

Abstract
Regio-, diastereo-, and enantioselective coupling reactions between imines and allylic alcohols have been developed. These coupling reactions deliver complex homoallylic amine products through a convergent C–C bond forming process that does not proceed through intermediate allylic organometallic reagents. In general, convergent coupling, by exposure of an allylic alkoxide to a preformed Ti–imine complex, occurs with allylic transposition in a predictable and stereocontrolled manner. While simple diastereoselection in these reactions is high, delivering anti-products with ≥ 20:1 selectivity, the organometallic transformation described is compatible with a diverse range of functionality and substrates (including: aliphatic and aromatic imines, allylic silanes, trisubstituted alkenes, vinyl- and aryl halides, trifluoromethyl groups, thioethers, and aromatic heterocycles). Alkene geometry of the products is a complex function of the allylic alcohol structure, and is consistent with a mechanistic proposal based on syn-carbometalation followed by syn-elimination by way of a boat-like transition state geometry. Single asymmetric coupling reactions provide a means to translate the stereochemical information of the allylic alcohol to the homoallylic amine with very high levels of fidelity, or to control diastereoselection in the coupling reactions of achiral allylic alcohols with chiral imines. Double asymmetric coupling reactions are also described that afford a unique means to control stereoselection in these complex convergent coupling processes. Finally, empirical models are proposed that are consistent with the observed stereochemical course of these coupling reactions en route to chiral homoallylic amines possessing di- or trisubstituted alkenes, and anti- or syn- relative stereochemistry at the allylic and homoallylic positions. Overall, the bond construction enabled by this Ti-mediated reductive cross-coupling is unmatched by available methods in organic chemistry.
Introduction
Methods that accomplish bimolecular (convergent) C–C bond formation define the backbone of organic synthesis.1 Despite their central role in the construction of natural products and synthetic small molecules of biological significance, the broad classes of reactions suitable for complex convergent C–C bond formation remain rather limited and include reactions based on: 1) nucleophilic substitution, 2) nucleophilic addition to polarized π-bonds, 3) cycloaddition, 4) metal-catalyzed “cross-coupling”, and 5) crossed olefin metathesis. Arguably, carbonyl addition chemistry is among the most powerful subclass of convergent coupling, offering opportunities for fragment union through the generation of a diverse range of stereodefined architecture (including di- and tri-substituted alkenes, β-hydroxy carbonyls, allylic- and homoallylic alcohols).2
Carbonyl allylation is a subset of carbonyl addition chemistry that, over the last thirty years, has emerged as a particularly attractive method for acyclic stereocontrol, offering highly stereoselective pathways to homoallylic alcohols and amines.3 The vast majority of established methods for carbonyl allylation, while powerful, depend on the use of allylic organometallic reagents. Despite the discovery of catalytic methods for the in situ generation of such reagents, the dependence on accessing an allylic organometallic species has generally limited the utility of such processes to the addition of simple unsaturated hydrocarbon fragments.4, 5 While the synthesis and application of more functionalized allylic organometallic reagents for complex fragment union is possible, as illustrated in Figure 1 such processes are often complicated by: 1) Functional group tolerance in the preparation of the allylic organometallic reagent (A → B1/B2), 2) challenges associated with the control of site-selectivity in the metalation step (A → B1/B2), 3) difficulties in controlling the regioselectivity of C–C bond formation [due to (a) the propensity for allylic isomerization of the intermediate allylorganometallic (B1 ⇌ B2), and (b) competition between α- vs γ- attack], and 4) problems associated with stereoselection [both tetrahedral stereochemistry and olefin geometry (i.e. C vs E, or D vs F)]. In cases where the allylic metal reagent is generated in a catalytic fashion, functional group tolerance is often enhanced, but complexities still remain due to competing isomerization of the allylic metal species, and the aforementioned issues with regio- and stereoselection in the C–C bond forming event.5 As such, the impact that carbonyl allylation has in complex convergent bond construction, independent of whether such processes are rendered catalytic in the metal, remains fundamentally limited.
Figure 1.

State-of-the-art methods for allyl transfer via the intermediacy of allylic organometallic reagents.
An orthogonal strategy for allyltransfer can be envisioned to proceed in an umpolung sense,6 with bond formation occurring between a carbonyl-derived anion and an allylic electrophile (Figure 2A). In the simplest of analyses, such a transformation has related challenges to the use of allylic organometallic reagents, where issues of regioselection derive instead from a competition between SN2 and SN2′ modes of addition, leading to the isomeric products of formal allyl-transfer (3 and 4). While the regiochemical course of transformations in this class have the potential to be influenced by the character of the groups flanking the allyl-unit (R3 and R4), and the nature of the metal counterion associated with 1, major barriers associated with the control of relative and absolute stereochemistry combine to further complicate this subset of potential coupling processes.7
Figure 2.

Umpolung-based strategy for carbonyl allylation.
As depicted in Figure 2B, a related reaction with a heteroatom-substituted metallacyclopropane 5 was reasoned to proceed through a mechanistically distinct process, where bimolecular C–C bond formation would result from carbometalation of 2 (via a formal metal-centered [2+2+1] process) in preference to simple nucleophilic addition (via SN2 or SN2′ reaction pathways). The resulting isomeric metallacyclopropanes 6 and 7 would differ with respect to stability and reactivity, affording a pathway to 3 by elimination of 6. Control of this process could derive from one of two basic mechanistic considerations. If the carbometalation process (5 + 2 → 6 or 7) is reversible, the elimination reaction (6 → 3) may represent a means to funnel the mixture of metallacycles (6 + 7) to the desired product 3, through a process that would have the potential to offer stereochemical control via selective elimination through the lowest energy transition state. Alternatively, if the carbometalation process were to proceed under kinetic control, then the stereochemical information derived from the carbometalation (en route to metallacyclopentane 6) would translate directly to the relative stereochemistry of the product, assuming that the elimination reaction would proceed in a mechanistically predictable fashion (syn- or anti-). While glimpses of this mode of reactivity with Cp2Zr–imine complexes were reported twenty years ago,8 the complex issues associated with selectivity that are critical to the development of a broadly useful convergent coupling reaction remain ill-defined.9
Here, we present a full account of the scope and limitations of a mild and general titanium alkoxide-mediated coupling reaction between allylic alcohols and imines as a route to stereochemically defined and highly functionalized homoallylic amines (Figure 3A).10 The scope of these reactions now encompasses: 1) the coupling of aromatic and aliphatic imines, 2) enantioselective allyltransfer through the use of chiral allylic alcohols, 3) double asymmetric coupling as a strategy to override inherent trends in selectivity of single asymmetric reactions, 4) the use of coupling partners bearing a diverse range of functionality (including di- and trisubstituted alkenes, thioethers, allylic silanes, aromatic heterocycles, and aliphatic, vinyl- and aromatic halides), and 5) stereodivergent coupling reactions to afford a range of substituted homoallylic amines (Figure 3B).
Figure 3.

