

Abstract
The Id1 and Id3 genes play major roles during cardiac development, despite their expression being confined to non-myocardial layers (endocardium – endothelium - epicardium). We previously described that Id1Id3 double knockout (dKO) mouse embryos die at mid-gestation from multiple cardiac defects, but early lethality precluded the studies of the roles of Id in the postnatal heart. To elucidate postnatal roles of Id genes, we ablated the Id3 gene and conditionally ablated the Id1 gene in the endothelium to generate conditional KO (cKO) embryos. We observed cardiac phenotypes at birth and at 6 months of age. Half of the Id cKO mice died at birth. Postnatal demise was associated with cardiac enlargement and defects in the ventricular septum, trabeculation and vasculature. Surviving Id cKO mice exhibited fibrotic vasculature, cardiac enlargement and decreased cardiac function. An abnormal vascular response was also observed in the healing of excisional skin wounds of Id cKO mice. Expression patterns of vascular, fibrotic and hypertrophic markers were altered in the Id cKO hearts, but addition of Insulin-Like Growth Factor binding protein-3 (IGFbp3) reversed gene expression profiles of vascular and fibrotic, but not hypertrophic markers. Thus, ablation of Id genes in the vasculature leads to distinct postnatal cardiac phenotypes. These findings provide important insights into the role/s of the endocardial network of the endothelial lineage in the development of cardiac disease, and highlight IGFbp3 as a potential link between Id and its vascular effectors.
Keywords: Id1, Id3, IGFbp3, Thrombospondin, Angiopoietin, conditional knockout, angiogenesis, fibrosis, cardiomyopathy, ventricular septal defects, trabeculation
Introduction
The Id proteins are dominant negative antagonists of basic helix-loop-helix (bHLH) transcription factors and regulate differentiation in multiple lineages (Ruzinova and Benezra, 2003). The Id1-4 proteins are highly expressed during embryogenesis in partially overlapping patterns, and play major roles during development, for example cardiac and brain development (Fraidenraich and Benezra, 2006; Fraidenraich et al., 2004; Jen et al., 1996; Jen et al., 1997; Lyden et al., 1999). In the developing heart Id1 to Id3 are expressed exclusively in nonmyocardial layers, for example in the epicardium, endocardium and endothelium (Fraidenraich et al., 2004).
Although ablation of 3 copies of Id1 and Id3 (Id1−/−Id3+/− or Id1+/−Id3−/−) genes does not produce an embryonic phenotype, full elimination of the 4 copies of Id1 and Id3 (Id1−/−Id3−/− or dKO) genes results in cardiac defects and mid-gestation lethality (Fraidenraich et al., 2004; Lyden et al., 1999). The cardiac phenotype is associated with ventricular septal (VS), trabeculation and proliferation defects that result in a marked thinning of the myocardial wall (Chien et al., 2004; Fraidenraich et al., 2004). The “thin myocardial wall” phenotype is shared by a number of mouse models that lack genes implicated in non-cell autonomous pathway deficiencies (Chien and Olson, 2002; Kastner et al., 1995; Sucov et al., 1994; Yoshida et al., 1996). Injection of WT embryonic stem cells (ESCs) into Id dKO blastocysts corrects the cardiac defects and the embryonic lethality of the resultant chimeric mice with just 20–40% chimerism (Fraidenraich et al., 2004). Gene expression profiles in the Id dKO hearts are restored to WT levels in the chimeras. In the Id dKO embryos, expression of insulin-like growth factor 1 (IGF1) and IGF binding protein 4 (which binds and sequesters IGF1) is down- and up-regulated respectively (Fraidenraich et al., 2004) resulting in a compromise of myocardial proliferation. Maternal injection of IGF1 rescues myocardial proliferation defects and extends the viability of the dKO embryos until birth (Fraidenraich et al., 2004; Schneider et al., 2009). The hearts of the partially rescued pups are enlarged, and display endothelial, endocardial and ventricular septal defects (VSDs)(Fraidenraich et al., 2004).
The early lethality of the Id dKO embryos precluded the studies of the roles of Id1 and Id3 genes in the postnatal heart. We reasoned that if we specifically ablated the Id genes in just one nonmyocardial layer, but left intact the rest of the nonmyocardium, the embryos would survive past mid-gestation. Because the Id genes play an important role in angiogenesis (Lyden et al., 1999), and because the impact of loss of Id-vasculature on the postnatal heart is not known, we ablated Id genes in the endothelium. This paradigm allows us to: 1) study the requirement for Id genes in the endothelium and endocardium; 2) study the role of Id genes in the postnatal heart; and 3) unmask novel vascular-related roles for Id genes in the adult mice.
