

Abstract
TRPML3 is an inward rectifying Ca2+ channel that is regulated by extracytosolic H+. Although gain-of-function mutation in TRPML3 causes the varitint-waddler phenotype, the role of TRPML3 in cellular physiology is not known. Here, we report that TRPML3 is a prominent regulator of endocytosis, membrane trafficking and autophagy. Gradient fractionation and confocal localization reveal that TRPML3 is expressed in the plasma membrane and multiple intracellular compartments. However, expression of TRPML3 is dynamic, with accumulation of TRPML3 in the plasma membrane upon inhibition of endocytosis, and recruitment of TRPML3 to autophagosomes upon induction of autophagy. Accordingly, overexpression of TRPML3 leads to reduced constitutive and regulated endocytosis, increased autophagy and marked exacerbation of autophagy evoked by various cell stressors with nearly complete recruitment of TRPML3 into the autophagosomes. Importantly, both knock-down of TRPML3 by siRNA and expression of the channel-dead dominant negative TRPML3(D458K) have a reciprocal effect, reducing endocytosis and autophagy. These findings reveal a prominent role for TRPML3 in regulating endocytosis, membrane trafficking and autophagy, perhaps by controlling the Ca2+ in the vicinity of cellular organelles that is necessary to regulate these cellular events.
Keywords: TRPML3, Ca2+ channel, recycles, membrane trafficking, autophagy
Introduction
TRPML3 is a member of the TRPML subfamily of the TRP (transient receptor potential) channel superfamily (1). The TRPML subfamily was established with identification of TRPML1 as the protein that is mutated in the lysosomal storage disease Mucolipidosis type IV (MLIV) (2). Subsequently, TRPML3 was found by positional cloning as the channel mutated in the mouse varitint-waddler phenotype (3) and TRPML2 was identified by database searches (1). The varitint-waddler phenotype is caused by the gain-of-function mutation A419P in TRPML3 (4-8). Members of the TRPML subfamily share the same basic structure with 6 transmembrane domains, a pore domain between transmembrane domains 5 and 6 and a unique large extracytosolic loop between transmembrane domains 1 and 2.
TRPML1 is a lysosomal channel (9) that functions to regulate lysosomal pH (10, 11) and thereby membrane trafficking (11, 12). TRPML1 was suggested to regulate interactions between late endosomes and lysosomes to control delivery of cellular material to lysosomes (12). However, a recent work used knock-down (KD) of TRPML1 to show that absence of TRPML1 causes loss of protein and lipid hydrolytic activity that led to lysosomal dysfunction (11). Lysosomal dysfunction appears to be associated with increased autophagy (13). TRPML1 can function as a Ca2+ channel (4). A new potential role for TRPML1 is to be the channel activated by the Ca2+ mobilizing messenger NAADP (14). Yet another potential function of TRPML1 is in iron transport by operating as endosomal iron channel (15).
Much less is known about the localization and function of TRPML3. It was suggested that TRPML3 localizes in the ER when expressed alone, but to be escorted to the lysosomes when co-expressed with TRPML1 or TRPML2 (16). However, the native TRPML3 is expressed in intracellular vesicular compartments and in the plasma membrane (3, 17). Accordingly, several recent reports also showed that the expressed TRPML3 is present in intracellular vesicular compartments and the plasma membrane (4-8), although the identity of the vesicular compartments and whether TRPML3 can translocate between compartments is not known. We have recently shown that TRPML3 functions as a Ca2+ channel that is regulated by Na+ (5) and extracytosolic H+ (H+e-cyto) (5, 8). Regulation by Na+ and H+ is mediated by a string of three histidines, with H283 conveying the regulation by Na+ and H+ and H252 and H273 tunneling the H+ to H283 (8).
Practically, no information is available on the nature of the organelles expressing TRPML3, let alone the cellular functions of TRPML3. Here, we show that TRPML3 is found in many cellular compartments. The localization of TRPML3 is dynamic and TRPML3 regulate endocytosis and membrane trafficking. Moreover, cell stressors that induce autophagy cause quantitative recruitment of TRPML3 to autophagosomes. Accordingly, TRPML3 is required for autophagy. These findings reveal a prominent role for TRPML3 in cellular trafficking and in the control of autophagy.
Results
Cellular localization of TRPML3
To understand the cellular function of TRPML3 it is necessary to determine its cellular localization. Although the native and recombinant TRPML3 are localized in intracellular vesicular compartments and in the plasma membrane (3-8), the identity of the compartments expressing TRPML3 is not known. In the present work, we performed extensive studies to determine the sites and dynamics of TRPML3 expression. Since all TRPML channels have targeting motifs in their C terminus (18), we tagged the N terminus of TRPML3 with HA (HA-TRPML3), GFP (GFP-TRPML3) or mCherry (mCherry-TRPML3). Previous work suggested that expressed TRPML3 is localized in the ER and is escorted to the lysosomes when co-expressed with TRPML1 (16). Fig. 1a shows the vesicular expression pattern of TRPML3. Effect of TRPML1 on the expression pattern of TRPML3 would predict different localization of TRPML3 in TRPML1+/- and TRPML1-/- cells. Fig. 1a shows that this is not the case. Fig. 1b shows that only partial overlap of TRPML1 and TRPML3 is observed when the channels are co-expressed in TRPML1-/- fibroblasts. Moreover, Fig. 1c shows minimal co-IP of the channels when co-expressed in HEK cells. Hence, the combined findings indicate that TRPML1 does not dictate localization of TRPML3, and for the most part the two channels reside in different compartments.
Fig. 1. Expressed TRPML3 is expressed in multiple intracellular compartments.

