

Abstract
Surface plasmon resonance (SPR) technology with biosensor surfaces has become a widely–used tool for the study of nucleic acid interactions without any labeling requirements. The method provides simultaneous kinetic and equilibrium characterization of the interactions of biomolecules as well as small molecule–biopolymer binding. SPR monitors molecular interactions in real time and provides significant advantages over optical or calorimetic methods for systems with strong binding coupled with small spectroscopic signals and/or reaction heats. A detailed and practical guide for nucleic acid interaction analysis using SPR–biosensor methods is presented. Details of the SPR technology and basic fundamentals are described with recommendations on the preparation of the SPR instrument, sensor chips and samples as well as extensive information on experimental design, quantitative and qualitative data analysis and presentation. A specific example of the interaction of a minor–groove–binding agent with DNA is evaluated by both kinetic and steady–state SPR methods to illustrate the technique. Since the molecules that bind cooperatively to specific DNA sequences are attractive for many applications, a cooperative small molecule–DNA interaction is also presented.
Keywords: Biosensor, surface plasmon resonance, small molecule–nucleic acid interaction, kinetics, steady–state analysis, cooperativity, Biacore, minor groove
I. Introduction
Biological systems function on a platform of complex and integrated biomolecular interactions (1–3). Signaling, transcription control of gene expression and a host of other complex processes frequently are built on sets of sequential biomolecular interactions and subsequent reactions (4–8). These sequential processes that control and direct cellular functions can generally be understood in terms of the interaction of a small number of molecules in each step. Appropriate assembly of the entire sequential array gives the beautifully intricate mechanism of cell function. The entire process can be understood and explained on the basis of thermodynamic theory that has been firmly established for over 100 years. The biomolecular associations involve macromolecular complexes as well as small molecule-macromolecule interactions that are essential for the control of cell processes. Most drugs, for example, are small molecules that must interact with a macromolecular receptor in order to affect the target cell function in a selective manner (5–12). The field of chemical biology is built around design and use of small molecules to control specific aspects of cell function through biopolymer interactions (13–15). In order to understand this intricate array of interactions and control systems, it is very informative to evaluate all of the specific sequential steps that can be isolated. This quantitative interaction information is essential to put available structural results into the appropriate context of cell function.
To establish a basic quantitative characterization of the interactions it is essential to determine a set of basic thermodynamic quantities (16–21). Two questions must be answered at the start of the investigations: (i) what do we want to know about the interactions and (ii) how can we accurately and with a reasonable effort determine the desired information? In the optimum case the binding affinity (the equilibrium constant, K, and Gibbs energy of binding, ΔG), stoichiometry (n, the number of compounds bound to the biopolymer), cooperative effects in binding, and binding kinetics (the rate constants, k, that define the dynamics of the interaction) should be determined. These fundamental parameters are the keys to a molecular understanding of the interactions and how they affect cellular functions. To determine these parameters an accurate method of determining the concentration of each component that is not bound and the concentration of their complex is required. The information must be determined as a function of concentration at equilibrium for accurate K and n, and as a function of time for k. For each system then the question becomes one of how to accurately determine the necessary concentrations as a function of time, reactant total concentrations, solution conditions, temperature, etc. The interacting systems can range from as little as two molecules to as many as are required to form the final complex. As described above, a more complete understanding of the biomolecular complexes requires determination of additional thermodynamic quantities that characterize complex formation in detail.
Because biological molecules have widely different molecular characteristics that can and generally do change on complex formation, it can be difficult to find methods to evaluate the full array of interactions under an appropriate variety of conditions. This is the “reasonable effort” part of question (ii) above. For many biologically important interactions binding is very strong and experiments must be conducted at very low concentrations, down to the nanomolar or lower levels. In a binding experiment the compound concentration should vary from below to above the Kd in order to obtain accurate binding results and this requires significant concentrations of both free and complexed molecules (16,22–24). In many methods, for example, those that involve some separation of free and bound forms such as dialysis, the unbound concentration has to be determined and if this is below around 100 nM, as expected for the large K values generally observed in biological systems and as generally required for effective drug molecules, measurement can be difficult or impossible. These concentrations fall below the detection limit for many systems and may require special methods such as radiolabels or fluorescent probes for added sensitivity in detection. The labels may significantly increase the sensitivity of detection, however, they may also perturb the interaction that is being investigated. An attractive alternative method, which is operational down to very low concentrations, is the use of biosensors with surface plasmon resonance (SPR) detection (17,25–30). SPR responds to the refractive index or mass changes at the biospecific sensor surface on complex formation (27–32). Since the SPR signal responds directly to the amount of bound compound in real time, as versus indirect signals at equilibrium that are obtained for many physical measurements, it provides a very powerful method to study biomolecular interaction thermodynamics and kinetics. Use of the SPR signal and direct mass response to monitor biomolecular reactions also removes many difficulties with labeling or characterizing the diverse properties of biomolecules (25–31).
To illustrate the power of the SPR method, small organic cation interactions with specific DNA sequences will be used as examples. Small molecule targeting of DNA has applications in therapeutics, from cancer treatment to antiparasitic applications, and in biotechnology, and development of such molecules for control of nucleic acid function is at the heart of chemical biology (9,5–10,16,25,29,32–47). The interaction of nucleic acids with small molecules has been a primary area of interest and research since before the discovery of the double helical structure of DNA. There are many successful anticancer agents that intercalate or alkylate DNA and they can have quite varied structure and properties (34–37). Dicationic minor groove binding heterocycles which target eukaryotic organisms that cause parasitic diseases, such as malaria and sleeping sickness, have also been known for many years and are used in humans and animals (9,10,38–40). Therapeutic targeting of nucleic acids recently entered a new and very exciting area with the discovery that four–stranded, quadruplex DNA structures can occur in important cellular DNA regions from chromosomal telomeres to oncogene promoters (34,35,41–46). Several studies have clearly shown that interactions of a range of small molecules with quadruplexes can yield anticancer activity. Although this discussion focuses on DNA, essentially all of the methods can be applied to RNA interactions. Additional treatment of the biosensor surfaces may be required when using RNA, however, due to possible hydrolysis of RNA by a number of agents.
Applications with small molecules can be very challenging in terms of signal obtained on binding in a biosensor–SPR experiment. The larger the bound molecule, the larger the SPR signal and macromolecule binding can give large signal to noise ratios (27,29,31). We will show, however, that the biosensor–SPR method with a macromolecular sensor surface can be used under appropriate conditions to investigate small molecule binding with current state of the art instruments. Because of the varied properties of the compounds, it is difficult and time–consuming to find other suitable methods that can quantitatively define their interactions with DNA (or RNA) under a variety of conditions.
As described above, in biosensor–SPR instruments and experiments it is the bound compound on the surface that is detected by the change in plasmon resonance angle and this can be determined very accurately from a low to a high fraction of surface binding sites covered. The unbound or free solution compound concentration is not measured but is simply prepared by dilution and is in a constant concentration in the solution that flows over the sensor surface. Since the SPR signal responds to the refractive index or mass changes at the biospecific sensor surface on complex formation, no specific labeling of detection ability for the compounds is required (25–27,31). For compounds that have molecular weights of approximately 300 or more, the SPR technology provides a very attractive method to study interactions with nucleic acids or proteins immobilized to form a biospecific target surface.
