

Abstract
The stability, activity, and solubility of a protein sequence are determined by a delicate balance of molecular interactions in a variety of conformational states. Even so, most computational protein design methods model sequences in the context of a single native conformation. Simulations that model the native state as an ensemble have been mostly neglected due to the lack of sufficiently powerful optimization algorithms for multistate design. Here, we have applied our multistate design algorithm to study the potential utility of various forms of input structural data for design. To facilitate a more thorough analysis, we developed new methods for the design and high-throughput stability determination of combinatorial mutation libraries based on protein design calculations. The application of these methods to the core design of a small model system produced many variants with improved thermodynamic stability and showed that multistate design methods can be readily applied to large structural ensembles. We found that exhaustive screening of our designed libraries helped to clarify several sources of simulation error that would have otherwise been difficult to ascertain. Interestingly, the lack of correlation between our simulated and experimentally measured stability values shows clearly that a design procedure need not reproduce experimental data exactly to achieve success. This surprising result suggests potentially fruitful directions for the improvement of computational protein design technology.
Keywords: protein engineering, high-throughput stability determination, library design, molecular dynamics, NMR ensemble
Protein-engineering efforts based on directed evolution have met with considerable success (1–3). In tandem, structure-based computational protein design (CPD) methods have been developed to allow screening for desirable sequences to be performed in silico (4–6). Despite a number of high-profile results that demonstrate the utility of CPD (7–12), the routine computational design of functional proteins remains elusive. Thus, many current efforts focus on the improvement of CPD methodology or on the synergistic application of CPD with experimental high-throughput screening or selection (13).
Although the stability, solubility, and activity of a protein depend on the relative energetic contributions of many conformational states, including ensembles of native, unfolded, and aggregated structures (14), most CPD methods evaluate sequences based on their energies in the context of one fixed-backbone structure. This simplification has made design results undesirably sensitive to slight changes in main-chain and side-chain conformation and has made difficult the selection of sequences with amino acid composition similar to naturally occurring proteins. These issues have been approached via the use of high-resolution structural templates, expanded rotamer libraries (15, 16), energy functions with softened repulsive terms (10, 17, 18), iteration between structural refinement and sequence design (10, 19), and amino acid reference energies (10, 20). Although these strategies can help to mitigate the impact of the fixed-backbone approximation, they do not address the fundamental reality that sequence fitness is a function of multiple conformational states.
In a handful of cases, multistate design (MSD) procedures have been used to find sequences that simultaneously stabilize or destabilize a combination of a few different conformational states (21–23). However, MSD techniques have not yet been applied to native ensembles with many conformational states that might better reflect the flexibility of real proteins. The degree to which various energy functions, rotamer libraries, and structural templates of single-state design (SSD) might be appropriate for this type of MSD calculation is, so far, unknown. We recently developed a framework for MSD that allows for efficient sequence optimization given hundreds of conformational states (24). Here, we have applied this framework to test the applicability of current CPD methods to large structural ensembles, and to investigate whether the use of such ensembles might result in the selection of more desirable sequences by CPD.
The most basic goal of CPD has been to optimize interactions between amino acid side chains to promote thermodynamic stability of the native state. Unfortunately, standard methods for the measurement of protein stability are too laborious to allow the testing of more than a few designed variants, and the top-scoring sequence produced by a new design procedure does not yet sufficiently reflect its general utility. Fortunately, recent progress in laboratory automation has allowed us to construct an efficient pipeline for the basic evaluation of new procedures in CPD. In our scheme, gene libraries are assembled from degenerate oligonucleotides, proteins are expressed and purified in microtiter plates, and liquid-handling robotics assist in the preparation of chemical denaturation series in a 96-well format for assay by tryptophan fluorescence. The integration of these technologies has allowed us to assess the stability of hundreds of designed protein variants with minimal experimenter intervention and limited incremental expense.
Given several design procedures to evaluate and a high-throughput experimental assay, we needed a general and rigorous method to choose a limited number of representative sequences to test from each design. Although several useful computational protein library design methods have been developed (25–28), none reported so far takes directly into account simulation energies, allows control over library size and possible sets of amino acids, and eschews heuristics that can introduce bias into the libraries it produces. So that our experimental results might better reflect the results of the underlying CPD calculations, we developed a unique library design procedure, called Combinatorial Libraries Emphasizing And Reflecting Scored Sequences (CLEARSS), which satisfies these criteria.
We used standard single-state design (SSD) and MSD to redesign the core of the small, stable domain Gβ1 based on several sources of structural information, including a crystal structure, an NMR structure, and MD simulations. Our efforts were motivated by a curiosity about the relative merits of different sources of structural data for design and the hypothesis that use of a structural ensemble might help to correct for design failures observed in SSD. Because the imperfect nature of CPD limits the conclusions that can be drawn from a comparison of single sequences, we developed previously undescribed methods for the computational design and high-throughput experimental stability determination of combinatorial protein libraries. The results we report here provide simultaneous experimental validation for (i) the application of multistate protein design methods to large conformational ensembles, (ii) the transformation of arbitrary CPD results into combinatorial mutation libraries, and (iii) the experimental stability determination of these libraries by high-throughput gene assembly, protein expression, purification, and screening.
Results and Discussion
Designed Libraries.
