

Abstract
Gram negative pathogens are protected against toxic electrophilic compounds by glutathione-gated potassium efflux systems (Kef) that modulate cytoplasmic pH. We have elucidated the mechanism of gating through structural and functional analysis of Escherichia coli KefC. The revealed mechanism can explain how subtle chemical differences in glutathione derivatives can produce opposite effects on channel function. Kef channels are regulated by potassium transport and NAD-binding (KTN) domains that sense both reduced glutathione, which inhibits Kef activity, and glutathione adducts that form during electrophile detoxification and activate Kef. We find that reduced glutathione stabilizes an interdomain association between two KTN folds, whereas large adducts sterically disrupt this interaction. F441 is identified as the pivotal residue discriminating between reduced glutathione and its conjugates. We demonstrate a major structural change on the binding of an activating ligand to a KTN-domain protein. Analysis of the regulatory interactions suggests strategies to disrupt pathogen potassium and pH homeostasis.
Keywords: channel regulation, KTN structure
Cellular homeostasis is central to life and in bacterial pathogens is attained by controlling the permeability of the cytoplasmic membrane to ions and solutes. Potassium ion (K+) transport is a critical determinant of growth and survival through its role in regulating cytoplasmic pH and cell turgor (1, 2). As a result, K+ pools are tightly regulated through controlled uptake and efflux. A common fold, the potassium transport and NAD-binding (KTN) domain, links uptake systems (Trk, Ktr), channels (Kch), and efflux systems (Kef) and is thus central to regulation of intracellular K+ (3–5). The best studied KTN-regulated system is KefC from Escherichia coli, a transporter with homologues in the majority of Gram negative pathogens. Kef systems aid E. coli survival during exposure to toxic electrophiles, including metabolic methylglyoxal (6–11). Cytoplasmic glutathione acts as a scavenger of electrophiles. Adducts thus formed are degraded during subsequent detoxification reactions. Activation of Kef systems by glutathione-S-conjugates integrates K+ efflux with detoxification, and the consequent cytoplasmic acidification protects against electrophile-mediated assault (11, 12). Large hydrophobic adducts fully activate all Kef transporters, whereas smaller adducts activate only a subset (7). In contrast, glutathione (GSH) and structurally related γ-linked peptides, such as ophthalmic acid, maintain Kef systems inactive (6, 7). Kef activity is therefore a reflection of electrophilic stress. For E. coli KefC, strongest activation is observed with hydrophobic conjugates such as N-ethylsuccinimido-S-glutathione (ESG) (7, 10). Previous mutational analysis suggested that GSH and its adducts bind directly to the carboxy-terminal KTN domain of KefC (13). Thus this domain appears critical to sensing cytoplasmic cues and gating the associated transmembrane pore-forming domain.
KTN domains are small (∼140 amino acids) and invariantly positioned near the base of pore-forming helices, appropriate for their central role in regulation of K+ flux across cellular membranes. The importance of this domain in channel and transporter regulation has led to the determination of several KTN structures (13–22). However, despite numerous studies, the conformational changes associated with ligand-mediated control of their associated channel or transporter has remained elusive (14, 15, 18). KefC consists of a membrane domain attached to a carboxy-terminal KTN domain via a flexible linker. Maximum activity of KefC is attained by formation of a complex with a small flavoprotein, KefF, which binds directly to the KTN domain (13, 23). KefC has proved to be amenable to genetic, physiological, and structural analysis making it one of the most completely described K+ efflux systems (7–11).
Here we report the structures of the KTN-bearing KefC C-terminal domain (KefC-CTD) in complex with KefF and either the inhibitory ligand GSH, or the activating glutathione adduct ESG. The structures reveal a distinct ligand-mediated conformational change within the KTN domain itself, resulting from disruption of a protein–protein association between KTN folds within a dimeric complex formed by two discreet KefC chains. Functional analysis of structure-led mutants and EPR measurements support the crystallographic data. The resulting model explains how KefC can respond to a variety of chemically diverse glutathione adducts and suggests a mechanism by which these ligands initiate K+ flux through the associated transmembrane domain.
Results
KefC Binds Glutathione in a Cleft Between Dimeric KTN Domains.