Panel (a): mCherry-TRPML3 was expressed in human skin fibroblasts obtained from TRPML1+/- relative (left image) and TRPML1-/- patient with MLIV (10) (right image) showing that the vesicular expression pattern of TRPML3 is independent of TRPML1. Bars in these and all images donates 10 μm. Panel (b): GFP-TRPML1 and mCherry-TRPML3 were expressed in TRPML1-/- cells and the overlap in expression was determined using imageJ. Panel (c) shows minimal co-IP of HA-TRPML1 and Myc-TRPML3 when co-expressed in HEK cells. The upper blots are the lysates and the lower blots are the co-IP with the indicated antibodies. Panel (d) shows the sensitivity of TRPML3 and TRPML1 to PNGase F and Endo H, and that TRPML3 is not cleaved. Panel (e) show expression of TRPML3 in different subcellular fractions separated on a percoll gradient and expression of TRPML1 only in the heavy fraction. Here and in all blots (ML1) refer to TRPML1 and (ML3) to TRPML3.
TRPML3 has two putative glycosylation sites in the large extracytosolic loop. Therefore, we assayed the sensitivity of the TRPML3 constructs to PNGase F and Endo H. The results with HA-TRPML3 are shown in Fig. 1d and similar results were obtained with GFP-TRPML3 and mCherry-TRPML3 (not shown). Unlike TRPML1 (19), TRPML3 does not undergo proteolytic cleavage. Treatment with PNGase F, which removes N-linked sugars, generates the core unglycosylated TRPML3. Importantly, treatment with Endo H had no effect on glycosylation of TRPML3 (n>20), indicating that TRPML3 must have exited the ER, since resistance to Endo H is acquired in the Golgi.
A more extensive analysis of TRPML3 cellular distribution was obtained by subcellular fractionation on a percoll gradient. The cells were transfected with HA-TRPML3 only to avoid complication from co-expression with TRPML1. TRPML3 was detected with anti-TRPML3 αC2 or anti-HA antibodies. Fig. 1e shows that the two antibodies similarly detected TRPML3 in fractions 2-7. On the other hand, TRPML1 was found predominantly in the heavy fraction 2. LAMP1 is a marker of late endosomes and lysosomes, and migrated mainly to fractions 2 and 3. The plasma membrane marker Na+ pump migrated to fractions 7-11 and the early endosomal marker EEA1 was found in fractions 5-8. The transferrin receptor was found mainly in fractions 5-10. Finally, the ER marker Bip migrated to fractions 6-9. Hence, while TRPML1 is found only in lysosomes, TRPML3 cofractionates with several cellular compartment, including the plasma membrane and early endosomes with larger fractions at late endosomes and lysosomes.
To further verify expression of TRPML3 in multiple cellular fractions we determined the distribution of the native TRPML3. Fig. 2a shows the specificity of the anti-TRPML3 antibodies from ProteinTec and the αc2 anti-TRPML3 antibodies (see methods). The TRPML3 bend is marked by arrowheads. Knockdown by siRNA markedly reduced expression of the TRPML3 protein in HeLa and HEK cells and the antibodies recognized expressed TRPML3. Fig. 2b shows that native TRPML3 is detected in multiple gradient fractions that do not express TRPML1 or LAMP1 and partially co-localize with transferrin receptors. Unfortunately, the two antibodies were not suitable for immunolocalization.
Fig. 2. Native TRPML3 is expressed in multiple cellular compartments.

In panel (a) HeLa or HEK cells were untreated (Extract), were treated with two different TRPML3 siRNA (si1ML3 and si2ML3), or were transfected with human (hML3) of mouse (mML3) TRPML3 cDNA constructs and were used to probe expression of TRPML3 by western blots with the ProteinTec antibodies (left blot) or the αC2 antibodies (right blot). The arrowheads show that both antibodies recognize the native TRPML3. In panel (b), fractions of untransfected HeLa cells were collected from a percoll gradient as in Fig. 1 and were probed for expression of native TRPML3 (ML3), TRPML1 (ML1), LAMP1 and Transferrin receptor (TrnfR).
Next, mCherry-TRPML3 was used to determine the extent of co-localization with well-defined organellar markers by confocal microscopy. The overlap of TRPML3 with these markers was quantified with ImageJ software (NIH). Consistent with the gradient results, the images in Fig. 3a and the summary of the overlaps in Fig. 3b show that significant amount of TRPML3 is found in several subcellular compartments. Thus, about 17% of TRPML3 co-localized with EEA1 and about 45% with LAMP1. Interestingly, only about 25% overlap was found between TRPML3 and lysotracker, which is statistically different from the 45% found for TRPML3 and LAMP1, further indicating that TRPML3 is expressed in both late endosomes and lysosomes.
Fig. 3. Subcellular localization of TRPML3.

Panel (a): HeLa cells were transfected with mCherry-TRPML3 and either fixed and stained for EEA1 (left images) or LAMP1 (middle images), or live cells were incubated with 10 nM lysotracker green for 10 min (right images) and imaged by confocal microscopy. Panel (b): Images similar to those in panel (a) from at least 10 cells in three different experiments were used to determine the staining overlap with ImageJ. The results are given as mean±s.e.m.
TRPML3 and endocytosis
The altered distribution of melanosomes and impaired vestibular hair cells function in the varitint-waddler mouse (4, 20), and the expression of TRPML3 in multiple cellular compartments (Figs. 1-3) are suggestive of a role of TRPML3 in membrane trafficking. Several trafficking assays were employed to probe for this possibility. In the first set of experiments, we determined the effect of over-expression and of knock-down (KD) of TRPML3 on the endocytosis of FITC-tagged transferrin and EGF. Endocytosis was measured for 5-45 min, but the largest differences are seen in the first 10 min of uptake. The images and the analysis in Fig. 4a-c are from the 10 min uptake time points. The uptake was quantified by puncta counting with ImageJ software. The blot in Fig. 2a shows that TRPML3 siRNA (siML3) effectively silenced expression of TRPML3 in HeLa and HEK cells. The images in Fig. 4a and 4b and the summary in Fig. 4c show that KD of TRPML3 nearly doubled the rate of transferrin and EGF uptake in the first 10 min. Notably, expression of TRPML3 has the opposite effect and significantly reduced the rate of transferrin and EGF uptake. In additional, complimentary experiments, we measured 125I-EGF uptake into control cells, cells transfected with TRPML3 and cells treated with siML3. Fig. 4d confirms the results obtained with the FITC-EGF and shows that expression of TRPML3 inhibited, whereas KD of TRPML3 enhanced 125I-EGF uptake.