Homoallylic amines by Ti-alkoxide-mediated reductive cross-coupling.
Background Associated with Ti-alkoxides in Azametallacyclopropane-mediated C–C Bond Formation
The discovery that Ti(IV) alkoxides can be employed in a simple experimental procedure for the generation of metallacyclopropanes represented perhaps an underappreciated advance in the chemistry of Group 4 metallacycles.11 While reaction development in this broad area of chemistry was initiated with cyclopentadienyl-based Zr- and Ti- reagents,12, 13 the observation that related reactivity was accessible from inexpensive and robust Ti-alkoxides, marked what could be referred to as a historic advance in the area. Early attention remained focused on the Kulinkovich reaction as a means to prepare functionalized cyclopropanes from the reaction of Grignard reagents with esters.14 Later, this general mode of reactivity was expanded to encompass intermolecular variations of this reaction that proceeded by transfer of Ti from a preformed metallacyclopropane to a synthetically valuable terminal alkene.15 Concurrent with these studies, the basic reactivity patterns of (RO)2Ti–alkyne and (RO)2Ti–imine complexes, reflecting the related chemistry of Cp2Ti– and Cp2Zr– complexes, were beginning to emerge.16
These early studies served to demonstrate that metallacyclopropanes/propenes, formed by the in situ reduction of Ti(IV) alkoxides, were competent organometallic intermediates for metal-centered [2+2+1] chemistry. However, barriers associated with the control of regioselectivity, stereoselectivity, and generally low levels of reactivity in these bimolecular C–C bond forming processes have broadly restricted this, and related areas of reaction development to a relatively modest collection of convergent coupling processes between a limited set of unsaturated substrates.17
Only recently have reaction strategies emerged that offer general and highly selective means for the control of intermolecular metal-centered [2+2+1] chemistry.17 The early observations of Rothwell and Kulinkovich validated that C–C bond formation with Ti-alkoxide-based metallacyclopropanes was viable. Given the well established propensity for Ti(IV) alkoxides to undergo rapid and reversible ligand exchange,18 and their ability to generate reactive metallacyclopropanes, we have been actively pursuing the development of alkoxide-directed metal-centered [2+2+1] chemistry based on these relatively benign, readily available, inexpensive, and easily handled metal alkoxides. The allylic alcohol–imine coupling reaction described here is a unique bimolecular bond construction that has surfaced from our studies in this area.
Results and Discussion
Our pursuit began with the proposal of a general pathway for Ti-alkoxide-mediated reductive cross-coupling of imines with allylic alcohols (Figure 4).10a Formation of a Ti–imine complex 9, followed by exposure to an allylic alkoxide 10 would lead to the transient mixed titanate ester 11 by rapid and reversible ligand exchange. Subsequent carbometalation, by way of 12, would deliver a strained bicyclic azametallacyclopentane (13), which has the potential to undergo a syn-elimination similar to that postulated in the Tebbe olefination.19
Figure 4.

General strategy for Ti-alkoxide-mediated coupling of allylic alcohols with imines.
If such a sequence were possible, we anticipated that carbometalation would proceed through a boat-like transition state geometry, as in 12, based on mechanistic assumptions that include: 1) alignment of the σTi–C bond with πC=C bond, and 2) association of the allylic alkoxide of 10 with Ti to render the carbometalation event intramolecular.20 Further assuming that the carbometalation (12 → 13) would be essentially irreversible,21 we anticipated that all aspects of stereochemistry associated with product (3) would derive from stereochemical control through minimization of non-bonded steric interactions in the boat-like transition state 12.22 Interestingly, complexities with this process emerge from a cursory inspection of this proposal, as the Ti–imine complex 9 is chiral, and the addition to substituted allylic alcohols (10) could be complicated by significant double asymmetric relationships.23
We initiated study of the proposed bond construction with the attempted coupling of an aromatic imine with simple allylic alcohols. As illustrated in eq 1 of Figure 5, coupling of imine 14 with allyl alcohol (15) delivers homoallylic amine 16 in 70% yield. While we were delighted to observe success in this initial coupling reaction, our enthusiasm was dampened by the host of other methods available for simple allylation of imines.24 We therefore decided to move on to explore more challenging and unprecedented bond constructions with this novel Ti-mediated coupling reaction.
Figure 5.

Allyl and prenyl transfer chemistry.
Imine prenylation remains a challenging problem in reaction methodology, as allylic organometallic reagents typically deliver homoallylic amine products derived from inverse prenylation.25 Interestingly, as depicted in eq 2 of Figure 5, Ti-alkoxide-mediated coupling of imine 14 with the tertiary allylic alcohol 17 proceeds with exquisite levels of regioselection to deliver the product of prenylation 18 in 83% yield. In this process, no evidence could be found for the production of isomeric homoallylic amine products (rs ≥ 20:1). This impressive level of regioselection can be extended to the production of homoallylic amines bearing a tetrasubstituted alkene. As depicted in eq 3, coupling of imine 14 with allylic alcohol 19 delivers 20 in 80% yield, again with high regioselection (≥ 20:1).
While initial reports documenting the conversion of imines to azatitanacyclopropanes with Ti(Oi-Pr)4 were limited to a small subset of aromatic imines, subtle perturbation of the established reaction conditions defined a pathway to employ aliphatic imines in this reductive cross-coupling reaction.26 Simply replacing the Grignard reagent typically employed to access the initial “low-valent” Ti-species with n-BuLi provides a set of conditions suitable to extend this mode of reactivity to aliphatic imines. While we are uncertain of the mechanistic underpinnings that allow for success in these transformations, imine prenylation of branched (21) and unbranched (23) imines proceed in ≥ 80% yield and deliver products as single regioisomers (eqs 4 and 5).
Next, we shifted our attention to the examination of relative stereochemistry in the coupling of achiral imines with achiral allylic alcohols bearing an internal alkene. Coupling of imine 14 with the (E)-allylic alcohol 25 delivers homoallylic amine 26 in 87% yield (Figure 6A, eq 6). Here, the regioselective allyl transfer reaction is coupled to very high levels of diastereoselection that favor the formation of the anti- product (ds ≥ 20:1). In the related example depicted in eq 7, coupling of imine 14 to the isomeric (Z)-allylic alcohol 27 also furnishes homoallylic amine 26 with very high levels of stereoselection, albeit with slightly diminished yield (68%). This stereoconvergence, whereby each alkene isomer 25 and 27 furnishes the same anti-product 26, is consistent with an empirical model that is based on the minimization of non-bonded steric interactions in the boat-like transition state geometries A and B (Figure 6B). The isomeric transition states C and D suffer from the buildup of significant non-bonded steric interactions as a result of the eclipsing of Me- and Ph- substituents about the developing C–C bond.
Figure 6.

Simple diastereoselection.
Moving on, we explored the potential of this allylic alcohol–imine coupling process for the establishment of homoallylic amines bearing stereodefined alkenes.27 As depicted in eq 8 of Figure 7A, coupling of imine 14 with allylic alcohol 28 provides the stereodefined homoallylic amine 29 in 87% yield. Here, the highly regioselective coupling process proceeds with exquisite levels of selectivity for the formation of a (Z)-trisubstituted alkene (Z:E ≥ 20:1). Unfortunately, union of 14 with 30 did not proceed in a similarly selective manner (eq 9). While this coupling reaction delivers homoallylic amines in 92% yield, with very high levels of regio- and anti-selectivity, the homoallylic amine products are formed as a 1.6:1 mixture of alkene isomers, favoring the (Z)-isomer 31. Interestingly, the related coupling reaction of 14 with the isomeric allylic alcohol 32 does proceed in a highly selective manner, and delivers homoallylic amine 33 in 57% yield (eq 10). Here, highly regioselective C–C bond formation occurs in concert with the establishment of an anti-homoallylic amine possessing an (E)-disubstituted alkene (anti:syn ≥ 20:1; E:Z ≥ 20:1).
Figure 7.