1.1. Fundamentals of molecular interactions by biosensor-SPR methods
For a single compound (C) binding to n equivalent sites on DNA (or other biopolymer) to give a complex (DNA•nCbound) the interaction process is described by kinetic and equilibrium equations as follows:
| 1 |
More complex models with nonequivalent sites are also well–known and are treated in a similar manner with more complex functions (27 and described below under Data Analysis, 3.2). Although a discussion of complex data fitting is beyond the scope of this article, the topic is covered in most biophysical chemistry texts and more complex models are included in commercial SPR instrument software packages as well as in software available from Myszka and coworkers (http://www.cores.utah.edu/interaction/software.html).
In the most common case it is of interest to study a variety of compounds for binding to a limited number of different DNAs. For this case, however, it is more convenient and efficient to immobilize the DNAs on the sensor surface to monitor complex formation. Biacore T100 and 2000/3000 instruments have sensorchips with four channels such that three DNAs can be immobilized with one flow cell left blank as a control for bulk refractive index subtraction. With a common sensor surface that has covalently attached streptavidin, a nucleic acid strand with biotin linked to either the 5′ or 3′ terminus can be captured to create the biospecific surface. The terminal attachment of biotin, through a flexible linker, leaves the nucleic acid binding sites open for complex formation. If streptavidin or the biotin linker creates problems with the interaction or nonspecific compound interactions, the nucleic acid can be synthesized with a terminal alkyl amine and the amine can be used to form a direct covalent amide bond with the surface through activation of carboxyl groups on the sensor surface. In Biacore instruments, which have been most widely used to date in the area of biosensor–SPR experiments, any molecule with free amino groups can be immobilized through amide bonds with activated carboxyl groups from sensor chip surface-linked carboxymethyl (CM) dextran. A range of other sensorchips surfaces and immobilization chemistries are also available and it is generally possible to find an appropriate surface for any biological interaction application (for Biacore instruments, see the web http://www.biacore.com/lifesciences/products/sensor_chips/guide/index.html).
The results from a biosensor–SPR experiment are typically presented as a set of sensorgrams, which plot a function that is directly related to the SPR angle versus time as shown in Figure 2. The angle change is reported in Biacore technology as resonance units (RU) where a 1000 RU response is equivalent to a change in surface concentration of about 1ng/mm2 of protein or DNA (the relationship between RU and ng of material bound will vary with the refractive index of the bound molecule) (27,29,31). With a DNA sequence immobilized on the chip surface, a compound solution is injected and as the solution flows over the surface, compound binding to the DNA is monitored by a change in SPR angle (as RU). After a selected time, buffer flow is restarted and dissociation of the complex can be monitored for an additional selected time period (Figure 2). Note that when the association phase collection time is long enough, a steady state plateau is reached such that the rate of binding of the small molecule equals the rate of dissociation of the complex and no change of signal with time is observed. The response of interest is the difference between the response in cells with immobilized DNA minus the response of the blank flow cell without DNA. If the added molecule does not bind to a target/receptor at the surface, the SPR angle change in the sample and reference flow cells will be the same in a properly functioning instrument and the signals, after subtraction, give a zero net RU response that is indicative of no binding. In the case where binding does occur, an extra amount, relative to the blank surface, of the added molecule is bound at the sensor surface and an additional SPR angle change is generated in the sample flow cell. Again, the amount of unbound compound in the flow solution is the same in the sample and reference flow cells and is subtracted so that only the bound molecule generates a positive SPR signal. The concentration of the unbound molecule is constant and is fixed by the concentration in the flow solution.
Fig. 2.
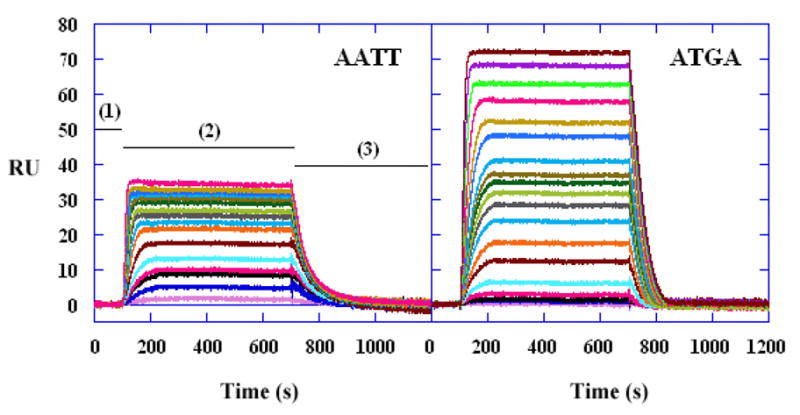
Representative SPR sensorgrams for the interaction of DB293 with AATT and ATGA oligomer hairpin duplexes. The DB293concentrations from bottom to top are 0 to 1μM.
Both the association and dissociation phases of the sensorgram can be simultaneously fit to a desired binding model in several sensorgrams at different concentrations with a global fit routine (27–32). Global fitting allows the most accurate determination of the kinetics constants as well as calculation of the equilibrium constant, Ka, from the ratio of kinetic constants (Equation 1). It is also possible to determine Ka independently of rate constants by fitting the steady–state response versus the concentration of the binding molecule in the flow solution over a range of concentrations. For the binding process in Equation 1 the steady–state RU at each compound concentration is determined and Ka can be obtained by fitting to the following equation:
| 2 |
where RUmax is the maximum change in RU for binding to a single DNA site. It can be calculated, determined experimentally at high compound concentrations or used in the above equation as a fitting parameter such that Ka, n and RUmax are determined by fitting RU versus Cf. Note that the common term “r”, the moles of compound bound per mole of DNA, is equal to RU/RUmax. At high Cf where all binding sites are filled with compound, RU = RUmax · n.
The refractive index change in SPR experiments generates essentially the same response for each bound molecule and can provide a direct determination of the stoichiometry, n, in equations 1 and 2 provided that the amount of immobilized nucleic acid is known (29,31). If the complex dissociates slowly, the surface can be regenerated before complete dissociation has occurred by a solution that causes rapid dissociation of the complex without irreversible damage to the immobilized DNA (29,47). For example, a solution at low or high pH can unfold DNA and cause complex dissociation. It should be noted, however, that accurate fitting of the dissociation part of the curve requires collection of as much of the total dissociation signal as possible. Additional injections of buffer at pH near 7 allow the DNA to refold for the next binding experiment. If the immobilized DNA is composed of separate strands, the duplex must be reformed by hybridization.
After the dissociation/regeneration phase is over and a stable baseline is re–established, a second concentration sample can be injected to generate a second sensorgram. This process can be repeated with as many concentrations as needed to obtain a broad coverage of the fraction of compound bound to the nucleic acid site or sites (see Figure 2). The kinetic and equilibrium constants that describe the reaction in Equation 1 are obtained by global fitting the sensorgrams with equations from a kinetic model or by fitting steady–state RU versus concentration plots to an appropriate binding model. The models are the same for all types of binding experiments and are not unique to SPR methods. It should be emphasized that to obtain accurate kinetic information for a binding reaction, it is essential that the kinetics for transfer of the binding molecule to the surface immobilized nucleic acid (mass transfer) be faster than the binding reaction. Equilibrium information can be obtained, however, by fitting sensorgrams even when mass transfer limitations prevent accurate determination of kinetics (48). It should be emphasized that, when properly conducted, biosensor–SPR kinetic and equilibrium results are in excellent agreement with other methods (26,27,29,30,48–50).