To simplify the validation of our multistate design methods, we applied them to a previously studied set of core positions (Fig. S1) in a small model system, protein Gβ1, and relied on a set of energy functions that previously found stabilized variants of this sequence (17). We assessed these methods by performing designs based on each of the following sources of structural information: a crystal structure (xtal-1), an NMR-constrained minimized average solution structure (NMR-1), an NMR ensemble (NMR-60), a constrained MD ensemble (cMD-128), and an unconstrained MD ensemble (uMD-128). Our algorithm for library design (Fig. 1) was then applied to produce degenerate oligonucleotide sequences that reflect quantitatively the amino acid preferences determined by the design calculations. Given the requirements for purified protein of our stability assay, we chose to design and screen a 24-member library based on each structural data source described above.
Fig. 1.

General scheme used to design combinatorial mutation libraries based on computational protein design calculations. A line of boxes indicates a protein sequence; each box represents a position in the protein chain. Different colored boxes represent different amino acids. The set of sequences on the far right corresponds to the expansion of a particular combinatorial library into the set of sequences it represents. The energies of the sequences in the expansions are used to decide which combinatorial library to test experimentally, as described in Materials and Methods.
All five designed libraries comprise relatively conservative sets of mutations away from the wild-type sequence (Table 1). The libraries other than uMD-128 share many characteristics in common. Each of these libraries chose only the wild-type amino acid at positions A20, A26, F30, and A34. Every member of each of these four libraries contained the single-mutant Y3F, which previous experiments have shown to be well tolerated by the structure. These four libraries all allowed the wild-type amino acid at every other position, and all contain the most stable Gβ1 core variant previously characterized, Y3F + L7I + V39I (17).
Table 1.
Combinatorial libraries designed from different sources of structural information

xtal-1: library based on single-state design of the crystal structure. NMR-1: library based on single-state design of the constrained minimized average NMR solution structure. NMR-60: library based on multistate design of the 60-member NMR structural ensemble. cMD-128: library based on multistate design of the constrained molecular dynamics ensemble. uMD-128: library based on multistate design of the unconstrained molecular dynamics ensemble.
The two NMR libraries were extremely similar to each other: Both chose the amino acids FILV at position 52 and directed the remaining diversity to positions 7 and 39. In contrast, xtal-1 and cMD-128 allowed only the wild-type Phe at position 52 and instead allocated diversity toward positions 7, 39, and 54. xtal-1 differs from cMD-128 in that it gave up L7F and V39L to allow L5I. The unconstrained MD ensemble library uMD-128 was the least conservative, specifying a size reversal of two nearby residues via mutations L5A and A34F, and diversity at residue 30, a position untouched in the other libraries.
As shown in Table S1, the designed libraries generally succeeded in representing the top-scoring sequences from each design calculation, given the constraints imposed by the genetic code. The exception was the uMD-128 library, which represented only three of the best 100 sequences from the original design calculation. This was caused by an unusual designed sequence list, in which the best-scoring sequence contained a feature (the size reversal mentioned above) that was very uncommon in the remainder of the list.
Experimental Characterization of Designed Libraries.
Experimental screening of the xtal-1 library (Fig. S2A) showed two distinct sets of variants. The 12 library members with wild-type Leu at position 5 all exhibited stabilities similar to or better than the wild-type sequence, while the 12 with Ile at position 5 were all significantly destabilized. Screening of the NMR-based libraries (Fig. S2 B and C) showed a similar dichotomy. In each case, the six library members with the wild-type Phe at position 52 exhibited wild-type-like stability or better. The remaining 18 variants from each NMR-based library were highly destabilized, and many lacked enough of a pretransition to be fit to the two-state unfolding model.
Evaluation of the MD libraries indicated that all 24 variants from the constrained library, cMD-128, had stability similar to the wild-type protein or better (Fig. S2D). In contrast, all 24 variants from the uMD-128 library failed to produce any significant change in fluorescence signal across the denaturation series and likely assume an alternative structure, as discussed below. Sorting the individual sequence members from every library except uMD-128 according to experimental stability (Fig. 2) shows that the cMD-128 input structural ensemble favored more high-quality sequences (better than wild type) than any other library. Every other designed library specified at least one problematic substitution that rendered many of its sequences destabilized or otherwise unlike the wild type.
Fig. 2.

Library mutants sorted by experimental stability. All sequences from the cMD-128, NMR-1, NMR-60, and xtal-1 libraries were named according to their designed positions (Sequence ID) and sorted by their experimentally determined Cm value. Some sequences have membership in more than one library. All sequences above the “wild-type stability” label are more stable than the wild-type sequence. No sequences below the “unfolded protein” label gave a measurable transition in the stability assay.
Impact of Input Structural Data on Designed Libraries.
Why were apparently destabilizing mutations such as L5I, F52ILV, and A34F chosen by the design procedure? These mutations were all present in high-scoring sequences from the original design calculations and thus reflect real preferences of the original design procedures, rather than artifacts introduced by the library design process.