Our previous genetic analysis of the KefC system predicted the presence of a single binding site for GSH, or its adducts, at the interface between dimeric KTN domains. To test this hypothesis and to understand how GSH and its adducts can elicit opposite responses, we sought to cocrystallize KefC-CTD with these ligands. We obtained crystals bound with chemically synthesized ESG. As we could not obtain crystals adequate for structural analysis in the presence of GSH, we exploited the ability of reducing agents to convert ESG back to GSH in crystal to obtain the structure of the inhibited conformation (see Materials and Methods). The resulting structures are globally similar to the ligand-free structure we reported previously (Fig. S1) (13). Detailed analysis of the nucleotide bound structures revealed the presence of adenosine monophosphate (AMP) in the GSH structure, replaced by a sulphate ion in the ESG structure. Previously we modeled NAD+ into the apostructure because this was the nucleotide included in the crystallization mix. At that time it was assumed that the nicotinamide ring was too mobile to form discrete density. However, appraisal of the structures indicates that NAD+ is unlikely to bind to these forms of the protein because a second phosphate group could not be built into the model without introducing substantial steric clashes and energetically unstable interactions. Positioning of the γ-glutamate carboxyl group of GSH and D472, which is at the equivalent position to a more accommodating glycine residue in other KTN structures with bound NAD(H) (15, 18), severely restricts association of nucleotides larger than AMP in the observed conformation of KefC (Fig. S2). Thus it appears likely that AMP, arising either from the breakdown of NAD+ or as a contaminant of the NAD+ stock, out-competes the supplemented NAD+ for the nucleotide binding pocket.
The two chains in the KefC-CTD dimer display conformational asymmetry owing to crystal contacts near a mobile region of one of the chains (chain B in the model). These contacts lead to disordering of α-helix 8 of one subunit. Consequently, the proximate GSH ligand binding site is invariably unoccupied. As a result, both finished models include only one molecule per dimer of either ESG or GSH. Analysis of the electron-density maps unequivocally show the position and orientation of the critical small molecule ligands (Fig. 1A; mean refined B factor for GSH and ESG backbone atoms are 45 Å2 and 39 Å2, respectively). GSH is bound within a crevice formed at the junction of the two KTN folds within a canonical dimer and as predicted from our earlier genetic and structural analysis. Residues from both KTN subunits contribute to the coordination of the molecule, including R416, R516, and N551, identified in functional assays as important for ligand sensitivity (9, 13). From the structures presented here, it is clear that these three residues coordinate the carboxylate of glycine in GSH (Fig. 1B). Interestingly, N551 contributes indirectly through a well-structured water molecule. The new structures also predicted other residues to be critical for ligand affinity, including Q412, R498, D499, and the backbone carbonyl of V500.
Fig. 1.

Glutathione binding to KefC. (A) GSH binds in a pocket formed at the interface of two KTN domains within a canonical dimeric assembly (green and white). Residues that coordinate glutathione include Q412 and R416 from one subunit, and R498-D499, the backbone amide of V500, as well as R516 and N551 indirectly through a water molecule, from the partnering fold. Electron density from a 2Fo-Fc map contoured at 1.5σ is shown for the ligand (blue wire). (B) Residues contributing to the coordination of GSH are shown with hydrogen bond lengths indicated in angstroms (red). (C) Equivalent schematic for the coordination of ESG by KefC.
To validate the biological relevance of the observed structure, residues Q412 and D499 were subjected to serial mutation and functional analysis in the full-length construct. In the native protein, the presence of GSH was sufficient to prevent a spontaneous leak of K+; rapid K+ efflux resulted from the formation and binding of ESG by the reaction of N-ethylmaleimide (NEM) with endogenous GSH (Fig. 2A). KefC proteins with mutations that disrupt GSH binding in the KTN domain lead to a rapid K+ leak from cells (9, 13). Progressively disruptive mutations of Q412 caused effects varying from reduced inhibition by GSH (Q412A) (Fig. 2B) to complete insensitivity to both GSH and glutathione adducts (Q412K) (Fig. 2C). A Q412K mutation was functionally equivalent to a triple mutation affecting all three GSH glycine carboxylate group coordinating residues, R416, R516, and N551 (Fig. 2D). Similar results were observed for D499, where substitution by a residue with a hydrophilic group (D499S) does not modify activity significantly, whereas substitution with either glycine or alanine led to substantial loss of activation by ESG, without affecting inhibition by GSH (Fig. 2E). These results corroborate that the crystal structure accurately portrays the in vivo binding of GSH to KefC.
Fig. 2.

Functional analysis of ligand binding residues. (A) WT KefC retains K+ when inhibited by GSH (red), leaks K+ in the absence of GSH (blue broken), and, when activated by NEM (added at arrow), mediates rapid K+ efflux (black). Functional analysis of the mutant Q412A (B), Q412K (C) and the triple mutant (R416A/R516A/N551A) (D). (E) Relative first-order rate constant for D499 mutants (mean and standard error).
Glutathione Adducts Break a Protein–Protein Interaction Between Adjoining KTN Domains.
The complementary structure of KefC-CTD with bound ESG was compared to the GSH-inhibited structure to elucidate the ligand gating mechanism. ESG occupies the same site as GSH, making contacts with the same residues in the KTN domain (Fig. 1C). However, in the GSH-bound structure, residue F441 on α-helix 2 of one subunit protrudes into a well-defined, hydrophobic pocket formed from α-helices 7 and 8 of the opposing KTN subunit (Fig. 3A). Comparison of the ligand-free (13) and GSH-bound structures revealed that the reduced thiol group of GSH stabilizes both the protein–ligand association and resulting protein conformation by displacing water from the cavity adjacent to the phenyl ring of F441. In stark contrast the ESG-bound structure revealed a dramatic conformational change in this region (Fig. 3B). Superimposition of the GSH and ESG structures shows that the N-ethylsuccinimide ring would overlap with the phenyl ring of F441. This steric clash forces the phenylalanine residue to be ejected from its original position in the pocket, observed in the GSH-bound structure. Instead, F441 repositions to an alternative conformation in which it, along with α-helix 2, has pulled away from the adjoining KTN fold and now associates solely with other amino acids from its own chain. An equally substantial effect is observed in the partnering KefC subunit, where due to loss of association with F441, α-helix 8 becomes completely disordered and could not be built into the electron–density maps.