Fig. 4. Overexpression of TRPML3 inhibits transferrin and EGF endocytosis.
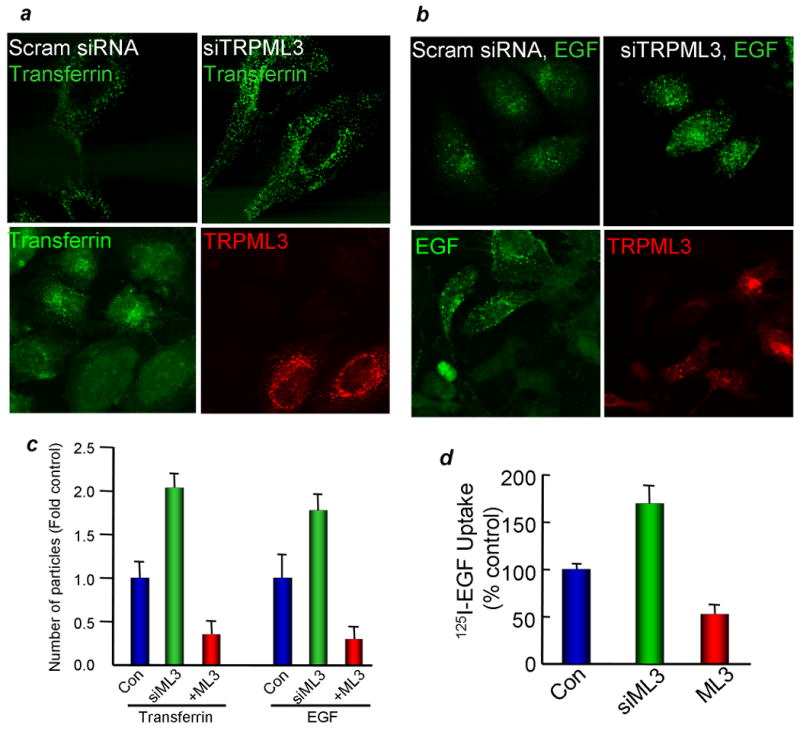
The blot in panel (a) shows the effect of treating HeLa cells for 48 hrs with TRPML3 siRNA (siTRPML3) on TRPML3 mRNA level. Panels (a, b): The upper images show the effect of siTRPML3 and the lower images the effect of expressing mCherry-TRPML3 on 10 min uptake of transferrin (a) and EGF (b). In panel (c), ImageJ was used to count the number of puncta in 17-19 cells from 4-5 experiments and the fold change is plotted as mean±s.e.m at the indicated conditions. Panel (d) shows the 125I-EGF uptake into cells transfected with TRPML3 or treated with siTRPML3. The results are mean±s.e.m of three experiments.
In a third, independent assay for endocytosis and membrane trafficking we determined the degradation of the native EGF receptor (EGFR) in response to cell stimulation with EGF. EGFR degradation entails endocytosis of the EGFR and its trafficking to and degradation in the lysosomes (21). Figs. 5a and 5b show that in control cells the EGFR is degraded within about 60 min of incubation with EGF. Fig. 5a shows that expression of TRPML3 reduced the rate of EGFR degradation (p=0.04, n=4). The reciprocal experiments in Fig. 5b show that KD of TRPML3 increased the rate of degradation of the EGFR (p=0.04, n=4). Moreover, Fig. 5c shows that inhibition of the native TRPML3 channel activity by expression of the dominant negative mutant TRPML3(D458,495K) similarly increased the rate of EGFR degradation (see Fig. 8d below for the effect of TRPML3(D458K) mutation on channel function). TRPML3(D458,459K) was very effective in accelerating the degradation of the EGFR, with increased degradation observed already after 1 min of stimulation with EGF. It is not clear at present how the EGFR can be degraded so rapidly. However, TRPML3(D458,495K) may increase both the rate of endocytosis and membrane trafficking and the EGFR may be degraded in the lysosomes and by other unidentified cellular hydrolyses. Nevertheless, the purpose of the experiment in Fig. 5c is to show that TRPML3(D458,495K) enhances endocytosis and this can be concluded from Fig. 5c since EGFR degradation requires endocytosis. In control experiments, Fig. 5d shows that deletion of the homologues TRPML1 does not affect EGFR degradation. Finally, Fig. 5e shows that the effects of expression and KD of TRPML3 on endocytosis and trafficking are not due to altered total of surface expression of EGFR.
Fig. 5. TRPML3 regulates endocytosis and membrane trafficking.

Panel (a): degradation of EGFR was measured by incubating control HeLa cells and HeLa cells transfected with TRPML3 with 100 ng/ml EGF for the indicated times and analysis of EGFRs by western blot. Tubulin levels were used to control for loading. The 10 min endocytosis averages are the mean±s.e.m. and the effect of TRPML3 is different from control at the p=0.041 level. Panel (b): degradation of EGFRs was measured by incubating control cells and cells treated with TRPML3 siRNA with 100 ng/ml EGF for the indicated times. The 10 min endocytosis averages are the mean±s.e.m. and the effect of TRPML3 siRNA is different from control at the p=0.043 level. Panel (c): degradation of EGFRs was measured in cells transfected with empty vector (control) or with TRPML3(D458,459K) and stimulated with 100 ng/ml EGF for 1 or 5 min. This is one of two experiments with similar results showing the marked increase in EGFR degradation by the DN TRPML3 mutant. Panet (d): degradation of EGFR was measured in control or TRPML1-/- fibroblasts stimulated with 100 ng/ml EGF for the indicated times. This is one of three similar experiments. Panel (e): HeLa cell treated with the two TRPML3 siRNA or transfected with empty vector, TRPML3 or TRPML3(D458K) were used to determine the effect of the various treatments on total and surface expression of EGFR. Total EGFRs (left columns) were determined by densitometry and intensities were first normalized relative to tubulin or actin that were used as loading controls and were then calculated as fold change relative to cells transfected with empty vector. Surface expression (right columns) was normalized relative to the calculated total EGFRs and were then calculated as fold change relative to cells transfected with empty vector. The columns show the m±s.e.m. of 4 experiments.