The selection of the amino acids FILV at position F52 in the two NMR-based libraries resulted in three quarters of each library being significantly destabilized. In the context of the NMR structures, no Phe rotamer in the library was able to fit perfectly at position 52, encouraging the selection of smaller amino acids. If the set of rotamers at this position is supplemented with the observed rotamer in each structure, the designs choose to allocate diversity to positions 7 and 39, resulting in libraries similar to xtal-1. This result highlights how dramatically the rotameric approximation can influence the results of a design, despite our biophysical intuition that a solution ensemble might better reflect protein structure than a single crystallographic snapshot. It suggests that, at the very least, rotamers optimized for the wild-type sequence should be included when the goal is to simply find desirable sequences. For this project, we omitted the structurally observed rotamer at each position in order to limit the significant bias toward the wild-type sequence that these rotamers tend to cause. In the context of a real-world protein-engineering project, including these rotamers would have considerably increased our chances of success. Interestingly, this failure of discrete rotamers, occurring as it did in the design of the NMR ensemble, indicates that continuous side-chain optimization may be useful during design, even when allowing conformational flexibility of the main chain.
The L5I mutation, which caused half of the xtal-1 library members to be destabilized relative to the wild-type sequence, may have been selected due to a failure of the softened repulsive contact potential that is used to counteract unrealistic rigidity introduced by the CPD model. The γ methyl group of Ile5 bumps into a Thr residue on an adjacent β strand and is scored as a serious clash using unscaled van der Waals radii but appears innocuous with the atomic radius scaling factor of α = 0.9 that we used for the designs evaluated here (17). Repeating the design calculations with radii scaled by intermediate values such as 0.925 and 0.95 prevents Ile from being chosen at position 5 but also increases the frequency with which smaller residues are chosen at position F52. Interestingly, the recommendation of α = 0.9 is derived from previous experiments based on the same set of Gβ1 core positions that were designed here. The earlier work drew conclusions based only on the best-scoring sequences produced by the design calculations and found no difference between scaling atomic radii by 0.9 or 0.95 (17). Our results indicate that the mutations produced by the design procedure vary significantly with values of α between 0.9 and 0.95 when more sequences are taken into account. Therefore, a more rigorous investigation of appropriate α values for design may be warranted. Although the L5I mutation might also be reasonably attributed to the fixed main-chain and discrete rotamers, several good-scoring libraries based on the constrained MD ensemble also contained this mutation (see below). Because the additional conformational diversity provided by the ensemble did not inhibit this design failure, we find explanations related to energy function more plausible.
To analyze the uMD-128 data, it is important to note that our stability assay reports on the environment of the single Trp residue of Gβ1. Changes in packing caused by substitutions at other positions could alter the native-state environment of Trp43 enough to flip its side chain out into solution or change its fluorescence properties, crippling our ability to monitor unfolding by fluorescence. This interpretation seems unlikely for the destabilized members of the crystal structure and NMR libraries, for which a partial unfolding transition is clearly indicated by the raw data. However, the members of the uMD-128 library fail to show any such a transition, rendering the validity of our assay suspect in this case.
A constant feature of the uMD-128 library is a size reversal specified by mutations A34F and L5A. The model structures produced by this design were well packed and contained no obvious flaws such as Trp43 flipping out into the solvent. Previous characterization of several Gβ1 variants that include mutation A34F has indicated that these sequences assume oligomeric structures and exhibit altered fluorescence properties (29–31). This suggests that the structural basis for our designs, as well as our fluorescence assay, may be inappropriate for sequences containing this mutation. When we reanalyzed a subset of the uMD-128 variants using circular dichroism, they uniformly displayed wild-type-like secondary structure but lower stability and low levels of protein expression. The previous reports and our new results indicate that the uMD-128 library sequences likely assume structures different from the design target. As target structures move away from experimentally determined structures and greater sequence diversity is enabled (32, 33), more effective negative design strategies may be required to exclude sequences that preferentially adopt alternative conformations.
A recent theoretical analysis of NMR and crystal structures as templates for design has suggested that some individual members of NMR ensembles might be more appropriate templates than others (34). To assess the impact this might have had on our results, we ranked the members of each structural ensemble by DREIDING energy (35) and separately by Rosetta energy (36). We then designed new libraries using only the top 16 energy-ranked structures from each ensemble using each energy ranking (Table S2). The two new libraries produced from the NMR structural ensemble were similar to those from the original design; both specified diversity at position 52 and contain destabilized sequences. The library based on the top 16 DREIDING-ranked sequences from the constrained MD ensemble only specifies known nondestabilizing substitutions, whereas the top 16 Rosetta-ranked structures again gave diversity at position 52. For the unconstrained MD ensemble, the top 16 Rosetta-ranked structures gave a library very similar to that produced by the entire ensemble, and the top 16 DREIDING-ranked structures gave a library of sequences that appear severely overpacked. In total, the libraries produced from the top-ranked sequences were similar to those produced from the full ensembles in four cases and were worse in the remaining two cases. Based on this post hoc analysis, our multistate library design procedure seems robust to the influence of poor templates within each ensemble. However, more sophisticated methods of template selection may ultimately prove more fruitful. For example, it might be interesting to choose a subset of a structural ensemble according to the degree to which individual members are able to recover wild-type-like sequences and apply MSD to this subset rather than the entire ensemble.
Influence of the Designed Library Selection Method.