Fig. 3.

Mechanism of glutathione adduct modulation of KTN conformation. (A) The reduced thiol group of glutathione (yellow sphere) stabilizes the binding of a phenylalanine residue (F441) from α-helix 2 from one KTN subunit (blue) into a hydrophobic pocket formed by residues from α-helices 7 (M558) and 8 (T565; A569; Y572) from the partnering KTN subunit (gray surface). (B) When the thiol group of glutathione is conjugated to form an adduct, the attached chemical group (both diastereomers of N-ethylsuccinimido-S-glutathione shown here) displaces F441 (red) to an alternative conformation 12 Å distant, where it forms new interactions with hydrophobic residues within its own chain. The disruption of the original protein–protein interaction also leads to repositioning of α8, which is no longer observed in the crystallographic data.
The importance of F441 in gating was investigated by mutagenesis and analysis of ESG-activated K+ efflux through KefC. This residue could be substituted with hydrophobic residues of similar size (tyrosine and tryptophan) without significant loss of activity (Fig. 4 A and B and Fig. S3). In contrast, ESG-elicited efflux was lowered in an F441L mutant, suggesting that the flexibility of this hydrophobic residue limits its effectiveness as a trigger of conformational change. Substitution by an even smaller residue, as in F441D, further reduced gating in the presence of ESG. Note that none of the substitutions affected regulation by GSH, suggesting that overall ligand binding was not changed by the mutations (Fig. S3). The wild-type protein was not significantly activated by S-lactoylglutathione (SLG), which is a strong activator of KefB (Fig. 4B). However, F441W, F441Y, and F441D were strongly activated by SLG, whereas F441L retained the nonactivating character of the wild type (Fig. 4B). It is notable that KefB, which is naturally strongly activated by SLG, has a tyrosine residue at the equivalent position of KefC F441. The current mutant data are consistent with activation by SLG (and related polar adducts) requiring a polar cavity, such as that provided by W, Y, and D residues. Native KefC activation is dependent upon the binding of relatively large hydrophobic glutathione-S conjugates (7, 8), and the structural data now fully explain this observation. These results confirm the critical role of F441 as a molecular sensor for glutathione adducts and as a trigger for channel activation.
Fig. 4.

The importance of F441 and the proposed activation mechanism are supported by in vivo functional analysis of mutants. The rate constant for electrophile-elicited K+ efflux was measured using a sensitive electrode-based method that allowed the initial activity to be determined accurately. (A) Electrode recordings for individual experiments in which ESG is generated by addition of NEM to cells. (B) Rate constants (mean and standard deviation) for KefC activity in the presence of the electrophile (open bars, ESG formed from N-ethylmaleimide; closed bars, SLG formed from methylglyoxal). All data represent multiple replicates, and the analytical method is described in SI Text. *No activity was observed for the F441L mutant in the presence of methylglyoxal.
Confirmation of Structural Changes During ESG-Mediated Gating.
Further evidence for structural changes of the regulatory assembly was obtained from analysis of the full-length transporter in solution using electron paramagnetic resonance (EPR). Several KefFC homologues that have low numbers of Cys residues were expressed from codon-optimized clones and purified. The Gloeobacter violaceus KefB homologue was taken for further study because it was found to express well and be stable during detergent extraction and purification (Fig. S4 A–C). A cysteine-free derivative was created and its in vivo activity verified (Fig. S4 D–F). The purified protein migrated in SDS-PAGE with an apparent mass of 50 kDa compared with ∼69 kDa predicted mass; however, membrane proteins frequently exhibit lower apparent masses by SDS-PAGE due to their high binding affinity for SDS and a similar effect has also been observed for E. coli KefC (24). The KefB protein was confirmed to be a dimer by Blue Native gel electrophoresis with an apparent mass of ∼320 ± 30 kDa, which once corrected for dye binding yields a mass ∼160 kDa, consistent with a dimeric assembly (25). Subsequently, a single cysteine residue was introduced (R528C) at a position equivalent to D501 in E. coli KefC, within the KTN protein interaction interface (Fig. 5 A and B). This protein exhibited the same properties as the wild-type transporter during purification and analysis.
Fig. 5.