Allylic alcohol–imine coupling reactions that establish stereodefined di- and tri-substituted alkenes.
In a similarly impressive example, coupling of imine 14 with the trisubstituted allylic alcohol 34 provides the stereodefined homoallylic amine 35 in 54% yield (eq 11). Here, regioselective coupling again occurs to deliver a homoallylic amine possessing an anti-stereochemical relationship, yet this time it does so while establishing a (Z)-trisbustituted alkene with high levels of stereocontrol (≥ 20:1).
The reactions depicted in Figure 7A are quite complex and deserve further comment. All allylic alcohols employed in these reactions were racemic, and formation of the Ti–imine complex of 14 occurs without stereochemical bias. As a result, all of these coupling reactions are complicated by the potential of diastereomeric combinations of metallacyclopropane and allylic alcohol [i.e. (R)-imine complex + (R)-28 vs (R)-imine complex + (S)-28]. Nevertheless, the results of these experiments are consistent with an empirical model that is based on: 1) in situ interconversion of azatitanacyclopropane enantiomers,28 2) kinetically controlled carbometalation,21 and 3) syn-elimination through a boat-like transition state geometry. As depicted in Figure 7B, coupling reactions of (Z)-allylic alcohols are reasoned to be (E)-selective via reaction by way of a transition state geometry E, where minimization of A-1,3 strain29 results in an equatorial disposition of the allylic substituent (R). If this stereoselective carbometalation is followed by syn-elimination, then the equatorial disposition of this group results in the formation of the (E)-alkene of 33. The high anti-selectivity observed in the formation of 33 is consistent with the minimization of eclipsing non-bonded steric interactions about the developing C–C bond.
Alternatively, in the (Z)-selective coupling reactions of 34, stereoselection is consistent with transition state F (Figure 7B). Here, minimization of A-1,2 interactions in a boat-like transition state geometry is reasoned to result in a pseudo-axial disposition of the allylic substituent. As discussed previously, stereoselective carbometalation followed by syn-elimination would then furnish the (Z)-anti-homoallylic amine 35.
The lack of stereoselection observed in the coupling reaction of the (E)-disubstituted allylic alcohol 30, likely results from the ability of the boat-like transition state geometry to accommodate pseudo-axial or pseudo-equatorial orientations of the allylic substituent without suffering destabilization from significant A-1,2 or A-1,3 strain.
Enantioselective coupling
The high anti-selectivities observed in the reactions of allylic alcohols 30, 32, and 34 point to the ability of these coupling reactions to accomplish kinetic resolution. In each case, one enantiomer of the allylic alcohol (32 or 34) preferentially reacts with one enantiomer of the Ti–imine complex to afford the anti- products.30 If this were incorrect, then one would anticipate the appearance of syn-homoallylic amine products. Their absence in these coupling reactions led us to conclude that coupling reactions of optically active allylic alcohols with racemic Ti–imine complexes would define a means to render this convergent coupling process enantioselective.
As depicted in Figure 8, eqs 12–14, we were delighted to find that coupling reactions of (R)-32 with aromatic and aliphatic imines proceed in a manner to effectively translate stereochemical information from the allylic alcohol starting material to the homoallylic amine products.31 While these observations are consistent with reasoning derived from analysis of the reactions depicted in Figure 7A, we realized that a central feature of the enantioselective reactions depicted in eqs 12–14 was the translation of stereochemical information from the allylic alcohol to the azametallacyclopropane through the minimization of eclipsing interactions about the developing C–C bond (see the positioning of the highlighted Me- and Ph substituents in E and F of Figure 7B). We wondered whether this control feature is necessary for the enantioselective version of this convergent coupling reaction. As such, we examined the coupling of the enantioenriched allylic alcohol (R)-28 (80% ee); a substrate that lacks terminal substitution of the alkene. While this coupling reaction did proceeded in an enantioselective fashion, significant erosion of optical purity was observed for homoallylic amine product 29 (46% ee).
Figure 8.

Enantioselective allylic alcohol–imine coupling.
These studies (focused on stereochemistry) provided support that the present Ti-mediated allylic alcohol–imine coupling can proceed in a highly controlled manner. Alongside these efforts, we have broadly explored functional group compatibility associated with this reductive cross-coupling reaction. Due to the wealth of reactivity available with vinyl- and aromatic halides, we initiated studies to explore the compatibility of the allylic alcohol–imine coupling reaction with substrates harboring this functionality. As depicted in Figure 9A, allylic alcohols that contain vinyl chlorides, bromides and iodides are all compatible with the coupling process. 10a, 32 Recently, the orthogonality of this Ti-mediated coupling process with vinyl halides was employed to define a simple one-pot process for the stereoselective synthesis of exo-alkylidene γ-lactams (Figure 9B).32
Figure 9.

A one-pot annulation for the synthesis of stereodefined exo-alkylidene γ-lactams.
This allylic alcohol–imine reductive cross-coupling reaction is also compatible with allylic silanes. As illustrated in Figure 10, orthogonality of this formal allyltransfer process to the chemistry of allylsilanes,10a, 33 was demonstrated in a reaction sequence of utility for the formation of stereodefined heterocycles. This complex example, also revealing the compatibility of indoles and acetals in the Ti-mediated coupling process, delivers the stereodefined polycyclic heterocycle 43 (dr ≥ 20:1), in just two steps from allylic alcohol 40 and imine 41.
Figure 10.

Convergent synthesis of complex heterocycles via Ti-mediated coupling of allylic alcohols with imines.
Double Asymmetric Coupling Processes
Overall, the Ti-mediated allylic alcohol–imine coupling process has been demonstrated to be a powerful convergent coupling reaction, being compatible with a range of functional groups and offering control over regioselection, relative stereochemistry (allylic and homoallylic positions), and olefin geometry. As depicted in Figure 11, coupling reactions have proven useful for the stereoselective synthesis of anti-homoallylic amines possessing (E)-disubstituted (eq 16), and “(Z)”-trisubstituted olefins (eq 17). While a related coupling reaction did produce an anti-homoallylic amine bearing a (Z)-disubstituted alkene (eq 18) (anti:syn ≥ 20:1), this process did not proceed with very high levels of stereochemical control (Z:E = 1.6:1). In addition to these observations concerning relative stereochemical control, we have been successful in defining a highly enantioselective cross-coupling reaction (eq 19). Unfortunately, asymmetric coupling of a simple 1,1-disubstituted olefin with an achiral imine does not proceed with good translation of optical purity (eq 20). In an effort to overcome the limitations in stereochemical control observed from these studies (i.e. eq 18 and 20), we directed our attention to the study of double asymmetric reductive cross-coupling reactions.
Figure 11.