2. Materials
2.1. Required Materials for Instrument Cleaning
These materials are for the Biacore T100, 3000 and 2000 research instruments but similar materials are required for other instruments.
2.2. Running Buffer for Immobilization of DNA
HBS–EP buffer: 10 mM HEPES pH 7.4, 150 mM NaCl, 3 mM EDTA, 0.005%, v/v polysorbate 20. (GE Healthcare Inc.)
HBS–N buffer: 10 mM HEPES pH 7.4, 150 mM NaCl. (GE Healthcare Inc.)
Filter and degas all solution quite thoroughly.
It should be emphasized that the internal flow system of the instrument has microcapillaries that can be damaged by particulate matter in any solution.
2.3. Sensorchip Preparation for DNA Immobilization: CM5 or CM4 chip
A CM5 or CM4 sensor chip that has been at room temperature for at least 30 min (all sensorchips are available from GE Healthcare Inc.).
100 mM N-hydroxsuccinimide (NHS) freshly prepared in water.
400 mM N-ethyl-N′-(dimethylaminopropyl) carbodiimide (EDC) freshly prepared in water.
10 mM acetate buffer pH 4.5 (immobilization buffer).
200~400 mg/ml streptavidin in immobilization buffer.
Amino-labeled nucleic acid solutions (~25 nM of single strand or hairpin dissolved in HBS–EP buffer). (5′-end modified DNA obtained from Integrated DNA Technologies, Coralville, IA)
1 M ethanolamine hydrochloride in water pH 8.5 (deactivation solution).
Dock the CM4 or CM5 chip, Prime with running buffer. Start a sensorgram in all flow cells with a flow rate of 5μl/min. “Dock” and “Prime” are Biacore software commands that instruct the instruments to carry out specific operations. The commands and operations are listed in Table 2.
With NHS (100 mM) in one vial and EDC (400 mM) in other, use the Dilute command to make a 1:1 mixture of NHS/EDC.
Inject NHS/EDC for 10 min (50 μl) to activate the carboxymethyl surface to reactive esters.
Using Manual Inject with a flow rate of 5μl/min, load the loop with ~100 μl of streptavidin in the appropriate buffer and inject streptavidin over all flow cells. Track the number of RUs immobilized, which is available in real time readout, and stop the injection after the desired level is reached (typically 2500~3000 RU for CM5 chip and 1000~1500 RU for CM4 chip).
Inject ethanolamine hydrochloride for 10 min (50 μl) to deactivate any remaining reactive esters.
Prime several times to ensure surface stability.
DNA is immobilized as described under 2.4.
To reduce the non specific binding to immobilized streptavidin by small molecules, covalent immobilization using amino-labeled DNA could also be used. In this way, DNA can be directly captured on the EDC/NHS activated carboxymethyl dextran surface of the sensor chip (from step 16 to step 19).
Start from step 8 to step 10 to activate the sensorchip surface. Then, start a new sensorgram with a flow rate of 2 μl/min and select one desired flow cell on which to immobilize the nucleic acid.
Use Manual Inject, load the injection loop with ~100 μl of a 25 nM nucleic acid solution and inject over the low cell. Track the number of RUs immobilized and stop the injection after a desired level is reached (see Note 3).
Inject ethanolamine hydrochloride for 10 min (50 μl) to deactivate any remaining reactive esters.
Prime several times to ensure surface stability.
Table 2.
Biacore instrument commands
| Software commands | Function |
|---|---|
| Dock | Docks the sensor chip |
| Undock | Undocks the sensor chip |
| Prime | Flushes the flow system with running buffer |
| Dilute | Diluting samples with buffer or for preparing a defined mixture of two samples |
| Manual Inject | Manually controlled injection |
| Desorb | Removes adsorbed samples from the autosampler and IFC using SDS and glycine |
| Sanitize | Cleans pumps, IFC and autosampler from micro organisms using BIAdisinfectant solution |
2.4. Sensorchip Preparation for DNA immobilization: SA chip
A streptavidin–coated sensor chip (SA chip or prepared as outlined above) that has been at room temperature for at least 30 min.
HBS–EP buffer is used as running buffer.
Activation buffer (1 M NaCl, 50 mM NaOH).
Biotin–labeled nucleic acid solutions (~25 nM of single strand or hairpin dissolved in HBS-EP buffer).
Dock a streptavidin–coated chip and start a sensorgram with a 20 μl/min flow rate.
Inject activation buffer (1 M NaCl, 50 mM NaOH) for 1 min (20 μl) five to seven times to remove any unbound streptavidin from the sensor chip.
Allow buffer to flow at least 5 min before immobilizing the nucleic acids.
Start a new sensorgram with a flow rate of 2 μl/min and select one desired flow cell on which to immobilize the nucleic acid. Take care not to immobilize nucleic acid on the flow cell chosen as the control flow cell. Generally, flow cell 1 (fc1) is used as a control and is left blank for subtraction. It is often desirable to immobilize different nucleic acids on the remaining three flow cells (see Note 4).
Wait for the baseline to stabilize which usually takes a few minitues. Use Manual Inject, load the injection loop with ~100 μl of a 25 nM nucleic acid solution and inject over the low cell. Track the number of RUs immobilized and stop the injection after a desired level is reached (see Note 3).
At the end of the injection and after the baseline has stabilized, use the instrument crosshair to determine the RUs of nucleic acid immobilized and record this amount. The amount of nucleic acid immobilized is required to determine the theoretical moles of small molecule binding sites for the flow cell.
Repeat steps 4 to 6 for another flow cell (e.g., fc3 or fc4) (see Note 5).
2.5. Sensorchip Preparation for DNA Immobilization: HPA chip
A HPA sensor chip that has been at room temperature for at least 30 min (see Note 6).
HBS–N buffer (10 mM HEPES pH 7.4, 150 mM NaCl) is used as running buffer (see Note 7).
40 mM n-octyl glucoside (Sigma Chemical Co.) water solution.
100 mM NaOH water solution.
Cholesterol conjugated nucleic acid solutions (~25 nM of hairpin dissolved in HBS-N buffer). (3′-end conjugated DNA obtained from Integrated DNA Technologies, Coralville, IA or another source)
0.1 mg/ml bovine serum albumin (BSA) prepared in HBS–N running buffer.
Before starting, the instrument must be kept scrupulously clean by running Desorb followed by Sanitize, then run on Standby or a low continuous flow rate overnight with distilled water. Switch to running buffer for the experiment. Make sure all solutions used are properly filtered and thoroughly degassed.
Liposomes for adsorption on HPA chip should be prepared in running buffer, using standard liposome preparation techniques (51). A liposome concentration of 0.5 mM with respect to phospholipids is usually sufficient.
Choose an appropriate experimental temperature based on the phase transition temperature (Tc) of liposome to be prepared. At temperatures below Tc, adsorption may be slow and the lipids may not form a monolayer on the surface (see Note 8).
Dock a HPA chip and start a sensorgram with a 20 μl/min flow rate using HBS–N running buffer. The chip was precleaned and conditioned by injecting twice with 40 mM n-octyl glucoside for 5 mim every time.
Start a new sensorgram with a flow rate of 2 μl/min and select one desired flow cell on which to immobilize liposome preparation. Inject 60~180 μl liposome preparation (~0.5 mM), depending on lipid composition, liposome size and experimental temperature. Adsorption is seen as a steady increase in response, which flattens out as the surface coverage approaches completion. Typically, the maximum responses reached are in the region of 1500~2000 RU.