KTN conformational changes correspond with K+ flux regulation. (A) Most KTN domains associate with other elements of their system through a flat protein interaction interface formed largely by α-helices 4 and 5 (15–19). The helix-turn-helix arm of KefC (α 7/8) protrudes adjacent to this surface, preventing the formation of protein–protein contacts similar to those seen in other KTN structures. This obstruction is stabilized by the presence of GSH in the ligand pocket (orange). In contrast, on binding a glutathione adduct, the structure of this regulatory element becomes entirely disrupted, fully exposing the protein interaction interface (red). This frees the region to bind gating components of the transporter in order to initiate ion flow through the pore. The location of D501 in the center of this surface is illustrated. (B) Sequence alignment of KefC from E. coli and KefB from G. violaceus show conservation in the region of spin labeling while the mutated and labeled residue R528C itself (blue) is not conserved. (C) A substantial conformational change is indicated by the power saturation π-values of EPR spectra measurements that show an increased accessibility for oxygen and NiEDDA in the presence of ESG. (D) The EPR spectra in the absence (black) or presence (green) of 1 mM ESG and their respective simulations (red) indicate that the mobility of the spin label is not changed by ESG addition. Note that physiological concentrations of ESG fall in the 0.5–10 mM range (29).
Purified KefB R528C protein was labeled with the spin probe, S-(2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-3-yl)methyl methanesulfonothioate (MTSL), and the EPR spectrum measured in the presence and absence of ESG (note that equivalent data with GSH were not possible due to the immediate reduction of the spin label Cys bond in the presence of this reducing agent). From comparison of the structures, we predicted D501 should become more exposed upon ESG association due to displacement of the α-8 helix. Binding of ESG caused a substantial change in the accessibility of this site to O2 and Ni2+ ethylenediamine-N, N2-diacetate (NiEDDA) (Fig. 5C). The shape of the spectra was not substantially affected by ESG binding, suggesting the structural constraint on mobility of the spin label is barely modified by ESG (Fig. 5D). The mobility can be quantified by the peak-to-peak first derivative width of the central line (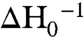 ; 2.8 mT-1 for the ESG-free form and 3.0 mT-1 for the ESG-bound form) or the spectral second moment (〈H2〉-1; 0.39 mT-2 for the ESG-free form and 0.38 mT-2 for the ESG-bound form). These values are consistent with an α-helix-located spin label with little or no tertiary interaction (26). The increased accessibility for both relaxation agents cannot be caused by the presence of the bound ESG directly, but must be due to a tertiary conformational change. Consistently, the mobility of the spin label does not change, which excludes direct contact of the spin label with ESG. These measurements are notable as they were recorded on a full-length protein and not just the isolated regulatory assembly. These data corroborate that the structural differences between the GSH-bound and ESG-bound forms are biologically significant and relevant to the in vivo gating movement in Kef transporters.
; 2.8 mT-1 for the ESG-free form and 3.0 mT-1 for the ESG-bound form) or the spectral second moment (〈H2〉-1; 0.39 mT-2 for the ESG-free form and 0.38 mT-2 for the ESG-bound form). These values are consistent with an α-helix-located spin label with little or no tertiary interaction (26). The increased accessibility for both relaxation agents cannot be caused by the presence of the bound ESG directly, but must be due to a tertiary conformational change. Consistently, the mobility of the spin label does not change, which excludes direct contact of the spin label with ESG. These measurements are notable as they were recorded on a full-length protein and not just the isolated regulatory assembly. These data corroborate that the structural differences between the GSH-bound and ESG-bound forms are biologically significant and relevant to the in vivo gating movement in Kef transporters.
Discussion
The binding and activation of KefFC, and related systems, by GSH adducts formed by reaction with electrophiles is a major mechanism of resistance in Gram negative bacterial pathogens, such as E. coli, Salmonella, and Pseudomonas. Here we carefully dissect the regulation of E. coli KefC by its C-terminal KTN domain using well-characterized, biologically relevant ligands and demonstrate significant conformational changes associated with the binding of the activating ligand. Cocrystallization of KefC-KTN-KefF with ESG, and derivation of the GSH-bound structure by in crystal reduction of ESG, clearly reveals the position and orientation of the GSH moiety within a cleft formed between two KTN domains in a canonically dimeric assembly. The GSH is anchored at both ends, through the α-carboxyl of the glycine (R416, R516, and N551 via H2O) and through both the α-amino and α-carboxyl groups of the glutamate residue (R498 and D499), thus maximizing the interactions possible from a γ-linked peptide (Fig. 1B). Further critical contacts are formed between Q412 and backbone carbonyls and amides (Figs. 1B and 2 B and C). These contacts are maintained in the binding of ESG (Fig. 1C). Analysis of the molecular contacts of the thiol group in GSH suggests that it stabilizes an intersubunit association between α-helix 2 from one KTN fold with α-helices 7/8 of the partnering protein chain. Comparison with the structure of ESG-bound KefC-CTD reveals that it is precisely this connection that GSH adducts disrupt as the first step in activating the KefC system. By displacing a critical phenylalanine (F441) from its position in a hydrophobic cavity created within α7/8, the bulky group conjugated to GSH triggers a separation of the two KTN subunits and exposure of the KTN protein interaction interface. This mechanism eloquently explains how Kef systems can respond to a chemically diverse array of electrophiles through a single ligand binding site. Strongest activation is observed with ESG, which can be seen to occupy the same physical space as F441 in the GSH-bound structure. Moreover, the structural data also explain why small adducts do not activate KefC, because they can fit within the pocket without displacing F441. This may occur either by slight repositioning of the F441 ring, or by the chemical group of the adduct occupying an alternate pocket adjacent to the thiol moiety of GSH. Such flexibility is not available for the more rigid ring structure of ESG. Finally, the structural data explain the observation that KefC is moderately active in cells that have been depleted genetically of their GSH pool. In the absence of GSH, the Kef system exhibits a partially active conformation intermediate between the closed and the fully activated states (7). The observation that, in the absence of GSH, water molecules fill the GSH binding pocket and make unfavorable interactions with F441, provides a mechanistic explanation for the observed partial activation of Kef in vivo in the absence of GSH.