At this point, it is important to address the degree to which the library design method might affect the conclusions we draw from our experiments. The CLEARSS library design procedure was developed with an understanding that many different combinatorial libraries may similarly represent a given list of scored sequences. It is intended to produce a list of the top-scoring designed combinatorial libraries that satisfy all constraints and to let the user choose between them. In general, this choice might be influenced by chemical intuition or prior mutational data and thus partially account for properties of the system that are not modeled during the design procedure. To make our evaluation of input structural data sources as fair as possible, we chose to ignore such influences and apply an objective strategy based on the energies of the sequences in the libraries. Still, we must ask how the other libraries generated by CLEARSS would have fared in our experimental assay.
Each of the top 20 designed libraries based on the NMR ensemble, and each based on the single average NMR structure, assigned smaller residues than the wild-type Phe to position 52. The remaining diversity of each library was occupied by various combinations of the other mutations present in the xtal-1, NMR-1, and NMR-60 libraries we screened in this work. It seems very likely, then, that the screening of any of the top NMR-based libraries from our designs would have resulted in stabilities similar to those we have reported here. Similarly, all of the top 20 designed libraries based on the unconstrained MD ensemble contained mutations L5A and A34F and would be expected to exhibit properties similar to uMD-128.
A more interesting case is provided by the designs based on the crystal structure and constrained MD ensemble. Our analysis of the libraries xtal-1 and cMD-128 produced by these designs seems to indicate that cMD-128 was more successful, because a much greater fraction of its members were shown to be highly stable. However, when the top 20 libraries from each design were inspected in aggregate, it became clear that the xtal-1 and cMD-128 designs had produced a variety of libraries, some featuring the destabilizing mutations described above. Both the xtal-1 library and the cMD-128 library were found in the top-20 set of libraries produced by each design. Furthermore, each design produced several libraries with diversity at position 52, like NMR-1 and NMR-60.
The influence of the library design procedure on a comparison between structural inputs can also be assessed by scoring the sequences from each library on each of the other input structures or ensembles. Histograms of these energies (Fig. S3) show that each structural input prefers the sequences from its own library over those from other libraries, though often by narrow margins.
These observations, taken in total, suggest that the library design method we used did not unduly influence our optimistic conclusions about the merits of high-quality structural ensembles as inputs for computational protein design.
Approximation in Computational Protein Design.
In addition to helping validate the use of multistate and combinatorial library design methods for computational protein design, our results also reflect unexpectedly on protein design itself. Plots of experimental stability versus simulation energy for the cMD-128 library (Fig. S4) failed to yield any correlation, despite the apparent success of this design calculation. Likewise, the design calculations for xtal-1 and the NMR libraries failed to predict the pronounced destabilizing effects of mutations L5I or F52L, even though these designs also found a variety of stabilized variants.
An intuitive perspective on the development of CPD methods is that improvements in designed sequences will follow from improvements in our ability to predict or rank experimental stabilities (37). However, recent advances in stability prediction procedures (38, 39) have not yet, to our knowledge, produced the expected benefits to combinatorial protein design. Our results are consistent with a recent assessment of stability prediction methods, which found that the ability to reproduce experimental stability rankings is unnecessary for useful CPD (40). These conclusions prompt a modified view of the factors that make structure-based design possible in the first place.
Protein structures relax to accommodate mutations, and the computational difficulty of simulating and scoring these relaxed structures has so far rendered intractable the accurate stability ranking of sequence variants with many mutations. Fortunately, this malleability also means that sequences chosen to fit into a rigid protein model, even using approximate energy functions, will likely be tolerated by whatever relaxed structure results from the mutations they contain. In this way, the soft material properties of proteins serve to impede the development of the accurate quantitative protein design methods but also enable the more qualitative methods we can apply today.
The standard view of CPD has been as a single, rigorously quantitative problem: Correct packing of amino acid side chains into a high-resolution template structure leads to a stable and well-behaved designed sequence. However, our analysis supports a revised view of CPD, comprising two distinct problems: (i) to find areas of sequence space that can favorably adopt the target structure, and (ii) to avoid areas of sequence space that might favorably adopt alternate structures. The first problem is simply an enhancement of the original formulation of CPD in which we admit that current methods for native-state sequence selection are approximate, and we focus on finding areas of sequence space enriched with variants that satisfy the target fold.
The second problem has typically been treated implicitly, as discussed above. The energy function used in this work applies a simple tripeptide model of the reference state for solvation energies and assumes that all other interactions average out in the unfolded ensemble. However, issues such as those encountered with the uMD-128 library likely cannot be addressed in a general way without the use of explicit competing state models. Such simulations are more difficult than those that model only the native state, in large part because few nonnative states have been characterized experimentally. In alpha helical peptide systems where large numbers of undesirable states are readily identifiable, explicit negative design has yielded improvements in structural specificity (41). We hope that general models of unfolded and aggregated states will lead to similar improvements in the design of globular proteins.
Conclusions
We enlisted previously undescribed methods for the design and screening of combinatorial libraries to test the application of multistate design procedures to several structural ensembles and to compare the resulting designs to those based on single structures. Single-state and multistate designs based on NMR data produced similar sets of libraries; likewise did those based on crystallographic data. Although an MD-based library gave superlative results, we cannot definitively conclude that the use of a structural ensemble provides any particular advantage over a single high-resolution structure for the purposes of design. Nevertheless, this initial success confirms that the energy functions and rotamer libraries developed for single-state modeling are equally applicable for the multistate design of large structural ensembles.