Summary of stereochemical observation.
While it was straightforward to prepare enantiodefined allylic alcohols suitable for these studies, the selection of an appropriate chiral imine was more complicated. As illustrated in Figure 12, chiral substituents could be introduced by modification of R3, R4, or both. We selected to pursue installation of a chiral auxiliary at R3 as a general means to control formation of the chiral azametallacyclopropane reaction partner. This choice was made based on previous success in Zr- and Ti-mediated coupling reactions of imine 44 with TMS-acetylene (Figure 13). First reported in the Zr-promoted reaction by Taguchi, imine 44 could be converted to the chiral allylic amine 45 with very high levels of stereoselection (ds = 95:5; obtained after heating the preformed azametallacyclopropane to 70 °C prior to addition of the alkyne).34 Later, in 2003, Sato demonstrated that bond construction can be promoted at low temperature with a Ti-alkoxide reagent, providing 45 with similarly high levels of stereoselection (dr > 97:3).35
Figure 12.

Potential double asymmetric coupling reactions.
Figure 13.

Use of a phenylglycine auxiliary in azametallacyclopropane-based coupling reactions.
As depicted in Figure 14, the double asymmetric reductive cross-coupling between imine 44 and a range of chiral (E)-allylic alcohols proved to be quite effective in preparing (Z)-anti-homoallylic amine products (eqs 24–27). This reaction is also compatible with chiral imines derived from aliphatic aldehydes. As illustrated in eq 28, reductive cross-coupling imine 55 with allylic alcohol 53 proceeds in 60% yield, and delivers the stereodefined hoimoallylic amine 56 with very high levels of stereocontrol (Z:E ≥ 95:5; anti:syn ≥ 95:5).
Figure 14.

Double asymmetric reductive cross-coupling.
Interestingly, changing the absolute stereochemistry of the imine coupling partner results in a unique stereoselective bond construction. As depicted in eq 29 (Figure 14), reaction of ent-44 with 47 delivers the syn-homoallylic amine 57, albeit with only modest levels of stereoselection (57:58 = 2.3:1).
The sum of the experiments depicted in Figure 14 defines a substantial double asymmetric relationship in these Ti-mediated allylic alcohol–imine reductive cross-coupling reactions. The stereochemical pairing in eqs 24–27 clearly represent matched double asymmetric reactions, while that depicted in eq 28 represents a mismatched double asymmetric relationship.
An empirical model for these complex coupling reactions is depicted in Figure 15. As has been previously proposed, chirality transfer from the phenylglycine auxiliary to the azametallacyclopropane likely results from two factors: 1) coordination of the pendant methyl ether to Ti, and 2) minimization of non-bonded steric interactions inside the resulting cis-fused bicyclo[3.1.0] system (Figure 15A). As depicted in Figure 15B, assuming that ligand exchange (Ti–O bond formation) is required prior to carbometalation, the matched and mismatched transition state geometries may resemble A and B. If these models were correct, then they would seem to imply that the alkyl substituent (R) is best accommodated in a position oriented toward the developing C–C bond. While it is not yet possible to conclude that this is a preferred orientation of the allylic substituent, we searched for an alternative explanation, as we could not find a compelling argument to support this proposition.
Figure 15.

Empirical model for double asymmetric allylic alcohol–imine reductive cross-coupling.
If instead, the carbometalation could proceed prior to ligand exchange, with association occurring by a different Lewis acid–Lewis base interaction, then one could imagine reaction through a conformation consistent with the “inside alkoxy effect.”36 As depicted in Figure 15C, the matched double asymmetric pairing could then occur through a geometry similar to C, where the allylic substituent (R) is preferentially disposed away from the developing σTi–C bond. In a similar fashion, the mismatched pair could react through a related geometry (D). Here, while the allylic moiety (R) is positioned anti to the developing σTi–C bond, this relationship requires that the transition state suffer from the build up of significant non-bonded steric interactions about the developing C–C bond. We propose these models to provide a tool to aid in the selection of appropriate reaction partners for this complex convergent coupling process and recognize that future computational studies may aid in both refining their structure and assessing their validity.
Finally, in an effort to overcome the low levels of stereoinduction in the asymmetric coupling of chiral 1,1-disubstituted olefins with achiral imines, we pursued a modified version of this process to deliver (Z)-homoallylic amines with high levels of stereocontrol. As illustrated in Figure 16, eq 30, coupling of the chiral imine ent-44 with the racemic allylic alcohol (+/−)-28 proceeded in a highly stereoselective fashion and delivered 59 in 89% yield. With (+/−)-28 as the limiting reagent, success in this process indicates the ability of each enantiomer to react with the chiral complex of ent-44 in a stereoconvergent manner.
Figure 16.

