

Abstract
Modulators of metabotropic glutamate receptor subtype 5 (mGluR5) may provide novel treatments for multiple central nervous system (CNS) disorders, including anxiety and schizophrenia. Although compounds have been developed to better understand the physiological roles of mGluR5 and potential usefulness for the treatment of these disorders, there are limitations in the tools available, including poor selectivity, low potency, and limited solubility. To address these issues, we developed an innovative assay that allows simultaneous screening for mGluR5 agonists, antagonists, and potentiators. We identified multiple scaffolds that possess diverse modes of activity at mGluR5, including both positive and negative allosteric modulators (PAMs and NAMs, respectively). 3-Fluoro-5-(3-(pyridine-2-yl)-1,2,4-oxadiazol-5-yl)benzonitrile (VU0285683) was developed as a novel selective mGluR5 NAM with high affinity for the 2-methyl-6-(phenylethynyl)-pyridine (MPEP) binding site. VU0285683 had anxiolytic-like activity in two rodent models for anxiety but did not potentiate phencyclidine-induced hyperlocomotor activity. (4-Hydroxypiperidin-1-yl)(4-phenylethynyl)phenyl)methanone (VU0092273) was identified as a novel mGluR5 PAM that also binds to the MPEP site. VU0092273 was chemically optimized to an orally active analog, N-cyclobutyl-6-((3-fluorophenyl)ethynyl)nicotinamide hydrochloride (VU0360172), which is selective for mGluR5. This novel mGluR5 PAM produced a dose-dependent reversal of amphetamine-induced hyperlocomotion, a rodent model predictive of antipsychotic activity. Discovery of structurally and functionally diverse allosteric modulators of mGluR5 that demonstrate in vivo efficacy in rodent models of anxiety and antipsychotic activity provide further support for the tremendous diversity of chemical scaffolds and modes of efficacy of mGluR5 ligands. In addition, these studies provide strong support for the hypothesis that multiple structurally distinct mGluR5 modulators have robust activity in animal models that predict efficacy in the treatment of CNS disorders.
Introduction
Glutamate, the major excitatory neurotransmitter in the central nervous system (CNS), exerts its effects through the activation of both glutamate-gated cation channels and eight distinct subtypes of G protein-coupled metabotropic glutamate receptors (mGluRs), termed mGluR1 to mGluR8 (Niswender and Conn, 2010). Previous studies suggest that selective agonists and antagonists of the mGluR5 subtype could be beneficial in the treatment of a number of CNS disorders (Gasparini et al., 2008; Conn et al., 2009a). For instance, a large number of preclinical studies and preliminary reports of clinical studies suggest that mGluR5 antagonists may be effective in the treatment of anxiety disorders (Porter et al., 2005; Swanson et al., 2005), Parkinson's disease (Marino and Conn, 2002a; Marino et al., 2002), and Fragile X syndrome (Bear et al., 2004; Gasparini et al., 2008).
In addition to mGluR5 antagonists, selective activators of mGluR5 have been proposed as having potential usefulness in the treatment of schizophrenia and disorders of cognitive function (Marino and Conn, 2002b; Kinney et al., 2003; Moghaddam, 2004; Conn et al., 2009b). This is largely based on a number of investigations suggesting that mGluR5 is a closely associated signaling partner with the N-methyl-d-aspartate (NMDA) subtype of ionotropic glutamate receptor that plays a key role in regulating NMDA receptor function in a variety of forebrain regions (Marino and Conn, 2002b; Lindsley et al., 2006). Based on this and a large number of cellular studies suggesting that activation of mGluR5 and NMDA receptors could have robust effects in forebrain circuits believed to be disrupted in schizophrenia, it has been postulated that activators of mGluR5 could provide novel therapeutic agents that may be useful for treating this disorder (Marino and Conn, 2002b; Moghaddam, 2004; Conn et al., 2009b).
A major breakthrough in the area of mGluR5 biology came with the discovery of highly selective allosteric antagonists of mGluR5, including 2-methyl-6-(phenylethynyl)-pyridine (MPEP) and related compounds (Gasparini et al., 1999, 2002). These compounds do not interact with the orthosteric glutamate binding site but bind to an allosteric site in the seven transmembrane-spanning domain of mGluR5 to inhibit coupling of the receptor to GTP binding proteins (Knoflach et al., 2001). These mGluR5-selective negative allosteric modulators (NAMs) have had a major effect on our understanding of the physiological roles of this receptor and have allowed studies that suggest that antagonists of mGluR5 have potential as novel therapeutic agents. We have reported discovery of compounds that act as highly selective positive allosteric modulators (PAMs) of mGluR5 (O'Brien et al., 2003, 2004; Lindsley et al., 2004; Kinney et al., 2005; Conn et al., 2009b; Engers et al., 2009). These compounds have no agonist activity by themselves but act at an allosteric site to potentiate glutamate-induced activation of mGluR5 in transfected cell lines. Two of these compounds, termed CDPPB and S-(4-fluoro-phenyl)-{3-[3-(4-fluoro-phenyl)-[1,2,4]-oxadiazol-5-yl]-piperidin-1-yl}-methanone (ADX-47273), display efficacy in animal models commonly used to test for potential antipsychotic-like activity and cognition-enhancing effects of novel compounds but have limited use when administered systemically (Lindsley et al., 2004; Kinney et al., 2005; Liu et al., 2008; Ayala et al., 2009; Gass and Olive, 2009).
Although progress in the discovery of novel mGluR5-selective PAMs and NAMs has been exciting, there are a number of limitations to the currently available mGluR5 modulators. For instance, prototypical mGluR5 NAMs such as MPEP and MTEP are inverse agonists (Gasparini et al., 1999; Roppe et al., 2004; Lea and Faden, 2006) and have disruptive effects on cognitive function and effects in animal models that predict potential psychotomimetic activity (Porter et al., 2005; Rodriguez et al., 2005). Furthermore, CDPPB and other mGluR5 PAMs such as ADX-47273 have limited aqueous solubility and relatively low potencies at the receptor (de Paulis et al., 2006; Liu et al., 2008; Engers et al., 2009). These deficiencies do not allow for full exploration of the functional effects of these compounds or sufficient dosing required for more extensive behavioral studies. To address these issues, we developed and used an innovative high-throughput screening assay that allows simultaneous identification of mGluR5 agonists, antagonists, and potentiators. We now report the discovery of multiple novel scaffolds possessing diverse activities as mGluR5 allosteric modulators. These novel molecules reveal a rich diversity of mGluR5 modulators, in terms of diverse chemical scaffolds, distinct modes of efficacy, and interactions with multiple allosteric sites. Furthermore, these novel compounds are active in vivo and show that some of the liabilities associated with previous mGluR5 modulators are not intrinsic to the target but can be avoided with compounds that provide efficacy in animal models.
Materials and Methods
Materials
The Vanderbilt High-Throughput Screening Center compound collection was obtained from ChemBridge Corporation (San Diego, CA) and ChemDiv, Inc. (San Diego, CA) and stored in bar-coded, 384-well, U-bottomed, standard volume polypropylene plates (Corning Life Sciences, Lowell, NY). The plates were thermally sealed with peelable seals using a PlateLoc (Velocity 11, Santa Clara, CA). Groups of 10 plates were vacuum-packed in thermally sealed freezer bags (FoodSaver, Jarden Corp., Rye, NY) and stored frozen at −80°C. l-Glutamate, DHPG, and MPEP were obtained from Tocris Bioscience (Ellisville, MO). [3H]MethoxyPEPy was obtained from American Radiolabeled Chemicals, Inc. (St. Louis, MO). CDPPB (Lindsley et al., 2004), CPPHA (Zhao et al., 2007), MTEP (Cosford et al., 2003b), and 5MPEP (Rodriguez et al., 2005) were synthesized as described previously. NADPH was purchased from Sigma-Aldrich (St. Louis, MO). Rat liver microsomes (20 mg/ml protein) were purchased from BD Biosciences (Woburn, MA). All the solvents were of either analytical or HPLC grade.
Chemistry
Full experimental details for mGluR5 allosteric modulators are available in the Supplemental Data section.
5-(3,5-Dimethoxyphenyl)-3-(pyridine-2-yl)-1,2,4-oxadiazole (VU0040228).
1H NMR (400 MHz, d4-MeOH) δ 8.77 (dd, J = 5.0, 1.0 Hz, 1H), 8.29 (d, J = 8.0 Hz, 1H), 8.07 (td, J = 8.0, 1.5 Hz, 1H), 7.64 (ddd, J = 8.0, 5.0, 1.0 Hz, 1H), 7.41 (d, J = 2.5 Hz, 1H), 6.82 (t, J = 2.5 Hz, 1H), 3.92 (s, 6H); liquid chromatography/mass spectrometry (214 nm) 1.45 min (>98%); MS (ESI) m/z = 284.1; HRMS = 284.1035 (calculated 284.1035), C15H14N3O3; for 3-fluoro-5-(3-(pyridine-2-yl)-1,2,4-oxadiazol-5-yl)benzonitrile (VU0285683): 1H NMR (400 MHz, CDCl3) δ 8.88 (d, J = 5 Hz, 1H), 8.43 (s, 1H), 8.29 to 8.24 (m, 2H), 7.94 (td, J = 8.0, 1.5 Hz, 1H), 7.65 (ddd, J = 8.0, 2.5, 1.5 Hz, 1H), 7.52 (ddd, J = 8.0, 5.0, 1.5 Hz, 1H); liquid chromatography/mass spectrometry (214 nm) 2.91 min (>98%); MS (ESI) m/z = 267.1; HRMS = 267.0679 (calculated 267.0682), C14H8N4O1F1.
4-Hydroxypiperidin-1-yl)(4-phenylethynyl)phenyl)methanone (VU0092273).
Melting point, 157.7°C; 1H NMR (400 MHz, CDCl3) δ 7.58 (d, J = 8.0 Hz, 2H), 7.56 to 7.52 (m, 2H), 7.44 to 7.34 (m, 5H), 4.21 to 4.08 (m, 1H), 4.03 to 3.96 (m, 1H), 3.81 to 3.48 (m, 1H), 3.47 to 3.16 (m, 2H), 2.08 to 1.79 (m, 3H), 1.71 to 1.42 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 169.7, 135.5, 131.6, 131.5, 128.5, 128.3, 126.9, 124.7, 122.8, 90.8, 88.5, 66.9, 44.8, 39.3, 34.4, 33.8; LC (214 nm) 2.86 min (>98%); MS (ESI) m/z = 306.1 ([M + 1]+); HRMS = 306.1496 (calculated 306.1494), C20H20N1O2; and for
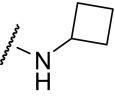
N-Cyclobutyl-6-((3-fluorophenyl)ethynyl)nicotinamide hydrochloride (VU0360172).
1H NMR (400 MHz, DMSO) δ 9.04 (d, J = 1.2 Hz, 1H), 8.93 (d, J = 7.8 Hz, 1H), 8.27 (dd, J = 7.8, 1.2 Hz, 1H), 7.78 (d, J = 8.4 Hz, 1H), 7.57 to 7.49 (overlapped, 3H), 7.37 (m, 1H), 4.43 (m, 1H), 2.25 (m, 2H), 2.11 (m, 2H), 1.70 (m, 2H); 13C NMR (100 MHz, DMSO) δ 163.08, 162.22 (d, J = 244.0 Hz, 1C), 148.82, 143.31, 136.89, 131.52 (d, J = 9.0 Hz, 1C), 129.72, 128.67 (d, J = 3.0 Hz, 1C), 127.68, 123.33 (d, J = 9.0 Hz, 1C), 120.35, 118.82 (d, J = 23.0 Hz, 1C), 117.64 (d, J = 21.0 Hz, 1C), 90.08 (d, J = 3.0 Hz, 1C), 89.01, 45.07, 30.38 (2C), 15.18; LC (214 nm) 1.35 min (>98%); MS (ESI), m/z = 295.10 ([M + 1]+); HRMS (ESI) m/z = 295.1248 ([M + 1]+, 100%) calculated for C18H16N2OF, 295.1247.
Fluorescence-Based Calcium Flux Assay.
Assays were performed within Vanderbilt University's High-Throughput Screening Center. HEK293 cells stably expressing rat mGluR5 were plated in black-walled, clear-bottomed, poly(d-lysine)-coated 384-well plates (Greiner Bio-One, Monroe, NC) in 20 μl of assay medium (DMEM containing 10% dialyzed FBS, 20 mM HEPES, and 1 mM sodium pyruvate) at a density of 20,000 cells/well. The cells were grown overnight at 37°C in the presence of 5% CO2. The next day, medium was removed using a VSpin (Velocity 11) fitted with a modified bucket allowing the 384-well plate to be mounted inverted over a catch basin and spun at 80g for 10 s with 40% acceleration and deceleration settings on the instrument. The medium was replaced using a Thermo Fisher Combi (Thermo Fisher Scientific, Waltham, MA), with 20 μl of 1 μM Fluo-4/acetoxymethyl ester (Invitrogen, Carlsbad, CA) prepared as a 2.3 mM stock in DMSO and mixed in a 1:1 ratio with 10% (w/v) Pluronic F-127 and diluted in assay buffer (Hanks' balanced salt solution, 20 mM HEPES, and 2.5 mM probenecid; Sigma-Aldrich) for 45 m at 37°C. Dye was removed using the VSpin and replaced, using a Combi, with 20 μl of assay buffer, and the plate was incubated for 10 min at room temperature. Single concentrations of test compounds (10 μM final) were transferred to daughter plates using the Echo acoustic plate reformatter (Labcyte, Sunnyvale, CA) and then diluted into assay buffer to a 2× stock using a Combi. Ca2+ flux was measured using the Functional Drug Screening System 6000 (Hamamatsu Corporation, Tokyo, Japan). For the primary screen, test compound was applied to cells at t = 3 s after baseline readings were taken. Cells were incubated with the test compounds for 140 s and then stimulated with an EC20 concentration of glutamate; 60 s later, an EC80 concentration of glutamate was added, and readings were taken for an additional 40 s. Data were collected at 1 Hz. The assay protocol was automated using the instruments noted above integrated with a Thermo Fisher F3 robotic arm under the control of a Polara scheduler (Thermo Fisher Scientific). All data were recorded to instruments' local drives and later migrated to a network drive. FDSS data were analyzed using a custom analysis application and were associated with unique compound identifiers based on liquid handler transfer logs and plate barcode readings captured by the Echo and by Polara. Agonist hits were selected by comparing the amplitude of the response at the time of compound addition plus and minus test compound; potentiator hits were selected by comparing the amplitude of the responses at the time of EC20 addition plus and minus test compound; antagonist hits were selected by comparing the amplitude of the responses at the time of EC80 addition plus and minus test compound (Niswender et al., 2008b; Marlo et al., 2009).
Putative hits from the primary screen were confirmed by testing for concentration-dependent activity on mGluR5 over a range of 4 log units. Compounds were serially diluted 1:3 into 10-point concentration-response curves (final concentration, 30 μM–1 nM), transferred to daughter plates using the Echo acoustic plate reformatter, and tested as described in the primary screen. Putative potentiators were applied for 300 s and followed by EC20 concentrations of glutamate while antagonists were applied for 300 s and followed by EC80 concentrations of glutamate. Concentration-response curves were generated using a four-parameter logistical equation in XLfit curve fitting software (IDBS, Bridgewater, NJ) for Excel (Microsoft, Redmond, WA) or Prism (GraphPad Software, Inc., San Diego, CA).
Selectivity Studies
Muscarinic Response.
HEK293 cells were plated as described above for mGluR5. Compounds were added 300 s before an EC20 or EC80 concentration of the muscarinic agonist carbachol. Raw data from the FDSS were imported into Microsoft Excel and analyzed as described above.
Rat mGluR1.
Baby hamster kidney cells expressing rat mGluR1 were cultured as described previously (Hemstapat et al., 2006). Calcium flux assays were used for counterscreening rat mGluR1 by measuring the glutamate concentration-response relationship in the presence and absence of a fixed concentration of test compound using a double-addition protocol adding test compound 2.5 min before varying concentrations of glutamate. mGluR1 cells were plated at 15 × 103 cells/well in black-walled, tissue culture-treated, 384-well plates (Greiner Bio-One, Monroe, NC) in assay medium, and calcium assays were performed as described above.
Rat mGluRs 3 and 4.
Compound activity at the rat group II and III mGluRs, mGluR3 and mGluR4, respectively, was assessed using thallium flux through G protein-coupled inwardly rectifying potassium channels, a method that has been described in detail in (Niswender et al., 2008a). These cell lines were grown in growth medium containing 45% DMEM, 45% Ham's F-12, 10% FBS, 20 mM HEPES, 2 mM l-glutamine, antibiotic/antimycotic, nonessential amino acids, 700 μg/ml G418, and 0.6 μg/ml puromycin at 37°C in the presence of 5% CO2. In brief, mGluR3 or 4 G protein-coupled inwardly rectifying potassium cells were plated into 384-well, black-walled, clear-bottomed poly(d-lysine)-coated plates at a density of 15 × 103 cells/20 μl/well in assay medium and incubated overnight at 37°C in the presence of 5% CO2. The following day, the medium was removed from the cells, and 20 μl/well of 1 μM concentration of the indicator dye FluxOR (Invitrogen, Carlsbad, CA) in assay buffer was added. Cells were incubated for 1 h at room temperature, and the dye was replaced with 20 μl/well of assay buffer. For these assays, compounds were added at 2× final concentration, and then 2.5 min later, varying concentrations of glutamate were added using the FDSS 6000. Glutamate was diluted in thallium buffer (125 mM sodium bicarbonate, 1 mM magnesium sulfate, 1.8 mM calcium sulfate, 5 mM glucose, 12 mM thallium sulfate, and 10 mM HEPES) at 5× the final concentration to be assayed. Data were analyzed as described previously (Niswender et al., 2008a).
Calcium Assays on the FlexStation.
Follow-up experiments with VU0285683 were performed using the FlexStation II (Molecular Devices, Sunnyvale, CA) using the following protocols. HEK cells stably expressing rat mGluR5 were plated at 6 × 104 cells per well in medium containing DMEM, 10% dialyzed FBS, 20 mM HEPES, 1 mM sodium pyruvate, and antibiotic/antimycotic in clear-bottomed, black-walled, poly(d-lysine)-coated 96-well plates (BD BioCoat, Bedford, MA) 24 h before assay and were incubated overnight at 37°C in 5% CO2. On the day of the assay, the medium was removed and replaced with Hanks' balanced salt solution containing 20 mM HEPES, 2.5 mM probenecid, and 2 μM Fluo-4/acetoxymethyl ester dye, pH 7.4. Cells were incubated for 45 min (37°C, 5% CO2) for dye-loading. This medium was removed and replaced with 40 μl of calcium assay buffer (Hanks' balanced salt solution, 20 mM HEPES, and 2.5 mM probenecid, pH 7.4). For measurement of antagonist potency by calcium fluorescence, vehicle or concentration-response curves (CRCs) of antagonist made in calcium assay buffer were added (40 μl) at the 17-s time point, and an EC80 concentration of glutamate (20 μl) was added at the 107-s time point via a FlexStation II (Molecular Devices). Fluorescence was monitored for a total of 137 s using an excitation wavelength of 488 nm, an emission wavelength of 525 nm, and a cutoff wavelength of 515 nm. For fold shift assays, vehicle or fixed concentrations of antagonist were added (40 μl), and a glutamate CRC (20 μl) was added via a FlexStation II. For 5MPEP assays, calcium assay buffer (20 μl) and 5MPEP (20 μl) were added by hand and allowed to incubate for 30 min before the standard calcium fluorescence assay protocol described above. All calcium response data were normalized to the EC80 response to a glutamate. Data were transformed and fit with GraphPad Prism to determine EC50 values.
