

Abstract
Protein stabilization was achieved through in vivo screening based on the thermodynamic linkage between protein folding and fragment complementation. The split GFP system was found suitable to derive protein variants with enhanced stability due to the correlation between effects of mutations on the stability of the intact chain and the effects of the same mutations on the affinity between fragments of the chain. PGB1 mutants with higher affinity between fragments 1 to 40 and 41 to 56 were obtained by in vivo screening of a library of the 1 to 40 fragments against wild-type 41 to 56 fragments. Colonies were ranked based on the intensity of green fluorescence emerging from assembly and folding of the fused GFP fragments. The DNA from the brightest fluorescent colonies was sequenced, and intact mutant PGB1s corresponding to the top three sequences were expressed, purified, and analyzed for stability toward thermal denaturation. The protein sequence derived from the top fluorescent colony was found to yield a 12 °C increase in the thermal denaturation midpoint and a free energy of stabilization of -8.7 kJ/mol at 25 °C. The stability rank order of the three mutant proteins follows the fluorescence rank order in the split GFP system. The variants are stabilized through increased hydrophobic effect, which raises the free energy of the unfolded more than the folded state; as well as substitutions, which lower the free energy of the folded more than the unfolded state; optimized van der Waals interactions; helix stabilization; improved hydrogen bonding network; and reduced electrostatic repulsion in the folded state.
Keywords: green fluorescent protein, protein stability, protein stabilization method, protein evolution, protein reconstitution
The fold of a protein and its stability toward denaturation are governed by the sequence of amino acids and the environment (solvent, salts, pH, T, crowding, etc.). The fold and stability of the protein are dependent on a multitude of noncovalent interactions (hydrogen bonds, electrostatic interactions, van der Waals interactions, hydrophobic effect) under the steric constraints imposed by the covalent chain. With protein stability defined as the free energy difference between the folded and unfolded state, the stability will increase with any factor that raises the free energy of the unfolded state more than the folded state and with any factor that lowers the free energy of the folded state more than the unfolded state.
A common problem with proteins used in therapeutic, diagnostic, or technical applications is their limited stability toward different forms of handling and storage, or after administration in the body. The proteins may be sensitive to denaturation and/or proteolytic degradation. The therapeutic and technical value of the proteins may therefore increase if their stability can be improved.
Several approaches have been used to stabilize proteins through formulation or through redesign of the amino acid sequence. Formulation with sugars, liquid crystals, polymeric nanoparticles, liposomes, emulsions, or microencapsulation, or by site-specific glycosylation, may protect the native protein toward unfolding and/or degradation (reviewed in refs. 1–3). Computational approaches to improve protein stability have focused on improving the packing of the hydrophobic core and on optimizing interactions in partially solvent exposed regions (4, 5). Screening methods with combinatorial approaches such as phage display have been applied to intact proteins and used increased protease resistance as a selection criterion leading to variants with increased thermal stability (6). There are many reported cases of stabilized proteins by directed evolution (reviewed in ref. 7) and rational design (reviewed in ref. 8). In directed evolution, most strategies in screening for stability depend on activity measurements during or after exposure to denaturing conditions (9).
Here we introduce an alternative approach to protein stabilization based on the thermodynamic linkage between fragment complementation and protein folding relying on in vivo screening under physiological conditions. Our approach is based on the correlation between mutational effects on the stability of the intact chain and the effects of the same mutations on the affinity between fragments of the chain (10–12) as illustrated in Fig. 1. Screening based on fragment complementation and phage display has previously been used to study the role of particular amino acids in the specificity of folding and stabilization of the corresponding intact proteins (13, 14).
Fig. 1.

Correlation between mutational effects on the stability of the intact chain and the effects of the same mutations on the affinity between fragments of the chain (10–12).
The combined interactions, which stabilize the protein fold, are often strong and specific enough to allow for reconstitution of the protein from fragments of the amino acid chain. Hence, an intact covalent chain of amino acids is not necessary to maintain the fold and function of a protein. This was found in the 1950s for ribonuclease (15) and in the 1970s–2000s for a number of other proteins (see, for example, refs. 16–24). The reconstituted complex has lower and concentration-dependent stability toward denaturation than the intact protein, but functions like receptor or ligand binding are often retained.
As illustrated in Fig. 1, there is a strong correlation between mutational effects on the stability of the intact chain and the effects of the same mutations on the affinity between fragments of the chain (10–12, 25). This implies that proteins may be stabilized using a method based on protein reconstitution from fragments and screening of a fragment library for enhanced affinity. Preliminary studies using the split GFP system and fragment complementation systems of widely different affinities and stabilities pointed to a markedly brighter green fluorescence in the higher affinity/stability system (26). Here we built on this finding to develop an in vivo screening method that can be used to identify reconstitution couples of improved affinities and subsequently to produce intact protein variants with increased stability toward unfolding. As a test case we use the 56-residue B1 domain of protein G (PGB1) with a β-grasp fold (27) that can reconstitute from fragments (28). We use a split GFP system, described in depth elsewhere (29–31), to screen for PGB1 mutants with higher affinity between two fragments comprising residues 1 to 40 and 41 to 56, respectively (Fig. 2) as visualized from brighter green fluorescence. After expression and purification of the corresponding intact mutants we show that this leads to the production of markedly stabilized protein variants. The protein sequence derived from the top fluorescent colony yields a 12 °C increase in the midpoint of thermal denaturation and a free energy of stabilization of -8.7 kJ/mol at 25 °C.