To understand the means by which local conformational changes in KTN structure might gate the associated transmembrane pore-forming domain, we analyzed the differences between the GSH-bound, the ESG-bound, and the ligand-free structures around the KTN interaction interface. This planar surface, formed primarily by α-helices 4 and 5, is implicated in the function of KTN subunits in other systems (15–17, 27, 28). In numerous structures, this surface is hydrophobic and forms large, stable protein–protein contacts with other KTN dimers within the crystal lattice, supporting models that this interface is critical to overall mechanism. In the KefC structure, however, this surface is more polar and partially obstructed by residues of α-helices 7 and 8 that form a protruding arm (Fig. 5A). This arm is stabilized by the binding of GSH to KefC. In contrast, in the ESG-bound structure, all potentially obstructing residues (residues 562–580, including all of α-helix 8) are too disordered to model in the experimentally derived electron–density maps. Thus there is a direct correlation between the activity of the KefC transporter and flexibility of the α7/8 regulatory arm.
It is noteworthy that this conformational change within a KTN subunit is directly attributable to the exchange of a known inhibitor for a known activator. Uncovering the mechanism by which KTN domains control their associated transmembrane pores is key to understanding many biophysical phenomena. All previously reported KTN structural transformations were in the relative positional relationship of crystallographic symmetry-related canonical dimers, leaving ambiguous whether the observed variations were biologically relevant or driven by crystal lattice requirements. In our crystal structures, supported by EPR, exchange of GSH for an activating glutathione adduct increases the solvent exposure of the protein interaction interfaces of the KTN dimer. The increased accessibility of these surfaces may be directly involved in initiating ion flux through the transporter.
The structures reported here give a molecular-level insight into a key mechanism regulating K+ flux in a specific system that in turn effects lowering in cytoplasmic pH that can enhance bacterial cell survival during electrophile exposure (Fig. S5). Clearly, the system has considerable potential as a target for therapeutics because inhibition of the system would sensitize cells to electrophiles. Additionally, because regulation of cytoplasmic pH is critical for cell growth, artificial activation with consequent inappropriate lowering of cytoplasmic pH could be an effective strategy. Analysis of the density for the N-ethylsuccinimido moiety of ESG is weak (with mean refined B factor of 58 Å2). The reaction of GSH with NEM in vivo is spontaneous and leads to an equal mixture of both diastereoisomers. Although the electron–density data did not allow us to determine the precise orientation of the moiety, the data indicate significant space in this region of the activated KTN conformation to allow a variety of ligands to be accommodated, which provides significant opportunities for chemical syntheses, applicable to both the development of antibiotics and the circumvention of drug resistance in certain pathogenic bacteria.
Materials and Methods
Protein Expression and Purification.
Production of KefC-CTD: KefF fusion protein followed standard laboratory protocols for recombinant bacterial protein expression and purification as described previously (13).
ESG Synthesis.
ESG was synthesized from the reaction of reduced L-GSH and NEM in water in the presence of base. Details of the synthesis are elaborated on in SI Text.
Crystallization.
Purified KefC-CTD:KefF fusion protein was supplemented with 1 mM GSH or ESG and subjected to crystal screening utilizing previously successful crystallization protocols (13) and the JCSG+ crystallization screen (Qiagen) with and without addition of 1 mM NAD+ or NADH. Although original crystallization conditions proved fruitless, a lead containing PEG 3350 and ammonium sulfate with NAD+ and ESG was optimized to produce three-dimensional crystals over a few days that exceeded 50 μm in each dimension. Data were collected on crystals grown for 2 d in 9–10% PEG 3350, 100 mM ammonium sulfate, 100 mM Bis-Tris buffer (pH 5.5), and 50 mM magnesium formate with 1 mM NAD+. Extensive crystallization trials of the KefC-CTD: KefF complex with GSH failed to produce crystals adequate for structural analysis. To overcome this obstacle, an alternative approach whereby crystallization was conducted under conditions described above in the presence of 1 mM ESG followed by supplementation of a strong reducing agent, 1 mM tris(2-carboxyethyl)phosphine hydrochloride (TCEP), that could convert the ESG back to GSH over time. This process led to the growth of large rod-shaped crystals after several weeks of incubation.