Mutagenesis and Transient Transfection.
HEK cells were plated on 100-mm cell culture dishes in media containing DMEM, 10% FBS, 20 mM HEPES, and antibiotic/antimycotic. Cells were transfected with cDNA for rat wild-type mGluR5 or A809V mutant mGluR5. cDNA (5 μg) and FuGENE 6 (25 μl) were added to 970 μl of Opti-MEM (Invitrogen) and allowed to incubate at room temperature for 15 min. After incubation, the cDNA/FuGENE mixture was applied dropwise to HEK cells. Twenty-four hours later, cells were plated and assayed using the same protocol described above.
Radioligand Binding Assays.
The allosteric antagonist MPEP analog [3H]methoxyPEPy was used to evaluate the ability of test compounds to interact with the MPEP site on mGluR5 (Cosford et al., 2003a). Membranes were prepared from rat mGluR5 HEK293 cells (Rodriguez et al., 2005). Compounds were diluted in assay buffer (50 mM Tris/0.9% NaCl, pH 7.4) to a 5× stock, and 100 μl of test compound was added to each well of a 96-deep-well assay plate. Aliquots (300 μl) of membranes diluted in assay buffer (40 μg/well) were added to each well. [3H]methoxyPEPy (100 μl; final concentration, 2 nM) was added, and the reaction was incubated at room temperature for 1 h with shaking. After the incubation period, the membrane-bound ligand was separated from free ligand by filtration through glass-fiber 96-well filter plates (Unifilter-96 GF/B; PerkinElmer Life and Analytical Sciences, Waltham, MA). The contents of each well were transferred simultaneously to the filter plate and washed three to four times with assay buffer using a cell harvester (Brandel Inc., Gaithersburg, MD). Scintillation fluid (40 μl) was added to each well, and the membrane-bound radioactivity was determined by scintillation counting (TopCount; PerkinElmer Life and Analytical Sciences). Nonspecific binding was estimated using 5 μM MPEP. Concentration-response curves were generated using a four-parameter logistical equation in GraphPad Prism.
Plasma Protein Binding.
Plasma protein binding assays on the test compounds were performed in a high-throughput mode. Plasma samples spiked with 5 μM concentration of the test compound in DMSO was added to the cis chambers of Rapid Equilibrium Dialysis plates (Thermo Fisher Scientific, Waltham, MA), and Dulbecco's phosphate-buffered saline was added to the corresponding trans sides. The samples were dialyzed for 4 h at 37°C with shaking. The dialyzed samples from the buffer and plasma compartments were extracted by a protein precipitation method using ice-cold acetonitrile containing 0.1% formic acid and an internal standard (VU0366031) having a final concentration of 50 ng/ml.
The extracts from the plasma and buffer compartments were analyzed by means of HPLC/MS/MS using a Thermo Finnigan TSQ Quantum Ultra (Thermo Fisher Scientific) mass spectrometer in the positive-ion mode by selective reaction monitoring. The chromatographic separation was achieved on an Acquity ultra-performance liquid chromatography BEH C18 column (1.7 μm; 2.1 × 50 mm) at a flow rate of 0.8 ml/min. A gradient program was used with the mobile phase, combining solvent A (95:5 0.1% formic acid in water/acetonitrile) and solvent B (95:5 acetonitrile/0.1% formic acid in water) as follows: 20% solvent B up to 0.5 min, ramped from 20 to 100% solvent B by 1 min, and held at 100% solvent B until 2 min. The composition was returned to 20% solvent B by 2.2 min. The total run time was 5 min. The column temperature was maintained at 50°C. The software Xcalibur version 2.2 was used to control the instrument and collect data. The electrospray ionization source was fitted with a stainless steel capillary (100 μm internal diameter). Nitrogen was used as both the sheath gas and the auxiliary gas. The ion-transfer tube temperature was maintained at 350°C. The spray voltage, tube lens voltage, and pressure of sheath gas and auxiliary gas were optimized to achieve maximal response using the test compounds mixing with the mobile phase A (50%) and B (50%) at a flow rate of 0.8 ml/min. Collision-induced dissociation was performed on test compounds and internal standard under 1.5 mTorr of argon. Percentage of unbound was calculated based on the percentage of compound in the phosphate-buffered saline chamber (trans) compared with that in the plasma chamber.
Metabolic Stability Assays.
Microsomal stability of the test compounds was also performed using an HTS platform. The test compounds (1 μM in DMSO) were incubated for 15 min at 37°C with shaking, in medium containing microsomes, phosphate buffer, and the cofactor NADPH. After incubation, the samples were extracted using ice-cold acetonitrile containing 0.1% formic acid and 50 ng/ml internal standard (VU0366031). The extracts were analyzed by means of HPLC/MS/MS using methods identical with those used for the plasma protein binding assay (detailed above). Percentage of test compound remaining after incubation was calculated based on the amount of compound in the incubated samples compared with similarly prepared unincubated controls.
In Vivo Pharmacokinetic Study.
Compound was formulated as 20% hydroxypropyl β-cyclodextrin in sterile water at the concentration of 1 mg/ml and administered orally to male Sprague-Dawley rats weighing 225 to 250 g (Harlan, Indianapolis, IN) at the dose of 10 mg/kg. The rat blood (hepatic portal vein and cardiac puncture) and brain were collected at 0.5, 1, 3, and 6 h. Animals were euthanized and decapitated, and the brains were removed, thoroughly washed in cold phosphate-buffered saline, and immediately frozen on dry ice. Plasma was separated by centrifugation and stored at −80°C until analysis.
On the day of analysis, frozen whole-rat brains were weighed and homogenized in 1:3 (w/w) parts of ice-cold phosphate-buffered saline, pH 7.4. The sample extraction of plasma (100 μl) and brain homogenate (100 μl) was performed by a method based on protein precipitation using three volumes of ice-cold acetonitrile containing 0.1% formic acid and an internal standard (VU0366031) having final concentration of 50 ng/ml. Extracts were vortex mixed for 5 min followed by centrifugation at 14,000 rpm for 10 min.
The supernatants of plasma and brain homogenate extracts were analyzed by means of HPLC/MS/MS as described above. Selected reaction monitoring was carried out using the transitions from m/z 295 to 194 for VU0360172 and m/z 310 to 223 for VU0366031 (internal standard). The calibration curves were constructed, and linear response was obtained in the range of 20 to 10,000 ng/ml by spiking known amounts of VU00360172 in blank brain homogenates and plasma. The final PK parameters were calculated by noncompartmental analysis using WinNonlin software (version 5.1; Pharsight Inc., Mountain View, CA).
In Vivo Behavioral Studies.
All experiments were conducted in accordance with the National Institutes of Health regulations of animal care covered in the Institute of Laboratory Animal Resources' (1996) Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee.
Conflict Paradigm
Subjects.
The subjects were 24 male Sprague-Dawley rats (225–249 g) (Harlan) individually housed and food-deprived to 90% of their free-feeding weight 1 week before the onset of training. The rats had continuous access to water, except during training, and were given enough food to maintain their weights at 90% of their expected nondeprived weights immediately after training and on weekends. All rats were maintained on a 12-h light/dark cycle with light onset at 6:00 AM.
Apparatus.
Six commercially available operant chambers (MED Associates, St. Albans, VT), each housed in a sound-attenuating chamber, were used. The operant chambers were equipped with two response levers, a pellet dispenser centered between the levers, stimulus lights, a tone generator, and a grid floor for delivery of foot shock. The start of the session was signaled by illumination of the house light (7.5-W bulb). All equipment was controlled by programs written with MED Associates software.
Training Procedure.
Rats were trained to lever-press for food reinforcement (Bioserv 45-mg pellets) on an FR-1 schedule that also included delivery of a free pellet every 60 s during daily 2 h sessions. All animals acquired the lever-press response within 3 days. After acquisition of the FR-1, the rats were switched to a variable interval (VI) schedule of reinforcement that began as a VI 10-s schedule and increased by 10 s each week until arriving at a final VI 30-s schedule of reinforcement. This phase of training was conducted with the house light on and later became the unpunished component of a multiple schedule. Once stable responding had occurred on the VI 30-s schedule, two components were added to training: a punished component, and a time-out. In the presence of a 70-dB tone and left-lever stimulus light, both of which remained on for the duration of the punished component, responses continued to be reinforced on a VI 30-s schedule; however, in addition, every 10th response was punished with a brief (500-ms) foot shock. The shock intensity was individually adjusted (0.25–0.5 mA) to produce an approximate 80% reduction in punished response rates relative to unpunished rates. The unpunished and punished components were 3 min in duration separated by 1-min time-outs, during which the lights and tone were extinguished and responses were without consequences. This multiple schedule was repeated four times for a total of 31 min of training. Training was given 5 days per week. Once stable rates of responding had been established in both the unpunished and punished components, testing began. Training required approximately 3.5 months.
Testing Procedure.
After stable response rates had been achieved, rats were tested either once or twice a week depending on whether the test was with vehicle or drug. Drug tests were only given once a week. Test sessions were identical with training sessions. Testing began with the mGluR5 receptor antagonist MTEP, a compound that has been shown previously to increase punished responding in the conflict paradigm (Busse et al., 2004). Forty-five minutes before testing, rats (n = 24) were given injections with MTEP (vehicle, 1, 3, 5.6, or 10 mg/kg i.p.) suspended in 10% Tween 80/90% water. All concentrations were injected in a volume of 1 ml/kg. Each animal was tested with all doses of MTEP and vehicle.
VU0285683 (vehicle, 1, 3, and 10 mg/kg i.p.), suspended in 20% β-cyclodextrin, was injected 45 min before placing the animal (n = 19) in the test chamber. All concentrations were injected in a volume of 1 ml/kg except for the 10-mg concentration, which was injected in a volume of 2 ml/kg. Each animal was tested on all doses of VU0285683 and vehicle.
Marble Burying
Compounds.
Doses of MTEP (positive control) and VU0285683 were dissolved in 10% Tween 80, vortexed vigorously, heated gently with a Master Heat Gun (Master Appliance Corporation, Racine, WI), and sonicated at 37°C for 30 min. The pH was checked using 0 to 14 EMD strips (EMD Biosciences, San Diego, CA) and adjusted to approximately 7. All doses were administered in a volume of 10 ml/kg i.p. Seven dose groups were administered: vehicle, 3, 5.6, 10, and 15 mg/kg MTEP, and 3, 5.6, and 10 mg/kg VU0285683.
Subjects.
This study was conducted using male Harlan CD-1 mice weighing 30 to 35 g. Subjects were housed in a large colony room under a 12-h light/dark cycle (lights on at 6:00 AM) with food and water provided ad libitum. Test sessions were performed between 10:00 AM and 4:00 PM. All dose groups consisted of 9 to 11 mice. All experiments were conducted in accordance with Institute of Laboratory Animal Resources' (1996) Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee.
Procedure.
Eight small Plexiglas cages (32 × 17 × 14 cm) were arranged in two rows of four cages on top of a large, round table. Mice were transported from the colony room to the testing room and were allowed to habituate for 30 min. Mice were pretreated with a dose of MTEP (15-min pretreatment) or VU0285683 (30-min pretreatment) and individually placed in the cages in which 12 black glass marbles (14 mm diameter) had been evenly distributed (spaced 6.4 cm vertically and 4.25 cm horizontally from each other and the walls of the cage) on top of 2.5 cm Diamond Soft Bedding (Harlan Teklad, Madison, WI). The compound and comparator were evaluated in a counterbalanced design, in which all doses of compounds were tested in each session. Mice receiving the same dose were placed in cages on opposite sides of the table to control for effects of lighting and context. Clear, perforated plastic lids were set on top of each cage, and the amount of marble burying was recorded over a 30-min interval. The mice were then removed from the cages, and the number of buried marbles was counted using the criteria of greater than two thirds covered by bedding. Each session was videotaped with a Sony MiniDV camcorder equipped with a Sony wide-angle lens mounted on a 1.5-m tripod (Sony, Tokyo, Japan).
Data Analysis.
The data for the dose-response studies were analyzed by a between-group analysis of variance. If there was a main effect of dose, then each dose group was compared with the vehicle control group using a Dunnett's comparison. The calculations were performed using JMP IN 8 (SAS Institute, Cary, NC) statistical software and graphed using SigmaPlot9 (Systat Software, San Jose, CA).
Amphetamine- and Phencyclidine-Induced Hyperlocomotion
Subjects.
All behavioral studies were conducted using male Sprague-Dawley rats (Harlan) weighing 270 to 300 g. Subjects were housed in pairs in a large colony room under a 12-h light/dark cycle (lights on at 6:00 AM) with food and water provided ad libitum. Test sessions were performed between 6:00 AM and 6:00 PM. Dose groups consisted of 8 to 16 rats per dose group. All doses of test compounds were injected in a 1.0 ml/kg volume. Test compound was dissolved in vehicle and double-deionized water with the pH adjusted to approximately 7.0 using 1 N NaOH.
Apparatus.
Amphetamine- and phencyclidine (PCP)-induced hyperlocomotor activity studies were conducted using a SmartFrame Open Field System (KinderScientific, San Diego, CA) equipped with 32 horizontal (x- and y-axes) infrared photobeams located 1 cm above the floor of the chamber. Changes in ambulation or locomotor activity were measured as the number of total photobeam breaks, expressed in 5-min intervals, and were recorded with a Pentium I computer equipped with the Motor Monitor System software (KinderScientific).
Procedure for Amphetamine-Induced Hyperlocomotor Activity.
Rats were placed in the open-field chambers for a 30-min habituation period, followed by a pretreatment with vehicle or an oral or intraperitoneal dose of test compound for an additional 30 min. Next, all rats received an injection of 1 mg/kg s.c. amphetamine or saline, and locomotor activity was measured for an additional 60 min.
PCP-Induced Hyperlocomotor Activity.
Rats were placed in the open-field chambers for a 30-min habituation period, followed by a pretreatment with vehicle or a dose of either 10 mg/kg i.p. VU0285683 or MTEP for an additional 30 min. Next, all rats received an injection of 2.5 mg/kg s.c. PCP or saline, and locomotor activity was measured for an additional 120 min. Data were analyzed by a one-way analysis of variance with comparison with the vehicle + amphetamine control group or vehicle + PCP control group using Dunnett's test. Calculations were performed using JMP version 8 (SAS Institute Inc.) statistical software.
Results
Novel Modulators of mGluR5 Are Identified via High-Throughput Screening.
A major challenge in the discovery of novel compounds that fall into each of the three major functional classes of mGluR5 modulators is that assays for each of these pharmacological modes often require different conditions that are optimized for each activity. However, we were interested in the discovery of novel compounds that act as mGluR5 agonists, antagonists, and allosteric potentiators. To accomplish this, we developed a three-part assay that includes multiple compound additions with defined intervals that allows simultaneous detection of compounds with each of these activities. We measured receptor-induced mobilization of intracellular calcium by using an imaging-based plate reader that makes simultaneous measurements of calcium levels in each well of a 384-well microplate. Either vehicle or a test compound (10 μM final concentration) was added to cells expressing rat mGluR5 that were loaded with calcium-sensitive fluorescent dye. After a 2.5-min incubation period, a submaximally effective (EC20) concentration of glutamate was added followed by a nearly maximal (EC80) concentration added 1 min later. These intervals were chosen after extensive systematic testing of different compound addition intervals and based on the time required for maximal effects of known allosteric ligands and lack of desensitization between additions of EC20 and EC80 glutamate. Using this triple-add protocol, we were able to screen for agonists, antagonists, and potentiators simultaneously, maximizing the efficiency of the screen. Figure 1 depicts representative traces showing the effect of the different categories of mGluR5 ligands. The addition of an EC20 concentration of glutamate followed by an EC80 glutamate addition with this interval did not lead to substantial desensitization of the EC80 glutamate response (Fig. 1A). As can be seen in Fig. 1B, the addition of a maximally effective concentration of the mGluR5 agonist DHPG as the test compound induced a robust response upon the addition and desensitized responses to EC20 and EC80 concentrations of glutamate. The mGluR5 NAM MPEP elicited no response alone but completely blocked responses to both EC20 and EC80 concentrations of glutamate (Fig. 1C). Finally, the known mGluR5 PAM CPPHA elicited no response when added alone but potentiated the response to EC20 glutamate application (Fig. 1D). These data suggest that this assay is capable of detecting compounds belonging to each of these categories. It is important to note that inverse agonist activity of mGluR5 NAMs that has been described in earlier studies has never been observed using the calcium fluorescence assay used here. Thus, this assay cannot be used to determine whether novel NAMs have inverse agonist activity.
Fig. 1.