Fig. 2.

(A) A cartoon model of the ribbon structure of split GFP with fused PGB1 fragments. The model is produced using MOLMOL (42) and the pdb files 1ema (43) and 1pgb (44). (B) Topology of PGB1 fragments fused to GFP fragments. (C) Comparison of fluorescence intensity of Top1 and parent construct as well as negative (noninduced) and positive (CBD9k) controls. (D) The sequences of the fused fragments where the underlined part represents the PGB1 fragments. (E) Outline of the library with 3,456 possible sequences. Previously determined mutants that are stabilizing (F3, I7, I16, I18, E25, F29, I39), destabilizing (L18, Q25, K29), and possible mutations arising from the nature of the genetic code are included in the library. (F) Structure of parent protein where the seven library positions are indicated. The structures are produced using MOLMOL (41).
Results
Nomenclature.
PGB1-QDD (also called parent) denotes a variant of PGB1 (protein G B1-domain from Streptococcus sp.) with the three substitutions T2Q (to avoid N-terminal processing) (32), N8D, and N37D (to avoid covalent rearrangement during deamidation) (33, 34). PGB1-40-CGFP denotes a fusion construct in which residues 1 to 40 of PGB1-QDD are followed by a seven-residue linker and then residues 158 to 238 of GFP. NGFP-PGB41-56 denotes a fusion construct in which a hexa-histidine tag is followed by residues 1 to 157 of GFP, then an eight-residue linker, and then residues 41 to 56 of PGB1 (Fig. 2).
Coexpression of Parent Constructs.
Green fluorescent was observed under inducing conditions after coexpression of NGFP-PGB41-56 and PGB1-40-CGFP (Fig. 2). Most colonies were found to be fluorescent after 2 days at room temperature. The fluorescence intensity is, however, much lower than for the very high affinity control case of calbindin D9k fragments fused to the GFP fragments (Fig. 2). No green fluorescence was observed under noninducing conditions nor from colonies on the negative control plate with cotransformed vectors carrying NGFP and CGPF devoid of fusion partners (Fig. 2).
Library Design.
A small library of 3456 variants of PGB1-40 was designed for proof of principle. The library is randomized over two to nine choices at positions 3, 7, 16, 18, 25, 29, and 39, while the other 33 positions are kept constant according to the PGB1-40-QDD sequence (Fig. 2). In addition to the wild-type residues (Y3, L7, T16, T18, T25, V29, and V39), the library contains seven alternatives (F3, I7, I16, I18, E25, F29, I39) previously found in different combinations in stabilized variants, three alternatives (L18, Q25, K29) found in destabilized variants (5, 6, 35, 36), as well as 11 novel alternatives (P18, L18, K25, A25, P25, E29, D29, N29, Y29, L29, and I29), which arise because some amino acid substitutions require more than one base change. The library was produced by overlapping PCR from degenerate oligonucleotides and cloned on the N-terminal side of the gene for residues 158 to 238 of GFP, to obtain PGB1-40lib-CGFP (see SI Text). The library is thus a collection of plasmids in which each plasmid has the same GFP158-238 sequence but a unique PGB1-40 sequence (out of 3,456 possibilities).
Library Coexpression, Screening, and Sequencing.
PGB1-40lib-CGFP was cotransformed with the constant construct NGFP-PGB41-56 and spread on plates with inducers to yield a total of 278 colonies. Colonies that were found to develop similar or brighter green fluorescence than parent were picked for individual overnight cultures, plasmid preparation, and DNA sequencing. Each single-clone plasmid was cotransformed with NGFP-PGB41-56 and plated on several agar plates with inducers. The plates were compared side by side with each other and with the parent constructs to obtain a fluorescence rank order. After this reassessment of fluorescence intensity, 25 clones were judged to have brighter green fluorescence than parent. The amino acid sequences of the PGB1-40-CGFP fragments from the top three fluorescent clones are listed in Table 1 and from all top 25 fluorescent clones in Table S1. The corresponding intact proteins based on the sequences from the three brightest colonies are named Top1, Top2, and Top3, with Top1 representing the clone with the brightest green fluorescence, Top2 the second brightest, and Top3 the third brightest.
Table 1.