Inject 10 μl 100 mM NaOH water solution to remove loosely bound structures such as partially fused liposomes and multi–layered structure.
Start a new sensorgram with a flow rate of 2 μl/min and select one desired flow cell (fc2 to fc4) on which to immobilize cholesterol conjugated DNA hairpin. Use Manual Inject, load the injection loop with ~100 μl of a 25 nM nucleic acid solution and inject over the low cell. Track the number of RUs immobilized and stop the injection after a desired level is reached (see Note 3).
Inject 10~50 μl 0.1 mg/ml BSA or another inert protein to block any exposed hydrophobic area on the sensor chip.
Prime several times to ensure surface stability.
2.6. Flow Solutions: Buffers and Samples
-
General buffers: (see Note 9)
10 mM HEPES pH 7.4, 150 mM NaCl, 3 mM EDTA, 0.005%, v/v polysorbate 20. (HBS–EP buffer) (GE Healthcare Inc.)
10 mM MES [2-(N-morpholino) ethanesulfonic acid] pH 6.25, 100 mM NaCl, 1 mM EDTA, 0.005%, v/v polysorbate 20 (MES10 buffer);
mM CCL [cacodylic acid] pH 6.25, 100 mM NaCl, 1 mM EDTA, 0.005%, v/v polysorbate 20 (CCL10 buffer);
10 mM Tris [(Hydroxymethyl) Aminomethane] pH 7.4, 100 mM NaCl, 1 mM EDTA, 0.005%, v/v polysorbate 20 (Tris10 buffer).
The sample solution must be prepared in the same buffer used to establish the baseline– the running buffer (see Note 10).
The sample concentration to be used depends on the magnitude of the binding constant (KA). With a single binding site, for example, concentrations at least 10 times above and below 1/KA should be used (i.e., a 100 fold difference between the lowest and highest concentrations). A larger concentration range above and below 1/KA will yield a more complete binding curve. For binding constants of 107–109 M−1, as observed with many nucleic acid/small molecule complexes, small molecule concentrations from 0.01 nM to 10 μM in the flow solution allow accurate determination of binding constants. Injecting samples from low to high concentration is useful for eliminating artifacts in the data from adsorption or carry over (see Note 11).
Possible problems at high sample concentrations: poor sensorgrams and non–specific binding may be obtained.
2.7. Regeneration Solution
Good regeneration conditions should remove the analyte completely from the surface without removing or damaging the immobilized ligand. The general used regeneration solutions are listed in Table 1. In general, mider conditions are initially used, but more stringent conditions are applied as need. Some other regeneration solutions for special sample are available from the Biacore website. In our studies, 10 mM Glycine/HCl (pH 2.5) is typically used as an efficient regeneration solution to remove small molecules from the DNA immobilized sensor chip surface.
Inject 10~20 μl regeneration solution twice consecutively to assure the efficient regeneration (see Note 12).
After injection of regeneration solution, a Mix command with excess volume of buffer may be used to rinse the injection needle and sample delivery tubes and this should be followed by two 1–minute injections of running buffer to reduce the carry–over of the regeneration solution.
At the end of each cycle, 5 minutes waiting with running buffer flowing is also set to ensure that the chip surface is re–equilibrated for binding (i.e., the dextran matrix is re-equilibrated with running buffer) and the baseline has stabilized before the next sample injection.
Table 1.
Choice of Regeneration Solutions.
| Type of Bond | Acidic | Basic | Hydrophobic | Ionic |
|---|---|---|---|---|
| Weak Strength | pH > 2.5 Formic acid HCl 10 mM Glycine/HCl | pH < 9 10 mM HEPES/NaOH | pH < 9 50% ethylene glycol | 1 M NaCl |
| Intermediate Strength | pH 2~2.5 Formic acid HCl 10 mM Glycine/HCl H3PO4 | pH 9~10 NaOH 10 mM Glycine/NaOH | pH 9~10 50% ethylene glycol | 2 M MgCl2 |
| Strong Strength | pH < 2 Formic acid HCl 10 mM Glycine/HCl H3PO4 | pH > 10 NaOH | pH > 10 25~50% ethylene glycol | 4 M MgCl2 6M guanidinechloride |
3. Methods
The Biacore software supplied with the instruments allows users to write a method or to use a software wizard to set up experiments. Several important factors, such as flow rate, association time and dissociation time, injection order and surface regeneration, must be considered in setting up experiments. A sample method used to collect small molecule binding results on nucleic acid surfaces is shown below. The structure of the compound (DB293) and the biotin–labled DNA sequences (ATGA, AATT, ATAT) used in this example are shown in Figure 1.
Fig. 1.
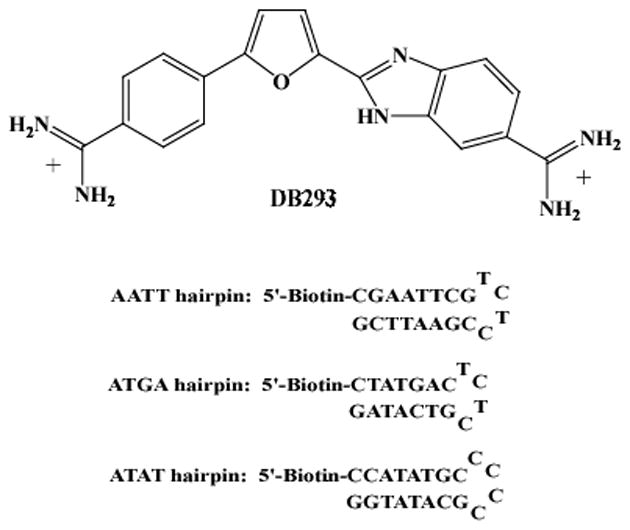
Structure of DB293 and sequences of 5′-biotin-labeled DNA hairpins.
3.1. Data Collection and Processing
A Biacore T100 instrument (GE Healthcare Inc.) is used in this study.
Three biotin-labled DNA hairpins are immobilized in different flow cells of a SA chip as described in Subheading 2.4. Approximately the same moles of each DNA oligomer were immobilized on the surface of these flow cells so that the sensorgram saturation levels can be compared directly for stoichiometry differences.
MES10 buffer (10 mM MES [2-(N-morpholino) ethanesulfonic acid] pH 6.25, 100 mM NaCl, 1 mM EDTA, 0.005%, v/v polysorbate 20) is used as running buffer.
10 mM Glycine/HCl (pH 2.5) is used as regeneration solution.
Serial dilutions (concentration range is from 1 nM to 1 μM) of DB293 compound are prepared with the running buffer as the diluent to minimize the effect of bulk refractive index changes.
The flow rate is set to 25 μl/min (see Note 13).
A wait period of 5 minutes is used with running buffer flowing at the beginning of each concentration injection cycle to give a very stable baseline that is essential for accurate small molecule binding analysis. Several buffer samples are injected at the start of each experiment and these indicate whether the instrument is performing within specifications as well as serving as controls for data processing.
Inject 250 μl of each concentration of the compound solutions and set 600 s as the dissociation time (see Note 14). Inject samples from low to high concentration to eliminate the artifacts in the data from adsorption carry over on the instrument flow system (see Note 15).
