-d5 (0.5 μL) was added, and the tube was heated at 60 °C for 72 h. Conversion was assessed by 1H NMR.
Molecular Modeling of Grassypeptolide C (3)
Because of the general similarity of proton and carbon chemical shifts of 1 and 3, the same distance and angle constraints were used as in the previously reported modeling of 1,(12) except that the amide bond between N-Me-Phe and Pro was set to cis due to a ROESY correlation between H-28 and H-38 in 3. Also, four additional distance constraints were added on the basis of ROESY correlations unique to 3. The modeling procedure used was identical to that previously used for 1,(12) with 10 random structures of 3 being subjected to (1) minimization, (2) distance geometry, (3) minimization, and (4) molecular dynamics in Sybyl 7.3. Constraints were applied at every step, and the distance geometry procedure consisted of bounds generation, bounds smoothening, and embedding of coordinates, followed by an optimization procedure where the structures were minimized and subjected to simulated annealing (SA, 2000 to 200 K over 100,000 fs, step time 0.3 fs) and then another round of minimization. Dynamics were run at 500 K for 1 ns, with a step size of 1 fs. Following simulation, structures were overlaid in PyMol by pair-fitting backbone carbons only.
Cell Viability Assays
HT29 and HeLa cells were cultured in Dulbecco’s modified Eagle medium (DMEM; Invitrogen) containing 10% fetal bovine serum (Hyclone) in a humidified atmosphere containing 5% CO2 at 37 °C. Cells were seeded into 96-well plates at a density of 10 000 cells/well (HT29) or 3 000 cells/well (HeLa) in 100 μL of medium. After 24 h, compounds 1−3 were added to wells at varying concentrations (as 1 μL stock solutions in DMSO). Taxol was used as a positive control for cytotoxicity, and DMSO alone was used as a negative control. After 48 h of treatment the plates were developed with MTT dye according to the manufacturer’s protocol (Promega). Taxol exhibited IC50s of 2.2 and 1.7 nM for HT29 and HeLa cells, respectively.
Cell Cycle Analysis
HT29 cells were seeded in 6 cm dishes (650 000 cells/dish in 3 mL of DMEM). After 24 h of incubation, various concentrations of 1 and 3 were added to the dishes, in 10 μL of DMSO. After a further 24 h, cells were washed with PBS and then detached with 600 μL of trypsin (Invitrogen). DMEM (2 mL) was added to each dish, and the cell suspensions were centrifuged at 650g for 10 min. The supernatant was discarded, and the cells were resuspended in 500 μL of PBS and centrifuged again at 650g for 10 min. The supernatant was discarded, and the cells were resuspended in 300 μL of PBS before 700 μL of ice-cold EtOH was added. Cells were incubated at −20 °C for 30 min, and then centrifuged at 890g for 10 min. The EtOH/PBS was removed, and then cells were resuspended in 500 μL of PBS containing 1 mM EDTA and 1 mg/mL RNase A (Sigma). The cells were incubated at 37 °C for 30 min, with shaking, and then 5 μL of propidium iodide (1 mg/mL, Invitrogen) was added to each tube. Fluorescence from propidium iodide−DNA complexes was quantified using FACScan (Becton Dickinson).
CD Spectra
Aliquots of 1 (200 μg) were added to various amounts of CuCl2 in CH2Cl2−MeOH (1:100, total volume 400 μL). Data were collected at 25 °C in the range 200−350 nm.
Detection of Metal Complexes by MS
Solutions of 1 (10 μg/mL) were made containing various amounts of Cu2+ or Zn2+ in CH2Cl2−MeOH (1:100) and directly infused the ESIMS at a rate of 10 μL/min. MS parameters were as follows: DP 113.5, EP 4.5, CEP 45, CUR 10, IS 5500, TEM 400, GS1 20, GS2 10.
Acknowledgments
This research was supported by the National Institutes of Health, NIGMS Grant P41GM806210. The 600 MHz 1 mm triple-resonance HTS cryogenic probe used for NMR of compounds 2 and 3 in CDCl3 was developed through collaboration between U.F., the National High Magnetic Field Laboratory, and Bruker Biospin.(41) K.A.A. wishes to acknowledge the National Science Foundation and the University of Florida for funding of the purchase of the X-ray equipment. This is contribution number 833 from the Smithsonian Marine Station. We thank Eusebio Juaristi for kindly providing standards for the Maba unit.