Fig. 8. Inhibition of TRPML3 channel activity inhibits cell-stress induced autophagy.

HeLa cells were treated with scrambled (a) or two different siTRPML3 (b) and were incubated in feeding media or were starved for 2 hrs. The average number of puncta is shown in (c) as the mean±s.e.m of at least 7 cells from 2 experiments. Panel (d) shows the Na+ current measured in cells expressing wild-type GFP-TRPML3, GFP-TRPML3(D458K) and TRPML3+ TRPML3(D458K). TRPML3 was activated by exposing the cells to Na+-free medium and then the Na+ current was measured by re-addition of Na+ to the medium. The procedure for current measurement is detailed in Methods and the properties of the TRPML3 current can be found in (8). Note the nearly complete inhibition of TRPML3 current by the DN TRPML3(D458K). In (e-h) the cells were transfected with eGFP-LC3 and the DN mCherry-TRPML3(D458K) and were starved for 2 hrs (e), treated with tunicamycin (f) or CPA (g). The mean±s.e.m of at least 9 cells from 3 experiments is given in (h).
Dynamic localization of TRPML3
Together, the results in Figs. 4 and 5 indicate that TRPML3 affects endocytosis and membrane trafficking, implying that the cellular localization of TRPML3 is dynamic and its subcellular distribution should be affected by altering membrane trafficking. The first indication of dynamic localization of TRPML3 was obtained by its recruitment into the autophagosomes during induction of autophagy (Fig. 6a-e). These results are discussed below in relation to the role of TRPML3 in autophagy. Another prediction of the dynamic localization of TRPML3 is that inhibition of endocytosis should alter TRPML3 localization. This prediction was tested by determining the effect of the dominant negative dynamin (K44A) (Dyn(K44A)) that inhibits endocytosis on surface expression of TRPML3. Fig. 6f shows that Dyn(K44A) has no effect on total expression of TRPML1 or TRPML3 or the cleavage and surface expression of TRPML1. On the other hand, Dyn(K44A) dramatically increased surface expression of TRPML3. Hence, it is clear that TRPML3 recycles between the plasma membrane and intracellular compartments by a dynamin-dependent endocytic pathway.
Fig. 6. Dynamic subcellular localization of TRPML3.

In panel (a) HeLa cells were transfected with TRPML3 and were kept in fed media or starved for 2 hrs in media without energy substrates. In panel (b) the cells were co-transfected with eGFP-LC3 and mCherry-TRPML3 and maintained in fed media. Panel (c) shows two examples of cells co-transfected with eGFP-LC3 and mCherry-TRPML3 and starved for 2 hrs. The upper images are of cells in which the autophagosomes are not coalesced into large macrophagosomes and the autophagosomes can be resolved and counted. The lower images show the coalesced autophagosomes. Only cells similar to the upper cells in (c) were used for autophagosomes counting, while the two cell types were used to calculate overlap. The number of puncta was counted (d) and the overlap between TRPML3 and LC3 was determined (e) with imageJ and are given as mean±s.e.m of at least 8 cells. Panel (f) shows total (input) and surface expression of TRPML1 and TRPML3 in control cells and cells transfected with the DN dynamin (K44A). Surface expression was determined by biotinylation.
TRPML3 and autophagy
A particularly dynamic cellular compartment is the autophagosomes that are formed during induction of autophagy in response to cellular stress (22, 23). As part of determining the dynamic localization of TRPML3, we asked whether TRPML3 is found in autophagosomes and whether TRPML3 plays a role in autophagy. The autophagosomes were detected by transfecting the cells with GFP-tagged autophagosomal light chain three (GFP-LC3). Autophagy was induced by various cell stressors that act by different mechanisms; 2 hrs cell starvation, 1 hr treatment with tunicamycin which inhibits N-glycosylation, and 3 hrs treatment with the SERCA pump inhibitor CPA to deplete ER Ca2+. When appropriate, autophagy was quantified by puncta counting with ImageJ software. The upper images in Fig. 6a and the summary in 6d show that the number of autophagosomes in fed cells is low and is markedly increased by cell starvation without a major change in particle size. Fig. 6b shows that expression of TRPML3 resulted in significant induction of autophagy in fed cells, indicating that expression of TRPML3 induces modest cell stress. Starvation of cells expressing TRPML3 resulted in a striking increase in the number of autophagosomes. Most remarkably, nearly all the autophagosomes contained TRPML3. Calculation of overlap between GFP-LC3 and mCherry-TRPML3 revealed 90-95% overlap in both fed and starved cells (Fig. 6e). These findings indicate that a) TRPML3 must be dynamically recruited into the newly formed autophagosomes, and b) the autophagosomes originates from organelles/membranes that express TRPML3 or that the autophagosomes fuse with organelles that express TRPML3.
The expression of TRPML3 exacerbated autophagy induced by the various cell stressors. The upper images in Figs. 7a and 7b show that treatment of control cells for 3 hrs with CPA (7a) and 1 hr with tunicamycin (7b) induced relatively modest autophagy (quantified in Fig. 8). However, the same treatment of cells expressing TRPML3 resulted in severe induction of autophagy, as reflected in coalescence of the autophagosomes into large macrophagosomes. TRPML3 was also recruited into the macrophagosomes, with the overlap between LC3 and TRPML3 remaining in the 95% range (Fig. 7). The rate of autophagy was measured in cells treated with tunicamycin for 0, 10 or 30 min (Fig. 7c). At longer incubation times only macrophagosomes were observed in cells transfected with TRPML3 and treated with tunicamycin, which were also apparent in many cells treated with tunicamycin for only 30 min. Therefore, the rate of autophagy in cells expressing TRPML3 and treated with tunicamycin for 30 min is under estimated. The dashed lines in Fig. 7c show the increase in the number of autophagosomes in the first 10 min of treatment with tunicamycin, indicating that TRPML3 increases, whereas inhibition of TRPML3 reduces the rate of autophagy.