At the end of the dissociation phase, inject two short pulses of 10 μl Glycine/HCl (10 mM, pH 2.5) solution, followed by a Mix command with excess volume of buffer and two 1–minute injections of running buffer (see Subheading 2.7. step3).
After the data are collected, open the experimental sensorgrams in the BIAevaluation software for processing (see Note 16). First, zero the sensorgrams on the y–axis (RU) to allow the responses of each flow cell to be compared. Generally the average of a stable time region of the sensorgram, prior to sample injection, should be selected and set to zero for each sensorgram. Then, zero on the x–axis (time) to align the beginnings of the injections with respect to each other.
Subtract the control flow cell (fc1) sensorgram from the reaction flow cell sensorgrams (i.e. fc2–fc1, fc3–fc1, and fc4–fc1). This removes any bulk shift contribution to the change in RUs.
Subtract a buffer injection, or better, an average of several buffer injections from the compound injections (different concentrations) on the same reaction flow cell (see Note 17). This is known as double subtraction and removes any flow cell specific baseline irregularities (27). At this point, the data should be of optimum quality and is ready for analysis as shown below.
3.2. Data Analysis
After the data are processed as described, kinetic and/or steady state analysis is performed. Both kinetic and steady–state fitting can be done in Biacore software or in other available software packages (27,29). As can be seen in Figure 2, DB293 binding results reach a steady–state plateau in the injection period so that both kinetic and steady analysis can be used. In this case the binding rate is not limited by mass transfer and the association and dissociation rate constants can be determined. The average of the data over a selected time period in the steady–state region of each sensorgram can be obtained, converted to r (r = RU/RUmax) and plotted as a function of compound concentration in the flow solution (see Note 18).
-
Equilibrium constants can be obtained by fitting the results to the equivalent site model in Equation 1. For two nonequivalent sites the following equation can be used:
3 where K1 and K2 are the macroscopic thermodynamic binding constants (for a single site K2 = 0) and Cfree is the constant concentration of the compound in the flow solution (see Note 19). As described above, the binding stoichiometry can also be obtained directly from comparing the maximum response with the predicted response per compound.
Since in this example, equal moles of DNA hairpin duplexes were immobilized, the difference in maximum responses among the sets of sensorgrams is readily seen and directly reflects the difference in binding stoichiometry. The differences in kinetics constants, binding constants, stoichiometry and cooperativity for binding of DB293 to two different DNA hairpins, AATT and ATGA, can now be obtained as illustrated in Figures 3 and 4. Under these experimental conditions, DB293 binds with a 1:1 ratio to the AATT site or the ATAT site (not shown), but with a 2:1 ratio to the ATGA site.
The sensorgram in Figure 2 contains several distinct regions. In region (1) buffer flows over all surfaces and a reference baseline is established. In region (2) DB293 is injected and the kinetics of association can be determined. With time a steady–state plateau region is established when binding and dissociation of DB293 are equal. In region (3) buffer flow is again started and the DB293–DNA complex dissociates until the baseline is reached. If complete dissociation does not occur in a time period, a surface regeneration solution can be introduced. The RU on the surface is directly related to the DB293 bound. Based on the RU at saturation we can determine that DB293 forms a 1:1 complex with the AATT DNA, as expected, and an unusual 2:1 complex with ATGA.
The steady state data for compound binding are fit with one–site (AATT) or two-site (ATGA) binding models (Figure 4 and Table 3). The relative values of the macroscopic equilibrium constants, K1 and K2, reflect a highly cooperativity interaction with ATGA. A cooperativity factor to assess the degree of cooperativity is defined as CF = (K2/K1) × 4. For interaction with no cooperativity, CF = 1, and CF >1 for positive cooperativity and <1 for negative cooperativity (52). The positive cooperativity in binding of DB293 to ATGA can be easily seen from Table 3, suggesting that DB293 interacts as a cooperative dimer stacked with the ATGA sequence.
In cases where a steady–state plateau is reached, the ratio of the rate constants (ka/kd) should be compared to the steady–state KA value. To illustrate a kinetic fit, the binding data for AATT with DB293 at low concentration is globally fitted with a single site kinetic model with a mass transport term. The results are shown in Figure 3 and Table 4. The binding constants obtained from kinetic and steady–state analyses are in excellent agreement, reflecting the high quality of data in the SPR experiments. In this case, the mass transfer effect is not significant and does not dominate the kinetics. The association rate constant ka is less than the transfer rate constant kt.
Kinetic analysis using global fitting of SPR data places great demand on obtaining high-quality data. Experimental design, analysis and optimization of kinetic studies have been described in detail elsewhere (27,29) (see Note 20). In general, low surface densities of the immoiblized reactant and high analyte flow rate should be used to minimize the effects of mass transfer. Several criteria must be satisfied when considering if a global kinetic fit is acceptable (48): (i) within experimental limits RUmax is the same as the predicted value or from the steady–state results for one binding molecule; (ii) the rate constants are within the range of small molecules; (iii) the mass transport constant kt is in the 107range; (iv) (ka × RUmax/kt) ≤ 5; (v) the half–life t1/2 from the dissociation phase of sensorgram is close to the calculated half–life using the fitted value (t1/2 = ln2/kd), suggesting the mass transport effect is minimized; (vi) the residuals are within the instrumental noise and there are no systematic deviations; (vii) a low chi–squared value is obtained at convergence.
Fig. 3.
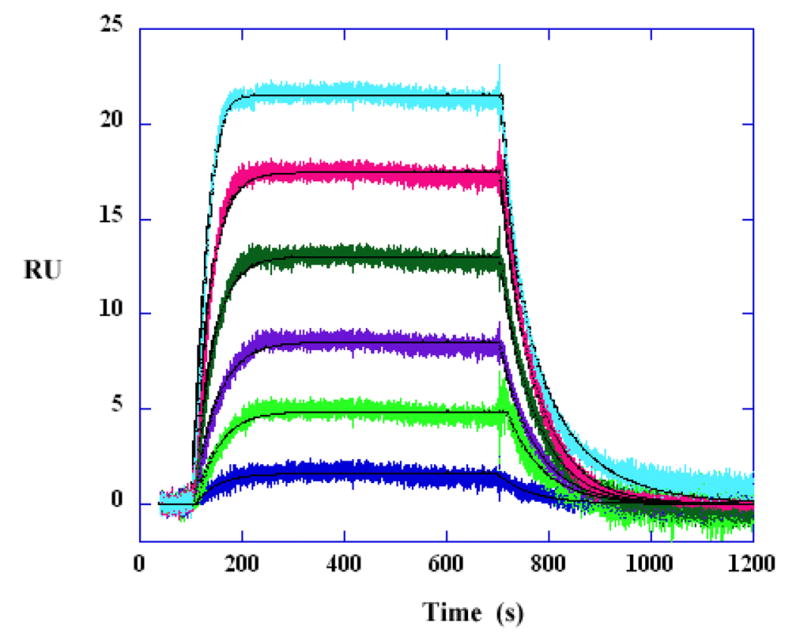
Kinetic fitting to the AATT DNA data at low compound concentrations. The DB293 concentrations from bottom to top are 1, 4, 6, 10, 15 and 20 nM. The kinetic analysis is performed with mass transport kinetic 1:1 binding model. The smooth lines are the best fit lines using global fitting.
Fig. 4.