Stereoselective coupling for the synthesis of chiral (Z)-trisubstituted homoallylic amines.
Conclusion
With our focus on the discovery and elaboration of unique metal-centered [2+2+1] chemistry for convergent C–C bond formation, we have discovered a series of stereoselective Ti-mediated reactions for the union of allylic alcohols with imines. During the course of our study, we have found that Ti-mediated coupling reactions of allylic alcohols with imines offers a family of bond constructions that are unrivaled by existing chemical methods. Moving beyond the study of simple allylation, our studies have realized: 1) a highly regioselective allylic alcohol-based coupling reaction, 2) a general pathway for imine prenylation, 3) a means for the establishment of anti-homoallylic amines in concert with the stereoselective generation of (E)- or (Z)-alkenes, and 4) enantioselective formation of complex homoallylic amines via single- or double-asymmetric reaction manifolds. Finally, a series of empirical models have been proposed for these complex bimolecular C–C bond forming processes that are consistent with the sense of regio- and stereoselection observed. Due to the low cost of the metal-containing reagents,37 the ready availability of the coupling partners, benign nature of the byproducts (TiO2 and Mg(II) salts), and substrate-controlled selectivity, the current fragment coupling process is anticipated to be of great utility in chemical synthesis. We look forward to advances in reaction development and chemical synthesis that follow from this report.
Experimental Section
General procedure for the reductive cross-coupling of aromatic imines with allylic alcohols
Synthesis of (±)-N-benzyl-1-phenylbut-3-en-1-amine (16)
To a solution of imine 14 (125 μL, 131 mg, 0.672 mmol) and Ti(Oi-Pr)4 (300 μL, 288 mg, 1.01 mmol) in Et2O (2.5 mL) at −78 °C was added dropwise c-C5H9MgCl (2.25 M in Et2O, 2.03 mmol) via a syringe. The mixture was warmed to −40 °C over 30 min and stirred for 1 h at this temperature. A solution of lithium alkoxide, prepared by the deprotonation of alcohol 15 (69 μL, 58.9 mg, 1.01 mmol) in THF (1.0 mL) at −78 °C with n-BuLi (2.56 M in hexanes, 1.08 mmol) followed by warming to 0 °C over 15 min, was added dropwise to the brown solution of imine–Ti complex at −40 °C via cannula. The mixture was warmed to 0 °C over 30 min then stirred at this temperature for 3 h. The reaction was quenched by addition of H2O (1.0 mL) followed by rapid stirring until the precipitate became white in color. The mixture was diluted with saturated aqueous NaHCO3 (20 mL) and extracted with EtOAc (3 × 20 mL). The organic extracts were combined, dried over MgSO4 then concentrated in vacuo. The crude product was purified by flash column chromatography on silica gel (10% EtOAc/hexanes) to afford homoallylic amine 16 as a colorless oil (112 mg, 70%). 1H NMR (500 MHz, CDCl3) δ 7.37-7.21 (m, 10H), 5.75-5.66 (m, 1H), 5.09-5.02 (m, 2H), 3.71-3.66 (m, 2H), 3.52 (d, J = 13.3 Hz, 1H), 2.46-2.36 (m, 2H), 1.75 (br, 1H); 13C NMR (125 MHz, CDCl3) δ 144.1, 140.9, 135.7, 128.6, 128.5, 128.3, 127.5, 127.3, 127.0, 117.7, 61.9, 51.7, 43.3; IR (thin film, NaCl) νmax 3063, 3026, 2835, 1639, 1603, 1585, 1493, 1453, 1356, 1326, 1307, 1198, 1117, 1070, 1028, 995, 916, 824, 759, 700 cm−1; HRMS (EI, H) m/z calc’d for C17H20N [M + H]+ 238.1590, found 238.1590.
General procedure for the reductive cross-coupling of aliphatic imines with allylic alcohols
Synthesis of (±)-N-benzyl-2-methylundec-2-en-5-amine (24)
To 6.0 mL of Et2O at −78 °C was added successively Ti(Oi-Pr)4 (300 μL, 288 mg, 1.0 mmol) and n-BuLi (2.4 M in hexanes, 2.0 mmol). The resulting orange solution was stirred for 10 min, and then imine 23 (203 mg, 1.0 mmol) in THF (1.0 mL) was added in one portion via a syringe. The bright orange solution was allowed to warm to room temperature over 1 h, during which it became deep red-brown. In a separate flask at −30 °C the lithium alkoxide of alcohol 17 (52 μL, 43.1 mg, 0.5 mmol) was generated in THF (0.5 mL) with an equimolar amount of n-BuLi, and allowed to warm slowly to 0 °C over 15 min. The orange solution of imine–Ti complex was recooled to −78 °C, and the alkoxide was added dropwise over 5 min. The resulting dark red solution was slowly warmed to room temperature over 12 h, and then saturated aqueous NH4Cl (0.8 mL) was added dropwise. The resulting blue suspension was stirred for 30 min, filtered, the flask rinsed with EtOAc (2 × 5 mL), and concentrated. The crude product was purified by flash column chromatography on silica gel (1/8 EtOAc/hexanes) to afford homoallylic amine 24 as a colorless oil (252 mg, 92%). 1H NMR (400 MHz, CDCl3) δ 7.32-7.16 (m, 5H), 5.11 (triplet of septets, J = 7.2, 1.2 Hz, 1H), 3.77 (s, 2H), 2.55 (m, 1H), 2.13 (t, J = 7.2 Hz, 2H), 1.72 (s, 3H), 1.63 (s, 3H), 1.45-1.26 (m, 15H), 0.89 (t, J = 7.2 Hz, 4H); 13C NMR (100 MHz, CDCl3) δ 141.1, 133.6, 128.3, 128.1, 126.7, 121.4, 57.3, 51.4, 34.2, 32.7, 31.9, 29.6, 25.9, 25.8, 22.7, 18.1, 14.1; IR (thin film, NaCl) νmax 3027, 2956, 2926, 2855, 1494, 1454, 1376, 1103, 1028, 729, 697 cm−1; HRMS (EI, H) m/z calc’d for C19H32N [M + H]+ 274.2529, found 274.2554.
General procedure for the one-pot synthesis of γ-lactam
Synthesis of (3S*,3aS*)-2-benyl-3-phenyl-2,3,3a,4,5,6-hexahydro-1H-isoindol-1-one (39)
To a solution of imine 14 (74 μL, 78 mg, 0.400 mmol) and ClTi(Oi-Pr)3 (1.0 M in Et2O, 0.500 mmol) in toluene (1.6 mL) at −70 °C was added c-C5H9MgCl (2.00 M in Et2O, 1.00 mmol) in a dropwise manner via a syringe. The orange-brown solution was slowly warmed to −40 °C over 30 min and stirred at this temperature for 1.5 h. A solution of sodium alkoxide, prepared by the deprotonation of alcohol 38 (106 mg, 0.600 mmol) in THF (1.6 mL) at 0 °C with NaH (60% dispersion in mineral oil, 30 mg, 0.750 mmol), was added dropwise via cannula to the brown solution of imine–Ti complex at −40 °C. The reaction was allowed to warm to room temperature and stirred overnight. To the reaction was added H2O (36 μL, 36 mg, 2.00 mmol), and the resultant mixture was rapidly stirred for 2 h.