High-throughput screen identifies compounds exhibiting agonist, antagonist, or potentiator activity. Traces show effect of either vehicle (A), a representative agonist (DHPG) (B), antagonist (MPEP) (C), or potentiator (CPPHA) (D) on mGluR5 calcium mobilization response to glutamate. Compounds (1 or 10 μM) were added to cells loaded with a calcium-sensitive dye and incubated for 2.5 min. A submaximal (EC20) followed by a nearly maximal (EC80) concentration of glutamate was added, and the calcium response was measured by the FDSS plate reader. Responses are expressed as a fluorescence ratio.
Raw kinetic data were normalized in a multistep process: 1) differences in cell number, nonuniform illumination/imaging, and dye-loading were controlled for based on the initial readings for the well by dividing each time point by the fluorescence reading at the initial time point; 2) three measurement windows were defined surrounding the addition of compound, glutamate EC20, and glutamate EC80; and 3) the signal amplitude for the minimum data point of each window was subtracted from each point on the trace. Wells in which responses that differed from vehicle wells by 3 S.D. were selected as hits for further study. In addition, data from each plate were visually inspected to ensure the selection of hits. This assay was used to screen an internal Vanderbilt library consisting of 160,000 small molecules selected based on maximal chemical diversity as reported for previous screens performed at Vanderbilt (Niswender et al., 2008b; Marlo et al., 2009; Sheffler et al., 2009). Compounds were screened (n = 1) at a nominal concentration of 10 μM diluted from 10 mM stocks that were maintained in 100% DMSO (final DMSO concentration, 0.1%).
Primary hits were selected for retest using full CRC analysis for the appropriate activity. Thus, CRC analysis for agonist hits was performed in the absence of glutamate, CRC analysis of potentiators was performed in the presence of an EC20 concentration of glutamate, and CRC analysis for antagonist hits was performed in the presence of an EC80 concentration of glutamate. Finally, all primary hits were screened for activity at an endogenous Gq-coupled muscarinic receptor present in a parental, untransfected HEK cell line not containing mGluR5 using an identical protocol except that EC20 and EC80 concentrations of carbachol were used in place of glutamate. The rationale for this counterscreen was that compounds acting by a nonspecific mechanism should have no effect in cells that do not express mGluR5. Thus, they should have no agonist activity in the parental cell line and should not potentiate or inhibit the response to an agonist of an unrelated G protein-coupled receptor. Compounds for which clear CRC relationships were demonstrated in mGluR5-expressing cells but which had no activity in the parental cell line were selected as verified hits.
Table 1 provides a summary of primary and secondary screening results. The primary screen of 160,000 compounds for activity at mGluR5 at 10 μM yielded 2877 (2403 available for retesting) potentiator hits, 784 (624 available for retesting) antagonist hits, and 22 agonist hits. A total of 1387 compounds were confirmed as having potentiator activity, producing a retest rate of approximately 60% and an overall hit rate of 0.9%. Likewise, 345 antagonists (55%) and all 22 agonists were confirmed. The industry standard is generally at or below 50% for retests and 0.1 to 0.5% for overall hit rate (Hodder et al., 2003). Thus, we had a hit rate for allosteric potentiators that was relatively high, a hit rate for antagonists that was consistent with this roughly defined industry standard, and a relatively low hit rate for mGluR5 agonists. Of the verified potentiator hits, 9 compounds had EC50 values lower than 100 nM, 63 compounds had EC50 values lower than 500 nM, and 156 compounds had EC50 values lower than 1 μM. The verified antagonists demonstrated increased potency compared with potentiators in which 33 compounds had EC50 values lower than 100 nM, 109 compounds had EC50 values lower than 500 nM, and 173 compounds had EC50 values lower than 1 μM. A wide range of efficacy values was present for potentiators, represented by the percentage of maximal glutamate values of a test compound at 30 μM, with some nearing that of the maximal glutamate response. Approximately 10% of verified hits belonging to each hit category affected the agonist response of carbachol or untransfected HEK cells, demonstrating initial selectivity for mGluR5 among the majority of the compounds.
TABLE 1.
Summary of primary and secondary screening results
| Primary Screen | Verification | Nonselective | |
|---|---|---|---|
| Agonist | 22 (0.01%) | 22 (100%) | 2 (9%) |
| Potentiator | 2403 (1.5%) | 1387 (60%) | 147 (11%) |
| Antagonist | 624 (0.39%) | 345 (55%) | 40 (12%) |
Compounds Discovered in the Screen Exhibit Multiple Functional Effects.
CRC analysis of the confirmed hits in each category revealed that these compounds exhibited a broad range of functional activities, including full and partial antagonism and allosteric potentiator activity. Figure 2 depicts the structures, concentration-response curves, and raw data traces of examples of hits discovered in the screen highlighting the various categories. Figure 2, A and B, illustrates examples of two structurally and pharmacologically distinct verified antagonist hits. The example shown, VU0040228, acts as a full antagonist, completely blocking the glutamate response to mGluR5 in a concentration-dependent manner (IC50 = 62 ± 28 nM, mean ± S.E.M., n = 3 experiments). In addition, 13 of the confirmed antagonist compounds (3.8%) proved to have “partial antagonist” activity when assessed for their ability to inhibit the response to an EC80 concentration of glutamate. We reported previously the discovery and detailed characterization of MPEP analogs that have partial antagonist activity (Rodriguez et al., 2005). However, it has not been clear whether it would be possible to identify partial antagonists based on other chemical scaffolds. In addition, compounds in this category had an unprecedented range of partial antagonism of mGluR5 ranging from 10 to 80%, with EC50 values in the range of 62 nM to 2 μM. This suggests that these compounds exhibit a range of negative cooperativities for regulating glutamate activity. Compound VU0029251 [2-(methylthio)-6,7-dihydro-5H-cyclopenta[4,5]thieno[2,3-d]pyrimidin-4-amine] exemplified this unique class of compounds (IC50 = 1.7 ± 0.6 μM, mean ± S.E.M., n = 3 experiments), with a maximal inhibition of the glutamate response of 50 ± 6.8% (mean ± S.E.M., n = 3 experiments) (Fig. 2, A and B).
Fig. 2.