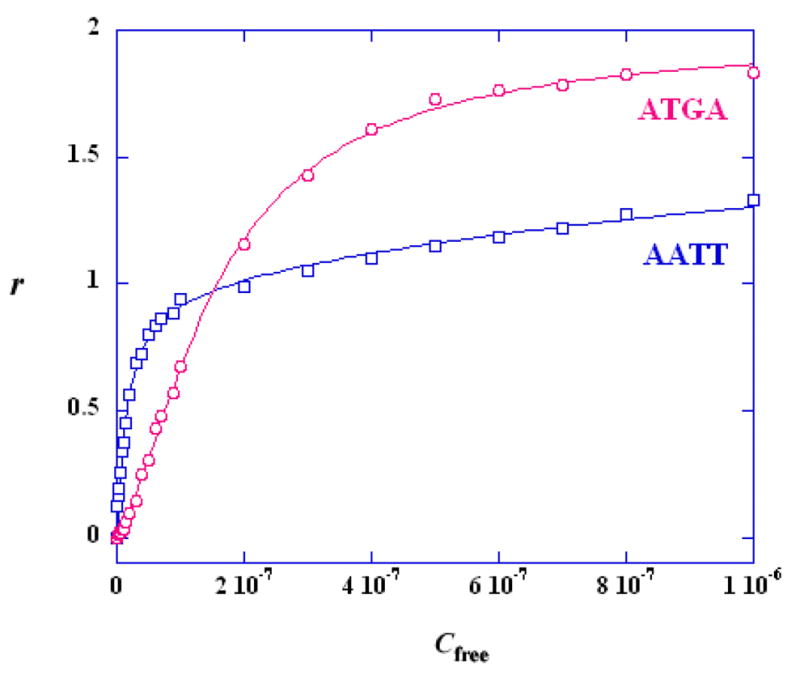
Comparison of the SPR binding affinity for AATT and ATGA. RU values from the steady–state region of SPR sensorgrams were converted to r (r = RU/RUmax) and are plotted against the unbound compound concentration (flow solution) for DB293 binding with AATT (squares) and ATGA (circles). The lines are the best fit values using appropriate binding models as described in the text.
Table 3.
Binding affinity comparison from steady–state fitting.
| DNA binding site | K1 ×107 (1/M) | K2 ×107 (1/M) | CF (K2/K1) × 4 | Binding mode |
|---|---|---|---|---|
| ATGA | 0.41 | 0.98 | 9.6 | Dimmer, Cooperative |
| AATT | 4.87 | — | — | Monomer |
| ATAT | 0.62 | — | — | Monomer |
Table 4.
Binding affinity comparison of kinetic and steady–state analysis for DB293 binding to the AATT DNA.
| Exp. | ka (M−1 s−1) | kd (s−1) | KA (M−1) | kt [RU/(Ms)] | Chi2 (RU)2 | ka×RUmax/kt |
|---|---|---|---|---|---|---|
| Kinetics | 6.75×105 | 0.0155 | 4.35×107 | 1.56×1010 | 0.278 | 1.5×10−3 |
| Steay-state | — | — | 4.87×107 | — | 0.22 | — |
Acknowledgments
We very much thank the NIH for funding the research that has made this review possible and the Georgia Research Alliance for funding of Biacore instruments. We also very much thank Professor David W. Boykin and his coworkers (Georgia State University, Atlanta, GA, USA) for supplying DB293 and many other DNA and RNA binding agents for SPR studies as well as for helpful discussions.
Footnotes
Maintenance chips are available from GE Healthcare Inc. For desorb running, 50% DMSO and 10% DMSO can be used instead of Biacore desorb solution 1 and Biacore desorb solution 2 to effectively remove sticky small molecules. For biological samples such as protein, Sanitize method is also used after Desorb to insure that no microbial growth is present in the liquid injection and flow system.
After running the regular Desorb, if the baseline is still not stable within ±1.0 RU/min (check with a new CM5 chip using running buffer), additional Super clean method may be used. First, run Desorb using 1% (v/v) acetic acid in place of Biacore desorb solution 1 and Biacore desorb solution 2, followed by one Prime to wash out the residual acetic acid. Then, run Desorb using 0.2 M sodium bicarbonate in place of SDS (solution 1) and glycine (solution 2), followed by one Prime to wash out the sodium bicarbonate residuals. Last, run Desorb using 6 M guanidine HCl for the SDS and 10 mM HCl for glycine. Prime the instrument a few times to thoroughly clean all residuals.
Typically, an immobilization amount of 300–450 RUs of hairpin nucleic acid (~20–30 bases in length) is immobilized for running steady–state experiments and a low amount of 100–150 RUs for kinetic experiments to minimize mass transfer effects.
A scrambled nonbinding sequence could be immobilized on flow cell 1 in some situations to provide a more similar control surface for subtraction.
To visualize and illustrate a difference in stoichiometry for different nucleic acid hairpins, equal moles of nucleic acid can be immobilized on different flow cells using the equation (RU = C× moles bound × MWnucleic acid), where C is a relatively constant conversion factor for nucleic acid, proteins, etc. A higher level of response units (RU) is required for a higher molecular weight hairpin.
Don’t open the package or dock the chip until the liposomes are prepared and ready for use to minimize adsorption of unwanted material from the air and running buffer on to the sensor chip.
Detergents shoud not be used.
Buffer degassing is very important and is particularly important if experiments are run at temperatures above 25 °C.
For the Biaore 2000 instrument, even 10 times lower surfactant concentration (0.0005%, v/v polysorbate 20) can be applied to the running buffer and it works very well. While for Biacore 3000 and Biacore T100, higher concentration (0.005%, v/v polysorbate 20) is needed in the running buffer. Depending on the binding compound, a little higher binding affinity may be obtained when increasing surfactant concentration in the running buffer. Salt concentration can be adjusted based on the experimental requirement. The higher the salt concentration, the lower the binding affinity that will be obtained when cations bind to nucleic acids.
If the small molecule requires the presence of a small amount of an organic solvent (e.g., <5% DMSO) to maintain solubility, the same amount of this organic solvent must be in the running buffer to minimize the refractive index difference.
If the KA is unknown, it is necessary to conduct a preliminary experiment with several concentrations spread over a broad range to obtain an estimate of KA. A more focused set of concentrations is then prepared to cover the specific binding range.
A short contact time, 30~60 s, is usually sufficient. Longer exposure to regeneration conditions involves greater risks for loss of binding activity on the surface, and often does not lead to improved regeneration.
For the steady–state method, equilibrium, but not kinetics, constants can be obtained even when mass transfer effects dominate the observed kinetics. Thus, higher flow rates are not required in steady-state experiments as long as a clear steady–state plateau is obtained for determining RU at the steady–state. Higher flow rates (> 50 μl/min) are used for kinetic experiments to minimize mass transport effects.
A sufficient association phase with a plateau region is needed for steady-state analysis. For the most accurate fitting of the dissociation phase it is best to allow sufficient time for the compound to dissociate at least 80% from the complex
Many organic small molecules are easily adsorbed nonspecifically to the tubing of the injection micro–fluidics and are slowly released over the course of the experiment.
Other software programs such as Scrubber2 and CLAMP are available for processing Biacore data. The results can also be exported and presented in graphing software such as KaleidaGraph for either PC or Macintosh computers. Although it is useful to experiment with different software packages, BIAevaluation (current version 4.1) is sufficient for most routine analyses of sensorgram data. For the Biacore T100 user, data processing can be performed automatically using the Biacore T100 evaluation software, which is much more convenient for new users.