Fig. 7. TRPML3 exacerbates cell-stress induced autophagy.

HeLa cells transfected with eGFP-LC3 (the upper image in a, b) or with eGFP-LC3 and mCherry-TRPML3 were treated with 25 μM CPA for 3 hrs or with 3 μM tunicamycin for 1 hr and used to determine the formation of macro-autophagosomes and recruitment of mCherry-TRPML3 into the macro-autophagosomes. Panel (c) shows the time course of autophagy induced by 3 μM tunicamycin in cells transfected with empty vector (blue), TRPML3 (red) or TRPML3(D458K) (green) and left untreated or were treated with tunicamycin for 10 or 30 min. The results are the mea±s.e.m. from at least 6 cells at each time. The dashed line show the change in the number of autophagosomes in the first 10 min of incubation with tunicamycin.
Exacerbation of autophagy by TRPML3 can be due to a combined effect of cell stress brought about by TRPML3 and the other stressors. However, the nearly complete recruitment of TRPML3 into the autophagosomes suggests that TRPML3 may have an active role in formation of the autophagosomes. To examine this possibility, we determined the effect of inhibiting the activity of the native TRPML3 by two independent means, KD with siRNA and expression of the DN TRPML3(D458K). Figs. 8a-8c show that two siRNA probes targeted to different sequences of TRPML3 reduced the starvation-induced autophagy by about 60%. Fig. 7d shows that TRPML3(D458K) functions as DN by inhibiting the current mediated by expressed wild-type TRPML3. At 1:1 expression ratio 75% of the current was inhibited, as expected from the tetrameric functional unit of TRP channels and inhibition of the channel if the tetramer contain at least one mutant subunit. Figs. 8e-8h show that the DN TRPML3(D458K) almost completely inhibited autophagy induced by starvation, tunicamycin or CPA. Hence, TRPML3 appears to be essential for induction of autophagy.
Discussion
To understand the function of TRPML3 in cellular physiology and in the varitint-waddler phenotype, in the present work we determined the cellular localization of TRPML3 and revealed its role in membrane trafficking and autophagy. Here, we show that TRPML3 is heavily glycosylated and its glycosylation is Endo H resistant, indicating that it must have exited the ER. In addition, cell fractionation and co-localization studies with established organellar markers revealed that TRPML3 can be found in several cellular sites and it is largely segregated from TRPML1. Expression of TRPML3 in multiple cellular compartment can be demonstrated for the native and expressed TRPML3. Most notably, expression of TRPML3 is dynamic, as indicated by its altered localization by inhibition of endocytosis and by its recruitment into autophagosomes upon induction of autophagy.
The multiple compartmental localization of TRPML3 is likely a consequence of its role in membrane trafficking. TRPML3 appears to regulate both constitutive (transferrin) and regulated (EGF and EGFR) endocytosis. This conclusion is based on the findings that KD of TRPML3 or expression of the DN TRPML3(D458K) increases, while overexpression of TRPML3 decreases endocytosis. The reciprocal effects of inhibition and increase of TRPML3 activity on both constitutive and regulated endocytosis suggests that TRPML3 regulates a step essential for both forms of endocytosis. At present, it is not known what endocytic step is regulated by TRPML3, since the two forms of endocytosis share many steps and regulatory events (24).
The role of TRPML3 in endocytosis is likely linked to its role in autophagy reported here. All autophagosomes, the sparse small ones found in fed cells and the larger or coalesced macrophagosomes found in stressed cells contain TRPML3. The striking massive recruitment of TRPML3 into the newly formed autophagosomes in stressed cells points to an essential role of TRPML3 in autophagy. Indeed, KD of TRPML3 and expression of the DN TRPML3(D458K) markedly reduced formation of autophagosomes in response to a variety of cell stressors. Based on the findings of a role of TRPML3 in membrane trafficking and in autophagy, a possible scenario is that overexpression of TRPML3 augments the autophagic response to result in accumulation of cellular material in the autophagosomes and lysosomes. This, in turn, results in a feedback response to slow endocytosis. On the other hand, KD of TRPML3 and expression of TRPML3(D458K) markedly reduced autophagy and delivery of cellular material to the lysosomes, allowing enhanced endocytosis and targeting of material to other cellular compartments.
How might TRPML3 regulate endocytosis, membrane trafficking and autophagy? TRPML3 functions as an inward rectifying, selective Ca2+ channel that is highly regulated by extracytosolic H+ (H+e-cyto) (5, 8). Endocytosis, membrane trafficking and autophagy involves many fusion and fission events, which are regulated by Ca2+. TRPML3 may provide the Ca2+ that is required for the fusion and fission reactions in endocytosis, membrane trafficking and autophagy. The selectivity of TRPML3 for Ca2+, the inward rectification and the regulation by H+e-cyto indicate that when TRPML3 is present in intracellular organelles, TRPML3 functions as a Ca2+ efflux channel that can be activated by membrane depolarization. Since the membrane potential of intracellular organelles and its fluctuations are not known with certainty, it is not possible to predict at what conditions TRPML3 will mediate Ca2+ efflux out of the organelles. However, under resting conditions cytoplasmic Ca2+ concentration ([Ca2+]i) is about 100 nM, while organellar Ca2+ concentration, such as in the Golgi, is in the 0.3-0.5 mM range (25). With this steep gradient and similar K+ concentration in the cytosol and organellar lumen, even minimal depolarization will results in a large Ca2+ efflux. The only available value of organellar membrane potential is a recent report that the phagosomes (lysosomes) membrane potential is about +28 mV (26). This translates to a lysosome to cytosol potential of about -30 mV, at which TRPML3 is active as a Ca2+ channel. Under these conditions TRPML3 activity is strongly affected by changes in organellar membrane potential. Large Ca2+ efflux due to changes in organellar membrane potentials or pH in the vicinity of the organelles can mediate fusion and/or fission events that are involved in endocytosis, membrane trafficking and autophagy.