In this study, we demonstrate that ablation of Id genes in the endocardium and endothelium circumvents embryonic lethality, but neonates develop a late gestational cardiac phenotype associated with defects in trabeculation, VS and vasculature that leads to 50% postnatal lethality. We also show that the mice that survive develop an adult cardiac phenotype associated with defects in the vasculature. The study determines the postnatal requirement for Id genes in the endocardium/endothelium, allowing a phenotypic characterization of the cardiac defects caused by absence of Id genes in the endothelium.
Methods
Mouse Colonies and Genotyping
Mice with a Flox insertion in the Id1 allele (Nam and Benezra, 2009) (generously provided by Drs. Nam and Benezra) and mice with a null mutation in the Id1 and Id3 genes (Fraidenraich et al., 2004) (generously provided by Dr. Benezra) were crossed to generate Id1F/FId3−/− or Id1F/−Id3−/− compound mice (referred as Id control mice). Tie2cre transgenic mice (Kisanuki et al., 2001) (generously provided by Dr. Yanagisawa) were subsequently crossed with Id control mice to generate Tie2cre+Id1F/FId3−/− and Tie2cre+Id1F/−Id3+/− mice (referred as Id cKO or Id1/3 cKO mice). Both genotypes displayed the same phenotype. Mst1 transgenic mice (Yamamoto et al., 2003) were generously provided by Dr. Sadoshima. Mice were genotyped by PCR using puRe-Taq Ready-To-Go PCR beads (GE Healthcare) from DNA obtained from tail tips using primers for Id1 Flox, Id1 wild type, Id1 mutant, Id3 wild type, Id3 mutant and Tie2cre according to published protocols (Fraidenraich et al., 2004; Kisanuki et al., 2001; Nam and Benezra, 2009). R26LacZR26 mice, B6.129S4-Gt(ROSA)26Sortm1Sor/J, for verification of Cre/loxP-mediated recombination were purchased from The Jackson Laboratory. All animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Medicine and Dentistry of New Jersey.
Histology, immuno-, Xgal-staining and Western Blot (WB)
Whole embryos were collected at embryonic day 11.5 (E11.5), E13.5, E15.5, E17.5 and whole hearts at postnatal day 1 (P1), P30, P60, P90 and P180. Placentas were collected at E17.5 Tissues were paraffin- or cryo-embedded and sectioned. Immunofluorescence was performed on paraffin sections using primary antibodies for Id1 (Biocheck), PECAM-1 (Santa Cruz), IGFbp3 (R&D) and Ki67 (abcam). Myosin heavy Chain MF20 (Developmental Studies Hybridoma Bank) was used to detect myogenic areas at embryonic and P1 and wheat germ agglutinin (WGA) was used to delineate cell membranes in adult hearts. Sections were pretreated with pressure cooker boiling for 5 minutes in citrate buffer (0.01M, pH 6.0, Polyscientifc), quenched of endogenous peroxidase in 3% H2O2, and blocked prior to primary antibody incubation. A biotin-streptavidin amplification procedure (Vector Labs) and tyramide signal amplification system (TSA) (Perkin Elmer) were used for Id1 and PECAM-1 staining while only biotin-streptavidin amplification was necessary for Ki67. Nuclei were identified with DAPI (Vector Labs). Visualization of fluorescence was performed using a Nikon Eclipse 80i microscope with NIS Elements Imaging Software. Nuclei positive for Ki67 were counted at 20x magnification at E13.5, E17.5, and P1 and divided by total number of nuclei to determine proliferation percentages. Hemotoxylin and Eosin staining was performed for morphological analysis and visualization of fibrosis was performed using Masson Trichrome Stain Kit (Richard-Allan Scientific). Whole mount X-gal staining was performed at E11.5 with X-gal (1 mg/mL) in PBS buffer containing 5 mM potassium ferricyanide, 5 mM potassium ferrocyanide, and 2 mM MgCl2 overnight at 37°C. X-gal wash buffer contained 0.02% Nonidet P-40. After staining embryos were cryo-embedded, sectioned and eosin counterstained before mounting. Adult hearts (p180) were harvested, cryo-embedded and sectioned before X-gal staining overnight at 37°C and subsequent eosin counterstain.