Data Collection and Processing.
Crystals of KefC-CTD: KefF with either GSH or ESG were cryoprotected in crystallization liquor supplemented with 25% glycerol and frozen by submersion in liquid nitrogen. X-ray diffraction data was collected at Stanford Synchrotron Radiation Laboratory (SSRL) beamlines 7-1 and 11-1. For the complex cocrystallized with ESG, a complete, high-quality dataset to 2.1-Å resolution was obtained (Table S1). These crystals were of the same orthogonal space group, P212121, as the original ligand-free structure with low mosaicity. One crystal grown with GSH diffracted X-rays particularly well, leading to the collection of a 1.75-Å resolution dataset, also in the P212121 space group. Collected data were processed and reduced by the HKL2000 package (30) with Denzo and Scalepack.
Structure Determination and Refinement.
Phasing was achieved by molecular replacement method employing Molrep (31), using the ligand-free KefC-CTD: KefF structure as the search model (PDB ID code 3EYW). The interactive modeling suite Coot (32) was used for building into Fourier electron–density maps. Iterative cycles of model building and refinement with Refmac (31) produced the final structures. Both models include two molecules of flavin mononucleotide in the KefF active site and two zinc atoms at the interface between KefC and KefF. The final ESG-bound structure also includes one molecule of ESG and two sulfate ions bound in the KTN-domain nucleotide binding pockets. The final GSH-bound structure includes one molecule of GSH, two molecules of AMP, and also a single sulfate ion, picked up from the crystallization solution, based on strong electron density near one R416 residue of KefC. Both models were validated using Procheck (33) and Molprobity (34). Note that the density for the N-ethylsuccinimido moiety of ESG is weak (with mean refined B factor of 58 Å2). Hence it is not possible to determine the precise orientation of this moiety, nor can we unambiguously determine whether one, or both, inseparable diastereomers of synthesized ESG are bound. Because the reaction of GSH with NEM in vivo is also spontaneous and likewise leads to an equal mixture of both diastereoisomers, it remains unclear whether one or both forms of ESG are the true physiological activators of Kef systems. Lacking contrary evidence, the structure is modeled with 50% occupancy of each isomer.
KefC Efflux Assays.
Site-directed mutants of Kef system-bearing plasmids (pSM12 and pKefFKefCHis6) (9) were generated using the Quickchange protocol (Stratagene) and transformed into MJF335 [kefB, kefC::Tn10, gshA::Tn10(Kan)], a glutathione-deficient strain of E. coli, allowing for control of the concentration of this ligand through the buffer; this strain also lacks KefB and KefC activity from the chromosome. Functional analysis of KefC K+ efflux was conducted as previously described (7, 9), but potassium concentrations were also detected directly with a sensitive ion-selective electrode to allow a higher time resolution (2-s intervals).
Purification, Spin Labeling, and EPR Measurements on KefB from G. violaceus.
The synthetic and sequence-optimized clone pTrcKefGBH6 (DNA2.0), containing the genes KefG and KefB from G. violaceus and a C-terminal His6 tag on KefB, was used to create a cysteine-free construct by mutating C152S in KefG and C450S in KefB. Then cysteine was reintroduced as R528C into KefB (using Stratagene Quickchange protocol). For purification, KefGB was expressed in BL21(DE3) induced with 0.8 mM IPTG for 4 h at 37 °C in LB medium. All used buffers contained 100 mM phosphate pH 7.5, 50 mM NaCl, 10% glycerol, and at least 0.05% dodecylmaltoside (DDM; Anatrace). The isolated bacterial membrane was solubilzed in 1.5% DDM for 1 h at 4 °C and applied to Ni+2-nitrilotriacetic acid (NTA) agarose column (Sigma). The column was washed in the presence of 20 mM imidazole and reduced with 3 mM TCEP in degassed buffer. Then 0.4 mM MTSL (about 10-fold excess over KefB) in degassed buffer was applied to the column and incubated overnight at 4 °C. The column was thoroughly washed the next morning and KefB was eluted with 300 mM imidazole. Characterization of the elution fraction by gel filtration (Superose 6, GE Healthcare), Blue Native PAGE, and SDS-PAGE indicated the presence of pure and homogeneous KefB with no detectable KefG. EPR spectroscopy was performed directly on the elution fraction using a Bruker ER-200 X-band spectrometer equipped with an ER 4123D resonator. Spectra were recorded with a modulation amplitude of 1G and a microwave power of 2 mW. They were simulated with the Matlab toolbox “Easyspin” (35). Power saturation curves were recorded using TPX capillary sample tubes equilibrated with nitrogen (in the presence or absence of 3 mM Ni2+ ethylenediamine-N,N′-diacetate) or air.