Methods
Plasmid Construction, mutagenesis and siRNA oligonucleotides
Human TRPML3 in pEGFPC1 and pCMV-HA vectors (clontech) was prepared as detailed before (8). To generate mCherry-TRPML3, mCherry was amplified by PCR from the pRSET-B mCherry vector and cloned ahead of TRPML3 into the pCMV-HA-TRPML3. The construct was used to prepare TRPML3(D458K) using QuikChange mutagenesis kit (Stratagene). All mutations were confirmed by sequencing the entire DNA insert to verify the presence of the desired mutation and the absence of extraneous mutations. The dominant negative dynamin construct pcDNA3-myc-DynK44A was kindly provided by Dr. Joe Albanesi (UT Southwestern medical center, Dallas) and pEGFP-LC3 was kindly provided by Dr. Tamotsu Yoshimori (Osaka University; Osaka, Japan). For knock-down experiments we used siRNA that targets the following human TRPML3 sequence siRNA 1: 5′-GGAUGGUACAUUAUGAUUAUU-3′; siRNA 2: 5′-GAACUUAACACUGGACUUC-3′ and siCONTROL Non-Targeting siRNA (Dharmacon). Unless otherwise stated, siRNA 1 was used since it was most effective in KD of TRPML3. siRNA 2 was used in selective experiments to exclude effects due to off target effects of the siRNA.
Antibodies and reagents
The H4A3 (Lamp1) antibody developed by J.T. August and J.E.K. Hildreth, and the α6F (Na+/K-ATPase) antibody developed by Douglas.M. Fambrough were obtained from the Developmental Studies Hydbridoma Bank, Iowa City, IA. EEA1 and GM130 antibodies were obtained from BD Transduction Laboratories. EGFR and α-tubulin antibodies were purchased from Cell Signaling. One anti-TRPML3 antibody was purchased from ProtenTec Inc. Anti-TRPML3 antibodies were also raised in two rabbits against the C-terminal sequence CKDLPNSGKYRLEDDPPVSLF to generate the anti-TRPML3 antibodies αC1 and αC2. Alexa-EGF and Alexa-transferrin were obtained from Invitrogen Molecular Probes. 125I-EGF was purchased from GE Healthcare. Anti-HA antibody was purchased from Covance Research Products Inc. Anti-Bip antibody was obtained from Stressgen Biotechnologies.
Cell culture, transfection and immunoblotting
HEK293 or HeLa cells were maintained in DMEM supplemented with 10% fetal bovine serum in a humidified incubator at 37 °C and 5% CO2/air. Cells were plated 1 day before transfection with Lipofectamine 2000 (Invitrogen) for plasmids or Dharmafect (Dharmacon) for siRNA according to the manufacturers' instructions. 24 hrs after transfections, cell extracts were prepared by sonication in homogenization buffer containing 50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 2 mM EDTA, 5 mM MgCl2, and complete protease inhibitor mixture tablet (Roche Applied Science). Microsomal pellets were extracted with 1% CHAPS or 1% Triton X-100 and subjected to SDS-PAGE and immunoblotted. Alternatively, the extracts were used to immunoprecipitate (IP) GFP-TRPML1 with anti-GFP and HA-TRPML3 with anti-HA and blotted for co-IP of, respectively, TRPML3 with anti-Ha and TRPML1 with the αN1 antibodies raised against a TRPML1 N terminal peptide (19).
Confocal microscopy and Immunocytochemistry
HeLa cells expressing TRPML3 or were grown on glass coverslips and fixed by methanol or 4 % paraformaldehyde and permeabilized by incubation with 0.1 % Triton X-100 at room temperature for 10 min. After fixation, nonspecific sites were blocked with 5 % goat serum. Cells were stained with primary antibody overnight and for 1 hour at room temperature with fluorescent secondary antibody. Cover slips were mounted on glass slides and analyzed using a Biorad 1024 confocal microscope. The images were recorded with a 40× objective and analyzed off-line using NIH ImageJ™ software. All images are maximum intensity z-projections and the bars in all images tonate 10 μm. Cell were transfected with eGFP-LC3 with or without mCherry-TRPML3 for 24 hrs and were treated as indicated and then fixed by incubation with 4 % paraformaldehyde for 15 min and imaged by confocal microscopy. Live cells transfected with mCherry-TRPML3 were incubated with 10 nM lysotracker Green at room temperature and images were recorded at different time points.
Cell Fractionation Studies
Untransfected HeLa cells or HeLa cells transfected with HA-TRPML3 for 24h, were washed twice with PBS and scraped into homogenization buffer (20 mM tricine, pH 7.6, 0.25 M sucrose, and 1 mM EDTA). Cells were lysed by passing through a tight-fitting Dounce homogenizer 20 times followed by passing 5 times through a 25-gauge needle. The homogenate was centrifuged for 10 min at 400 × g, and 1 mg of each post-nuclear supernatant was layered over a discontinuous gradient consisting of a 1.2-ml cushion of 2.5 M sucrose and 10 ml of an 18% Percoll in homogenization buffer. The gradient was centrifuged for 60 min at 28,000 RPM in a Beckman Type 90Ti rotor. The sucrose cushion, 11 0.25-ml and 4 1-ml fractions were collected from the bottom of the tube and the Percoll was removed by centrifugation for 40 min at 70,000 rpm in a Beckman TLA 100.3 rotor. The samples were adjusted to 0.5% Triton X-100, passed five times through a 25-gauge needle, and incubated on ice for 30 min. Fractions were prepared for immunoblots of TRPML3 and marker proteins distribution.