This work also provides further support in favor of rigorously screening an area of sequence space discovered by simulation and has helped in vetting our unique, general method for library design. For some designs that specified undesired destabilizing mutations, library screening suggested underlying causes for design failure that would not have been apparent via the ad hoc testing of individual sequences. Because our library design procedure is specifically intended to faithfully represent its input scored sequence list and is indifferent to the origin of the list, it should be more useful for the evaluation of new design procedures than its predecessors.
Current design procedures seem to find stable sequences by selecting mutations that are likely to be accommodated by a relaxed version of the template structure and not by accurately ranking the mutations relative to each other. Given that protein stability and function depend on competing states as well as the native state, the poor agreement we observed between simulated and experimental energies in our successful libraries suggests that future effort toward explicit negative design is warranted.
Materials and Methods
Input Structural Data.
Input atomic coordinates for the β1 domain of Streptococcal protein G (Gβ1) were taken from the 2.2 Å crystal structure 1pga (42), the 60-member NMR structural ensemble 1gb1, and a constrained, minimized average structure generated from the ensemble 2gb1 (43). Hydrogens (if any) were stripped from each structure, and new hydrogen positions were optimized along with side-chain amide and imidazolium group flips using Reduce (44). Each structure was then standardized with 50 steps of conjugate gradient minimization using the DREIDING force field (35). An unconstrained 128-member molecular dynamics (MD) ensemble was generated from the minimized crystal structure by running a 12.8 ps MD trajectory at 300 K in vacuum using the DREIDING force field and saving the coordinates every 0.1 ps. The constrained MD trajectory was generated by the same procedure, using an additional harmonic point restraint with a force constant of 100 kcal/mol/Å2 applied to keep Cα atoms near their initial positions. Each MD snapshot was standardized as described above. After standardization, the NMR, unconstrained MD and constrained MD ensembles exhibited average pairwise main-chain rmsds of 0.25, 0.84, and 0.12 Å, respectively.
Sequence Design Specifications and Energy Calculations.
In the sequence designs, 10 core positions of Gβ1 (3, 5, 7, 20, 26, 30, 34, 39, 52, and 54), were allowed to assume any of the hydrophobic amino acids A, V, L, I, F, Y, and W. Tryptophan 43 was allowed to change conformation but not amino acid type, so that our fluorescence-based stability assay would not be compromised. Allowed side-chain conformations at the variable positions were taken from the Dunbrack backbone-dependent rotamer library with expansions of ± 1 standard deviation around χ1 and χ2 (15). To avoid bias toward the wild-type sequence, this set was not supplemented with the side-chain coordinates from the input structure, except at position 43. All other side chains and the main chain were fixed in the input conformation. Pairwise energies were computed for each structure or ensemble member using energy functions described previously (45, 46), with the polar hydrogen burial term omitted.
Sequence Optimization.
Fast and accurate side-chain topology and energy refinement (FASTER) was used to find optimized sequences in the single-state design of the crystal structure and the NMR-constrained minimized average (47). Multistate sequence optimization of each ensemble was performed as described (24). The energies of a sequence in the context of an ensemble member were combined into a single score by computing the free energy of the ensemble system at 300 K:
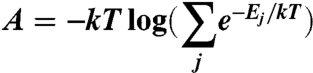 |
where each Ej is the energy of the sequence when threaded on member j of the ensemble. While various functions could be used to combine the state energies into a single score, we chose the free energy function over other averaging schemes because it prefers sequences that satisfy multiple states in a physically reasonable way that does not require any particular number of states to be satisfied.
Combinatorial Library Design.
To choose combinatorial sequence libraries for experimental screening, we used a new algorithm reported here (Fig. 1 and SI Text). Given a list of scored sequences, a list of allowed sets of amino acids, and a range of desired library sizes, the method evaluates all possible combinations of sets of amino acids at different positions that lead to a library with a size in the desired range. Each position in each library is scored by summing the Boltzmann weights of the sequences in the list that contain a library-specified amino acid at that position. The position scores are then summed to give an overall library score. Our algorithm is able to consider all possible libraries because it treats positions independently, and because it ignores amino acid sets that are unnecessarily large in the context of a given position. In this work, we allowed only those sets of amino acids that can be specified by degenerate codons that do not include codons observed with low frequency in Escherichia coli. A temperature of 300 K was used in the Boltzmann weighting, and the target library size was 24. Setting the desired library size to other values, such as 12 or 48, gave libraries composed of the same mutations found in the 24-member libraries.
After applying this algorithm to the lists of sequences produced by the computational designs, we instantiated the 20 best-scoring libraries from each design and rescored all of the amino acid sequences in each library by rotamer optimization. Each library we inspected contained the best-scoring sequence from the design it was based on, although this is not required by the method. From each design, we chose for experimental testing the library in the top 20 with the smallest energy spread between its best-scoring and worst-scoring sequence.
Library Construction, Expression, and Purification.