The three top fluorescent sequences ranked by estimated intensity
| Sequence |
||
| 10 20 30 40 | ||
| Rank Order | Colony | ....|....|....|....|....|....|....|....| |
| - | parent | MQYKLILDGKTLKGETTTEAVDAATAEKVFKQYANDDGVD |
| 1 | Top1 | ......I.................A...F.........I. |
| 2 | Top2 | ......I.................A...F........... |
| 3 | Top3 | ......I..........I......A..T.....T....I. |
Protein Expression and Purification.
PGB1-QDD parent and three variants (Top1, Top2, and Top3) were expressed in Escherichia coli (De3 pLysS BL21 star) and purified by ion exchange and gel filtration chromatography. Excellent yields of highly pure protein (around 150 mg/L culture) were obtained for all three variants. The proteins were judged homogeneous based on analysis by SDS/PAGE, analytical gel filtration chromatography, and mass spectrometry (see Materials and Methods and Figs. S1, S2 and S3). None of PGB1-QDD or variants migrate in SDS/PAGE according to their expected molecular weights but instead migrate similarly to one another (Fig. S1), probably because of poor SDS binding. In gel filtration, the elution volume and profile for PGB1-QDD and mutants was found to be independent of concentration (Fig. S2) and in agreement with monomer based on earlier reports (37). By mass spectrometry, a singly charged species with the expected monomer Mw was found to be strongly dominating for PGB1-QDD and the three variants (Fig. S3). Altogether our analyses indicate that PGB1-QDD and the three variants are monomeric.
Secondary Structure.
Far-UV CD spectra were recorded to evaluate the secondary structure of the three variants in comparison to the parent PGB1-QDD. The spectra (Fig. 3A) suggest that Top1, Top2, and Top3 have highly similar structures compared to the parent protein. The small changes in intensity are most likely due to concentration differences or minor structural rearrangements.
Fig. 3.

Normalized thermal unfolding as observed from loss in CD signal at 218 nm. Parent (solid line and diamonds), Top1 (dotted line and triangles), Top2 (dashed line and squares), Top3 (long dashed line and circles). The normalized fit to data is shown as solid line in all cases. Data are shown as lines with symbols at every tenth data point. Inset: Far-UV CD spectra of Top1–3 and parent.
Thermal Denaturation by CD.
The CD signal at 218 nm was monitored during thermal unfolding of PGB1-QDD parent and variants. Heating of Top1, Top2, and Top3 leads to a loss of CD signal. The denaturation data follows a sigmoidal curve indicative of cooperative denaturation events (Fig. 3B). Reverse thermal scans reveal reversible denaturation processes (Fig. S4). All three variants appear more stable than parent as their denaturation curves are shifted to higher temperature. In order to obtain melting temperatures and thermodynamic parameters, Eq. 1 was fitted to the data (Table 2). This shows that the midpoint of thermal denaturation has increased by 12 °C for Top1, 9 °C for Top2, and 8 °C for Top3 relative to parent. The extrapolated standard free energies of unfolding, ΔG°, at 25 °C have increased by 8.7 (Top1), 7.2 (Top2), and 3.2 (Top3) kJ/mol relative to parent. The values for ΔG°, however, contain greater uncertainty compared to Tm due to the lengthy extrapolation and the combination of uncertainties in 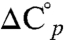 , ΔG° and Tm. The error is not easily determined and is left out.
, ΔG° and Tm. The error is not easily determined and is left out.
Table 2.
Thermodynamic parameters of parent and Top1–3 where standard errors are displayed
| Protein |
Tm (°C) |
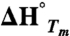 (kJ/mol) (kJ/mol) |
ΔG° (25 °C) (kJ/mol) |
| Top1 | 61.9 ± 0.40 | 236 ± 8 | 22.2 |
| Top2 | 59.4 ± 0.08 | 233 ± 2 | 20.7 |
| Top3 | 58.0 ± 0.13 | 199 ± 4 | 16.7 |
| parent | 50.3 ± 0.22 | 196 ± 5 | 13.5 |
Fragment Complementation.
The affinity between complementary fragments comprising residues 1 to 41 (with parent, Top1, Top2, or Top3 sequence) and 41 to 56 (WT) was measured using CD spectroscopy of fragment mixtures (as described in SI Text). Top1, Top2, and Top3 were found to have ca. one order of magnitude higher affinity than parent. However, the precision of the measurements did not allow ranking of the variants relative to one another.
Discussion
In nature, proteins are evolved for optimal function, not maximal stability. This means that there is room for significant stability improvement, and this may be achieved by any combination of substitutions that lowers the free energy of the folded more than unfolded state or raises the free energy of unfolded more than folded state. In line with this, each of the stabilized variants found here contains a different combination of substitutions even though a relatively small and confined library was explored. With a wider and more disparate library, e.g., produced by error-prone PCR, gene shuffling or degenerate oligonucleotides, one may foresee selection of an even larger repertoire of stabilizing substitutions.