Surface Biotinylation Assay
Cells were washed with PBS and incubated in 1 mg/ml Sulfo-NHS-LC-Biotin (Pierce) in PBS for 30 min on ice. Free biotin was quenched by the addition of 100 mM glycine in PBS and the cells were washed once with PBS. Lysates were prepared in lysis buffer by passing 7-10 times through a 27-gauge needle after sonication. The lysates were centrifuged at 14,000 rpm for 10 min at 4 °C, and protein concentration in the supernatants was determined. Volume and protein content were adjusted to be the same in all samples and 12.5 % avidin beads (Pierce) were added to each sample. After incubated for 2 hours at 4 °C, the beads were collected by centrifugation, washed three times with 0.5 % triton X-100 in PBS and the proteins were extracted in sample buffer. Collected proteins were analyzed by Western blot.
Uptake of fluorescent EGF and Transferrin
HeLa cells were grown on glass coverslips and transfected with TRPML3 or treated with TRPML3 siRNA. The cells were washed twice with PBS and incubated with 40 ng/ml labeled EGF for 5, 10, and 15 min at 37 °C. After a PBS wash, cells were incubated with unlabeled EGF for 1-2 min at 4 °C and fixed with cold MeOH. For transferrin uptake, 1 μg/ml labeled transferrin was used and incubated at different time points. Cells were mounted on glass slides, fixed with cold MeOH and analyzed using the confocal microscope.
EGFR endocytosis and degradation Assay
HeLa cells transfected with TRPML3 or TRPML3(D458/459K) or treated with TRPML3 siRNA were serum-starved for 16 hrs and stimulated with 100 ng/ml EGF in the presence of cyclohexamide (10 μg/ml). At each indicated times, cells were washed with cold PBS, lysed with M-PER mammalian protein extraction reagent (Pierce) and immunoblotted. To quantify the degradation of EGFRs, the intensity of EGFR immunoreactive bands was measured using NIH ImageJ™ software.
125I-EGF Internalization
HeLa cells transfected with TRPML3 or TRPML3 siRNA were serum-starved in binding medium (DMEM, 20 mM Hepes, 1 % bovine serum albumin) for 1 h. The control group was treated with monensin (100 μM) and nigericin (5 μM) in binding media for 20 min. The cells were then incubated with 1 ng/ml of 125I-EGF and 39 ng/ml of unlabeled EGF in binding media at 37 °C for 5 min. Uptake was terminated by transferring the cells to ice-water bath, washed 3 times with ice-cold PBS and lysed with 1 % NP-40 in 1 N NaOH. Cell lysates were counted in a Beckman scintillation counter.
Current recordings and solutions
The tight seal, whole-cell configuration was used to record the whole cell current at room temperature exactly as detailed in (8). Currents were measured in response to voltage ramps (-100 to +100 mV, 100 ms) applied every 5 second from a holding potential of 0 mV. The pipette solution contained (in mM) 150 KCl, 10 HEPES, 1.13 MgCl2, 5 ATP and 10 BAPTA adjusted to pH 7.3 with KOH. The bath solution consisted of (in mM) 140 NaCl, 5 KCl, 1 MgCl2, 1 CaCl2, 10 glucose and 10 HEPES, adjusted to pH 7.4 with NaOH. Na+-free solution contained (in mM) 150 NMDG-Cl and 10 HEPES, adjusted to pH 7.4.
Acknowledgments
This work was supported in part by National Institutes of Health Grants DE12309 and DK38938 and the Ruth S. Harrell Professorship in Medical Research.
Abbreviations
- TRP
transient receptor potential
- MLIV
Mucolipidosis type IV
- LSD
Lysosomal Storage Disease
- GOF
Gain-of-Function
- KD
knock-down
- [Ca2+]i
free cytoplasmic Ca2+
- EGFR
epidermal growth factor receptor
- DN
dominant negative
Footnotes
All authors declare that they do not have any conflict of interest to disclose.
References
- 1.Nilius B, Owsianik G, Voets T, Peters JA. Transient receptor potential cation channels in disease. Physiol Rev. 2007;87(1):165–217. doi: 10.1152/physrev.00021.2006. [DOI] [PubMed] [Google Scholar]
- 2.Bargal R, Avidan N, Ben-Asher E, Olender Z, Zeigler M, Frumkin A, Raas-Rothschild A, Glusman G, Lancet D, Bach G. Identification of the gene causing mucolipidosis type IV. Nature genetics. 2000;26(1):118–123. doi: 10.1038/79095. [DOI] [PubMed] [Google Scholar]
- 3.Di Palma F, Belyantseva IA, Kim HJ, Vogt TF, Kachar B, Noben-Trauth K. Mutations in Mcoln3 associated with deafness and pigmentation defects in varitint-waddler (Va) mice. Proc Natl Acad Sci U S A. 2002;99(23):14994–14999. doi: 10.1073/pnas.222425399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xu H, Delling M, Li L, Dong X, Clapham DE. Activating mutation in a mucolipin transient receptor potential channel leads to melanocyte loss in varitint waddler mice. Proc Natl Acad Sci U S A. 2007;104(46):18321–18326. doi: 10.1073/pnas.0709096104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim HJ, Li Q, Tjon-Kon-Sang S, So I, Kiselyov K, Muallem S. Gain-of-function mutation in TRPML3 causes the mouse varitint-waddler phenotype. J Biol Chem. 2007 doi: 10.1074/jbc.C700190200. [DOI] [PubMed] [Google Scholar]