To the yellow solution of the reaction mixture was added PdCl2 (1 mg, 0.008 mmol), t-Bu3P (1.0 M in toluene, 0.024 mmol), and Et3N (223 μL, 161 mg, 1.60 mmol). The reaction was placed under an atmosphere of carbon monoxide using a balloon, and heated at 70 °C for 12 h. After cooling down to room temperature, the mixture was diluted with EtOAc (10 mL) and filtered through a pad of Celite. The filtrate was further diluted with EtOAc (75 mL) and washed successively with 1.0 N HCl aqueous solution (75 mL), saturated aqueous NaHCO3 (75 mL), and brine (100 mL). The organic layer was dried over MgSO4 and concentrated in vacuo. The crude product was purified by flash column chromatography on silica gel (1/10 → 1/5 EtOAc/hexanes) to afford γ-lactam 39 as a white solid (89 mg, 73%, dr ≥ 20:1). 1H NMR (400 MHz, CDCl3) δ 7.34-7.22 (m, 3H), 7.18-7.10 (m, 3H), 7.10-7.05 (m, 2H), 6.94-6.86 (m, 2H), 6.54 (dt, J = 3.3, 3.3 Hz, 1H), 5.04 (d, J = 14.4 Hz, 1H), 3.78 (d, J = 7.6 Hz, 1H), 3.50 (d, J = 14.4 Hz, 1H), 2.54-2.40 (m, 1H), 2.28-2.14 (m, 1H), 1.85-1.66 (m, 2H), 1.43-1.26 (m, 1H), 1.16-0.99 (m, 1H); NMR (100 MHz, CDCl3) δ 169.2, 139.3, 136.5, 134.9, 129.0, 128.8, 128.5, 128.3, 127.6, 127.4, 66.7, 45.3, 44.4, 25.1, 24.8, 21.5; IR (thin film, NaCl) νmax 3026, 2929, 2862, 1688, 1494, 1455, 1393, 1272, 736, 701 cm−1; HRMS (EI, H) m/z calc’d for C21H22NO [M + H]+ 304.1696, found 304.1666.
General procedure for the synthesis of quinolizidines and indolizidines
Synthesis of (8S*,11S*,11aS*)-5,11-dimethyl-10-methylene-8-(thiophen-2-yl)-5,6,8,9,10,11,11a,12-octahydroindolo[3,2-b]quinolizine (43)
To a solution of imine 41 (295 mg, 0.86 mmol) in Et2O (10 mL) was added Ti(Oi-Pr)4 (390 μL, 366 mg, 1.29 mmol). After stirring for 10 min, the solution was cooled down to −78 °C and c-C5H9MgCl (2.0 M in Et2O, 2.58 mmol) was added via a syringe over 2 min. The solution turned from yellow to dark brown after stirring at −78 °C for 1.5 h. Next, a solution of lithium alkoxide, prepared by the deprotonation of alcohol 40 (201 mg, 1.29 mmol) in THF (2.0 mL) at −78 °C with n-BuLi (2.5 M in hexanes, 1.37 mmol) followed by stirring for 10 min, was added dropwise to the brown solution of imine–Ti complex via cannula. The mixture was slowly warmed to room temperature, and stirred for 16 h. The reaction was quenched by sequential addition of Et2O (10 mL) and saturated aqueous NaHCO3 (5 mL), followed by vigorous stirring for 1 h. The aqueous phase was extracted with Et2O (2 × 10 mL). the organic extracts were combined, dried over MgSO4 and concentrated in vacuo. The crude product was purified by flash column chromatography on silica gel (1/20 EtOAc/hexanes) to afford homoallylic amine 42 as a yellow oil (290 mg, 70%).
To a solution of homoallylic amine 42 (56 mg, 0.12 mmol) in THF (5 mL) was added 1.0 N HCl aqueous solution (0.5 mL, 0.5 mmol). After stirring for 16 h, the reaction was quenched by addition of pulverized K2CO3 (150 mg, 1.1 mmol). The neutralized mixture was extracted with Et2O (2 × 10 mL). The organic extracts were dried over MgSO4 and concentrated in vacuo. The crude product was purified by flash column chromatography on silica gel (1/40 EtOAc/hexanes) to afford quinolizidine 43 as a white solid (26 mg, 75%). 1H NMR (400 MHz, CDCl3) δ 7.38 (d, J = 7.2 Hz, 1H), 7.18 (m, 1H), 7.14 (m, 1H), 6.98 (m, 1H), 6.91 (m, 2H), 6.89 (m, 1H), 4.73 (s, 1H), 4.60 (s, 1H), 3.99 (ABq, J = 14.8 Hz, 1H), 3.61 (dd, J = 11.6, 3.6 Hz, 1H), 3.12 (ABq, J = 14.8 Hz, 1H), 3.04 (dd, J = 12.4, 10.4 Hz, 1H), 2.80 (dd, J = 13.6, 12.0 Hz, 1H), 2.75 (m, 1H), 2.70 (dd, J = 14.8, 3.6 Hz, 1H), 2.55 (qd, J = 6.4, 2.8 Hz, 1H), 2.29 (dd, J = 13.6, 2.8 Hz, 1H), 1.29 (d, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 150.7, 148.6, 137.2, 132.4, 126.4, 126.2, 124.7, 124.2, 120.7, 118.8, 117.8, 108.6, 106.2, 68.1, 62.5, 50.4, 42.2, 41.3, 29.1, 26.0, 14.8; IR (thin film, NaCl) νmax 3585, 2919, 1660, 1471 cm−1; LRMS (ESI, H) m/z calc’d for C22H25NOS [M + H]+ 412.2, found 412.5.
General procedure for the double asymmetric cross-coupling of phenylglycine-derived imines with chiral allylic alcohols
Synthesis of (1R,2R,Z)-N-((R-2-methoxy-1-phenylethyl)-2-methyl-1-phenylhex-3-en-1-amine (48)
To a solution of imine 44 (239 mg, 1.0 mmol) and Ti(Oi-Pr)4 (444 μL, 426 mg, 1.5 mmol) in Et2O (4 mL) at −78 °C was added dropwise c-C5H9MgCl (2.0 M in Et2O, 3.0 mmol) via a syringe. The mixture was warmed to −30 °C over 30 min and stirred at this temperature for another 3 h. A solution of lithium alkoxide 47 in Et2O (0.5 mL), generated in situ via deprotonation of the corresponding alcohol (50 mg, 0.5 mmol) with n-BuLi (2.54 M in hexanes, 0.55 mmol) at −78 °C followed by warming to 0 °C over 20 min, was added dropwise to the brown solution of imine–Ti complex at −30 °C via cannula. The resulting mixture was warmed to ambient temperature overnight. The reaction was quenched with saturated aqueous NaHCO3 (5 mL), and the resulting biphasic mixture was rapidly stirred until the precipitate became white in color. The mixture was further diluted with saturated aqueous NaHCO3 (10 mL) and extracted with EtOAc (3 × 20 mL). The combined organic extracts were washed with brine, dried over MgSO4, and concentrated in vacuo. The crude product was purified by flash column chromatography on silica gel (3% EtOAc/hexanes) to afford homoallylic amine 48 as a pale yellow oil (dr ≥ 20:1, Z:E ≥ 20:1, 126 mg, 78%). 1H NMR (400 MHz, CDCl3) δ 7.11-6.96 (m, 10H), 5.45 (app dt, J = 10.8, 7.3 Hz, 1H), 5.13 (dd, J = 10.8, 10.8 Hz, 1H), 3.67 (dd, J = 6.7, 4.7 Hz, 1H), 3.44-3.34 (m, 2H), 3.28 (d, J = 8.5 Hz, 1H), 3.26 (s, 3H), 2.72-2.60 (m, 1H), 2.35 (br, 1H), 2.08-1.97 (m, 2H), 0.90 (t, J = 7.5 Hz, 3H), 0.63 (d, J = 6.7 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 143.5, 142.2, 133.3, 133.1, 128.7, 128.0, 127.8, 127.7, 126.8, 126.7, 76.6, 67.5, 61.0, 59.1, 38.8, 21.2, 18.1, 14.6; IR (thin film, NaCl) νmax 3320, 3063, 2962, 2874, 2823, 1946, 1806, 1602, 1493, 1455, 1194, 1110, 757, 698 cm−1; HRMS (ESI, H) m/z calc’d for C22H30NO [M + H]+ 324.2327, found 324.2317; [α]D20 +19.8 (c 0.69, CHCl3).
Supplementary Material
Acknowledgments
We gratefully acknowledge financial support of this work by the National Institutes of Health (GM80266 and GM80266-S1).