Wound Healing
6 WT and cKO (P60) mice were anesthetized by isoflurane inhalation. At time=0 days, two circular 7mm-diameter wounds were inflicted using an Acupunch disposable sterile 7 mm skin biopsy punch (Accuderm Inc) and images were recorded. Wounds were dressed with a transparent dressing (Tegaderm; 3M) following initial wounding and again redressed at day 3. WT and cKO mice were sacrificed at day 6 and the remaining mice were sacrificed at day 8. Wounds with ~2mm surrounding skin were excised at time of sacrifice and bisected to provide cross-sections through the full width of the wounds. These samples were fixed and processed for paraffin embedding and sectioned from both faces of the bisected tissue for histological analysis. H&E and immunofluorescence for Id1 and PECAM-1 were performed as described above.
Semi qRT-PCR
RNA was extracted (RNeasy, QIAGEN) and 1ug total RNA was reverse transcribed using Superscript III reverse transcriptase (Invitrogen). PCR was performed with the following primers and expected product size: Tbp1, GTCACATCGCCAACACAATC and AGACCAAAGCCTGCAAGAAA, 169bp; Tbp4, GCAGGGATGGTATTTGCACT and TTCAGTCCCCAACTCCAAAC, 209bp; Ang-1, TCTGCACAGTCTCGAAATGG and AGGCTTGGTTTCTCGTCAGA, 204bp; IGFBP3, AATGCTGGGAGTGTGGAAAG and CTGTCTCCCGCTTAGACTCG, 193bp; G3PDH primers (Clontech) for internal control, 983bp. To produce bands in the log phase, PCR parameters were adjusted individually. Annealing temperatures: 55 °C 30sec for Tbp1, Tbp4, Ang1 and 60 °C 30sec for G3PDH; reaction cycle numbers: 24 for Tbp1; 29 for Tbp4 and Ang-1 and 22 for G3PDH.
IGFbp3 incubation
Hearts were harvested from Id cKO mice at P180. Adjacent, 1mm-thick transverse sections at mid-level were used for the experiment. One section was used for semi qRT-PCR without incubation. Two sections were incubated at 37 °C for 4 hours under rocking conditions in DMEM + 1mg/mL fatty acid-free BSA. Recombinant mouse IGFbp3 (1μg/mL) (R&D Systems) was added to one section. No recombinant protein was added to another section (untreated control). Id cKO mice with EF<50% were used. Experiments were performed in triplicate. For IGFbp3 antibody incubation, hearts were harvested from P180 WT mice. Adjacent, 1mm-thick transverse sections at mid-level were used for the experiment. One section was used for semi qRT-PCR without incubation. Three sections were incubated at 37 °C for 5 hours under rocking conditions with DMEM and fatty acid-free BSA. An antibody reactive with IGFbp3 (80 μg/ml) (AF775, R&D Systems) was added to one section. No antibody was added to another section (untreated control). An unrelated antibody reactive with Fst1 (80 μg/ml)(K19, Santa Cruz) was added to another section (negative control). Both negative controls provided identical results. Experiments were performed in triplicate.
Myocyte cross-sectional area
Myocyte cross-sectional area was determined on digitized images of rhodamine-labeled wheat germ agglutinin-stained sections of paraffin-embedded samples (Peter et al., 2007). Hearts from 6 WT (EF>60%) and 6 Id cKO (EF<50%) were used in this study. The myocyte outlines were traced and the cell areas measured with Image-Pro Plus (Media Cybernetics, Inc, Silver Spring, MD). In cross sections of LV, the measurable cross sections of the myocytes are found in the endocardial one third and in epicardial one third of myocardium. At least 150 myocytes were routinely measured in each region.