Supporting Information Available
CIF structure file for compound 2, Tables S1−S3, energies and constraint violations of compound 3 models (Table S4), Figure S1, atomic coordinates of compound 3 models (Tables S5−S14), and NMR spectra for compounds 2 and 3. This material is available free of charge via the Internet at http://pubs.acs.org.
Note Added in Proof
While this manuscript was under review, the first total synthesis of grassypeptolide A (1) was published, see: [Liu, H.; Liu, Y.; Xing, X.; Xu, Z.; Ye, T. Chem. Comm. 2010, 46, 7486−7488], confirming the structure and bioactivity.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Cragg G. M.; Grothaus P. G.; Newman D. J. Chem. Rev. 2009, 109, 3012–3043. [DOI] [PubMed] [Google Scholar]
- Newman D. J.; Cragg G. M. J. Nat. Prod. 2007, 70, 461–477. [DOI] [PubMed] [Google Scholar]
- Molinski T. F.; Dalisay D. S.; Lievens S. L.; Saludes J. P. Nat. Rev. Drug Discovery 2009, 8, 69–85. [DOI] [PubMed] [Google Scholar]
- Tan L. T. Phytochemistry 2007, 68, 954–979. [DOI] [PubMed] [Google Scholar]
- Jenke-Kodama H.; Müller R.; Dittmann E. Prog. Drug Res. 2008, 65, 119–140. [DOI] [PubMed] [Google Scholar]
- Matthew S.; Ross C.; Rocca J. R.; Paul V. J.; Luesch H. J. Nat. Prod. 2007, 70, 124–127. [DOI] [PubMed] [Google Scholar]
- Taori K.; Matthew S.; Rocca J. R.; Paul V. J.; Luesch H. J. Nat. Prod. 2007, 70, 1593–1600. [DOI] [PubMed] [Google Scholar]
- Kwan J. C.; Taori K.; Paul V. J.; Luesch H. Mar. Drugs 2009, 7, 528–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ploutno A.; Shoshan M.; Carmeli S. J. Nat. Prod. 2002, 65, 973–978. [DOI] [PubMed] [Google Scholar]
- Reshef V.; Carmeli S. Tetrahedron 2001, 57, 2885–2894. [Google Scholar]
- For clarity, hereafter grassypeptolide (1) will be referred to as grassypeptolide A.
- Kwan J. C.; Rocca J. R.; Abboud K. A.; Paul V. J.; Luesch H. Org. Lett. 2008, 10, 789–792. [DOI] [PubMed] [Google Scholar]
- Schmidt E. W.; Nelson J. T.; Rasko D. A.; Sudek S.; Eisen J. A.; Haygood M. G.; Ravel J. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 7315–7320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long P. F.; Dunlap W. C.; Battershill C. N.; Jaspars M. ChemBioChem 2005, 6, 1760–1765. [DOI] [PubMed] [Google Scholar]
- Donia M. S.; Ravel J.; Schmidt E. W. Nat. Chem. Biol. 2008, 4, 341–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh J. A.; Donia M. S.; Schmidt E. W. Nat. Prod. Rep. 2009, 26, 537–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan J. C.; Eksioglu E. A.; Liu C.; Paul V. J.; Luesch H. J. Med. Chem. 2009, 52, 5732–5747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W.-T.; Ng J.; Meluzzi D.; Bandeira N.; Gutierrez M.; Simmons T. L.; Schultz A. W.; Linington R. G.; Moore B. S.; Gerwick W. H.; Pevzner P. A.; Dorrestein P. C. Anal. Chem. 2009, 81, 4200–4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngoka L. C. M.; Gross M. L. J. Am. Soc. Mass Spectrom. 1999, 10, 732–746. [DOI] [PubMed] [Google Scholar]
- Plietker B. Synthesis 2005, 2453–2472. [Google Scholar]
- Salvatella X.; Caba J. M.; Albericio F.; Giralt E. J. Org. Chem. 2003, 68, 211–215. [DOI] [PubMed] [Google Scholar]