Footnotes
Supporting Information Available: Experimental procedures, compound characterization, copies of spectral data is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For a discussion of strategy in complex molecule synthesis, along with detailed case studies, see: Corey EJ, Cheng X-M. The Logic of Chemical Synthesis. John Wiley & Sons, Inc; New York: 1989. Nicolaou KC, Sorensen EJ. Classics in Total Synthesis. Wiley VCH; New York: 1996.
- 2.For reviews, see: Otera J, editor. Modern Carbonyl Chemistry. Wiley-VCH; Weinheim: 2000. p. 613.Takeda T, editor. Modern Carbonyl Olefination: Methods and Application. Wiley-VCH; Weinheim: 2004. p. 365.
- 3.For a current review of allylation chemistry, see: Denmark SE, Almstead NG. In: Modern Carbonyl Chemistry. Otera J, editor. Wiley-VCH; New York: 2000. p. 299.For a current review on the application of the allylation reaction to the synthesis of natural products, see: Chemler SR, Roush WR. In: Modern Carbonyl Chemistry. Otera J, editor. Wiley-VCH; New York: 2000. p. 403.Zanoni G, Pontiroli A, Marchetti A, Vidari G. Eur J Org Chem. 2007:3599.
- 4.Notable exceptions include bifunctionalized allylmetal reagents and complex crotylsilanes. For recent discussions, see: Flamme EM, Roush WR. J Am Chem Soc. 2002;124:13644. doi: 10.1021/ja028055j.and references therein; and Masse CE, Panek JS. Chem Rev. 1995;95:1293.
- 5.For a review of catalytic enantioselective addition of allylic organometallic reagents to aldehydes and ketones, see: Denmark SE, Fu J. Chem Rev. 2003;103:2763. doi: 10.1021/cr020050h.For a review of catalytically generated allylic metal reagents that serve as electrophiles, see: Lu Z, Ma S. Angew Chem Int Ed. 2008;47:258. doi: 10.1002/anie.200605113.
- 6.Seebach D. Angew Chem Int Ed Engl. 1979;18:239. [Google Scholar]
- 7.For recent reviews that describe allylic substitution chemistry, see: Alexakis A, Bäckvall JE, Krause N, Pámies O, Diéguez M. Chem Rev. 2008;108:2796. doi: 10.1021/cr0683515.Trost BM, Crawley ML. Chem Rev. 2003;103:2921. doi: 10.1021/cr020027w.
- 8.For the coupling of alkynes with allylic ethers, see: Suzuki N, Kondakov DY, Takahashi T. J Am Chem Soc. 1993;115:8485.Suzuki N, Kondakov DY, Kageyama M, Kotora M, Hara R, Takahashi T. Tetrahedron. 1995;51:4519.Takai K, Yamada M, Odaka H, Utimoto K, Fujii T, Furukawa I. Chem Lett. 1995:315.Barluenga J, Rodríguez F, Álvarez-Rodrigo L, Zapico JM, Fañanás FJ. Chem Eur J. 2004;10:109. doi: 10.1002/chem.200305374.
- 9.For related chemistry of azametallacyclopropanes, see: Buchwald SL, Watson BT, Woods Wannamaker M, Dewan JC. J Am Chem Soc. 1989;111:4486.Coles N, Whitby RJ, Blagg J. Synlett. 1990:271.Grossman RB, Davis WM, Buchwald SL. J Am Chem Soc. 1991;113:2321.Gao Y, Yoshida Y, Sato F. Synlett. 1997:1353.
- 10.Takahashi M, McLaughlin M, Micalizio GC. Angew Chem Int Ed. 2009;48:3648. doi: 10.1002/anie.200900236.Concurrent with our studies, Professor Cha recently reported a related process: Lysenko IL, Lee HG, Cha JK. Org Lett. 2009;11:3132. doi: 10.1021/ol901006c.
- 11.Kulinkovich OG, Sviridov SV, Vasilevsky DA, Prityckaja TS. Zh Org Khim. 1989;25:2245.For chemistry related to Ti-aryloxides, see: Chamberlain LR, Durfee LD, Fanwick PE, Kobriger L, Latersky SL, McMullen AK, Rothwell IP, Folting K, Huffman JC, Streib WE, Wang R. J Am Chem Soc. 1987;109:390. doi: 10.1021/ja00236a017.Chamberlain LR, Durfee LD, Fanwick PE, Kobriger LM, Latesky SL, McMullen AK, Steffey BD, Rothwell IP, Folting K, Huffman JC. J Am Chem Soc. 1987;109:6068. doi: 10.1021/ja00236a017.Thorn MG, Hill JE, Waratuke SA, Johnson ES, Fanwick PE, Rothwell IP. J Am Chem Soc. 1997;119:8630.
- 12.For reviews, see: Marek I, editor. Titanium and Zirconium in Organic Synthesis. Wiley-VCH-Weinheim; 2002. p. 512.Buchwald SL, Nielsen RB. Chem Rev. 1988;88:1047.Negishi E. J Chem Soc, Dalton Trans. 2005:827. doi: 10.1039/b417134a.
- 13.For early examples, see: McDermott JX, Whitesides GM. J Am Chem Soc. 1974;96:947.McDermott JX, Wilson ME, Whitesides GM. J Am Chem Soc. 1976;98:6529.Grubbs RH, Miyashita A. J Chem Soc, Chem Commun. 1977:864.Erker G, Kropp K. J Am Chem Soc. 1979;101:3659.Cohen SA, Auburn PR, Bercaw JE. J Am Chem Soc. 1983;105:1136.Nugent WA, Calabrese JC. J Am Chem Soc. 1984;106:6422.Cohen SA, Bercaw JE. Organometallics. 1985;4:1006.Negishi E, Holmes SJ, Tour JM, Miller JA. J Am Chem Soc. 1985;107:2568.Negishi E, Cederbaum FE, Takahashi T. Tetrahedron Lett. 1986;27:2829.Buchwald SL, Lum RT, Dewan JC. J Am Chem Soc. 1986;108:7441.Buchwald SL, Watson BT, Huffman JC. J Am Chem Soc. 1986;108:7411.Buchwald SL, Watson BT, Huffman JC. J Am Chem Soc. 1987;109:2544.Buchwald SL, Watson BT, Lum RT, Nugent WA. J Am Chem Soc. 1987;109:7137.Fagan PJ, Nugent WA. J Am Chem Soc. 1988;110:2310.RajanBabu TV, Nugent WA, Taber DF, Fagan PJ. J Am Chem Soc. 1988;110:7128.Hoveyda AH, Xu Z. J Am Chem Soc. 1991;113:5079.Hoveyda AH, Morken JP, Houri AF, Xu Z. J Am Chem Soc. 1992;114:6692.
- 14.For a recent review, see: Kulinkovich O. Eur J Org Chem. 2004:4517.
- 15.Lee J, Kim H, Cha JK. J Am Chem Soc. 1996;118:4198. [Google Scholar]
- 16.For reviews, see: Olan A, Six Y. Tetrahedron. 2010;66:15.Wolan A, Six Y. Tetrahedron. 2010;66:3097.Sato F, Urabe H, Okamoto S. Chem Rev. 2000;100:2835. doi: 10.1021/cr990277l.Kulinkovich OG, de Meijere A. Chem Rev. 2000;100:2789. doi: 10.1021/cr980046z.
- 17.For a recent discussion on a subset of such limitations (with respect to alkynes), see: Reichard HA, McLaughlin M, Chen MZ, Micalizio GC. Eur J Org Chem. 2010:391. doi: 10.1002/ejoc.200901094.For a review of metallacycle-mediated cross-coupling with substituted and electronically unactivated alkenes, see: Reichard HA, Micalizio GC. Chem Sci. doi: 10.1039/C0SC00394H.
- 18.Woodard SS, Finn MG, Sharpless KB. J Am Chem Soc. 1991;113:106. [Google Scholar]
- 19.Related processes for the stereoselective coupling of internal alkynes or vinylsilanes with allylic alcohols, see: Kolundzic F, Micalizio GC. J Am Chem Soc. 2007;129:15112. doi: 10.1021/ja075678u.Belardi JK, Micalizio GC. J Am Chem Soc. 2008;130:16870. doi: 10.1021/ja8074242.