Microarray analysis
1 mm thick transverse sections at mid-level from 2 hearts per group (WT, Id1 KO, Id3 KO, Id cKO, IGFbp3-treated Id cKO and IGFbp3-untreated Id cKO) were used for microarray analysis. In the case of Id cKO groups, enlarged heart tissue from mice with EF<50% were collected. Sections used for microarrays were also used for semi qRT-PCR validation and were adjacent to those used for histology. Total RNA from heart tissue was isolated (RNeasy, QIAGEN). Gene expression was analyzed by the GeneChip Mouse Gene 1.0 ST Array (Affymetrix). Microarray data were processed by the RMA (robust multi-chip analysis) method, and expressed genes were selected by the detection above background (DABG) method. Hierarchical clustering with Pearson correlation was employed to cluster genes and samples using Cluster 3.0 and Tree View. Gene Ontology (GO) analysis was carried out by hypergeometric test. Significantly regulated genes used in GO analysis were those with expression change greater than 1x standard deviation of all genes. For each GO term, its associations with up- and down-regulated genes were examined, resulting in two p-values. The more significant one was used to indicate the significance of the GO term. To report GO terms, we removed redundant ones, which contained more than 75% of genes associated with a GO term with higher significance.
Data sets have been deposited in Gene Expression Omnibus (GEO), with accession number GSE21924. Temporary link (access link to reviewers): http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=zfgvtusgmowiwdu&acc=GSE219 24.
Echocardiography
Echocardiographs were performed on WT, Id control and Id cKO mice at stages ranging from P30–P180 to determine left ventricular (LV) systolic fraction. Mice were anesthetized by intraperitoneal injection 2.5% Avertin 290mg/kg. Transthoracic echocardiography (Sequoia C256; Acuson, Mountain View, CA) was performed using a 13-MHz linear ultrasound transducer. The chest was shaved. Mice were placed on a warm saline bag in a shallow left lateral position and warm coupling gel applied to the chest. Small-needle electrocardiographic leads were attached to each limb. Two-dimensional images and LV M-mode tracing (sweep speed = 100–200 mm/s) were recorded from the parasternal short-axis view at the mid papillary muscle level. M-mode measurements of LV internal diameter (LVID) and wall thicknesses were made from 3 consecutive beats and averaged using the leading edge-to-leading edge convention adopted by the American Society of Echocardiography. End-diastolic measurements were taken at the peak of R wave of EKG. End-systolic measurements were made at the time of the most anterior systolic excursion of the posterior wall. LV ejection fraction (EF) was calculated by the cubed methods as follows: LVEF (%)=100*[(LVIDd)3− LVIDs)3]/(LVIDd)3, LV fractional shortening: (LVFS %)=100*(LVIDd-LVIDs)/LVIDd, where d indicates diastolic and s indicates systolic. Heart rate was determined from at least three consecutive RR intervals on the LV M-mode tracing. Following echocardiography at P180, mice were sacrificed and hearts harvested for histological analyses as described above.
Data analysis
Results are presented as mean ± s.e.m. or as a range. Statistical comparison was performed with nonparametric two-tailed unpaired analysis of variance. A probability value of <0.05 was considered to be statistically significant
Results
Id1 is compromised in the endothelial lineage of Id cKO hearts
To study a requirement for Id1 and Id3 genes in the adult heart, we analyzed Id compound mice with a Flox insertion in the Id1 allele (Nam and Benezra, 2009) and with a null mutation in the Id3 gene (Id1F/FId3−/− or Id1F/−Id3−/−, referred to as Id control) mice. We incorporated a Tie2Cre transgene into Id control mice to generate Tie2Cre+Id1F/FId3−/− or Tie2Cre+Id1F/−Id3−/− (referred to as Id cKO) mice. Tie2 is expressed in endothelial cells (Kisanuki et al., 2001), which highly express Id genes (Fraidenraich et al., 2004; Ruzinova and Benezra, 2003). The Tie2 promoter, therefore, drives expression of the Cre recombinase to the endothelial and endothelial-derived structures.
To verify that the recombination was layer-specific, Tie2Cre+Id1F/+ mice were crossed to R26LacZR26 mice, which reveal tissues (via X-gal staining) that undergo Cre/loxP-mediated recombination (Soriano, 1999). The reporter LacZ showed that the recombination occurred in Tie2Cre+Id1F/+R26LacZR26 compound mice in most of the endocardial/endothelial cells and their derivatives like the atrio-ventricular endocardial cushion (AV EC) (Fig. 1A). The epicardium and myocardium from atria and ventricles did not undergo recombination, and therefore did not stain blue (Fig. 1A). Accordingly, the epicardium was Id1 positive (Fig 1E, compare with 1D, see 1B, C for landmarks). Id1 was detected in the epicardium of both atria and ventricles, and in a few endothelial cells of the outer layer of the epi-myocardium (Fig 1E). In contrast, negligible levels of Id1 were observed in endothelial cells of the inner most portion of the endomyocardium and of the VS, as well as of the endocardium proper (Fig 1E). These observations suggest that Tie2Cre/LoxP-mediated recombination is more efficient in the inner myocardium, which is influenced by the endocardium, and may express higher levels of Tie2 (Cre). Alternatively, a portion of the epi-endothelium may originate from endothelial-independent structures (Tie2 negative = no Cre expression), such as from the epicardium. Thus, Tie2Cre/loxP-mediated recombination resulted in reduced Id1 production, especially in the most internal portions of the heart.