Supplementary Information Available.
A more detailed description of the chemical synthesis of ESG, protein purification, and efflux assays is presented in SI Text.
Supplementary Material
Acknowledgments.
We thank C. Smith, L. Dunn, and other members of the staff of SSRL for assistance in crystallographic data collection and D. Chong and D. Lurie for their contributions to the EPR experiments. This work was conducted in part at the SSRL, which is funded by the Department of Energy, Office of Biological and Environmental Research. This work was supported by funds from the Institutional Development Award for Networks of Biomedical Research Excellence Program of the National Center for Research Resources (P20 RR-016464) (to T.P.R.), from the Wellcome Trust (GR040174 and 086903) (to I.R.B. and S.M.), from the University of Aberdeen (to J.H.), and from St. Hugh’s College, Oxford (to S.J.C.).
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
Data deposition: The coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org [(PDB ID codes 3L9W (GSH-bound KefC-CTD:KefF) and 3L9X (ESG-bound KefC-CTD)].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1012716107/-/DCSupplemental.
References
- 1.Booth IR. Regulation of cytoplasmic pH in bacteria. Microbiol Rev. 1985;49:359–378. doi: 10.1128/mr.49.4.359-378.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Epstein W. Osmoregulation by potassium transport in Escherichia coli. FEMS Microbiol Lett. 1986;39:73–80. [Google Scholar]
- 3.Bossemeyer D, et al. K+-transport protein TrkA of Escherichia coli is a peripheral membrane protein that requires other trk gene products for attachment to the cytoplasmic membrane. J Biol Chem. 1989;264:16403–16410. [PubMed] [Google Scholar]
- 4.Derst C, Karschin A. Evolutionary link between prokaryotic and eukaryotic K+ channels. J Exp Biol. 1998;201:2791–2799. [PubMed] [Google Scholar]
- 5.Nakamura T, Yuda R, Unemoto T, Bakker EP. KtrAB, a new type of bacterial K+-uptake system from Vibrio alginolyticus. J Bacteriol. 1998;180:3491–3494. doi: 10.1128/jb.180.13.3491-3494.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meury J, Lebail S, Kepes A. Opening of potassium channels in Escherichia coli membranes by thiol reagents and recovery of potassium tightness. Eur J Biochem. 1980;113:33–38. doi: 10.1111/j.1432-1033.1980.tb06135.x. [DOI] [PubMed] [Google Scholar]
- 7.Elmore MJ, et al. Activation of potassium efflux from Escherichia coli by glutathione metabolites. Mol Microbiol. 1990;4:405–412. doi: 10.1111/j.1365-2958.1990.tb00607.x. [DOI] [PubMed] [Google Scholar]
- 8.Ferguson GP, Munro AW, Douglas RM, McLaggan D, Booth IR. Activation of potassium channels during metabolite detoxification in Escherichia coli. Mol Microbiol. 1993;9:1297–1303. doi: 10.1111/j.1365-2958.1993.tb01259.x. [DOI] [PubMed] [Google Scholar]
- 9.Miller S, Douglas RM, Carter P, Booth IR. Mutations in the glutathione-gated KefC K+ efflux system of Escherichia coli that cause constitutive activation. J Biol Chem. 1997;272:24942–24947. doi: 10.1074/jbc.272.40.24942. [DOI] [PubMed] [Google Scholar]
- 10.Ness LS, Booth IR. Different foci for the regulation of the activity of the KefB and KefC glutathione-gated K+ efflux systems. J Biol Chem. 1999;274:9524–9530. doi: 10.1074/jbc.274.14.9524. [DOI] [PubMed] [Google Scholar]
- 11.Ferguson GP, McLaggan D, Booth IR. Potassium channel activation by glutathione-S-conjugates in Escherichia coli: Protection against methylglyoxal is mediated by cytoplasmic acidification. Mol Microbiol. 1995;17:1025–1033. doi: 10.1111/j.1365-2958.1995.mmi_17061025.x. [DOI] [PubMed] [Google Scholar]
- 12.Krymkiewicz N. Reactions of methylglyoxal with nucleic acids. FEBS Lett. 1973;29:51–54. doi: 10.1016/0014-5793(73)80013-4. [DOI] [PubMed] [Google Scholar]
- 13.Roosild TP, et al. KTN (RCK) domains regulate K+ channels and transporters by controlling the dimer-hinge conformation. Structure. 2009;17:893–903. doi: 10.1016/j.str.2009.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jiang Y, Pico A, Cadene M, Chait BT, MacKinnon R. Structure of the RCK domain from the E coli K+ channel and demonstration of its presence in the human BK channel. Neuron. 2001;29:593–601. doi: 10.1016/s0896-6273(01)00236-7. [DOI] [PubMed] [Google Scholar]