Diverse structural scaffolds have a variety of functional activities on mGluR5-mediated calcium mobilization. Raw traces (A and C) and concentration-response curves (B and D) were generated for each compound in the presence of glutamate. Varying concentrations of test compound were added to calcium-sensitive dye-loaded cells and incubated for 5 min. A nearly maximal (EC80) or suboptimal (EC20) concentration of glutamate was added, and the calcium response was measured. A and B, full antagonist VU0040228 (▴) completely blocks the response to an EC80 concentration of glutamate, whereas partial antagonist VU0029251 (■) blocks the response to 50% of that of EC80 glutamate. C and D, PAMs VU0028316 (▴) and VU0092273 (■) enhance the response to an EC20 concentration of glutamate in a concentration-dependent manner. VU0028316 exhibits no intrinsic agonist activity, whereas VU0092273 induces a small sustained response when added alone. Data represent the mean ± S.E.M. of at least three independent experiments performed in triplicate. Data are plotted as a percentage of the EC80 (B) or maximum (D) response to glutamate.
Approximately 1400 compounds were discovered that potentiated a suboptimal concentration of glutamate. Of the mGluR5 PAMs identified, 63 had EC50 values lower than 500 nM with a wide range of efficacies, some potentiating the response to an EC20 glutamate concentration to near a maximal glutamate response. Compounds VU0092273 and VU0028316 [(E)-3-(3-(2-methyl-5-phenylfuran-3-carboxamido)phenyl)acrylic acid] (Fig. 2, C and D) demonstrate some of the structural and functional diversity exhibited by these novel mGluR5 PAMs. VU0028316 is a pure potentiator in the calcium mobilization assay (EC50 = 139 ± 26 nM, mean ± S.E.M., n = 3 experiments) and exhibited no intrinsic agonist activity. In contrast, VU0092273 acts as a potent agonist-potentiator and induces a small sustained response when added alone and potentiates the response to an EC20 concentration of glutamate in HEK cells expressing rat mGluR5. The potency of this compound as an allosteric potentiator is 10 ± 5 nM (mean ± S.E.M., n ≥ 3 experiments), making it one of the most potent mGluR5 PAMs identified to date.
Novel Modulators Exhibit Multiple Binding Profiles at mGluR5.
Competition binding studies were performed with the allosteric antagonist [3H]methoxyPEPy to determine whether the compounds discovered in the screen interact with the well characterized allosteric binding site for the mGluR5 NAM MPEP (Fig. 3). Both the mGluR5 NAM VU0040228 (Ki = 82 ± 12 nM, mean ± S.E.M., n = 3 experiments; Fig. 3A) and the PAM VU0092273 (Ki = 970 ± 140 nM, mean ± S.E.M., n = 3 experiments; Fig. 3B) fully compete with the equilibrium binding of the radioligand, suggesting an interaction with the MPEP allosteric site. In contrast, another mGluR5 PAM, VU0028316, does not compete for binding to the MPEP site at concentrations up to 100 μM (Fig. 3B), a concentration well above that at which the compound reaches a maximum response in the calcium assay. These data suggest that this compound does not directly interact with the MPEP binding site and instead acts via an alternate site not yet elucidated. The partial antagonist VU0029251 has a modest effect on radioligand equilibrium binding (Fig. 3A), reducing maximal [3H]methoxyPEPy binding to a level of approximately 50% of the total binding (Ki = 1.07 ± 0.15 μM, mean ± S.E.M., n = 3 experiments). The variety of binding modes exemplified by these compounds provides strong support for the growing view that allosteric modulators can act at multiple sites on mGluR5 to have a range of profiles in terms of functional modulation of receptor activity.
Fig. 3.

Novel mGluR5 modulators exhibit multiple binding profiles to mGluR5. Competition binding curves for allosteric modulators were obtained in the presence of 2 nM [3H]methoxyPEPy using membranes harvested from mGluR5-expressing HEK293 cells. A, antagonist VU0040228 (▴) fully competes with the equilibrium of [3H]methoxyPEPy (Ki = 82 ± 12 nM), whereas partial antagonist VU0029251 (■) only reaches the level of approximately 50% total binding (Ki = 1.07 ± 0.15 μM). B, PAM VU0092273 (■) fully competes with the equilibrium of radioligand (Ki = 970 ± 140 nM), whereas PAM VU0028316 (▴) has no effect. Data represent the mean ± S.E.M. of three independent experiments performed in triplicate. Data are plotted as a percentage of total [3H]methoxyPEPy binding.
Novel mGluR5 NAMs and PAMs Are Selective for mGluR5 Relative to Other mGluR Subtypes.
One of the most exciting aspects of the current findings is that they provide multiple allosteric modulators of mGluR5 that are structurally distinct from previous compounds. For instance, the majority of mGluR5 NAMs reported previously are based on the biaryl acetylene scaffold represented by MPEP. These novel compounds provide tools to allow further testing of the hypothesis that structurally diverse molecules exerting different modes of efficacy on mGluR5 have predicted effects in animal models. For a compound to be useful as a proof of concept tool in behavioral models relevant to mGluR5, it must be selective for this receptor relative to other mGluR subtypes. We examined the selectivity profiles of mGluR5 NAM, VU0040228, and PAM, VU0092273, by determining the activities of each compound at a representative example of each of the three subgroups of mGluRs: group I (mGluR1), group II (mGluR3), and group III (mGluR4). We initially measured the agonist concentration-response relationship of the other mGluRs in the presence and absence of 10 μM NAM or PAM. In this manner, potential negative or positive allosteric modulator activity could both be assessed in a single assay. VU0040228 (10 μM), a concentration capable of producing maximal effect on mGluR5 responses, had no significant effect on the agonist response of mGluRs 1 and 4 but was found to slightly decrease the maximal effect of the agonist response of mGluR3 whereas having no significant effect on the potency of glutamate (see Supplemental Fig. S1). The mGluR5 PAM, VU0092273, also had no effect on the agonist response to mGluRs 1 and 4 but was found to significantly block the mGluR3 response to glutamate at 10 μM compound (see Supplemental Fig. S2). We then determined the potency of VU0092273 at mGluR3 by examining the effects of a range of concentrations of compound in the presence of an EC80 concentration of glutamate. VU0092273 inhibited the mGluR3 EC80 response to glutamate with an IC50 value of 6.3 ± 1.6 μM (mean ± S.E.M., n = 3 experiments), a value significantly higher than the potency at which the compound potentiates the mGluR5 EC20 response to glutamate (EC50 = 10 ± 5 nM, mean ± S.E.M., n ≥ 3 experiments). Based on their selectivity profiles, interaction with the MPEP binding site, and potency values, VU0040228 and VU0092273 were selected for further optimization and characterization.
Chemical Optimization of Novel mGluR5 NAM VU0040228.
In an effort to optimize the HTS hit VU0040228 for enhanced functional potency and binding affinity, and to dial out undesirable mGluR3 antagonist activity, we prepared a small library that held the pyridyl-oxadiazole moiety constant while varying the phenyl region of the molecule. The chemistry was carried out in a single reaction by coupling 2-pyridyl amidoxime with various substituted benzoic acids to generate the oxadiazole library (see Supplemental Data for syntheses). 3-(Pyridine-2-yl)-5-(3-(trifluoromethyl) phenyl)-1,2,4-oxadiazole (VU0255038) and 3-cyano-4-fluoro (VU0255036) analogs retained activity, but these substitutions led to a significant loss in potency (IC50 values of 2.4 and 6.3 μM, respectively; Table 2). Introduction of a halide in the 3-position was well tolerated but led to a slight loss in potency [5-(3-chlorophenyl)-3-(pyridine-2-yl)-1,2,4-oxadiazole (VU0067144), IC50 = 240 nM; and 5-(3-bromophenyl)-3-(pyridine-2-yl)-1,2,4-oxadiazole (VU0255037), IC50 = 215 nM]. As described previously (Kulkarni et al., 2009), both potency and affinity were enhanced with the introduction of a 3-cyano-5-fluoro substitution pattern (VU0285683). VU0285683 acts as a full antagonist, completely blocking the glutamate response to mGluR5 in a concentration-dependent manner (IC50 = 24.4 ± 3.6 nM, mean ± S.E.M., n = 3 experiments) and was found to fully compete with the equilibrium binding of [3H]methoxyPEPy (Ki = 16.9 ± 1.1 nM, mean ± S.E.M., n = 3 experiments; see Supplemental Fig. S5). This provides a nonacetylene mGluR5 NAM with an in vitro potency that is comparable with that of the prototypical biaryl acetylene mGluR5 NAM, MPEP. As in the case of VU0040228, VU0285683 had no significant effect on the agonist response of mGluRs1 and 4, and in contrast to VU0040228 had no effect on the agonist response of mGluR3, indicating that this optimized NAM was selective for mGluR5 compared with these other mGluR subtypes (see Supplemental Fig. S3).
TABLE 2.
Chemical optimization of VU0040228
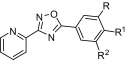
| R | R1 | R2 | IC50 | |
|---|---|---|---|---|
| nM | ||||
| VU0255038 | H | H | CF3 | 2360 ± 156 |
| VU0255036 | H | F | CN | 6310 ± 329 |
| VU0067144 | H | H | Cl | 240 ± 54 |
| VU0255037 | H | H | Br | 215 ± 29 |
| VU0285683 | F | H | CN | 24.4 ± 3.6 |
The Newly Identified mGluR5 NAM VU0285683 Induces Rightward Shifts in the Glutamate Concentration-Response Curves and Reduces the Maximal Effect of Glutamate.
We further evaluated the question of whether VU0285683 inhibits mGluR5 via a noncompetitive mechanism by examining the effects of this antagonist on the activation of mGluR5 by measuring the concentration-response relationship of glutamate in the presence and absence of a fixed concentration of test compound. We predicted that if the antagonist acted via a site other than the glutamate binding site, increasing concentrations of the compound would shift the curve to the right and decrease the maximal signal of glutamate, as opposed to inducing a parallel rightward shift seen in the presence of an orthosteric antagonist. Preincubation of mGluR5 cells with increasing concentrations (10, 30, and 100 nM) of VU0285683 induced robust rightward shifts of the glutamate concentration response curve and decreased the maximal response to glutamate, a pattern suggesting noncompetitive inhibition (Fig. 4). These data are consistent with the hypothesis that VU0285683 does not interact with the glutamate binding site and instead acts as a noncompetitive allosteric antagonist.
Fig. 4.

VU0285683 induces a rightward shift of the glutamate concentration-response curve and decreases the maximal response. DMSO-matched vehicle or 10, 30, or 100 nM VU0285683 was applied to mGluR5-expressing HEK293 cells before the addition of a range of glutamate concentrations, and the response was measured via calcium mobilization. Incremental rightward shifts of the glutamate CRC and a decrease of the maximal glutamate signal were observed. Data represent the mean ± S.E.M. of three independent experiments performed in triplicate. Data are plotted as a percentage of maximal response to glutamate.
5MPEP Blocks VU0285683 Inhibition of mGluR5 Activity in a Competitive Manner.
The finding that VU0285683 fully competes with [3H]methoxyPEPy for binding to mGluR5 and inhibits the mGluR5 concentration-response relationship in a noncompetitive manner suggests that it interacts with the MPEP site of mGluR5. The neutral site ligand 5MPEP is a positional isomer of MPEP that binds to the MPEP site in a competitive manner but does not alter the glutamate concentration-response relationship (Rodriguez et al., 2005). Therefore, if VU0285683 acts by binding to the MPEP site, 5MPEP should compete with VU0285683 and induce a parallel rightward shift of the concentration-response curve. Figure 5A shows the effect of a range of concentrations of VU0285683 alone and in the presence of 5MPEP (10 μM). Consistent with our predictions, the neutral MPEP site ligand 5MPEP induces a rightward shift in the VU0285683 CRC (11.8 ± 0.9-fold, mean ± S.E.M., n = 3 experiments), suggesting that 5MPEP blocks the inhibitory effect of VU0285683 in a competitive manner.
Fig. 5.