The stabilized variants are novel combinations of the mutations present in the library, although several of the mutations have been seen before in other combinations (5, 6, 35, 36). In addition, the stabilized variants found here contain novel substitutions representing the extra amino acids in the library arising from the genetic code, as well as spontaneous mutations not in the library design. The two most stable proteins, Top1 (12 °C Tm increase) and Top2 (9 °C Tm increase), contain the substitutions L7I, T25A, and V29F, while Top1 has the additional substitution V39I. Residues 7 and 39 interact with each other in the hydrophobic core. Introduction of both I7 and I39 as in Top1 occurs in 10 of the top 25 clones (Table S2) and improves the hydrophobic core packing (4). In the parent protein, V29 is located at a cleft between residues T16, T18, V29, F30, and Y33. The larger side chain of F29 may fill the space of the pocket better (35) to provide additional van der Waals’ interactions (decreasing the free energy of the folded state more than the unfolded state) and increased hydrophobic effect (increasing the free energy of the unfolded state more than the folded state). T25A is a novel stabilizing residue, not observed in previous studies. A25 is one turn away from F29 in the helix and may contribute further hydrophobic stabilization of this region. In addition, Ala is a better helix stabilizer than Thr, especially when found in the first turn of the helix, as is the case for position 2. The substitutions T25A, as well as L7I and V39I, are found in Top3 also (8 °C Tm increase), but instead of V29F we see two spontaneous mutations (K28T and A34T) that were not in the library design but have occurred during PCR amplification. Both residues are in the helix, and in the wild-type protein the side chain of A34 is completely buried, so the substitution to Thr is somewhat surprising. However, the Ala34 side chain packs against the Trp43 side chain, and the hydrogen bonding capacity of the Trp43 side chain may be better satisfied in the folded state by the introduction of Thr34, while in the unfolded state there may be little or no change as hydrogen bonds to solvent may dominate. Thus, the A34T substitution may lower the free energy of the folded state more than the unfolded state. K28 is located on the solvent exposed side of the helix between Thr25 (Ala25 in Top 3) and Lys31. The substitution K28T may stabilize the helix by removing a local electrostatic repulsion. The beneficial effect of the substitution would be smaller in the unfolded state where the two charges are further apart. In summary, Top1, Top2, and Top3 seem to be stabilized due to increased hydrophobic effect as well as increased van der Waals interactions, increased helix stability, improved hydrogen bonding network, and reduced electrostatic repulsion in the folded state.
The thermodynamic linkage between fragment complementation and protein stability was successfully exploited for in vivo screening for stabilized proteins. The approach relies on the correlation between mutational effects on the affinity of association between protein fragments and the effects on the stability of the corresponding intact protein. This correlation allows protein variants with enhanced stability to be derived from in vivo selection of higher affinity couples based on fluorescence intensity in a split GFP system. The increased affinity may arise from mutations that favor the associated state more than the dissociated state or mutations that disfavor the dissociated state more than the associated state. An advantage of in vivo screening is that the design of the library does not have to rely on one or the other option, and both options are explored in parallel to increase the likelihood of stability improvement of the corresponding intact protein. The sequence substitutions derived from the three most brightly fluorescent colonies yield fragment couples with increased affinity and also greatly improved stability toward denaturation when incorporated in intact protein variants, and the stability rank order follows the fluorescence rank order. This remarkable finding supports the idea that increased fluorescence is related to increased affinity, which in turn is related to increased stability.
The success of the current study relies on the ability to detect small changes in fluorescence intensity from E. coli colonies or cells in liquid culture. In an earlier work we showed that large changes in fluorescence intensity can be quantified during incubation and growth in a fluorescence plate reader (26). In the present work, the small differences in fluorescence intensity among different members of the library were not captured using the plate reader and were instead ranked by human eye twice. First colonies judged to have brighter green fluorescence than parent were picked for cultivation and preparation of a collection of plasmids each containing one mutant of PGB1-40-CGFP. Then each one of these PGB1-40-CGFP plasmids was cotransformed with the plasmid coding for NGFP-PGB41-56 and plated on several plates for a second round of fluorescence intensity ranking in comparison with the parent construct. The production of greatly stabilized proteins based on the sequences found in the top three fluorescent colonies underscores the ability of the human eye to rank small differences in fluorescence intensity by integrating over a large number of colonies and normalizing against differences in colony size and background intensity.
The affinity between the two GFP fragments in the absence of a fusion partner is too low to allow for maturation of the chromophore, and colonies display no detectable green fluorescence (30). However, the rather low affinity between the two fragments of PGB1 (KD = 200 μM at physiological salt concentration) (38) is sufficient to promote association, folding, and chromophore maturation of the fused GFP parts, and colonies develop green fluorescence (Fig. 2). As expected the fluorescence intensity is much lower than for the very high affinity case of calbindin D9k fragments fused to the GFP fragments (Fig. 2) (26).