- Wipf P.; Fritch P. C.; Geib S. J.; Sefler A. M. J. Am. Chem. Soc. 1998, 120, 4105–4112. [Google Scholar]
- Kwan J. C.; Luesch H.. Chem. Eur. J. 2010, published online ahead of print; DOI: 10.1002/chem.201001562. [Google Scholar]
- Vögeli B.; Ying J.; Grishaev A.; Bax A. J. Am. Chem. Soc. 2007, 129, 9377–9385. [DOI] [PubMed] [Google Scholar]
- Bertram A.; Pattenden G. Nat. Prod. Rep. 2007, 24, 18–30. [DOI] [PubMed] [Google Scholar]
- Morris L. A.; Jaspars M.; Kettenes-van den Bosch J. J.; Versluis K.; Heck A. J. R.; Kelly S. M.; Price N. C. Tetrahedron 2001, 57, 3185–3197. [Google Scholar]
- van den Brenk A. L.; Byriel K. A.; Fairlie D. P.; Gahan L. R.; Hanson G. R.; Hawkins C. J.; Jones A.; Kennard C. H. L.; Moubaraki B.; Murray K. S. Inorg. Chem. 1994, 33, 3549–3557. [Google Scholar]
- Morris L. A.; Milne B. F.; Thompson G. S.; Jaspars M. J. Chem. Soc., Perkin Trans. 2 2002, 1072–1075. [Google Scholar]
- Latifi R.; Bagherzadeh M.; Milne B. F.; Jaspars M.; de Visser S. P. J. Inorg. Biochem. 2008, 102, 2171–2178. [DOI] [PubMed] [Google Scholar]
- Turner J.; Koumenis C.; Kute T. E.; Planalp R. P.; Brechbiel M. W.; Beardsley D.; Cody B.; Brown K. D.; Torti F. M.; Torti S. V. Blood 2005, 106, 3191–3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris L. A.; Milne B. F.; Jaspars M.; Kettenes-van den Bosch J. J.; Versluis K.; Heck A. J. R.; Kelly S. M.; Price N. C. Tetrahedron 2001, 57, 3199–3207. [Google Scholar]
- Sreerama N.; Woody R. W. In Circular Dichroism: Principles and Applications, 2nd ed.; Berova N., Nakanishi K., Woody R. W., Eds.; Wiley-VCH: New York, 2000; pp 601−620. [Google Scholar]
- Perczel A.; Hollósi M. In Circular Dichroism and the Conformational Analysis of Biomolecules; Fasman G. D., Ed.; Plenum Press: New York, 1996; pp 285−380. [Google Scholar]
- Le N. T. V.; Richardson D. R. Biochim. Biophys. Acta 2002, 1603, 31–46. [DOI] [PubMed] [Google Scholar]
- Yu Y.; Kovacevic Z.; Richardson D. R. Cell Cycle 2007, 6, 1982–1994. [DOI] [PubMed] [Google Scholar]
- Renton F. J.; Jeitner T. M. Biochem. Pharmacol. 1996, 51, 1553–1561. [DOI] [PubMed] [Google Scholar]
- Huang P.; Feng L.; Oldham E. A.; Keating M. J.; Plunkett W. Nature 2000, 407, 390–395. [DOI] [PubMed] [Google Scholar]
- Juarez J. C.; Betancourt O.; Pirie-Shepherd S. R.; Guan X.; Price M. L.; Shaw D. E.; Mazar A. P.; Doñate D. Clin. Cancer Res. 2006, 12, 4974–4982. [DOI] [PubMed] [Google Scholar]
- Shackelford R. E.; Innes C. L.; Sieber S. O.; Heinloth A. N.; Leadon S. A.; Paules R. S. J. Biol. Chem. 2001, 276, 21951–21959. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Lucena D.; Gaboriau F.; Rivault F.; Schalk I. J.; Lescoat G.; Mislin G. L. A. Bioorg. Med. Chem. 2010, 18, 689–695. [DOI] [PubMed] [Google Scholar]
- Brey W. W.; Edison A. S.; Nast R. E.; Rocca J. R.; Saha S.; Withers R. S. J. Magn. Reson. 2006, 179, 290–293. [DOI] [PubMed] [Google Scholar]
- Jayaraman M.; Srirajan V.; Deshmukh A. R. A. S.; Bhawal B. M. Tetrahedron 1996, 52, 3741–3756. [Google Scholar]
- Corresponding in tR to its enantiomer (2S,3S)-Maba-l-FDLA.
- Corresponding in tR to its enantiomer (2S,3R)-Maba-l-FDLA.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.