Figure 1. Id1 expression is compromised in the endothelial lineage of Id cKO mice.

Specific recombination is visualized in endocardial/endothelial layers by Xgal staining (A). As a result, Id1 is reduced in the endocardium and endothelium of Id cKO developing hearts (D–E, see B–C for landmarks). Id1 expression normally persists in the endothelium, but not in the endocardium or epicardium of WT adult (P180) mice (F–K), but the adult (P180) heart of Id cKO mice is devoid of Id1 (L–N). Accordingly, the heart of Id cKO LacZ reporter compound mice displays endothelial Xgal staining (N, inset). Abbreviations: Vt: ventricle; At: atrium; AV EC: atrio-ventricular endocardial cushion, Tb: trabeculae; VS: ventricular septum; epic: epicardium; end: endocardium; Xgal: X-galactosidase staining; MF20, CD31, SMA and WGA demarcate myocardium, endocardium/endocardium, muscle vasculature and cell periphery respectively; blue, black, yellow and red arrowheads (A): Xgal-positive endocardial, Xgal-positive endocardial-derived (AV EC), Xgal-positive endothelial and Xgal-negative epicardial cells respectively. Yellow and orange arrows (D and E): endocardial/endothelial- and epicardial-Id1 positive cells respectively. Yellow arrowhead (F, F inset, G and J): Id1 positive cells. Green arrows and red arrowhead (H, H inset and I): CD31 (endothelial) and SMA (vascular smooth muscle) positive staining respectively. Note that I and J are adjacent areas displaying expression of Id1 in a subset of the endothelial lining. Red and green arrowheads (K): co-cellular localization of Id1 (nuclear) and CD31 (cytoplasmic) staining. Light blue arrowhead (K): a cell nucleus showing no Id1/CD31 staining. Yellow arrowheads (L–N): Id1-positive cells in small and large caliber vessels. Note that Id staining is not apparent in N (Cre/lox recombination in the Tie2+ compartment). Magnification: A–E: 40X; F–H: 100X; I–J: 200X, K: 400X; L–N: 100X; N, inset: 200X.
Id1 persisted in a fraction of the endothelium of small capillaries and large vessels in the heart of WT mice at postnatal day 180 (P180) (Fig 1F, F inset, G, J, K, L), but did not persist in the epicardium or endocardium at P180 (Fig 1F for epicardium, G for endocardium). In Id cKO; R26LacZR26 compound mice, Tie2Cre/loxP-mediated recombination resulted in Xgal staining of the endothelial compartment (Fig. 1N inset), coincident with absence of Id1 staining there (Fig. 1N, compare with 1L, M). Therefore, the only Id1-expressing cells in the WT heart are Tie2Cre+ cells, which are endothelial and endothelial-derived cells. Absence of Id1 in the epicardium and endocardium at P180, combined with Tie2Cre-ablation of Id1 in the endothelium leaves the heart of an adult Id cKO with no Id1 production
Id cKO neonates develop a cardiac phenotype
We examined the phenotypic consequences of Id loss in Id cKO embryos. In most of the experiments (except when noted), we generated embryos from crosses of Id cKO males and Id control females. The two genotypes that form the Id cKO group produced identical phenotypes. Despite the fact that Tie2Cre/loxP-mediated recombination takes place as early as E11.5, and that Id1-endothelium expression is compromised in Id cKO embryos at E13.5 and E15.5, their hearts did not display a phenotype (n = 25)(Fig. 2A–D for E13.5 and data not shown for E15.5). At E13.5 (n = 14) and E15.5 (n = 11), the thickness of the myocardial wall of the Id cKO hearts did not differ from that of WT hearts, the trabeculation network was not compromised, the endothelial lining was continuous and the VS complete, with no VSDs (Fig. 2A, B). Concordant with normal size and thickness, no myocardial proliferation defects were apparent (n=3)(% Ki67 positive cells: 46 ± 5 and 44 ± 5 for WT and cKO respectively)(Fig. 2C, D). These experiments suggest that partial (Tie2Cre+Id1F/−Id3−/− or Tie2Cre+Id1F/FId3−/−) but not complete (Id1−/−Id3−/−) absence (Fraidenraich et al., 2004) of Id does not lead to mid-gestation demise. These experiments also suggest that Id1 signals from the non-endocardium/endothelium (i.e. epicardium and epicardial-derived structures) suffice for maintaining normal development of the embryonic heart. This is particularly interesting in areas of the heart that are internal and that do not appose the epicardium. These areas, like the VS, are devoid of short-range Id-dependent signals, and yet, display no apparent phenotype at mid-gestation.