The current work relies on the thermodynamic link between effects on folding of an intact protein chain and on the association of fragments to reconstitute the protein. In addition, there is a link between fragment complementation and three dimensional domain swapping, in which two protein monomers dimerize via subdomain interchange so that the native fold is reconstituted twice in the dimer from segments of both chains (39, 40). Indeed, a switch between monomer and domain swapped dimer is a small evolutionary leap that can involve single amino acid substitutions (reviewed in ref. 40). Directed evolution of intact proteins to improve stability may therefore pick up substitutions that promote dimerization rather than stabilization of the monomer per se. This is avoided by the current approach, which relies on fragment complementation. The affinity between the interacting subdmomain fragments (and the stability of the corresponding intact protein) is enhanced either through substitutions that increase the prefolding of the isolated subdomains or strengthen the intramolecular interactions between them compared to the dissociated state, whereas substitutions that promote dimerization of the intact protein may not affect the affinity between the fragments.
PGB1-QDD, with the T2Q, N8D, and N37D substitutions, was chosen as a starting point for the present study because it lacks two deamidation sites and is protected against covalent rearrangement of the chain. The two Asn->Asp substitutions change the net charge at neutral pH from -4 to -6, leading to lower thermal stability; however, this is not a disadvantage in a study aiming at method development. The previously stabilized and destabilized variants were based on PGB1 with the T2Q substitution, PGB1-Q, but we argued that it is highly likely that some of these substitutions also stabilize PGB1-QDD.
The fragment library used in the present study is a small focused library of 3456 variants designed for proof-of-principle. We argued that to develop and evaluate a method for protein stabilization, a small library is sufficient if it contains both stabilizing and destabilizing amino acids. The library thus contains 10 point mutations that are previously found in different combinations to stabilize or destabilize PGB1 (5, 6, 35, 36). The use of a random library allowed for the emergence of new stabilizing combinations that also encompass some of the 11 novel substitutions in the library. In future studies, beyond proof of principle, larger libraries would yield more candidates and there may be no previous knowledge to base libraries on. It may also be desirable in future studies to vary residues in both complementary fragments. Error-prone PCR, gene shuffling, or degenerate oligonucleotides with a low frequency of random substitutions over the whole sequence may then be chosen for library generation with the advantage that any amino acid residue in a protein may change and no previous knowledge is needed about which positions are likely to play a role. However, to retain protein identity, library generation is preferably performed at a level that changes a small fraction of residues in every individual sequence. A general method for protein stabilization based on the current results is outlined in SI Text.
Conclusion
The present results show that the thermodynamic link between factors governing fragment complementation and protein stability can be exploited for in vivo screening for stabilized proteins. Protein variants with enhanced stability can be evolved based on fluorescence intensity in a split GFP system. Enhanced green fluorescence of E. coli cells or colonies reflects increased affinity between the protein fragments, which correlates with higher stability toward unfolding of the corresponding intact protein chain. The stabilized proteins are monomeric because the reconstitution approach avoids substitutions that improve stability through dimerization. The protein variants are stabilized through different combinations of substitutions that decrease the free energy of the folded more than the unfolded state, or increase the free energy of the unfolded more than the folded state: optimized van der Waals interactions, hydrophobic effect, helix stability, hydrogen bonding network, and reduced electrostatic repulsion in the folded state.
Materials and Methods
Cloning of NGFP-PGB41-56 and PGB1-40-CGFP.
The cloning procedure and primers used are described in SI Text and Table S2. The pET11a-link-GFP carrying the gene for GFP residues 1 to 157 (NGFP; ampicillin resistant) and pMRBAD-link-CGFP carrying the gene for GFP residues 158 to 238 (CGFP¸ kanamycin resistant) vectors were kind gifts from professor Lynne Regan, Yale University (31).
Library Cloning.
A PGB1-40 library was produced by overlap extension PCR of degenerate oligonucleotides to yield a DNA library that translates into a protein fragment library PGB1-40lib-CGFP (Fig. 2E). The PCR and cloning procedures are described in SI Text.
Coexpression, Screening for High Affinity Variants, and DNA Sequencing.
The plasmid coding for NGFP-PGB41-56 was cotransfomed with PGB1-40-CGFP or PGB1-40lib-CGFP in E. coli ER2566 and spread on LB agar plates with 100 μg/mL ampicillin, 35 μg/mL kanamycin, 10 μM isopropyl-β-D-1-thiogalactosid (IPTG), and 0.2% (w/v) arabinose (inducing plates) or with 100 μg/mL ampicillin and 35 μg/mL kanamycin (noninducing plate) and imaged as described in SI Text. The screening procedure to find for fragments with improved affinity and DNA sequencing is described in SI Text.
Cloning, Expression, and Purification of Intact Protein of Selected Variants.
Intact PGB1 variants were expressed in E. coli, purified using anion exchange and gel filtration chromatography, and judged homogeneous by SDS/PAGE, analytical gel filtration chromatography, and mass spectrometry, as described in SI Text.
Circular Dichroism (CD) Spectroscopy.