These two data processing steps are referred to as “double referencing”. Typically, multiple buffer injections are performed and averaged before subtraction. In double referencing, plots are made for each flow cell separately overlaying the control flow cell– corrected sensorgrams from buffer and all sample injections. The buffer sensorgram is then subtracted from the sample sensorgrams. “Double referencing” removes the systematic drifts and shifts in baseline and is helpful to minimize offset artifacts and also to correct the bulk shift that results from slight differences in injection buffer and running buffer (27).
In some instances at low concentrations where the response does not reach the steady-state, the equilibrium responses can be obtained from kinetic fits of the sensorgrams utilizing the known RUmax from the higher concentration sensorgrams. This extrapolation method works well with sensorgrams where the observed response is at least 50% of the equilibrium RU.
Various fitting models are included in the Biacore evaluation software and users can also write other models to be used with the program. While more complex binding models should only be used if the materials are pure, the experiment has been conducted properly, and the more complex model improves the fit significantly above the experimental error.
In cases where a steady–state plateau is reached, the ratio of the rate constants (ka/kd) should be compared to the steady–state KA value. An agreement between the two methods suggests that the binding constant, KA, is correct but does not necessarily mean that the ka and kd values are correct due to possible mass transfer effects and possible correlation of constants (29,47).
References
- 1.Charbonnier S, Gallego O, Gavin AC. The social network of a cell: recent advances in interactome mapping. Biotechnol Annu Rev. 2008;14:1–28. doi: 10.1016/S1387-2656(08)00001-X. [DOI] [PubMed] [Google Scholar]
- 2.Figeys D. Mapping the human protein interactome. Cell Res. 2008;18:716–724. doi: 10.1038/cr.2008.72. [DOI] [PubMed] [Google Scholar]
- 3.Reed JW, Bartel B. Cell signaling and gene regulation. Curr Opin Plant Biol. 2008;11:471–473. doi: 10.1016/j.pbi.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 4.Kornberg R. The molecular basis of eukaryotic transcription. Angew Chem Int Ed Engl. 2007;46:6956–6965. doi: 10.1002/anie.200701832. [DOI] [PubMed] [Google Scholar]
- 5.Majmudar CY, Lum JK, Prasov L, Mapp AK. Functional specificity of artificial transcriptional activators. Chem Biol. 2005;12:313–321. doi: 10.1016/j.chembiol.2005.01.014. [DOI] [PubMed] [Google Scholar]
- 6.Berg T. Inhibition of transcription factors with small organic molecules. Curr Opin Chem Biol. 2008;12:464–471. doi: 10.1016/j.cbpa.2008.07.023. [DOI] [PubMed] [Google Scholar]
- 7.Xiao X, Yu P, Lim HS, Sikder D, Kodadek T. Design and synthesis of a cell-permeable synthetic transcription factor mimic. J Comb Chem. 2007;9:592–600. doi: 10.1021/cc070023a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burnett R, Melander C, Puckett JW, Son LS, Wells RD, Dervan PB, et al. DNA sequence-specific polyamides alleviate transcription inhibition associated with long GAA. TTC repeats in Friedreich’s ataxia. Proc Natl Acad Sci U S A. 2006;103:11497–11502. doi: 10.1073/pnas.0604939103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wilson WD, Tanious FA, Mathis A, Tevis D, Hall JE, Boykin DW. Antiparasitic compounds that target DNA. Biochimie. 2008;90:999–1014. doi: 10.1016/j.biochi.2008.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tidwell RR, Boykin DW. Dicationic DNA minor-groove binders as antimicrobial agents. In: Demeunynck M, Bailly C, Wilson WD, editors. DNA and RNA Binders: From Small Molecules to Drugs. Wiley-VCH; 2003. pp. 414–460. [Google Scholar]
- 11.Cai Z, Greene MI, Berezov A. Modulation of biomolecular interactions with complex-binding small molecules. Methods. 2008 doi: 10.1016/j.ymeth.2008.05.008. [DOI] [PubMed] [Google Scholar]
- 12.Dominy BN. Molecular recognition and binding free energy calculations in drug development. Curr Pharm Biotechnol. 2008;9:87–95. doi: 10.2174/138920108783955155. [DOI] [PubMed] [Google Scholar]
- 13.Seiler KP, George GA, Happ MP, Bodycombe NE, Carrinski HA, Norton S, et al. ChemBank: a small-molecule screening and cheminformatics resource database. Nucleic Acids Res. 2008;36:351–359. doi: 10.1093/nar/gkm843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Butcher RA, Schreiber SL. Using genome-wide transcriptional profiling to elucidate small-molecule mechanism. Curr Opin Chem Biol. 2005;9:25–30. doi: 10.1016/j.cbpa.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 15.Mapp AK, Ansari AZ. A TAD further: exogenous control of gene activation. ACS Chem Biol. 2007;2:62–75. doi: 10.1021/cb600463w. [DOI] [PubMed] [Google Scholar]
- 16.Chaires JB. Calorimetry and thermodynamics in drug design. Annu Rev Biophys. 2008;37:135–151. doi: 10.1146/annurev.biophys.36.040306.132812. [DOI] [PubMed] [Google Scholar]
- 17.Papalia GA, Giannetti AM, Arora N, Myszka DG. Thermodynamic characterization of pyrazole and azaindole derivatives binding to p38 mitogen-activated protein kinase using Biacore T100 technology and van’t Hoff analysis. Anal Biochem. 2008 doi: 10.1016/j.ab.2008.08.010. [DOI] [PubMed] [Google Scholar]
- 18.Perozzo R, Folkers G, Scapozza L. Thermodynamics of protein-ligand interactions: history, presence, and future aspects. J Recept Signal Transduct Res. 2004;24:1–52. doi: 10.1081/rrs-120037896. [DOI] [PubMed] [Google Scholar]
- 19.Velazquez Campoy A, Freire E. ITC in the post-genomic era...? Priceless. Biophys Chem. 2005;115:115–124. doi: 10.1016/j.bpc.2004.12.015. [DOI] [PubMed] [Google Scholar]
- 20.Li L, Dantzer JJ, Nowacki J, O’Callaghan BJ, Meroueh SO. PDBcal: a comprehensive dataset for receptor-ligand interactions with three-dimensional structures and binding thermodynamics from isothermal titration calorimetry. Chem Biol Drug Des. 2008;71:529–532. doi: 10.1111/j.1747-0285.2008.00661.x. [DOI] [PubMed] [Google Scholar]
- 21.Privalov PL, Dragan AI. Microcalorimetry of biological macromolecules. Biophys Chem. 2007;126:16–24. doi: 10.1016/j.bpc.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 22.Plotnikov VV, Brandts JM, Lin LN, Brandts JF. A new ultrasensitive scanning calorimeter. Anal Biochem. 1997;250:237–244. doi: 10.1006/abio.1997.2236. [DOI] [PubMed] [Google Scholar]
- 23.Ciulli A, Williams G, Smith AG, Blundell TL, Abell C. Probing hot spots at protein-ligand binding sites: a fragment-based approach using biophysical methods. J Med Chem. 2006;49:4992–5000. doi: 10.1021/jm060490r. [DOI] [PubMed] [Google Scholar]