- 6.Grimm C, Cuajungco MP, van Aken AF, Schnee M, Jors S, Kros CJ, Ricci AJ, Heller S. A helix-breaking mutation in TRPML3 leads to constitutive activity underlying deafness in the varitint-waddler mouse. Proc Natl Acad Sci U S A. 2007 doi: 10.1073/pnas.0709846104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nagata K, Zheng L, Madathany T, Castiglioni AJ, Bartles JR, Garcia-Anoveros J. The varitint-waddler (Va) deafness mutation in TRPML3 generates constitutive, inward rectifying currents and causes cell degeneration. Proc Natl Acad Sci U S A. 2008;105(1):353–358. doi: 10.1073/pnas.0707963105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim HJ, Li Q, Tjon-Kon-Sang S, So I, Kiselyov K, Soyombo AA, Muallem S. A novel mode of TRPML3 regulation by extracytosolic pH absent in the varitint-waddler phenotype. The EMBO journal. 2008;27(8):1197–1205. doi: 10.1038/emboj.2008.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Manzoni M, Monti E, Bresciani R, Bozzato A, Barlati S, Bassi MT, Borsani G. Overexpression of wild-type and mutant mucolipin proteins in mammalian cells: effects on the late endocytic compartment organization. FEBS Lett. 2004;567(2-3):219–224. doi: 10.1016/j.febslet.2004.04.080. [DOI] [PubMed] [Google Scholar]
- 10.Soyombo AA, Tjon-Kon-Sang S, Rbaibi Y, Bashllari E, Bisceglia J, Muallem S, Kiselyov K. TRP-ML1 regulates lysosomal pH and acidic lysosomal lipid hydrolytic activity. J Biol Chem. 2006;281(11):7294–7301. doi: 10.1074/jbc.M508211200. [DOI] [PubMed] [Google Scholar]
- 11.Miedel MT, Rbaibi Y, Guerriero CJ, Colletti G, Weixel KM, Weisz OA, Kiselyov K. Membrane traffic and turnover in TRP-ML1-deficient cells: a revised model for mucolipidosis type IV pathogenesis. The Journal of experimental medicine. 2008;205(6):1477–1490. doi: 10.1084/jem.20072194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fares H, Greenwald I. Regulation of endocytosis by CUP-5, the Caenorhabditis elegans mucolipin-1 homolog. Nat Genet. 2001;28(1):64–68. doi: 10.1038/ng0501-64. [DOI] [PubMed] [Google Scholar]
- 13.Vergarajauregui S, Connelly PS, Daniels MP, Puertollano R. Autophagic dysfunction in mucolipidosis type IV patients. Human molecular genetics. 2008;17(17):2723–2737. doi: 10.1093/hmg/ddn174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang F, Li PL. Reconstitution and characterization of a nicotinic acid adenine dinucleotide phosphate (NAADP)-sensitive Ca2+ release channel from liver lysosomes of rats. J Biol Chem. 2007;282(35):25259–25269. doi: 10.1074/jbc.M701614200. [DOI] [PubMed] [Google Scholar]
- 15.Dong XP, Cheng X, Mills E, Delling M, Wang F, Kurz T, Xu H. The type IV mucolipidosis-associated protein TRPML1 is an endolysosomal iron release channel. Nature. 2008;455(7215):992–996. doi: 10.1038/nature07311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Venkatachalam K, Hofmann T, Montell C. Lysosomal localization of TRPML3 depends on TRPML2 and the mucolipidosis-associated protein TRPML1. J Biol Chem. 2006;281(25):17517–17527. doi: 10.1074/jbc.M600807200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van Aken AF, Atiba-Davies M, Marcotti W, Goodyear RJ, Bryant JE, Richardson GP, Noben-Trauth K, Kros CJ. TRPML3 mutations cause impaired mechano-electrical transduction and depolarization by an inward-rectifier cation current in auditory hair cells of varitint-waddler mice. The Journal of physiology. 2008 doi: 10.1113/jphysiol.2008.156992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Song Y, Dayalu R, Matthews SA, Scharenberg AM. TRPML cation channels regulate the specialized lysosomal compartment of vertebrate B-lymphocytes. European journal of cell biology. 2006;85(12):1253–1264. doi: 10.1016/j.ejcb.2006.08.004. [DOI] [PubMed] [Google Scholar]
- 19.Kiselyov K, Chen J, Rbaibi Y, Oberdick D, Tjon-Kon-Sang S, Shcheynikov N, Muallem S, Soyombo A. TRP-ML1 is a lysosomal monovalent cation channel that undergoes proteolytic cleavage. J Biol Chem. 2005;280(52):43218–43223. doi: 10.1074/jbc.M508210200. [DOI] [PubMed] [Google Scholar]
- 20.Cable J, Steel KP. Combined cochleo-saccular and neuroepithelial abnormalities in the Varitint-waddler-J (VaJ) mouse. Hearing research. 1998;123(1-2):125–136. doi: 10.1016/s0378-5955(98)00107-5. [DOI] [PubMed] [Google Scholar]
- 21.Ceresa BP. Regulation of EGFR endocytic trafficking by rab proteins. Histology and histopathology. 2006;21(9):987–993. doi: 10.14670/HH-21.987. [DOI] [PubMed] [Google Scholar]
- 22.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132(1):27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ferraro E, Cecconi F. Autophagic and apoptotic response to stress signals in mammalian cells. Archives of biochemistry and biophysics. 2007;462(2):210–219. doi: 10.1016/j.abb.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 24.Vassilieva EV, Nusrat A. Vesicular trafficking: molecular tools and targets. Methods in molecular biology (Clifton, NJ. 2008;440:3–14. doi: 10.1007/978-1-59745-178-9_1. [DOI] [PubMed] [Google Scholar]
- 25.Dolman NJ, Tepikin AV. Calcium gradients and the Golgi. Cell calcium. 2006;40(5-6):505–512. doi: 10.1016/j.ceca.2006.08.012. [DOI] [PubMed] [Google Scholar]
- 26.Steinberg BE, Touret N, Vargas-Caballero M, Grinstein S. In situ measurement of the electrical potential across the phagosomal membrane using FRET and its contribution to the proton-motive force. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(22):9523–9528. doi: 10.1073/pnas.0700783104. [DOI] [PMC free article] [PubMed] [Google Scholar]