CD spectra were recorded using a JASCO J-815 CD spectrometer (Jasco Corporation) with a JASCO PTC- 423S/15 Peltier type thermostated cell holder. CD spectra were recorded from 250 to 190 nm at 20 °C in a 1 mm cuvette at a protein concentration of 42 μM with an average of 3 scans (scan rate 20 nm/ min, response 8 s, bandwidth 1 nm, resolution 1 nm).
Thermal Denaturation by CD Spectroscopy.
The CD signal at 218 nm was monitored during thermal unfolding of the PGB1-QDD variants from 5–95 °C or 20–80 °C at a scan rate of 0.5° C/ min or 1.0° C/ min response of 16 s and bandwith of 1 nm. The protein concentration was 0.36 or 0.18 mg/ml in 10 mM sodium phosphate, 150 mM NaF pH 7.2, and a quartz cuvette with 1 mm pathlength was used. To investigate the reversibility, a thermal scan from 80–20 °C was monitored after the upward scan. Structural changes were determined from far-UV CD spectra, at 20 or 80 °C, before and after each thermal scan. Spectra were recorded between 250 and 190 nm, the scan rate was 20 nm/ min, the response 8 s, accumulations 3, and the bandwidth 1 nm. Assuming a two-state reversible unfolding Tm, 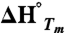 and ΔG° were obtained by fitting Eq. 1 to CD data.
and ΔG° were obtained by fitting Eq. 1 to CD data.
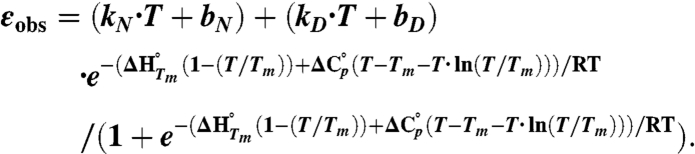 |
[1] |
In Eq. 1, εobs is the observed ellipticity at 218 nm, kN, bN, kD and bD define the baselines of the native and denatured states, respectively. 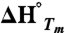 is the enthalpy of unfolding, T is the temperature in Kelvin, and R is the gas constant.
is the enthalpy of unfolding, T is the temperature in Kelvin, and R is the gas constant. 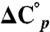 is the difference in heat capacity between the denatured and native states and is assumed to be constant in the studied temperature interval. The previously determined value for
is the difference in heat capacity between the denatured and native states and is assumed to be constant in the studied temperature interval. The previously determined value for 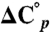 of PGB1-QDD (1.8 kJ/(K·mol) was used (41). To obtain ΔG° (25 °C) the standard free energy of unfolding was extrapolated from Tm to 25 °C:
of PGB1-QDD (1.8 kJ/(K·mol) was used (41). To obtain ΔG° (25 °C) the standard free energy of unfolding was extrapolated from Tm to 25 °C:
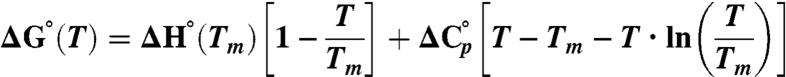 |
[2] |
where the weighted averages for Tm and 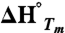 from 3 to 6 measurements were used.
from 3 to 6 measurements were used.
Supplementary Material
Acknowledgments.
The authors thank Professor Lynne Regan, Yale University, for providing plasmids, and Dr. David A Schultz, University of California, San Diego, for scientific discussions. This work was supported by the Swedish Research Council and its Linneaus OMM center, Carl Trygger Foundation, Royal Physiographic Society, Crafoord Foundation, and the PhD research school FLÄK.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1005689107/-/DCSupplemental.