- 20.As a result of the low reactivity of substituted alkenes in intermolecular carbometalation chemistry, we reasoned that pre-association of the allylic alkoxide with the Ti–imine complex would be required for productive C–C bond formation. For a discussion of directed reactions in organic chemistry, see: Hoveyda AH, Evans DA, Fu GC. Chem Rev. 1993;93:1307.
- 21.For a recent study that explores the potential of reversibility in related Ti-promoted coupling reactions of homoallylic alcohols with imines, see: Takahashi M, Micalizio GC. Chem Commun. 2010;46:3336. doi: 10.1039/b927430h.
- 22.An empirical model based on related boat-like transition states is consistent with the stereochemical results of Ti-alkoxide-promoted vinylsilane–allylic alcohol (ref 19b), and alkyne–allylic alcohol (ref 19a) coupling reactions.
- 23.Masamune S, Choy W, Petersen JS, Sita LR. Angew Chem Int Ed Engl. 1985;24:1. [Google Scholar]
- 24.For recent examples of imine allylation using preformed allylic silanes, see: Huber JD, Perl NR, Leighton JL. Angew Chem Int Ed. 2008;47:3037. doi: 10.1002/anie.200705621.Berger R, Rabbat PMA, Leighton JL. J Am Chem Soc. 2003;125:9596. doi: 10.1021/ja035001g.Schaus JV, Jain N, Panek JS. Tetrahedron. 2000;56:10263.For recent examples using preformed allylic stannanes, see: Gastner T, Ishitani H, Akiyama R, Kobayashi S. Angew Chem Int Ed. 2001;40:1896.Hallett DJ, Thomas EJ. Tetrahedron: Asymmetry. 1995;6:2575.Keck GE, Enholm EJ. J Org Chem. 1985;50:146.For recent examples employing preformed allylic boron reagents, see: Wu TR, Chong JM. J Am Chem Soc. 2006;128:9646. doi: 10.1021/ja0636791.Li S, Batey RA. Chem Commun. 2004:1382. doi: 10.1039/b403759f.Lou S, Moquist PM, Schaus SE. J Am Chem Soc. 2007;129:15398. doi: 10.1021/ja075204v.Itsuno S, Watanabe K, Ito K, El-Shehawy AA, Sarhan AA. Angew Chem Int Ed Engl. 1997;36:109.For recent examples using in situ generated allylic boron reagents, see: Selander N, Kipke A, Sebelius S, Szabó KJ. J Am Chem Soc. 2007:13723. doi: 10.1021/ja074917a.Shimizu M, Kimura M, Watanabe T, Tamaru Y. Org Lett. 2005;7:637. doi: 10.1021/ol047609f.For examples using in situ generated allyl palladium reagents, see: Fernandes RA, Stimac A, Yamamoto Y. J Am Chem Soc. 2003;125:14133. doi: 10.1021/ja037272x.Hopkins C, Malinakova HC. Org Lett. 2006;8:5971–5974. doi: 10.1021/ol0624528.For recent examples employing allylic zinc reagents, see: Wipf P, Kendall C. Org Lett. 2001;3:2773. doi: 10.1021/ol0163880.Wipf P, Pierce JG. Org Lett. 2005;7:3537. doi: 10.1021/ol051266j.For a recent example using in situ generated allylic titanium reagents, see: Gao Y, Sato F. J Org Chem. 1995;60:8136.For reviews on the synthesis of homoallylic amines via allylic metal reagents, see: Ramachandran PV, Burghardt TE. Pure Appl Chem. 2006;78:1397.Kouznetsov VV, Méndez LYV. Synthesis. 2008:491.Yamamoto T, Asao N. Chem Rev. 1993;93:2207.
- 25.For a rare example of imine prenylation, see: Yanagisawa A, Ogasawara K, Yasue K, Yamamoto H. J Chem Soc, Chem Commun. 1996:367.
- 26.Tarselli MA, Micalizio GC. Org Lett. 2009;11:4596. doi: 10.1021/ol901870n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Few reactions are broadly useful for the formation of homoallylic amines that establish both an allylic stereocenter and a stereodefined alkene. Examples include: Kazmaier U, Zumpe FL. Eur J Org Chem. 2001:4067.Kojima K, Kimura M, Tamaru Y. Chem Commun. 2005:4717. doi: 10.1039/b507229h.Kojima K, Kimura M, Ueda S, Tamaru Y. Tetrahedron. 2006;62:7512.Garigipati RS, Cordova R, Parvez M, Weinreb SM. Tetrahedron. 1986;42:2979.Bartlett PA, Barstow JF. J Org Chem. 1982;47:3933.See also Ref 24b and 24e.
- 28.For a recent evaluation of the rate of interconversion of zirconaaziridine enantiomers, see: Cummings SA, Tunge JA, Norton JR. J Am Chem Soc. 2008;130:4669. doi: 10.1021/ja0757935.
- 29.Hoffmann RW. Chem Rev. 1989;89:1841. [Google Scholar]
- 30.In these experiments, 2 equivalents of the imine were employed. While it is likely that the chiral Ti–imine complex rapidly undergoes inversion, these experiments cannot provide support that the current process defines a dynamic kinetic resolution.
-
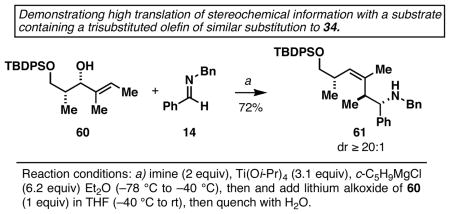
31.Note that the %ee of homoallylic amine products (eqs 12–14) reflect the %ee of the allylic alcohol starting materials. While we were also successful at accomplishing asymmetric coupling between a chiral allylic alcohol possessing a trisubstituted alkene [(R)- 34] and imine 14, we were unable to establish the ee of the homoallylic amine product by HPLC or derivatization (this coupling reaction proceeded in 77% yield). Given the likely mechanistic course of this coupling reaction, and our inability to identify a minor isomer, the transfer of stereochemical information in this process is reasoned to be as high as that depicted in eq 12. This expectation is in accord with a the highly diastereoselective coupling of imine 14 with the chiral allylic alcohol 96 which occurred with superb levels of diastereoselectivity:10a
- 32.Umemura S, McLaughlin M, Micalizio GC. Org Lett. 2009;11:5402. doi: 10.1021/ol9022134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang D, Micalizio GC. J Am Chem Soc. 2009;131:17548. doi: 10.1021/ja908504z. During the course of these studies with allylsilanes, we observed that a simple allylsilane related to 73, but bearing a primarly allylic alcohol, undergoes reductive cross-coupling with aromatic imines in a uniquely regioselective manner. Here, C–C bond formation occurs primarily at the internal carbon of the alkene, generating a 1,3-aminoalcohol product. Studies aimed at understanding this shift in site-selectivity are ongoing. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ito H, Taguchi T, Hanzawa Y. Tetrahedron Lett. 1992;33:4469. [Google Scholar]
- 35.Fukuhara K, Okamoto S, Sato F. Org Lett. 2003;5:2145. doi: 10.1021/ol034599u. [DOI] [PubMed] [Google Scholar]
- 36.Houk KN, Moses SR, Wu YD, Rondan NG, Jäger V, Schohe R, Fronczek FR. J Am Chem Soc. 1984;106:3880. [Google Scholar]
- 37.For a discussion in the context of regioselective reductive cross-coupling reactions of disubstituted alkynes, see ref 17.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.