VU0285683 interacts with the MPEP binding site. A, 5MPEP is a neutral antagonist of VU0285683. mGluR5-expressing HEK293 cells were preincubated with 5MPEP (10 μM, ▴) or vehicle (■) for 30 min; varying concentrations of VU0285683 were added, followed by an EC80 concentration of glutamate 90 s later, and the response was measured via calcium mobilization. 5MPEP shifted the VU0285683 concentration-response curve 11.8 ± 0.9-fold. B, a single point mutation that abolishes [3H]methoxyPEPy binding also blocks VU0285683-induced antagonism of mGluR5-mediated calcium mobilization in transiently transfected HEK293 cells. Introduction of A809V mutation shifted the concentration-response curve of VU0285683 5.6 ± 0.1-fold (▴) compared with wild-type mGluR5 (■). Data represent the mean ± S.E.M. of three independent experiments performed in triplicate. Data are plotted as a percentage of the EC80 response to glutamate.
A Mutation that Reduces the Effect of MPEP at mGluR5 Also Blocks VU0285683 Inhibition of mGluR5 Activity.
Mutation of alanine to valine at the 809 position of mGluR5 (A809V) significantly reduces the ability of MPEP to bind to the receptor and diminishes the potency of MPEP as a negative allosteric modulator of mGluR5 (Pagano et al., 2000; Malherbe et al., 2003). If our novel allosteric antagonist VU0285683 acts via the MPEP binding site, we would expect the A809V mutation to significantly reduce the activity of this compound in a similar manner. Figure 5B depicts the concentration-response relationship of VU0285683 in the presence of an EC80 concentration of glutamate on wild-type mGluR5 and on the A809V mutant receptor. The MPEP-sensitive mutant clearly reduces the potency of VU0285683 inhibition as seen by the parallel rightward shift of the concentration-response curve of the A809V mutant 5.6 ± 0.1-fold (mean ± S.E.M., n = 3 experiments) compared with wild type, consistent with the hypothesis that binding to the MPEP site is necessary for the antagonist activity of VU0285683. Taken together, these radioligand binding, mutagenesis, and molecular pharmacology data provide strong evidence that VU0285683 inhibits mGluR5 responses by binding to the same allosteric site as the prototypical mGluR5 antagonist MPEP.
The Novel mGluR5 NAM VU0285683 Produces Dose-Dependent Effects in Rodent Models of Anxiolytic Activity.
Previous studies have shown that the mGluR5 antagonists MPEP and MTEP produce anxiolytic effects in a wide variety of animal models (Spooren et al., 2000; Busse et al., 2004; Gasparini et al., 2008), including a modified version of the Geller-Seifter conflict paradigm, a classic measure of anxiolytic activity in rodents (Busse et al., 2004). To determine whether our conflict paradigm was sensitive to the anxiolytic effects of mGluR5 antagonists, we first tested MTEP as a positive control. Pretreatment with MTEP at 1, 3, 5.6, and 10 mg/kg produced a significant dose-related increase in punished responding (F4,23 = 24.8, p < 0.0001) (Fig. 6A) and a significant decrease in unpunished responding (F4,23 = 15.4, p < 0.0001) (Fig. 6C), suggesting anxiolytic effects. Post hoc analysis indicated that punished responses were significantly higher relative to vehicle treatment at the 3, 5.6, and 10 mg/kg doses of MTEP (p < 0.05, Newman-Keuls). Unpunished response rates declined significantly at the 10 mg/kg dose of MTEP (p < 0.05, Newman-Keuls). The results of this study indicated that the modified conflict paradigm is sensitive to the anxiolytic effects of mGluR5 antagonists. We then investigated the novel mGluR5 antagonist, VU0285683, for anxiolytic activity. Rats were pretreated with 1, 3, and 10 mg/kg VU0285683 45 min before being tested in the conflict paradigm. VU0285683 produced a significant dose-dependent increase in punished responding (F3,18 = 9.63, p < 0.0001) (Fig. 6B), as well as a significant decrease in unpunished responding (F3,18 = 16.78 p < 0.0001) (Fig. 6D), suggesting anxiolytic effects. Post hoc analysis indicated that both the 3 and 10 mg/kg doses of VU0285683 significantly increased punished responding relative to vehicle treated animals (p < 0.05, Newman-Keuls), whereas unpunished responding was significantly decreased at the 10 mg/kg dose (p < 0.05, Newman-Keuls).
Fig. 6.

VU0285683 dose-dependently increases punished responding to a level comparable with that of MTEP. Dose-response curves for the effects of MTEP on punished (A) and unpunished (C) responding. The data are the mean number of punished and unpunished responses that animals made when tested with 1, 3, 5.6, and 10 mg/kg MTEP and vehicle during 30-min test sessions. Each value represents the mean ± S.E.M. for 24 animals. Animals tested with 3, 5.6, and 10 mg/kg MTEP made a significantly greater number of punished responses than animals treated with vehicle (p < 0.05). Unpunished responding was significantly reduced at 10 mg/kg MTEP (p < 0.05). Dose-response curves for the effects of VU0285683 on punished (B) and unpunished (D) responding. The data are the mean number of punished and unpunished responses that animals made when tested with 1, 3, and 10 mg/kg VU0285683 and vehicle. Each value represents the mean ± S.E.M. for 19 animals. Animals tested with 3 and 10 mg/kg VU0285683 made a significantly greater number of punished responses than animals treated with vehicle (p < 0.05). Unpunished responding was significantly reduced at the 10 mg/kg dose (p < 0.05).
The effect of VU0285683 was also evaluated in marble burying, a second rodent behavioral model predictive of anxiolytic activity. Previous studies reveal that MPEP decreases a rodent's tendency to bury marbles, and this is believed to be indicative of the compound's anxiolytic activity (Spooren et al., 2000). We again validated our assay using MTEP as a positive control (Fig. 7A). Treatment with MTEP (10 and 15 mg/kg) produced a significant dose-dependent decrease in marbles buried compared with treatment with vehicle, suggesting anxiolytic effects (p < 0.005 and p < 0.0001, respectively). These results validated that our marble-burying protocol was sensitive to the effects of mGluR5 antagonists. Mice were then treated with 3, 5.6, and 10 mg/kg doses of VU0285683 as seen in Fig. 7B. Marble burying was significantly decreased relative to vehicle-treated animals at the 10 mg/kg dose (p = 0.0001), suggesting anxiolytic activity in a second model predictive of such an effect.
Fig. 7.

VU0285683 dose-dependently decreases marble-burying to a level comparable with that of MTEP. Dose-response curves for the effects of MTEP (A) and VU0285683 (B). The data are the mean number of marbles buried when tested with vehicle and 3, 5.6, 10, and 15 mg/kg MTEP or 3, 5.6, and 10 mg/kg VU0285683 during a 30-min test session. Each value represents the mean + S.E.M. for 9 to 11 animals. The vehicle-treated group buried approximately nine marbles over the 30-min time course. The positive comparator MTEP produced a significant inhibition of marble-burying behavior at a dose of 10 mg/kg (p < 0.005) and 15 mg/kg (p < 0.0001). VU0285683 dose-dependently inhibited marble burying over the course of the 30-min interval, significant at a dose of 10 mg/kg VU0285683 (p = 0.0001).
Next we evaluated the effects of VU0285683 on potentiation of PCP-induced hyperlocomotion, a preclinical model of psychotomimetic-like activity. Previous studies have demonstrated that MPEP and MTEP potentiate the psychotomimetic-like effects of PCP in animals over a dose range that overlaps with the anxiolytic effects of these compounds. It is believed that this may predict an adverse effect of mGluR5 NAMs based on the MPEP scaffold. In the present study, we evaluated the effects of a 10 mg/kg dose of VU0285683 or MTEP alone or in combination with a subthreshold dose of PCP (2.5 mg/kg) on locomotor activity. As shown in Fig. 8A, VU0285683 did not potentiate the effects of PCP-induced hyperlocomotion at a dose that produced robust anxiolytic-like effects in the punished responding and marble-burying assays. In contrast, MTEP produced a robust potentiation of PCP-induced hyperlocomotion over the time course tested (Fig. 8B).
Fig. 8.

VU0285683 does not potentiate the psychotomimetic-like effects of PCP on locomotor activity in contrast to MTEP. The effects of VU0285683 (A) and MTEP (B) on locomotor activity were evaluated at a dose of 10 mg/kg alone or in combination with a 2.5 mg/kg dose of PCP. Data are expressed as mean ± S.E.M. of the number of beam breaks per 5 min; S.E.M. are not shown if less than the size of the point (n = 6–8 animals per dose). *, p < 0.05 versus vehicle + PCP, Dunnett's test. Note, the Veh/Veh, VU0285683, and MTEP-alone dose groups were significantly different from the Veh + PCP group across the 70- to 120-min interval; * are not shown for clarity.
Chemical Optimization of Positive Allosteric Modulator VU0092273 Reveals Interesting Structure Activity Relationships.
In addition to providing a major advance in identifying and establishing a unique behavioral profile of structurally novel mGluR5 NAMs, discovery of novel mGluR5 PAMs provides an important advance. One mGluR5 PAM, VU0092273, bears close structural resemblance to the prototypical mGluR5 NAM MPEP (Fig. 9A). Although we reported previously that allosteric modulators that occupy the MPEP binding site can display a range of activities from PAM to NAM to neutral cooperativity with slight structural modifications (O'Brien et al., 2003; Rodriguez et al., 2005; Sharma et al., 2008), in previous studies, individual scaffolds tend to be strongly biased toward dominant PAM or NAM activity. Previous allosteric modulators based on the MPEP scaffold are predominantly biased toward robust and potent NAMs. Thus, it was interesting to identify VU0092273 as one of the most potent mGluR5 PAMs identified to date.
Fig. 9.

A, structures of phenyl acetylene mGluR5 NAMs and PAMs. B, regions of VU0092273 targeted for chemical optimization. C, optimization of phenyl acetylene VU0366026 to nicotinamide VU0360172.
Structure-activity relationship (SAR) development of allosteric potentiators has proven extremely challenging, with many chemical series displaying no tractable SAR (Lindsley et al., 2004, 2005; Kinney et al., 2005; Zhao et al., 2007). An iterative analog library synthesis approach was used to quickly establish SAR, determine chemical tractability for VU0092273, and eliminate the significant unwanted mGluR3 activity exhibited by the lead. In preliminary SAR studies, we found that the 3′F, 5′CN substitution that was useful in the NAM series leads to complete loss of activity of the mGluR5 PAMs. Thus, our focus, as outlined in Fig. 9B was the evaluation of alternative aryl/heteroaryl rings for the phenyl rings, diverse amide analogs for the piperidine nucleus, and alternative linkers to replace the acetylenic spacer.
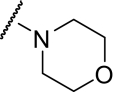
To accomplish this, we synthesized several libraries of functionalized aryl and heteroaryl analogs of VU0092273. In parallel, we focused on exploring a diverse array of amide analogs to diminish both oxidative metabolism and P-glycoprotein susceptibility. The scaffolds for conducting the amide library synthesis were generated via Sonogashira coupling to install the bi-phenyl or nicotinamide acetylene backbone, followed by hydrolysis of the ethyl ester to afford the acid. Standard amide formation chemistry was used to generate a library using polymer-supported reagents and scavengers (see Supplemental Data for representative syntheses). Table 3 highlights selected examples from this effort, which afforded a number of highly active mGluR5 PAMs. However, the efficacy of some compounds was relatively low in which the percentage of the maximal glutamate response was typically less than 50%. In addition, as observed with CDPPB (Lindsley et al., 2004; Chen et al., 2007), a degree of partial agonism was observed at higher compound concentrations when tested in our calcium assay using HEK293 cells expressing rat mGluR5. This is similar to what we reported previously for mGluR5 PAMs in the CDPPB series (Chen et al., 2007). Initially, morpholino amide derivative (4-morphonyl)(4-phenylethynyl)phenyl)methanone (VU0240381) provided an interesting profile (EC50 = 2.3 ± 1.0 nM, mean ± S.E.M., n = 3 experiments; cLog P = 3.6) relative to previous mGluR5 PAMs in that this compound has substantially improved potency and anticipated increased solubility in aqueous vehicles that could be used for in vivo studies. Alternative linker elements, such as amides and oxadiazoles, were evaluated as replacements for the acetylene moiety in an effort to avoid potential metabolic liabilities of the acetylene linker. Unfortunately, data from 2 24-member libraries demonstrated that these alternative linkers were generally inactive (data not shown).
TABLE 3.
Chemical optimization of VU0092273
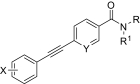
| Compound |
|
X | Y | Potency (EC50) | Efficacy | K i |
|---|---|---|---|---|---|---|
| nM | % Glu Max | nM | ||||
| VU0092273 |

|
H | CH | 10 ± 5 | 72 ± 7 | 970 ± 140 |
| VU0366025 |
|
H | CH | 98 ± 4 | 90 ± 9 | 1480 ± 100 |
| VU0366026 |
|
3-F | CH | 5.2 ± 1.7 | 84 ± 3 | 1120 ± 170 |
| VU0366024 |
|
H | CH | 19 ± 6 | 72 ± 11 | 1530 ± 320 |
| VU0240381 |