References
- 1.Jorgensen L, Hostrup S, Moeller EH, Grohganz H. Recent trends in stabilising peptides and proteins in pharmaceutical formulation-considerations in the choice of excipients. Expert Opin Drug Del. 2009;6:1219–1230. doi: 10.1517/17425240903199143. [DOI] [PubMed] [Google Scholar]
- 2.Solá RJ, Griebenow K. Effects of glycosylation on the stability of protein pharmaceuticals. J Pharm Sci. 2009;98:1223–1245. doi: 10.1002/jps.21504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.van der Walle CF, Sharma G, Ravi Kumar MNV. Current approaches to stabilising and analysing proteins during microencapsulation in PLGA. Expert Opin Drug Del. 2009;6:177–186. doi: 10.1517/17425240802680169. [DOI] [PubMed] [Google Scholar]
- 4.Dahiyat BI, Mayo SL. Probing the role of packing specificity in protein design. Proc Natl Acad Sci USA. 1997;94:10172–10177. doi: 10.1073/pnas.94.19.10172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Malakauskas SM, Mayo SL. Design, structure and stability of a hyperthermophilic protein variant. Nat Struct Mol Biol. 1998;5:470–475. doi: 10.1038/nsb0698-470. [DOI] [PubMed] [Google Scholar]
- 6.Wunderlich M, Martin A, Staab CA, Schmid FX. Evolutionary protein stabilization in comparison with computational design. J Mol Biol. 2005;351:1160–1168. doi: 10.1016/j.jmb.2005.06.059. [DOI] [PubMed] [Google Scholar]
- 7.Eijsink VGH, Gåseidnes S, Borchert TV, van den Burg B. Directed evolution of enzyme stability. Biomol Eng. 2005;22:21–30. doi: 10.1016/j.bioeng.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 8.Eijsink VGH, et al. Rational engineering of enzyme stability. J Biotechnol. 2004;113:105–120. doi: 10.1016/j.jbiotec.2004.03.026. [DOI] [PubMed] [Google Scholar]
- 9.Robertson DE, Steer BA. Recent progress in biocatalyst discovery and optimization. Curr Opin Chem Biol. 2004;8:141–149. doi: 10.1016/j.cbpa.2004.02.010. [DOI] [PubMed] [Google Scholar]
- 10.Berggård T, Julenius K, Ogard A, Drakenberg T, Linse S. Fragment complementation studies of protein stabilization by hydrophobic core residues. Biochemistry. 2001;40:1257–1264. doi: 10.1021/bi0014812. [DOI] [PubMed] [Google Scholar]
- 11.Ruiz-Sanz J, Prat-Gay Gd, Otzen DE, Fersht AR. Protein-fragments as models for events in protein-folding pathways—protein engineering analysis of the association of 2 complementary fragments of the barley chymotrypsin inhibitor-2 (Ci-2) Biochemistry. 1995;34:1695–1701. doi: 10.1021/bi00005a026. [DOI] [PubMed] [Google Scholar]
- 12.Xue WF, Szczepankiewicz O, Bauer MC, Thulin E, Linse S. Intra- versus intermolecular interactions in monellin: Contribution of surface charges to protein assembly. J Mol Biol. 2006;358:1244–1255. doi: 10.1016/j.jmb.2006.02.069. [DOI] [PubMed] [Google Scholar]
- 13.Linse S, Voorhies M, Norström E, Schultz DA. An EF-hand phage display study of calmodulin subdomain pairing. J Mol Biol. 2000;296:473–486. doi: 10.1006/jmbi.1999.3452. [DOI] [PubMed] [Google Scholar]
- 14.Schultz DR, Ladbury JE, Smith GP, Fox RO. Interactions of ribonuclease S with ligands from random peptide libraries. In: Ladbury JEaC BZ, editor. Applications of Calorimetry in the Biological Sciences. New York: John Wiley and Sons, Ltd; 1998. pp. 123–138. [Google Scholar]
- 15.Richards FM. On the enzymatic activity of subtilisin-modified ribonuclease. Proc Natl Acad Sci USA. 1958;44:162–166. doi: 10.1073/pnas.44.2.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Andria G, Taniuchi H, Cone JL. Specific binding of 3 fragments of staphylococcal nuclease. J Biol Chem. 1971;246:7421–7428. [PubMed] [Google Scholar]
- 17.Berggård T, Thulin E, Åkerfeldt KS, Linse S. Fragment complementation of calbindin D28k. Protein Sci. 2000;9:2094–2108. doi: 10.1110/ps.9.11.2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Finn BE, Kördel J, Thulin E, Sellers P, Forsén S. Dissection of calbindin D9k into two Ca(2+)-binding subdomains by a combination of mutagenesis and chemical cleavage. FEBS Lett. 1992;298:211–214. doi: 10.1016/0014-5793(92)80059-p. [DOI] [PubMed] [Google Scholar]
- 19.Gan ZR, Lewis SD, Stone JR, Shafer JA. Reconstitution of catalytically competent human zeta-thrombin by combination of zeta-thrombin residues A1-36 and B1-148 and an Escherichia coli expressed polypeptide corresponding to zeta-thrombin residues B149-259. Biochemistry. 1991;30:11694–11699. doi: 10.1021/bi00114a013. [DOI] [PubMed] [Google Scholar]
- 20.Juillerat M, Parr GR, Taniuchi H. A biologically active, three-fragment complex of horse heart cytochrome c. J Biol Chem. 1980;255:845–853. [PubMed] [Google Scholar]
- 21.Linse S, et al. Domain organization of calbindin D(28k) as determined from the association of six synthetic EF-hand fragments. Protein Sci. 1997;6:2385–2396. doi: 10.1002/pro.5560061112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Permyakov EA, Medvedkin VN, Mitin YV, Kretsinger RH. Noncovalent complex between domain AB and domains CD* EF of parvalbumin. Biochim Biophys Acta. 1991;1076:67–70. doi: 10.1016/0167-4838(91)90220-t. [DOI] [PubMed] [Google Scholar]