Figure 2. Newborn Id cKO pups develop multiple cardiac defects.

E13.5 Id cKO embryos do not develop a cardiac phenotype; accordingly, cell proliferation is not compromised (A–D, see also Fig 1, B and C). However, P1 Id cKO pups display enlarged hearts, with VSDs (E, F and K–N), dilated VS vasculature (K, L insets), trabeculation defects (G, H) and distended epicardial vasculature (I, J). Black dotted line (A, B, E and F): VS thickness; white line (A and B): myocardial wall thickness; green, black and orange arrows (F): VS hollow cavity, myocardial wall interruption and red blood cells in the VS cavity respectively; yellow arrowheads (H): interrupted endocardial lining; white arrowheads (J) endothelialized epicardial vasculature; light blue arrowheads (L inset): dilated VS vasculature. Magnification: A–F and M, N: 40X; E and F, insets: 10X; G and H: 200X, I and J: 400X; K and L: 100X.
At E17.5, Id cKO hearts exhibited VSDs (n=15) (SFig 1). The endocardial lining remained unaffected however a compromise in myocardial proliferation was observed (% Ki67 positive cells: 4.9 ± 1.0 and 0.9 ± 0.3 for WT and cKO respectively) (n = 15 P<0.05) (SFig 1A–D). E17.5 embryos (n=33) were collected with no resorption sites. No change in apoptosis was observed (n=5)(data not shown). All the embryos showed beating hearts, including the Id cKO embryos (n=15, expected n=17).
Because most of the Id cKO embryos survived past late gestation (88%, 15 out of 17 expected), we studied the postnatal requirement of Id1-endothelium in the Id cKO pups. Notably, only 46% of the P1 Id cKO mice (56 out of 120 expected) escaped postnatal lethality. The phenotype of P1 Id cKO pups (n = 11) was more pronounced to those of E17.5 embryos (Fig. 2E–J). The heart of the P1 Id cKO pups was enlarged (n=11)(Fig. 2F inset, compare with 2E inset) and displayed VSDs (n=11)(Fig. 2F, L, compare with 2E, K) associated with impaired ventricular trabeculation (n=11) (Fig. 2H, compare with 2G). Trabeculae were surrounded by discontinuous endocardial cell lining (Fig. 2H). The VS contained one main hollow cavity, surrounded by a thin wall of VS myocytes. Red blood cells were observed within the VS cavity, suggesting a connection to the ventricular chambers. Accordingly, interruption of the precarious VS wall was apparent (Fig. 2F). The intramyocardial vasculature of the VS was dilated (Fig. 2L and 2L inset). In the VS, no difference in apoptosis was apparent (n=5)(data not shown), and negligible proliferation was observed (n=5)(% Ki67 positive cells: 2.3 ± 1.0 and 0.5 ± 0.3 for WT and cKO respectively, P<0.05). The epicardial vasculature displayed endothelial lining but was dilated (Fig. 2J, compare with 2I). As expected, negligible levels of Id1 were observed in the heart of the Id cKO neonates (Fig. 2N, compare with 2M).
Id cKO mice develop an adult cardiac phenotype
We determined the requirement for Id genes in the adult heart of the surviving Id cKO mice at the functional and histological levels (n = 56) (Fig. 3 and Table I).
Figure 3. P180 Id cKO mice develop cardiac defects.