Oligonucleotides (Integrated DNA Technologies) containing approximately 18 bp overlapping segments were assembled via a modified Stemmer method (48) using KOD Hot Start Polymerase (Novagen) to generate full-length streptococcal Gβ1 with an N-terminal His6 tag. Secondary structure content and annealing temperatures were verified by nucleic acid package (NUPACK) (49, 50). For each library, oligonucleotides containing the desired single mutation or degenerate codon were swapped into the assembly mixture. Standard subcloning techniques were performed to first insert the library into the frameshift selection plasmid pInSALect (51) and finally into an expression plasmid (pET11a). The library was transformed into BL21 Gold DE3 cells (Stratagene), and colonies were picked into 96-well plates for plasmid miniprepping and sequencing (Beckman Genomics). Any missing library members were generated by standard quick-change protocols. Sequence-verified library members were pulled from replicated glycerol stocks and inoculated into Instant TB media (Novagen) in 24-well plates. After overnight incubation at 37 °C, cells were pelleted by centrifugation. Pellets were freeze/thawed once and resuspended in 1× CelLytic B (Sigma-Aldrich) lysis buffer before another identical centrifugation step. Cell lysates were loaded onto an equilibrated HIS-Select filter plate (Sigma-Aldrich), washed twice, and eluted with buffer containing 250 mM imidazole, pH 8.
Microtiter Plate-Based Stability Determination.
Appropriate amounts of GdmCl (Sigma-Aldrich), Milli-Q water, eluted protein, and NaPO4 buffer, pH 6.5, were added to maintain a fixed volume in each well of 96-well Costar UV transparent flat bottom plates by a Freedom EVO liquid-handling robot (Tecan). Adapting a previously reported stability assay, mutant proteins were subjected to a 12-point GdmCl gradient across the columns of the plate where each row contained a separate denaturation experiment (52). Accuracy and precision controls are described in SI Text. The plates were equilibrated for at least 1 h and shaken at 900 rpm on a microtiter plate shaker (Heidolph).
Tryptophan fluorescence measurements were taken on a fluorescence plate reader (Tecan) with a plate stacker attachment. Parameters empirically determined for wild-type Gβ1 were later used for each library assayed. Excitation was performed at 295 nm and emission measured at 341 nm with 10 nm bandwidths. Data were fit as a two-state unfolding transition using the linear extrapolation method (53) in Pylab. The GdmCl concentration at the midpoint of denaturation, Cm, was estimated numerically based on the fraction-unfolded curve fit.
Supplementary Material
Acknowledgments.
We thank Barry Olafson for preparation of the MD structural ensembles, Christina Vizcarra for the pInSALect plasmid, and Jost Vielmetter for useful discussions. This work was supported by the Howard Hughes Medical Institute, the Defense Advanced Research Projects Agency, and the National Security Science and Engineering Faculty Fellowship.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1012985107/-/DCSupplemental.
References
- 1.Arnold FH. Combinatorial and computational challenges for biocatalyst design. Nature. 2001;409(6817):253–257. doi: 10.1038/35051731. [DOI] [PubMed] [Google Scholar]
- 2.Bershtein S, Tawfik DS. Advances in laboratory evolution of enzymes. Curr Opin Chem Biol. 2008;12(2):151–158. doi: 10.1016/j.cbpa.2008.01.027. [DOI] [PubMed] [Google Scholar]
- 3.Jackel C, Kast P, Hilvert D. Protein design by directed evolution. Ann Rev Biophys. 2008;37:153–173. doi: 10.1146/annurev.biophys.37.032807.125832. [DOI] [PubMed] [Google Scholar]
- 4.Schueler-Furman O, Wang C, Bradley P, Misura K, Baker D. Progress in modeling of protein structures and interactions. Science. 2005;310(5748):638–642. doi: 10.1126/science.1112160. [DOI] [PubMed] [Google Scholar]
- 5.Alvizo O, Allen BD, Mayo SL. Computational protein design promises to revolutionize protein engineering. Biotechniques. 2007;42(1):31–35. doi: 10.2144/000112336. [DOI] [PubMed] [Google Scholar]
- 6.Lippow SM, Tidor B. Progress in computational protein design. Curr Opin Biotech. 2007;18(4):305–311. doi: 10.1016/j.copbio.2007.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dahiyat BI, Mayo SL. De novo protein design: Fully automated sequence selection. Science. 1997;278(5335):82–87. doi: 10.1126/science.278.5335.82. [DOI] [PubMed] [Google Scholar]
- 8.Malakauskas SM, Mayo SL. Design, structure and stability of a hyperthermophilic protein variant. Nat Struct Biol. 1998;5(6):470–475. doi: 10.1038/nsb0698-470. [DOI] [PubMed] [Google Scholar]