- 24.Turnbull WB, Daranas AH. On the value of c: can low affinity systems be studied by isothermal titration calorimetry? J Am Chem Soc. 2003;125:14859–14866. doi: 10.1021/ja036166s. [DOI] [PubMed] [Google Scholar]
- 25.Wilson WD. Analyzing biomolecular interactions. Science. 2002;295:2103–2105. doi: 10.1126/science.295.5562.2103. [DOI] [PubMed] [Google Scholar]
- 26.Rich RL, Myszka DG. Survey of the year 2006 commercial optical biosensor literature. J Mol Recognit. 2007;20:300–366. doi: 10.1002/jmr.862. [DOI] [PubMed] [Google Scholar]
- 27.Myszka DG. Kinetic, equilibrium, and thermodynamic analysis of macromolecular interactions with BIACORE. Methods Enzymol. 2000;323:325–340. doi: 10.1016/s0076-6879(00)23372-7. [DOI] [PubMed] [Google Scholar]
- 28.Nagata KaHH. Real–time analysis of biomolecular interactions: applications of Biacore. Springer; 2000. [Google Scholar]
- 29.Nguyen B, Tanious FA, Wilson WD. Biosensor-surface plasmon resonance: quantitative analysis of small molecule-nucleic acid interactions. Methods. 2007;42:150–161. doi: 10.1016/j.ymeth.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 30.Jason-Moller L, Murphy M, Bruno J. Overview of Biacore systems and their applications. Curr Protoc Protein Sci. 2006;Chapter 19 doi: 10.1002/0471140864. [DOI] [PubMed] [Google Scholar]
- 31.Davis TM, Wilson WD. Determination of the refractive index increments of small molecules for correction of surface plasmon resonance data. Anal Biochem. 2000;284:348–353. doi: 10.1006/abio.2000.4726. [DOI] [PubMed] [Google Scholar]
- 32.Davis TM, Wilson WD. Surface plasmon resonance biosensor analysis of RNA-small molecule interactions. Methods Enzymol. 2001;340:22–51. doi: 10.1016/s0076-6879(01)40416-2. [DOI] [PubMed] [Google Scholar]
- 33.Koh JT, Zheng J. The new biomimetic chemistry: artificial transcription factors. ACS Chem Biol. 2007;2:599–601. doi: 10.1021/cb700183s. [DOI] [PubMed] [Google Scholar]
- 34.Rezler EM, Bearss DJ, Hurley LH. Telomere inhibition and telomere disruption as processes for drug targeting. Annu Rev Pharmacol Toxicol. 2003;43:359–379. doi: 10.1146/annurev.pharmtox.43.100901.135733. [DOI] [PubMed] [Google Scholar]
- 35.Neidle S, Parkinson GN. Quadruplex DNA crystal structures and drug design. Biochimie. 2008;90:1184–1196. doi: 10.1016/j.biochi.2008.03.003. [DOI] [PubMed] [Google Scholar]
- 36.Hampshire AJ, Fox KR. The effects of local DNA sequence on the interaction of ligands with their preferred binding sites. Biochimie. 2008;90:988–998. doi: 10.1016/j.biochi.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 37.Chaires JB. Competition dialysis: an assay to measure the structural selectivity of drug-nucleic acid interactions. Curr Med Chem Anticancer Agents. 2005;5:339–352. doi: 10.2174/1568011054222292. [DOI] [PubMed] [Google Scholar]
- 38.Nguyen B, Neidle S, Wilson WD. A Role for Water Molecules in DNA-Ligand Minor Groove Recognition. Acc Chem Res. 2008 doi: 10.1021/ar800016q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wilson WD, Nguyen B, Tanious FA, Mathis A, Hall JE, Stephens CE, et al. Dications that target the DNA minor groove: compound design and preparation, DNA interactions, cellular distribution and biological activity. Curr Med Chem Anticancer Agents. 2005;5:389–408. doi: 10.2174/1568011054222319. [DOI] [PubMed] [Google Scholar]
- 40.Werbovetz K. Diamidines as antitrypanosomal, antileishmanial and antimalarial agents. Curr Opin Investig Drugs. 2006;7:147–157. [PubMed] [Google Scholar]
- 41.De Cian A, Lacroix L, Douarre C, Temime-Smaali N, Trentesaux C, Riou JF, et al. Targeting telomeres and telomerase. Biochimie. 2008;90:131–155. doi: 10.1016/j.biochi.2007.07.011. [DOI] [PubMed] [Google Scholar]
- 42.White EW, Tanious F, Ismail MA, Reszka AP, Neidle S, Boykin DW, et al. Structure-specific recognition of quadruplex DNA by organic cations: influence of shape, substituents and charge. Biophys Chem. 2007;126:140–153. doi: 10.1016/j.bpc.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 43.Parkinson GN, Cuenca F, Neidle S. Topology conservation and loop flexibility in quadruplex-drug recognition: crystal structures of inter- and intramolecular telomeric DNA quadruplex-drug complexes. J Mol Biol. 2008;381:1145–1156. doi: 10.1016/j.jmb.2008.06.022. [DOI] [PubMed] [Google Scholar]
- 44.Huppert JL. Four-stranded nucleic acids: structure, function and targeting of G-quadruplexes. Chem Soc Rev. 2008;37:1375–1384. doi: 10.1039/b702491f. [DOI] [PubMed] [Google Scholar]
- 45.Qin Y, Hurley LH. Structures, folding patterns, and functions of intramolecular DNA G-quadruplexes found in eukaryotic promoter regions. Biochimie. 2008;90:1149–1171. doi: 10.1016/j.biochi.2008.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lane AN, Chaires JB, Gray RD, Trent JO. Stability and kinetics of G-quadruplex structures. Nucleic Acids Res. 2008 doi: 10.1093/nar/gkn517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tanious FA, Nguyen B, Wilson WD. Biosensor-surface plasmon resonance methods for quantitative analysis of biomolecular interactions. Methods Cell Biol. 2008;84:53–77. doi: 10.1016/S0091-679X(07)84003-9. [DOI] [PubMed] [Google Scholar]
- 48.Karlsson R. Affinity analysis of non-steady-state data obtained under mass transport limited conditions using BIAcore technology. J Mol Recognit. 1999;12:285–292. doi: 10.1002/(SICI)1099-1352(199909/10)12:5<285::AID-JMR469>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 49.Navratilova I, Dioszegi M, Myszka DG. Analyzing ligand and small molecule binding activity of solubilized GPCRs using biosensor technology. Anal Biochem. 2006;355:132–139. doi: 10.1016/j.ab.2006.04.021. [DOI] [PubMed] [Google Scholar]
- 50.Day YS, Baird CL, Rich RL, Myszka DG. Direct comparison of binding equilibrium, thermodynamic, and rate constants determined by surface- and solution-based biophysical methods. Protein Sci. 2002;11:1017–1025. doi: 10.1110/ps.4330102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cooper MA, Hansson A, Lofas S, Williams DH. A vesicle capture sensor chip for kinetic analysis of interactions with membrane-bound receptors. Anal Biochem. 2000;277:196–205. doi: 10.1006/abio.1999.4389. [DOI] [PubMed] [Google Scholar]
- 52.Peixoto P, Liu Y, Depauw S, Hildebrand MP, Boykin DW, Bailly C, et al. Direct inhibition of the DNA-binding activity of POU transcription factors Pit-1 and Brn-3 by selective binding of a phenyl-furan-benzimidazole dication. Nucleic Acids Res. 2008;36:3341–3353. doi: 10.1093/nar/gkn208. [DOI] [PMC free article] [PubMed] [Google Scholar]