|
H | CH | 2.3 ± 1.0 | 84 ± 8 | 55 ± 4 |
| VU0361747 |

|
3-F | N | 214 ± 89 | 107 ± 8 | N.D. |
Finally, we synthesized a two-dimensional library to combine the optimal findings from each of the libraries prepared using the synthetic routes described herein to produce derivatives including compounds shown in Table 4. This library generated the most potent and efficacious (reaching a maximum response of 72 to 105% maximal glutamate) mGluR5 PAMs ever described. Of particular interest once again were the morpholino amide congeners such as (4-morphonyl)(4-(3-fluorophenyl)ethynyl)phenyl)methanone (VU0366031), which structurally represents an added 3-fluoro substituent on the pendant aryl group relative to VU0240381. The incorporation of the 3-fluoro substituent in particular improves both maximal glutamate response and affinity at the MPEP site. In addition, it was anticipated that fluorination of the pendant aryl ring may have a beneficial effect on metabolic stability based on metabolite structure identification studies using related nonfluorinated compounds (data not shown). Overall, VU0366031 displays the best combination of potency (EC50 = 3.8 ± 1.1 nM, mean ± S.E.M., n = 3 experiments) and efficacy (glutamate max = 98 ± 8%, mean ± S.E.M., n = 3 experiments) among all of the mGluR5 PAMs generated in this effort and generally provides a major improvement in affinity at the MPEP site. As mentioned earlier, the prototypical mGluR5 PAM CDPPB has an EC50 of 113 nM and a Ki at the MPEP site of 2.6 μM. VU0366031 (EC50 = 3.8 ± 1.1 nM, mean ± S.E.M., n = 3 experiments) has a Ki of 40 ± 12 nM (mean ± S.E.M., n = 3 experiments), and VU0240381 (EC50 = 2.3 ± 1.0 nM, mean ± S.E.M., n = 3 experiments) has a Ki of 55 ± 4 nM (mean ± S.E.M., n = 3 experiments). Thus, the biphenyl acetylene mGluR5 PAMs described herein represent a significant advance over CDPPB in terms of functional potency and binding affinity for mGluR5. Efforts to incorporate polarity in the core aryl group resulted in identification of (6-((3-fluorophenyl)ethynyl)pyridin-3-yl)(4-hydroxypiperidin-1-yl)methanone (VU0361747), however, at a potency loss of ∼50-fold (Fig. 9C and Table 3).
TABLE 4.
Chemical optimization of VU0092273 combining optimal findings from each library
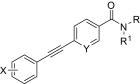
| Compound |
|
X | Y | Potency (EC50) | Efficacy | K i |
|---|---|---|---|---|---|---|
| nM | % Glu Max | nM | ||||
| VU0366030 |

|
3-F | CH | 23 ± 6 | 82 ± 8 | 1440 ± 150 |
| VU0366027 |
|
3-F | CH | 7.1 ± 2.1 | 72 ± 6 | 1580 ± 210 |
| VU0366031 |

|
3-F | CH | 3.8 ± 1.1 | 98 ± 8 | 40 ± 12 |
| VU0366028 |
|
3,4-difluoro | CH | 25 ± 5 | 103 ± 6 | 390 ± 50 |
| VU0366029 |
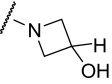
|
3-F | CH | 65 ± 16 | 104 ± 3 | 980 ± 20 |
| VU0360175 |

|
3-F | N | 49 ± 7 | 105 ± 4 | N.D. |
| VU0360172 |
|
3-F | N | 16 ± 6 | 87 ± 3 | 195 ± 20 |
Despite the apparent loss in potency upon incorporation of the nicotinamide nitrogen, this modification allowed for the first time the preparation of a salt form of the final compound that has not been possible for previously investigated mGluR5 PAMs. The ability to generate a salt can be an important factor in enhancing drug solubility and its dissolution rate for in vivo studies using neutral to weakly acidic nontoxic vehicles. As such, we incorporated the potency- and efficacy-enhancing morpholino amide substructure within the nicotinamide core to arrive at compound (6-((3-fluorophenyl)ethynyl)pyridin-3-yl)(morpholino)methanone (VU0360175). Although a potency loss was noted for VU0360175 relative to VU0366031 (EC50 = 49 versus 3.8 nM), the magnitude was 5-fold less than that observed for (4-hydroxypiperidin-1-yl)(4-(3-fluorophenyl)ethynyl)phenyl)methanone (VU0366026) and VU0361747. Further modification identified that small branched secondary amides and in particular, VU0360172 offers excellent potency (EC50 = 16 ± 6 nM, mean ± S.E.M., n = 3 experiments) and efficacy (glutamate maximum = 87 ± 3%, mean ± S.E.M., n = 3 experiments). More importantly, as described previously, we anticipate the presence of the nicotinamide functionality to be advantageous in terms of having optimal physiochemical properties for in vivo studies. It is noteworthy that VU0360172 had no significant effect on the agonist response of mGluRs1, 3, or 4, indicating that this optimized PAM was selective for mGluR5 compared with these other mGluR subtypes (see Supplemental Fig. S4). This represents a significant advance over HTS hit VU0092273, which exhibited considerable mGluR3 antagonist activity.
VU0360172 In Vitro and In Vivo Pharmacokinetic Profile.
A significant drawback of CDPPB and other mGluR5 PAMs are poor pharmacokinetic and physiochemical properties that limit in vivo dosing. Free fraction of a compound in the presence of plasma proteins and the stability of a compound in liver microsomes are important qualities in a desirable drug metabolism and pharmacokinetic profile. VU0360172 was tested for plasma protein binding in the presence of rat plasma proteins and was found to be 98.9% protein-bound. Although 4% free fraction is desirable, >1% demonstrates a degree of free fraction and is therefore greater than a minimum cutoff that we typically apply for a proof-of-concept compound. The compound was also tested for stability in both rat and human liver microsomes and was found to have excellent stability with 87 and 86% parent compound remaining after a 15-min incubation period (data not shown). In vivo pharmacokinetics of VU0360172 were studied in male Sprague-Dawley rats after oral administration of 10 mg/kg dose in 20% hydroxypropyl β-cyclodextrin (BCD) solution. At different time points including 0.5, 1, 3, and 6 h after dosing, the concentrations of VU0360172 were measured in hepatic portal vein plasma, systemic plasma (cardiac puncture), and whole-brain tissues. The compound was rapidly and very significantly absorbed, as evident from systemic plasma concentrations (see Supplemental Data, Fig. S6). The Cmax of 7432.98 ng/ml (∼21 μM) was achieved in systemic plasma within 1 h of dosing. There is very little or no first-pass hepatic effect, as indicated by the AUCsysplasma/AUChpvplasma ratio of 0.93. Although characterized by low to moderate CNS penetration (AUCbrain/AUCsysplasma = 0.13), more than acceptable levels (Cmax ∼2 μM) were achieved in brain after oral dosing of this compound.
Novel mGluR5 PAM Produced Dose-Dependent Activity in a Rodent Model Predictive of Antipsychotic-Like Activity.
Both typical and atypical antipsychotic drugs are known to reduce amphetamine-induced hyperlocomotion; this effect is believed to have predictive value for determining the antipsychotic efficacy of a compound (Kinney et al., 2003). Previous studies with CDPPB and ADX-47273 have demonstrated that these mGluR5 PAMs also have efficacy in this behavioral model (Kinney et al., 2005; Liu et al., 2008). However, although these compounds provided an important advance, they are not highly aqueous soluble and are therefore not optimal for in vivo dosing. Thus, previous studies with these earlier mGluR5 PAMs required dosing in vehicles containing DMSO, which is unpleasant for animals and makes more extensive behavioral studies difficult. Discovery of VU0360172 as a potent and efficacious mGluR5 PAM with improved aqueous solubility and a favorable PK profile represents a significant advance and may provide the first mGluR5 PAM that could be used for in vivo studies using standard aqueous vehicles. Thus, we determined the effect of the optimized mGluR5 PAM, VU0360172, on amphetamine-induced hyperlocomotor activity to determine whether this novel mGluR5 PAM has antipsychotic-like activity in this animal model. Administration of VU0360172 significantly reversed amphetamine-induced hyperlocomotion when analyzed across the 5-min intervals from the time of amphetamine delivery to the end of testing (t = 60–120 min). In the study using injections with the nontoxic vehicle 20% BCD, post hoc analysis revealed that doses of 30, 56.6, and 100 mg/kg i.p. VU0360172 produced significantly fewer ambulations than the group receiving vehicle and amphetamine across the time course (Fig. 10A). Again, the Veh/Veh and 56.6 mg · kg−1/Veh were also significantly different from the Veh/amphetamine-treated rats; however, the 56.6 mg · kg−1/Veh group was not different from the Veh/Veh group. When dosed orally in the 20% BCD vehicle, doses of 56.6 and 100 mg/kg VU0360172 also significantly reduced amphetamine-induced hyperlocomotor activity with no effect when administered alone 30 min before amphetamine addition (Fig. 10B). These data provide further support for the hypothesis that multiple structurally distinct mGluR5 PAMs can have antipsychotic-like activity in a rodent preclinical model and represent the first example of efficacy of an mGluR5 PAM when dosed in a vehicle that lacks short-term adverse effects. In addition, this provides a major advance in demonstration of an orally active mGluR5 PAM.
Fig. 10.