- 9.Bolon DN, Mayo SL. Enzyme-like proteins by computational design. Proc Natl Acad Sci USA. 2001;98(25):14274–14279. doi: 10.1073/pnas.251555398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kuhlman B, et al. Design of a novel globular protein fold with atomic-level accuracy. Science. 2003;302(5649):1364–1368. doi: 10.1126/science.1089427. [DOI] [PubMed] [Google Scholar]
- 11.Jiang L, et al. De novo computational design of retro-aldol enzymes. Science. 2008;319(5868):1387–1391. doi: 10.1126/science.1152692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rothlisberger D, et al. Kemp elimination catalysts by computational enzyme design. Nature. 2008;453(7192):190–195. doi: 10.1038/nature06879. [DOI] [PubMed] [Google Scholar]
- 13.Chica RA, Doucet N, Pelletier JN. Semi-rational approaches to engineering enzyme activity: Combining the benefits of directed evolution and rational design. Curr Opin Biotech. 2005;16(4):378–384. doi: 10.1016/j.copbio.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 14.Shortle D. The denatured state (the other half of the folding equation) and its role in protein stability. FASEB J. 1996;10(1):27–34. doi: 10.1096/fasebj.10.1.8566543. [DOI] [PubMed] [Google Scholar]
- 15.Dunbrack RL, Cohen FE. Bayesian statistical analysis of protein side-chain rotamer preferences. Protein Sci. 1997;6(8):1661–1681. doi: 10.1002/pro.5560060807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lassila JK, Privett HK, Allen BD, Mayo SL. Combinatorial methods for small-molecule placement in computational enzyme design. Proc Natl Acad Sci USA. 2006;103(45):16710–16715. doi: 10.1073/pnas.0607691103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dahiyat BI, Mayo SL. Probing the role of packing specificity in protein design. Proc Natl Acad Sci USA. 1997;94(19):10172–10177. doi: 10.1073/pnas.94.19.10172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grigoryan G, Ochoa A, Keating AE. Computing van der Waals energies in the context of the rotamer approximation. Proteins. 2007;68(4):863–878. doi: 10.1002/prot.21470. [DOI] [PubMed] [Google Scholar]
- 19.Hu X, Wang H, Ke H, Kuhlman B. High-resolution design of a protein loop. Proc Natl Acad Sci USA. 2007;104(45):17668–17673. doi: 10.1073/pnas.0707977104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pokala N, Handel TM. Energy functions for protein design: Adjustment with protein-protein complex affinities, models for the unfolded state, and negative design of solubility and specificity. J Mol Biol. 2005;347(1):203–227. doi: 10.1016/j.jmb.2004.12.019. [DOI] [PubMed] [Google Scholar]
- 21.Havranek JJ, Harbury PB. Automated design of specificity in molecular recognition. Nat Struct Biol. 2003;10(1):45–52. doi: 10.1038/nsb877. [DOI] [PubMed] [Google Scholar]
- 22.Ambroggio XI, Kuhlman B. Computational design of a single amino acid sequence that can switch between two distinct protein folds. J Am Chem Soc. 2006;128(4):1154–1161. doi: 10.1021/ja054718w. [DOI] [PubMed] [Google Scholar]
- 23.Boas FE, Harbury PB. Design of protein-ligand binding based on the molecular-mechanics energy model. J Mol Biol. 2008;380(2):415–424. doi: 10.1016/j.jmb.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Allen BD, Mayo SL. An efficient algorithm for multistate protein design based on FASTER. J Comput Chem. 2010;31:904–916. doi: 10.1002/jcc.21375. [DOI] [PubMed] [Google Scholar]
- 25.Kono H, Saven JG. Statistical theory for protein combinatorial libraries. Packing interactions, backbone flexibility, and the sequence variability of a main-chain structure. J Mol Biol. 2001;306(3):607–628. doi: 10.1006/jmbi.2000.4422. [DOI] [PubMed] [Google Scholar]
- 26.Hayes RJ, et al. Combining computational and experimental screening for rapid optimization of protein properties. Proc Natl Acad Sci USA. 2002;99(25):15926–15931. doi: 10.1073/pnas.212627499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mena MA, Daugherty PS. Automated design of degenerate codon libraries. Protein Eng Des Sel. 2005;18(12):559–561. doi: 10.1093/protein/gzi061. [DOI] [PubMed] [Google Scholar]
- 28.Treynor TP, Vizcarra CL, Nedelcu D, Mayo SL. Computationally designed libraries of fluorescent proteins evaluated by preservation and diversity of function. Proc Natl Acad Sci USA. 2007;104(1):48–53. doi: 10.1073/pnas.0609647103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kirsten Frank M, Dyda F, Dobrodumov A, Gronenborn AM. Core mutations switch monomeric protein GB1 into an intertwined tetramer. Nat Struct Biol. 2002;9(11):877–885. doi: 10.1038/nsb854. [DOI] [PubMed] [Google Scholar]
- 30.Byeon IJ, Louis JM, Gronenborn AM. A protein contortionist: Core mutations of GB1 that induce dimerization and domain swapping. J Mol Biol. 2003;333(1):141–152. doi: 10.1016/s0022-2836(03)00928-8. [DOI] [PubMed] [Google Scholar]
- 31.Jee J, Byeon IJ, Louis JM, Gronenborn AM. The point mutation A34F causes dimerization of GB1. Proteins. 2008;71(3):1420–1431. doi: 10.1002/prot.21831. [DOI] [PubMed] [Google Scholar]