- 15.Roosild TP, Miller S, Booth IR, Choe S. A mechanism of regulating transmembrane potassium flux by a ligand-mediated switch. Cell. 2002;109:781–791. doi: 10.1016/s0092-8674(02)00768-7. [DOI] [PubMed] [Google Scholar]
- 16.Jiang Y, et al. Crystal structure and mechanism of a calcium-gated potassium channel. Nature. 2002;417:515–522. doi: 10.1038/417515a. [DOI] [PubMed] [Google Scholar]
- 17.Dong J, Shi N, Berke I, Chen L, Jiang Y. Structures of the MthK RCK domain and the effect of Ca2+ on gating ring stability. J Biol Chem. 2005;280:41716–41724. doi: 10.1074/jbc.M508144200. [DOI] [PubMed] [Google Scholar]
- 18.Albright RA, Ibar JL, Kim CU, Gruner SM, Morais-Cabral JH. The RCK domain of the KtrAB K+ transporter: multiple conformations of an octameric ring. Cell. 2006;126:1147–1159. doi: 10.1016/j.cell.2006.08.028. [DOI] [PubMed] [Google Scholar]
- 19.Ye S, Li Y, Chen L, Jiang Y. Crystal structures of a ligand-free MthK gating ring: insights into the ligand gating mechanism of K+ channels. Cell. 2006;126:1161–1173. doi: 10.1016/j.cell.2006.08.029. [DOI] [PubMed] [Google Scholar]
- 20.Joint Center for Structural Genomics. Crystal structure of (tm1088a) from Thermatoga maritima at 1.50 Å resolution. 2006 RCSB Protein Data Bank PDB ID code 2g1u. [Google Scholar]
- 21.Wu R, Abdullah J, Joachimiak A. The crystal structure of TrkA domain of putative glutathione-regulated potassium-efflux KefB from Vibrio parahaemolyticus. 2008 RCSB Protein Data Bank PDB ID code 3c85. [Google Scholar]
- 22.Chang C, Bigelow L, Buck K, Joachimiak A. Crystal structure of TrkA-N domain of inner membrane protein ybaL from Escherichia coli. 2009 RCSB Protein Data Bank PDB ID code 3fwz. [Google Scholar]
- 23.Miller S, Ness LS, Wood CM, Fox C, Booth IR. Identification of an ancillary protein, YabF, required for activity of the KefC glutathione-gated potassium efflux system in Escherichia coli. J Bacteriol. 2000;182:6536–6540. doi: 10.1128/jb.182.22.6536-6540.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Munro AW, Ritchie GY, Lamb AJ, Douglas RM, Booth IR. The cloning and DNA sequence of the gene for the glutathione-regulated potassium-efflux system KefC of Escherichia coli. Mol Microbiol. 1991;5:607–616. doi: 10.1111/j.1365-2958.1991.tb00731.x. [DOI] [PubMed] [Google Scholar]
- 25.Heuberger EH, Veenhoff LM, Duurkens RH, Friesen RH, Poolman B. Oligomeric state of membrane transport proteins analyzed with blue native electrophoresis and analytical ultracentrifugation. J Mol Biol. 2002;317:591–600. doi: 10.1006/jmbi.2002.5416. [DOI] [PubMed] [Google Scholar]
- 26.McHaourab HS, Lietzow MA, Hideg K, Hubbell WL. Motion of spin-labeled side chains in T4 lysozyme. Correlation with protein structure and dynamics. Biochemistry. 1996;35:7692–7704. doi: 10.1021/bi960482k. [DOI] [PubMed] [Google Scholar]
- 27.Kim HJ, Lim HH, Rho SH, Eom SH, Park CS. Hydrophobic interface between two regulators of K+ conductance domains critical for calcium-dependent activation of large conductance Ca2+-activated K+ channels. J Biol Chem. 2006;281:38573–38581. doi: 10.1074/jbc.M604769200. [DOI] [PubMed] [Google Scholar]
- 28.Qian X, Niu X, Magleby KL. Intra- and intersubunit cooperativity in activation of BK channels by Ca2+ J Gen Physiol. 2006;128:389–404. doi: 10.1085/jgp.200609486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McLaggan D, Rufino H, Jaspars M, Booth IR. Glutathione-dependent conversion of N-ethylmaleimide to the maleamic acid by Escherichia coli: An intracellular detoxification process. Appl Environ Microbiol. 2000;66:1393–1399. doi: 10.1128/aem.66.4.1393-1399.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 31.Collaborative Computational Project Number 4. The CCP4 suite: Programs for protein crystallography. Acta Crytallogr D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 32.Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 33.Laskowski RA, Moss DS, Thornton JM. Main-chain bond lengths and bond angles in protein structures. J Mol Biol. 1993;231:1049–1067. doi: 10.1006/jmbi.1993.1351. [DOI] [PubMed] [Google Scholar]
- 34.Davis IW, et al. MolProbity: All-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stoll S, Schweiger A. EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J Magn Reson. 2006;178:42–55. doi: 10.1016/j.jmr.2005.08.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.