The mGluR5 PAM VU0360172 produced a dose-dependent reduction of amphetamine-induced hyperlocomotion. Rats were placed in the open-field chambers for a 30-min habituation interval, followed by a pretreatment with vehicle or a 10, 30, 56.6, or 100 mg/kg dose of test compound in 20% BCD vehicle for an additional 30 min. All rats then received an injection of 1 mg/kg s.c. amphetamine, and locomotor activity was measured for an additional 60 min. A, VU0360172 produced a significant decrease in amphetamine-induced hyperlocomotion after administration at doses of 30, 56.6, and 100 mg/kg i.p. and had no effect when administered alone at 56.6 mg/kg dose. B, VU0360172 produced a significant decrease in amphetamine-induced hyperlocomotion when administered orally at doses of 56.6 and 100 mg/kg and again had no effect when administered alone at the 56.6 mg/kg dose. Data are expressed as mean ± S.E.M. of the number of total beam breaks per 5-min intervals; S.E.M. are not shown if less than the size of the point (n = 6–8 per dose). Comparisons of treatment group effects relative to the Veh/amphetamine group were completed across the time interval from t = 60 to 120 min. *, p < 0.0001 versus vehicle/amphetamine group, Dunnett's test for both the intraperitoneal and oral administration studies.
Discussion
Using an innovative method of functional high-throughput screening capable of detecting multiple categories of activity, we have greatly expanded the structural and functional diversity of mGluR5 allosteric modulators. Structurally distinct novel molecules were discovered for each class of mGluR5 modulators, including NAMs with full antagonist activity (VU0285683), partial antagonists (VU0029251), agonist-potentiators (VU0092273), and pure potentiators (VU0028316). One of the advances provided by these studies is the finding that some of liabilities of previously identified mGluR5 modulators are not intrinsic to the target or mechanism and can be avoided with new chemical scaffolds. We and others reported previously that mGluR5 NAMs in the MPEP class induce significant potentiation of behavioral effects of NMDA receptor antagonists that are believed to reflect the psychotomimetic and cognition-impairing effects of these compounds (Henry et al., 2002; Kinney et al., 2003). This, coupled with findings that a member of a different chemical class of mGluR5 NAM, fenobam, induced adverse effects that seem to be similar to those of NMDA receptor antagonists in patients (Friedmann et al., 1980; Pecknold et al., 1982), led to a concern that mGluR5 NAMs may have psychotomimetic effects that could limit their clinical use. However, the mechanism of this effect of mGluR5 NAMs is not known and may not be intrinsic to all mGluR5 NAMs. Indeed, MPEP is known to interact with NMDA receptors, and this could contribute to this action in animal models (Homayoun et al., 2004). The finding that the novel mGluR5 NAM VU0285683 has anxiolytic-like activity in two rodent models but does not potentiate PCP-induced hyperlocomotor activity provides an exciting advance and suggests that it is possible to achieve full efficacy of mGluR5 NAMs in animal models of anxiolytic activity in the absence of potentiation of responses to NMDA receptor antagonists. In addition, the current finding that both MTEP and VU0285683 have efficacy in reducing marble-burying provides another model, lending support to the hypothesis that mGluR5 PAMs have anxiolytic-like activity.
In addition to the advance provided by discovery of this novel mGluR5 NAM, these studies represent a major step forward in the discovery of VU0360172 as an mGluR5 PAM with greatly improved properties relative to previous mGluR5 PAMs. A significant drawback of available mGluR5 PAMs is a lack of physiochemical and PK properties suitable for optimal in vivo dosing. Extensive medicinal chemistry efforts based on the CDPPB, ADX-47273, and CPPHA scaffolds (Lindsley et al., 2004; de Paulis et al., 2006; Zhao et al., 2007; Engers et al., 2009) have failed to produce compounds with the aqueous solubility and PK profiles required for optimal in vivo studies. Although limited in vivo studies have been performed with these earlier compounds, they have required dosing in DMSO, which can compromise the interpretation of in vivo data. Chemical optimization of HTS lead PAM VU0092273 led to the discovery of nicotinamide VU0360172 and the preparation of a salt form of the final compound, an important factor in enhancing drug solubility for in vivo studies. VU0360172 has activity in the animal model of psychosis, amphetamine-induced hyperlocomotion, validating the potential usefulness of mGluR5 potentiators in the treatment of schizophrenia. VU0360172 exhibited activity in this model not only when dosed intraperitoneally, but when dosed orally as well, providing the first orally bioavailable mGluR5 PAM.
The current studies also provide important advances in our understanding of the extent of structural diversity and modes of efficacy of mGluR5 allosteric modulators. VU0029251 provides the first example of a compound exhibiting mGluR5 partial antagonist activity that is not based on the MPEP scaffold (Rodriguez et al., 2005). Partial antagonists have potential as therapeutic agents in a setting in which there is a need to block receptor function but maintain some level of receptor activity. VU0029251 represents the first departure of partial antagonists from MPEP-related scaffolds. Both MPEP and structurally related MTEP function as inverse agonists, blocking all mGluR5 activity, including constitutive activity, a quality that could contribute to adverse side effects such as cognitive deficits and psychotomimetic effects. Divergence from this scaffold while maintaining partial antagonist activity may lead to an improved drug profile in the treatment of chronic disorders including pain and anxiety. However, examples of partial antagonists are rare, and the majority of MPEP-related mGluR5 NAMs that have been characterized fully block or act as inverse agonists at mGluR5. Thus, discovery of novel structural classes of mGluR5 NAMs that possess this profile provides further support for the potential usefulness of this approach.
It is noteworthy that although mGluR5 PAMs are known to interact at multiple allosteric sites, most NAMs identified to date are believed to act at the MPEP site (Gasparini et al., 2008). However, Zhang et al. (2010) recently identified a novel mGluR5 NAM with partial antagonist activity that did not fully displace [3H]MPEP binding. Likewise, it was interesting to find that another partial antagonist reported here, VU0029251, does not completely displace [3H]methoxyPEPy binding. This may suggest that VU0029251 does not compete directly for binding to this site but instead inhibits MPEP site binding by an allosteric mechanism. Previously identified partial antagonists that interact with the MPEP site fully displace binding and their apparent Ki values are consistent with the IC50 values in functional assays (Rodriguez et al., 2005). Furthermore, a broad range of structurally distinct mGluR1 NAMs all interact with a single allosteric site that is homologous with the MPEP site on mGluR5 (Pagano et al., 2000; Lavreysen et al., 2003), although mGluR1 PAMs have been identified that do not interact with this site (Chen et al., 2008). Based on this, there has been a growing view that multiple allosteric sites may exist but that it may be difficult to identify mGluR NAMs that do not act by competitive interactions with the MPEP site or a homologous site on other mGluR subtypes. The present data provide the first evidence that it may be possible to develop mGluR5 NAMs that interact with multiple allosteric sites.
Both the full antagonist VU0285683 and the agonist-potentiator VU0092273 fully compete with the equilibrium binding of the MPEP site ligand. However, reduction of MPEP binding does not necessarily imply a competitive interaction. We have reported previously that CDPPB and related compounds act by binding in a competitive manner at the MPEP site (Chen et al., 2007), and our current studies provide evidence suggesting that the novel NAM VU0285683 also acts competitively at the MPEP site. Based on the structural similarities of the new PAMs derived from VU0092273 and MPEP, along with the potent and full displacement of MPEP ligand binding, it is likely that these novel PAMs also act at the MPEP site. However, previous studies suggest that the structurally distinct mGluR5 PAM CPPHA does not act at this site (Chen et al., 2008), and in this report we have also identified VU0028316 as another example of an mGluR5 PAM that does not inhibit MPEP site binding. It is noteworthy that VU0028316 bears no obvious structural resemblance to either MPEP or CPPHA. In future studies, it will be important to determine whether this compound is likely to act at the same site as CPPHA or at a new distinct PAM site.
Chemical optimization of VU0092273 has provided us with clear advances over available PAMs such as CDPPB and ADX-47273 in terms of potency and binding affinity. However, it is important to note that although both potency and affinity values are left-shifted compared with CDPPB and ADX-47273, the binding affinity of these novel PAMs to the MPEP site is still considerably lower than the potencies of the compounds when assessed as PAMs in our functional assay. We have also observed this phenomenon in the CDPPB series, and this disconnect suggests a strong positive cooperativity between the allosteric modulator and glutamate when both ligands are present (Chen et al., 2007; Conn et al., 2009a). A greater leftward shift of potency compared with binding affinity implies a larger degree of positive cooperativity, as seen in the case of VU0092273.
In summary, we developed a screening strategy that allowed us to identify potent and efficacious mGluR5 modulators that possessed diverse chemical scaffolds. These modulators exhibited unique binding modes to the receptor, providing support for mGluR5 modulator activity acting via both the MPEP site and a distinct site. These ligands showed selectivity for mGluR5 versus other mGluR5 subtypes and belonged to scaffolds distinct from known compounds. Finally, both NAM VU0285683 and PAM VU0360172 demonstrated in vivo activity in rodent models of anxiety and antipsychotic activity, respectively, validating the potential of mGluR5 NAMs and PAMs for the treatment of CNS disorders, including anxiety and schizophrenia.
Supplementary Material
Acknowledgments
We thank Daryl F. Venable, Rocio Zamorano, Qingwei Luo, and Kiran K. Gogi for invaluable technical assistance.
The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.
This work was supported by the National Institutes of Health National Institute of Neurological Disorders and Stroke [Grants 1F32-NS049865, R01-NS031373]; the National Institutes of Health National Institute of Mental Health [Grants R01-MH062646, R01-MH074953]; and the National Institutes of Health National Institute on Drug Abuse [Grant 1R01-DA023947-01]. Vanderbilt is a center in the National Institutes of Health-supported Molecular Libraries Screening Centers Network.
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
doi:10.1124/mol.110.067207.
- CNS
- central nervous system
- mGluR
- metabotropic glutamate receptor
- PAM
- positive allosteric modulator
- ESI
- electrospray ionization
- NAM
- negative allosteric modulator
- MS
- mass spectrometry
- MS/MS
- tandem mass spectrometry
- MPEP
- 2-methyl-6-(phenylethynyl)pyridine hydrochloride
- NMDA
- N-methyl-d-aspartate
- DHPG
- dihydroxyphenylglycine
- SAR
- structure-activity relationship
- methoxyPEPy
- 3-methoxy-5-(pyridin-2-ylethynyl)pyridine
- CDPPB
- 3-cyano-N-(1,3-diphenyl-1H-pyrazol-5-yl)benzamide
- HRMS
- high-resolution mass spectrometry
- CPPHA
- N-{4-chloro-2-[(1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl)methyl]phenyl}-2-hydroxybenzamide
- 5MPEP
- 5-methyl-6-(phenylethynyl)pyridine
- HPLC
- high-performance liquid chromatography
- HEK
- human embryonic kidney
- DMEM
- Dulbecco's modified Eagle's medium
- PK
- pharmacokinetic
- PCP
- phencyclidine
- DMSO
- dimethyl sulfoxide
- FDSS
- functional drug screening system
- M1
- muscarinic receptor subtype 1
- Veh
- vehicle
- VI
- variable interval
- FBS
- fetal bovine serum
- CRC
- concentration-response curve
- MTEP
- 3-((2-methyl-4-thiazolyl)ethynyl)pyridine
- HTS
- high-throughput screening
- BCD
- β-cyclodextrin
- ADX-47273
- S-(4-fluoro-phenyl)-{3-[3-(4-fluoro-phenyl)-[1,2,4]-oxadiazol-5-yl]-piperidin-1-yl}-methanone
- VU0366025
- (3-hydroxyazetidin-1-yl)(4-phenylehtynyl)phenyl)methanone
- VU0366028
- (4-((3,4-difluorophenyl)ethynyl)phenyl) (morpholino)methanone
- VU0366029
- (3-hydroxyazetidin-1-yl)(4-(3-fluorophenyl)ethynyl)phenyl)methanone
- VU0366024
- (4-methanolpiperidin-1-yl)(4-phenylethynyl)phenyl)methanone
- VU0366027
- (4-((3-fluorophenyl)ethynyl)phenyl)(4-hydroxymethyl)piperidin-1-yl)methanone
- VU0366030
- (4-((3-fluorophenyl)ethynyl)phenyl)(4-hydroxy-4-methylpiperidin-1-yl)methanone
- VU0040228
- 5-(3,5-dimethoxyphenyl)-3-(pyridine-2-yl)-1,2,4-oxadiazole
- VU0285683
- 3-fluoro-5-(3-(pyridine-2-yl)-1,2,4-oxadiazol-5-yl)benzonitrile
- VU0255037
- 5-(3-bromophenyl)-3-(pyridine-2-yl)-1,2,4-oxadiazole
- VU0067144
- 5-(3-chlorophenyl)-3-(pyridine-2-yl)-1,2,4-oxadiazole
- VU0255038
- 3-(pyridine-2-yl)-5-(3-(trifluoromethyl)phenyl)-1,2,4-oxadiazole
- VU0092273
- (4-hydroxypiperidin-1-yl)(4-phenylethynyl)phenyl)methanone
- VU0240381
- (4-morphonyl)(4-phenylethynyl)phenyl) methanone
- VU0366026
- (4-hydroxypiperidin-1-yl)(4-(3-fluorophenyl)ethynyl)phenyl)methanone
- VU0366031
- (4-morphonyl)(4-(3-fluorophenyl) ethynyl)phenyl)methanone
- VU0360175
- (6-((3-fluorophenyl)ethynyl)pyridin-3-yl)(morpholino)methanone
- VU0361747
- (6-((3-fluorophenyl)ethynyl)pyridin-3-yl)(4-hydroxypiperidin-1-yl)methanone
- VU0360172
- N-cyclobutyl-6-((3-fluorophenyl)ethynyl)nicotinamide
- VU0029251
- 2-(methylthio)-6,7-dihydro-5H-cyclopenta[4,5]thieno[2,3-d]pyrimidin-4-amine
- VU0028316
- (E)-3-(3-(2-methyl-5-phenylfuran-3-carboxamido)phenyl)acrylic acid.
References
- Ayala JE, Chen Y, Banko JL, Sheffler DJ, Williams R, Telk AN, Watson NL, Xiang Z, Zhang Y, Jones PJ, et al. (2009) mGluR5 positive allosteric modulators facilitate both hippocampal LTP and LTD and enhance spatial learning. Neuropsychopharmacology 34:2057–2071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bear MF, Huber KM, Warren ST. (2004) The mGluR theory of fragile X mental retardation. Trends Neurosci 27:370–377 [DOI] [PubMed] [Google Scholar]