- 32.Larson SM, England JL, Desjarlais JR, Pande VS. Thoroughly sampling sequence space: Large-scale protein design of structural ensembles. Protein Sci. 2002;11(12):2804–2813. doi: 10.1110/ps.0203902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fu X, Apgar JR, Keating AE. Modeling backbone flexibility to achieve sequence diversity: The design of novel alpha-helical ligands for Bcl-xL. J Mol Biol. 2007;371(4):1099–1117. doi: 10.1016/j.jmb.2007.04.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schneider M, Fu X, Keating AE. X-ray vs.NMR structures as templates for computational protein design. Proteins. 2009;77(1):97–110. doi: 10.1002/prot.22421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mayo SL, Olafson BD, Goddard WA. Dreiding—a generic force-field for molecular simulations. J Phys Chem. 1990;94(26):8897–8909. [Google Scholar]
- 36.Rohl CA, Strauss CE, Misura KM, Baker D. Protein structure prediction using Rosetta. Methods Enzymol. 2004;383:66–93. doi: 10.1016/S0076-6879(04)83004-0. [DOI] [PubMed] [Google Scholar]
- 37.Mendes J, Guerois R, Serrano L. Energy estimation in protein design. Curr Opin Struct Biol. 2002;12(4):441–446. doi: 10.1016/s0959-440x(02)00345-7. [DOI] [PubMed] [Google Scholar]
- 38.Guerois R, Nielsen JE, Serrano L. Predicting changes in the stability of proteins and protein complexes: A study of more than 1000 mutations. J Mol Biol. 2002;320(2):369–387. doi: 10.1016/S0022-2836(02)00442-4. [DOI] [PubMed] [Google Scholar]
- 39.Yin S, Ding F, Dokholyan NV. Eris: An automated estimator of protein stability. Nat Methods. 2007;4(6):466–467. doi: 10.1038/nmeth0607-466. [DOI] [PubMed] [Google Scholar]
- 40.Potapov V, Cohen M, Schreiber G. Assessing computational methods for predicting protein stability upon mutation: Good on average but not in the details. Protein Eng Des Sel. 2009;22(9):553–560. doi: 10.1093/protein/gzp030. [DOI] [PubMed] [Google Scholar]
- 41.Grigoryan G, Reinke AW, Keating AE. Design of protein-interaction specificity gives selective bZIP-binding peptides. Nature. 2009;458(7240):859–864. doi: 10.1038/nature07885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gallagher T, Alexander P, Bryan P, Gilliland GL. 2 crystal-structures of the B1 immunoglobulin-binding domain of streptococcal protein-G and comparison with Nmr. Biochemistry. 1994;33(15):4721–4729. [PubMed] [Google Scholar]
- 43.Gronenborn AM, et al. A novel, highly stable fold of the immunoglobulin binding domain of streptococcal protein G. Science. 1991;253(5020):657–661. doi: 10.1126/science.1871600. [DOI] [PubMed] [Google Scholar]
- 44.Word JM, Lovell SC, Richardson JS, Richardson DC. Asparagine and glutamine: Using hydrogen atom contacts in the choice of side-chain amide orientation. J Mol Biol. 1999;285(4):1735–1747. doi: 10.1006/jmbi.1998.2401. [DOI] [PubMed] [Google Scholar]
- 45.Gordon DB, Marshall SA, Mayo SL. Energy functions for protein design. Curr Opin Struct Biol. 1999;9(4):509–513. doi: 10.1016/s0959-440x(99)80072-4. [DOI] [PubMed] [Google Scholar]
- 46.Gordon DB, Hom GK, Mayo SL, Pierce NA. Exact rotamer optimization for protein design. J Comput Chem. 2003;24(2):232–243. doi: 10.1002/jcc.10121. [DOI] [PubMed] [Google Scholar]
- 47.Allen BD, Mayo SL. Dramatic performance enhancements for the FASTER optimization algorithm. J Comput Chem. 2006;27(10):1071–1075. doi: 10.1002/jcc.20420. [DOI] [PubMed] [Google Scholar]
- 48.Stemmer WP, Crameri A, Ha KD, Brennan TM, Heyneker HL. Single-step assembly of a gene and entire plasmid from large numbers of oligodeoxyribonucleotides. Gene. 1995;164(1):49–53. doi: 10.1016/0378-1119(95)00511-4. [DOI] [PubMed] [Google Scholar]
- 49.Dirks RM, Pierce NA. A partition function algorithm for nucleic acid secondary structure including pseudoknots. J Comput Chem. 2003;24(13):1664–1677. doi: 10.1002/jcc.10296. [DOI] [PubMed] [Google Scholar]
- 50.Dirks RM, Pierce NA. An algorithm for computing nucleic acid base-pairing probabilities including pseudoknots. J Comput Chem. 2004;25(10):1295–1304. doi: 10.1002/jcc.20057. [DOI] [PubMed] [Google Scholar]
- 51.Gerth ML, Patrick WM, Lutz S. A second-generation system for unbiased reading frame selection. Protein Eng Des Sel. 2004;17(7):595–602. doi: 10.1093/protein/gzh068. [DOI] [PubMed] [Google Scholar]
- 52.Aucamp JP, Cosme AM, Lye GJ, Dalby PA. High-throughput measurement of protein stability in microtiter plates. Biotechnol Bioeng. 2005;89(5):599–607. doi: 10.1002/bit.20397. [DOI] [PubMed] [Google Scholar]
- 53.Santoro MM, Bolen DW. Unfolding free energy changes determined by the linear extrapolation method.1. Unfolding of phenylmethanesulfonyl alpha-chymotrypsin using different denaturants. Biochemistry. 1988;27(21):8063–8068. doi: 10.1021/bi00421a014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.