- 23.Shaw GS, Hodges RS, Sykes BD. Calcium-induced peptide association to form an intact protein domain: 1H NMR structural evidence. Science. 1990;249:280–283. doi: 10.1126/science.2374927. [DOI] [PubMed] [Google Scholar]
- 24.Holmgren A, Slaby I. Thioredoxin-C': Mechanism of noncovalent complementation and reactions of the refolded complex and the active-site containing fragment with thioredoxin reductase. Biochemistry. 1979;18:5591–5599. doi: 10.1021/bi00592a011. [DOI] [PubMed] [Google Scholar]
- 25.Carey J, Lindman S, Bauer M, Linse S. Protein reconstitution and three-dimensional domain swapping: Benefits and constraints of covalency. Protein Sci. 2007;16:2317–2333. doi: 10.1110/ps.072985007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lindman S, Johansson I, Thulin E, Linse S. Green fluorescence induced by EF-hand assembly in a split GFP system. Protein Sci. 2009;18:1221–1229. doi: 10.1002/pro.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gronenborn AM, et al. A novel, highly stable fold of the immunoglobulin binding domain of streptococcal protein G. Science. 1991;253:657–661. doi: 10.1126/science.1871600. [DOI] [PubMed] [Google Scholar]
- 28.Kobayashi N, Honda S, Yoshii H, Uedaira H, Munekata E. Complement assembly of two fragments of the streptococcal protein G B1 domain in aqueous solution. FEBS Lett. 1995;366:99–103. doi: 10.1016/0014-5793(95)00503-2. [DOI] [PubMed] [Google Scholar]
- 29.Ghosh I, Hamilton AD, Regan L. Antiparallel leucine zipper-directed protein reassembly: Application to the green fluorescent protein. J Am Chem Soc. 2000;122:5658–5659. [Google Scholar]
- 30.Magliery TJ, et al. Detecting protein-protein interactions with a green fluorescent protein fragment reassembly trap: Scope and mechanism. J Am Chem Soc. 2005;127:146–157. doi: 10.1021/ja046699g. [DOI] [PubMed] [Google Scholar]
- 31.Wilson CG, Magliery TJ, Regan L. Detecting protein-protein interactions with GFP-fragment reassembly. Nat Methods. 2004;1:255–262. doi: 10.1038/nmeth1204-255. [DOI] [PubMed] [Google Scholar]
- 32.Smith CK, Withka JM, Regan L. A thermodynamic scale for the beta-sheet forming tendencies of the amino acids. Biochemistry. 1994;33:5510–5517. doi: 10.1021/bi00184a020. [DOI] [PubMed] [Google Scholar]
- 33.Lindman S, et al. Salting the charged surface: pH and salt dependence of protein G B1 stability. Biophys J. 2006;90:2911–2921. doi: 10.1529/biophysj.105.071050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vanbelle C, et al. Deamidation and disulfide bridge formation in human calbindin D28k with effects on calcium binding. Protein Sci. 2005;14:968–979. doi: 10.1110/ps.041157705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wunderlich M, et al. Optimization of the Gb1 domain by computational design and by in vitro evolution: Structural and energetic basis of stabilization. J Mol Biol. 2007;373:775–784. doi: 10.1016/j.jmb.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 36.Wunderlich M, Schmid FX. In vitro evolution of a hyperstable GB1 variant. J Mol Biol. 2006;363:545–557. doi: 10.1016/j.jmb.2006.08.034. [DOI] [PubMed] [Google Scholar]
- 37.Jee JG, Byeon IJL, Louis JM, Gronenborn AM. The point mutation A34F causes dimerization of GB1. Proteins. 2008;71:1420–1431. doi: 10.1002/prot.21831. [DOI] [PubMed] [Google Scholar]
- 38.Bauer MC, Xue WF, Linse S. Protein GB1 folding and assembly from structural elements. Int J Mol Sci. 2009;10:1552–1566. doi: 10.3390/ijms10041552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bennett MJ, Choe S, Eisenberg D. Domain swapping: Entangling alliances between proteins. Proc Natl Acad Sci USA. 1994;91:3127–3131. doi: 10.1073/pnas.91.8.3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Håkansson M, Linse S. Protein reconstitution and 3D domain swapping. Curr Protein Pept Sc. 2002;3:629–642. doi: 10.2174/1389203023380459. [DOI] [PubMed] [Google Scholar]
- 41.Lindman S, et al. Salting the charged surface: pH and salt dependence of protein G B1 stability. Biophys J. 2006;90:2911–2921. doi: 10.1529/biophysj.105.071050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Koradi R, Billeter M, Wüthrich K. MOLMOL: A program for display and analysis of macromolecular structures. J Mol Graphics. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- 43.Ormö M, et al. Crystal structure of the Aequorea victoria green fluorescent protein. Science. 1996;273:1392–1395. doi: 10.1126/science.273.5280.1392. [DOI] [PubMed] [Google Scholar]
- 44.Gallagher T, Alexander P, Bryan P, Gilliland GL. Two crystal structures of the B1 immunoglobulin-binding domain of streptococcal protein G and comparison with NMR. Biochemistry. 1994;33:4721–4729. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.