- Busse CS, Brodkin J, Tattersall D, Anderson JJ, Warren N, Tehrani L, Bristow LJ, Varney MA, Cosford ND. (2004) The behavioral profile of the potent and selective mGlu5 receptor antagonist 3-[(2-methyl-1,3-thiazol-4-yl)ethynyl]pyridine (MTEP) in rodent models of anxiety. Neuropsychopharmacology 29:1971–1979 [DOI] [PubMed] [Google Scholar]
- Chen Y, Goudet C, Pin JP, Conn PJ. (2008) N-{4-Chloro-2-[(1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl)methyl]phenyl}-2-hydroxybenzamide (CPPHA) acts through a novel site as a positive allosteric modulator of group 1 metabotropic glutamate receptors. Mol Pharmacol 73:909–918 [DOI] [PubMed] [Google Scholar]
- Chen Y, Nong Y, Goudet C, Hemstapat K, de Paulis T, Pin JP, Conn PJ. (2007) Interaction of novel positive allosteric modulators of metabotropic glutamate receptor 5 with the negative allosteric antagonist site is required for potentiation of receptor responses. Mol Pharmacol 71:1389–1398 [DOI] [PubMed] [Google Scholar]
- Conn PJ, Christopoulos A, Lindsley CW. (2009a) Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nat Rev Drug Discov 8:41–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn PJ, Lindsley CW, Jones CK. (2009b) Activation of metabotropic glutamate receptors as a novel approach for the treatment of schizophrenia. Trends Pharmacol Sci 30:25–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosford ND, Roppe J, Tehrani L, Schweiger EJ, Seiders TJ, Chaudary A, Rao S, Varney MA. (2003a) [3H]Methoxymethyl-MTEP and [3H]methoxy-PEPy: potent and selective radioligands for the metabotropic glutamate subtype 5 (mGlu5) receptor. Bioorg Med Chem Lett 13:351–354 [DOI] [PubMed] [Google Scholar]
- Cosford ND, Tehrani L, Roppe J, Schweiger E, Smith ND, Anderson J, Bristow L, Brodkin J, Jiang X, McDonald I, et al. (2003b) 3-[(2-Methyl-1,3-thiazol-4-yl)ethynyl]-pyridine: a potent and highly selective metabotropic glutamate subtype 5 receptor antagonist with anxiolytic activity. J Med Chem 46:204–206 [DOI] [PubMed] [Google Scholar]
- de Paulis T, Hemstapat K, Chen Y, Zhang Y, Saleh S, Alagille D, Baldwin RM, Tamagnan GD, Conn PJ. (2006) Substituent effects of N-(1,3-diphenyl-1H-pyrazol-5-yl)benzamides on positive allosteric modulation of the metabotropic glutamate-5 receptor in rat cortical astrocytes. J Med Chem 49:3332–3344 [DOI] [PubMed] [Google Scholar]
- Engers DW, Rodriguez AL, Williams R, Hammond AS, Venable D, Oluwatola O, Sulikowski GA, Conn PJ, Lindsley CW. (2009) Synthesis, SAR and unanticipated pharmacological profiles of analogues of the mGluR5 ago-potentiator ADX-47273. Chem Med Chem 4:505–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedmann CT, Davis LJ, Ciccone PE, Rubin RT. (1980) Phase II double-blind controlled study of a new anxiolytic, fenobam (McN-3377) vs. placebo. Curr Ther Res 27:144–151 [Google Scholar]
- Gasparini F, Bilbe G, Gomez-Mancilla B, Spooren W. (2008) mGluR5 antagonists: discovery, characterization and drug development. Curr Opin Drug Discov Devel 11:655–665 [PubMed] [Google Scholar]
- Gasparini F, Kuhn R, Pin JP. (2002) Allosteric modulators of group I metabotropic glutamate receptors: novel subtype-selective ligands and therapeutic perspectives. Curr Opin Pharmacol 2:43–49 [DOI] [PubMed] [Google Scholar]
- Gasparini F, Lingenhöhl K, Stoehr N, Flor PJ, Heinrich M, Vranesic I, Biollaz M, Allgeier H, Heckendorn R, Urwyler S, et al. (1999) 2-Methyl-6-(phenylethynyl)-pyridine (MPEP), a potent, selective and systemically active mGlu5 receptor antagonist. Neuropharmacology 38:1493–1503 [DOI] [PubMed] [Google Scholar]
- Gass JT, Olive MF. (2009) Positive allosteric modulation of mGluR5 receptors facilitates extinction of a cocaine contextual memory. Biol Psychiatry 65:717–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemstapat K, de Paulis T, Chen Y, Brady AE, Grover VK, Alagille D, Tamagnan GD, Conn PJ. (2006) A novel class of positive allosteric modulators of metabotropic glutamate receptor subtype 1 interact with a site distinct from that of negative allosteric modulators. Mol Pharmacol 70:616–626 [DOI] [PubMed] [Google Scholar]
- Henry SA, Lehmann-Masten V, Gasparini F, Geyer MA, Markou A. (2002) The mGluR5 antagonist MPEP, but not the mGluR2/3 agonist LY314582, augments PCP effects on prepulse inhibition and locomotor activity. Neuropharmacology 43:1199–1209 [DOI] [PubMed] [Google Scholar]
- Hodder P, Cassaday J, Peltier R, Berry K, Inglese J, Feuston B, Culberson C, Bleicher L, Cosford ND, Bayly C, et al. (2003) Identification of metabotropic glutamate receptor antagonists using an automated high-throughput screening system. Anal Biochem 313:246–254 [DOI] [PubMed] [Google Scholar]
- Homayoun H, Stefani MR, Adams BW, Tamagan GD, Moghaddam B. (2004) Functional Interaction Between NMDA and mGlu5 Receptors: Effects on Working Memory, Instrumental Learning, Motor Behaviors, and Dopamine Release. Neuropsychopharmacology 29:1259–1269 [DOI] [PubMed] [Google Scholar]
- Institute of Laboratory Animal Resources (1996) Guide for the Care and Use of Laboratory Animals 7th ed Institute of Laboratory Animal Resources, Commission on Life Sciences, National Research Council, Washington DC [Google Scholar]
- Kinney GG, Burno M, Campbell UC, Hernandez LM, Rodriguez D, Bristow LJ, Conn PJ. (2003) Metabotropic glutamate subtype 5 receptors modulate locomotor activity and sensorimotor gating in rodents. J Pharmacol Exp Ther 306:116–123 [DOI] [PubMed] [Google Scholar]
- Kinney GG, O'Brien JA, Lemaire W, Burno M, Bickel DJ, Clements MK, Chen TB, Wisnoski DD, Lindsley CW, Tiller PR, et al. (2005) A novel selective positive allosteric modulator of metabotropic glutamate receptor subtype 5 has in vivo activity and antipsychotic-like effects in rat behavioral models. J Pharmacol Exp Ther 313:199–206 [DOI] [PubMed] [Google Scholar]
- Knoflach F, Mutel V, Jolidon S, Kew JN, Malherbe P, Vieira E, Wichmann J, Kemp JA. (2001) Positive allosteric modulators of metabotropic glutamate 1 receptor: characterization, mechanism of action, and binding site. Proc Natl Acad Sci USA 98:13402–13407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni SS, Zou MF, Cao J, Deschamps JR, Rodriguez AL, Conn PJ, Newman AH. (2009) Structure-activity relationships comparing N-(6-methylpyridin-yl)-substituted aryl amides to 2-methyl-6-(substituted-arylethynyl)pyridines or 2-methyl-4-(substituted-arylethynyl)thiazoles as novel metabotropic glutamate receptor subtype 5 antagonists. J Med Chem 52:3563–3575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavreysen H, Janssen C, Bischoff F, Langlois X, Leysen JE, Lesage AS. (2003) [3H]R214127: a novel high-affinity radioligand for the mGlu1 receptor reveals a common binding site shared by multiple allosteric antagonists. Mol Pharmacol 63:1082–1093 [DOI] [PubMed] [Google Scholar]
- Lea PM, 4th, Faden AI. (2006) Metabotropic glutamate receptor subtype 5 antagonists MPEP and MTEP. CNS Drug Rev 12:149–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsley CW, Shipe WD, Wolkenberg SE, Theberge CR, Williams DL, Jr., Sur C, Kinney GG. (2006) Progress towards validating the NMDA receptor hypofunction hypothesis of schizophrenia. Curr Top Med Chem 6:771–785 [DOI] [PubMed] [Google Scholar]
- Lindsley CW, Wisnoski DD, Leister WH, O'Brien JA, Lemaire W, Williams DL, Jr, Burno M, Sur C, Kinney GG, Pettibone DJ, et al. (2004) Discovery of positive allosteric modulators for the metabotropic glutamate receptor subtype 5 from a series of N-(1,3-diphenyl-1H- pyrazol-5-yl)benzamides that potentiate receptor function in vivo. J Med Chem 47:5825–5828 [DOI] [PubMed] [Google Scholar]
- Lindsley CW, Zhao Z, Leister WH, Robinson RG, Barnett SF, Defeo-Jones D, Jones RE, Hartman GD, Huff JR, Huber HE, et al. (2005) Allosteric Akt (PKB) inhibitors: discovery and SAR of isozyme selective inhibitors. Bioorg Med Chem Lett 15:761–764 [DOI] [PubMed] [Google Scholar]
- Liu F, Grauer S, Kelley C, Navarra R, Graf R, Zhang G, Atkinson PJ, Popiolek M, Wantuch C, Khawaja X, et al. (2008) ADX47273 [S-(4-fluoro-phenyl)-{3-[3-(4-fluoro-phenyl)-[1,2,4]-oxadiazol-5-yl]-piper idin-1-yl}-methanone]: a novel metabotropic glutamate receptor 5-selective positive allosteric modulator with preclinical antipsychotic-like and procognitive activities. J Pharmacol Exp Ther 327:827–839 [DOI] [PubMed] [Google Scholar]
- Malherbe P, Kratochwil N, Zenner MT, Piussi J, Diener C, Kratzeisen C, Fischer C, Porter RH. (2003) Mutational analysis and molecular modeling of the binding pocket of the metabotropic glutamate 5 receptor negative modulator 2-methyl-6-(phenylethynyl)-pyridine. Mol Pharmacol 64:823–832 [DOI] [PubMed] [Google Scholar]
- Marino MJ, Awad H, Poisik O, Wittmann M, Conn PJ. (2002) Localization and physiological roles of metabotropic glutamate receptors in the direct and indirect pathways of the basal ganglia. Amino Acids 23:185–191 [DOI] [PubMed] [Google Scholar]
- Marino MJ, Conn JP. (2002a) Modulation of the basal ganglia by metabotropic glutamate receptors: potential for novel therapeutics. Curr Drug Targets CNS Neurol Disord 1:239–250 [DOI] [PubMed] [Google Scholar]
- Marino MJ, Conn PJ. (2002b) Direct and indirect modulation of the N-methyl D-aspartate receptor. Curr Drug Targets CNS Neurol Disord 1:1–16 [DOI] [PubMed] [Google Scholar]
- Marlo JE, Niswender CM, Days EL, Bridges TM, Xiang Y, Rodriguez AL, Shirey JK, Brady AE, Nalywajko T, Luo Q, et al. (2009) Discovery and characterization of novel allosteric potentiators of M1 muscarinic receptors reveals multiple modes of activity. Mol Pharmacol 75:577–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moghaddam B. (2004) Targeting metabotropic glutamate receptors for treatment of the cognitive symptoms of schizophrenia. Psychopharmacology (Berl) 174:39–44 [DOI] [PubMed] [Google Scholar]
- Niswender CM, Conn PJ. (2010) Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu Rev Pharmacol Toxicol 50:295–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niswender CM, Johnson KA, Luo Q, Ayala JE, Kim C, Conn PJ, Weaver CD. (2008a) A novel assay of Gi/o-linked G protein-coupled receptor coupling to potassium channels provides new insights into the pharmacology of the group III metabotropic glutamate receptors. Mol Pharmacol 73:1213–1224 [DOI] [PubMed] [Google Scholar]
- Niswender CM, Johnson KA, Weaver CD, Jones CK, Xiang Z, Luo Q, Rodriguez AL, Marlo JE, de Paulis T, Thompson AD, et al. (2008b) Discovery, characterization, and antiparkinsonian effect of novel positive allosteric modulators of metabotropic glutamate receptor 4. Mol Pharmacol 74:1345–1358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien JA, Lemaire W, Chen TB, Chang RS, Jacobson MA, Ha SN, Lindsley CW, Schaffhauser HJ, Sur C, Pettibone DJ, et al. (2003) A family of highly selective allosteric modulators of the metabotropic glutamate receptor subtype 5. Mol Pharmacol 64:731–740 [DOI] [PubMed] [Google Scholar]
- O'Brien JA, Lemaire W, Wittmann M, Jacobson MA, Ha SN, Wisnoski DD, Lindsley CW, Schaffhauser HJ, Rowe B, Sur C, et al. (2004) A novel selective allosteric modulator potentiates the activity of native metabotropic glutamate receptor subtype 5 in rat forebrain. J Pharmacol Exp Ther 309:568–577 [DOI] [PubMed] [Google Scholar]
- Pagano A, Ruegg D, Litschig S, Stoehr N, Stierlin C, Heinrich M, Floersheim P, Prezèau L, Carroll F, Pin JP, et al. (2000) The noncompetitive antagonists 2-methyl-6-(phenylethynyl)pyridine and 7-hydroxyiminocyclopropan[b]chromen-1a-carboxylic acid ethyl ester interact with overlapping binding pockets in the transmembrane region of group I metabotropic glutamate receptors. J Biol Chem 275:33750–33758 [DOI] [PubMed] [Google Scholar]
- Pecknold JC, McClure DJ, Appeltauer L, Wrzesinski L, Allan T. (1982) Treatment of anxiety using fenobam (a nonbenzodiazepine) in a double-blind standard (diazepam) placebo-controlled study. J Clin Psychopharmacol 2:129–133 [PubMed] [Google Scholar]
- Porter RH, Jaeschke G, Spooren W, Ballard TM, Büttelmann B, Kolczewski S, Peters JU, Prinssen E, Wichmann J, Vieira E, et al. (2005) Fenobam: a clinically validated nonbenzodiazepine anxiolytic is a potent, selective, and noncompetitive mGlu5 receptor antagonist with inverse agonist activity. J Pharmacol Exp Ther 315:711–721 [DOI] [PubMed] [Google Scholar]
- Rodriguez AL, Nong Y, Sekaran NK, Alagille D, Tamagnan GD, Conn PJ. (2005) A close structural analog of 2-methyl-6-(phenylethynyl)-pyridine acts as a neutral allosteric site ligand on metabotropic glutamate receptor subtype 5 and blocks the effects of multiple allosteric modulators. Mol Pharmacol 68:1793–1802 [DOI] [PubMed] [Google Scholar]
- Roppe JR, Wang B, Huang D, Tehrani L, Kamenecka T, Schweiger EJ, Anderson JJ, Brodkin J, Jiang X, Cramer M, et al. (2004) 5-[(2-Methyl-1,3-thiazol-4-yl)ethynyl]-2,3′-bipyridine: a highly potent, orally active metabotropic glutamate subtype 5 (mGlu5) receptor antagonist with anxiolytic activity. Bioorg Med Chem Lett 14:3993–3996 [DOI] [PubMed] [Google Scholar]
- Sharma S, Rodriguez AL, Conn PJ, Lindsley CW. (2008) Synthesis and SAR of a mGluR5 allosteric partial antagonist lead: unexpected modulation of pharmacology with slight structural modifications to a 5-(phenylethynyl)pyrimidine scaffold. Bioorg Med Chem Lett 18:4098–4101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheffler DJ, Williams R, Bridges TM, Xiang Z, Kane AS, Byun NE, Jadhav S, Mock MM, Zheng F, Lewis LM, et al. (2009) A novel selective muscarinic acetylcholine receptor subtype 1 antagonist reduces seizures without impairing hippocampus-dependent learning. Mol Pharmacol 76:356–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spooren WP, Vassout A, Neijt HC, Kuhn R, Gasparini F, Roux S, Porsolt RD, Gentsch C. (2000) Anxiolytic-like effects of the prototypical metabotropic glutamate receptor 5 antagonist 2-methyl-6-(phenylethynyl)pyridine in rodents. J Pharmacol Exp Ther 295:1267–1275 [PubMed] [Google Scholar]
- Swanson CJ, Bures M, Johnson MP, Linden AM, Monn JA, Schoepp DD. (2005) Metabotropic glutamate receptors as novel targets for anxiety and stress disorders. Nat Rev Drug Discov 4:131–144 [DOI] [PubMed] [Google Scholar]
- Zhang P, Zou MF, Rodriguez AL, Conn PJ, Newman AH. (2010) Structure–activity relationships in a novel series of 7-substituted-aryl quinolines and 5-substituted-aryl benzothiazoles at the metabotropic glutamate receptor subtype 5. Bioorg. Med. Chem 18:3026–3035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Wisnoski DD, O'Brien JA, Lemaire W, Williams DL, Jr., Jacobson MA, Wittman M, Ha SN, Schaffhauser H, Sur C, Pettibone DJ, Duggan ME, Conn PJ, Hartman GD, Lindsley CW. (2007) Challenges in the development of mGluR5 positive allosteric modulators: the discovery of CPPHA. Bioorg Med Chem Lett 17:1386–1391 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.