Hearts from adult Tie2Cre+Id1F/FId3−/− or Tie2Cre+Id1F/−Id3−/− but not from Tie2Cre+Id1F/−Id3+/− or WT mice are dilated (A–D). The endo-myocardium of Id cKO mice displays disorganized areas of low or no cellularity filled with fibrotic tissue (E–H). The Id cKO mice have decreased cardiac function (I, J). The endocardial lining of Id cKO mice is interrupted (K, L and K, L insets). Fibrosis is apparent in the large vessels of the heart of Id cKO but not of WT of Mst1 Tg mice (M–O). N inset, shows enlargement in Id cKO hearts (relative to M inset). Abbreviations: end (E–H): endocardium; LV (E–H): left ventricle; sep and post (I): interventricular septal wall and posterior wall of the left ventricle respectively. Red arrows (F, H): disorganized areas of low cellularity in the endomyocardium; yellow arrows (L): regions of endocardial interruption, white arrows (K inset): typical elongated shape of endocardial nuclei (note CD31 staining), absent in L inset; light blue arrowheads (M–O): large vessels; yellow arrowheads (O): fibrotic myocardial interstitium. Magnification: A–D: 10X; E–H: 100X; K and L: 100X; K and L, insets: 400X; M–O: 200X; M and N, insets: 10X.
Table I.
Cardiac dilation, fibrosis and dysfunction in P180 Id cKO mice
| WT | Id cKO | |
|---|---|---|
| Body weight (g)* | ||
| Females | 26.2 ± 2.6 (n=6) | 22.3 ± 2.6 (n=30) |
| Males | 29.3 ± 2.2 (n=9) | 24.3 ± 2.4 (n=24) |
| Heart weight (mg)†# | ||
| Females | 105 ± 16 (n=6) | 118 ± 26 (n=9) |
| Males | 118 ± 23 (n=5) | 140 ± 38 (n=11) |
| Heart/Body weight (mg/g)* | 3.87 ± 0.62 (n=11) | 5.75 ± 1.10 (n=20) |
| Fibrosis (Masson Trichrome staining) | ||
| Epimyocardium | 0 (n=5) | 0 (n=14) |
| Endomyocardium | 0 (n=5) | 12 (n=14) |
| Left Ventricular End Diastolic Dimension (LVEDD) * | ||
| mm | 3.8 ± 0.3 (n=14) | 4.3 ± 0.7 (n=56) |
| Left Ventricular End Systolic Dimension (LVESD) * | ||
| mm | 2.7 ± 0.3 (n=14) | 3.3 ± 0.7 (n=56) |
| Fractional Shortening (FS) * | ||
| % | 29.4 ± 4.6 (n=14) | 23.3 ± 7.0 (n=56) |
| Left Ventricular Ejection Fraction (LVEF) * | ||
| % | 64.3 ± 6.8 (n=14) | 56.2 ± 13.2 (n=56) |
| Number of mice with LVEF < 50 % | 0 (n=14) | 11 (n=56) |
| Heart rate (HR)† | ||
| Beats/min (bpm) | 450 ± 55 (n=14) | 440 ± 62 (n=56) |
P<0.05
P>0.05
In parenthesis: number of mice
dry weight
At P30 (n=6), P60 (n=5), P90 (n=8) and P180 (n=28) the Id cKO mice did not display VSDs or trabeculation defects (Fig. 3 and data not shown). The hearts of P1, P30, P60, P90 and P180 Id cKO mice were dilated (Fig 3, Table I and data not shown). The heart/body (dry) weight ratio of the adult P180 cKO mice was 48% higher than that of WT mice (n=20)(Fig. 3C, D, compare with 3A, and Table I, P<0.05). The myocyte cross-sectional area of P180 Id cKO ([552 ± 85] μm2, n=6) was increased relative to WT ([314 ± 17]μm2, n=6)(P<0.01), The endo-myocardium of the left ventricle (apposing the Id-negative endocardium) was disorganized, displayed areas with low or no cellularity and signs of fibrosis (n=14)(Fig 3F, H, compare with 3E, G). The endocardium of the adult cKO mice was partially interrupted (n=8)(absence of CD31 positivity)(Fig. 3L, compare with 5K), with no visible endocardial nuclei (n=8)(Fig. 3L inset, compare with 3K inset). Because the Id genes are not produced in the P180 endocardium (Fig. 1G), developmental ablation of Id genes may have had a long-term impact on the phenotype of the adult endocardium.
Figure 5. Vascular markers are dysregulated.
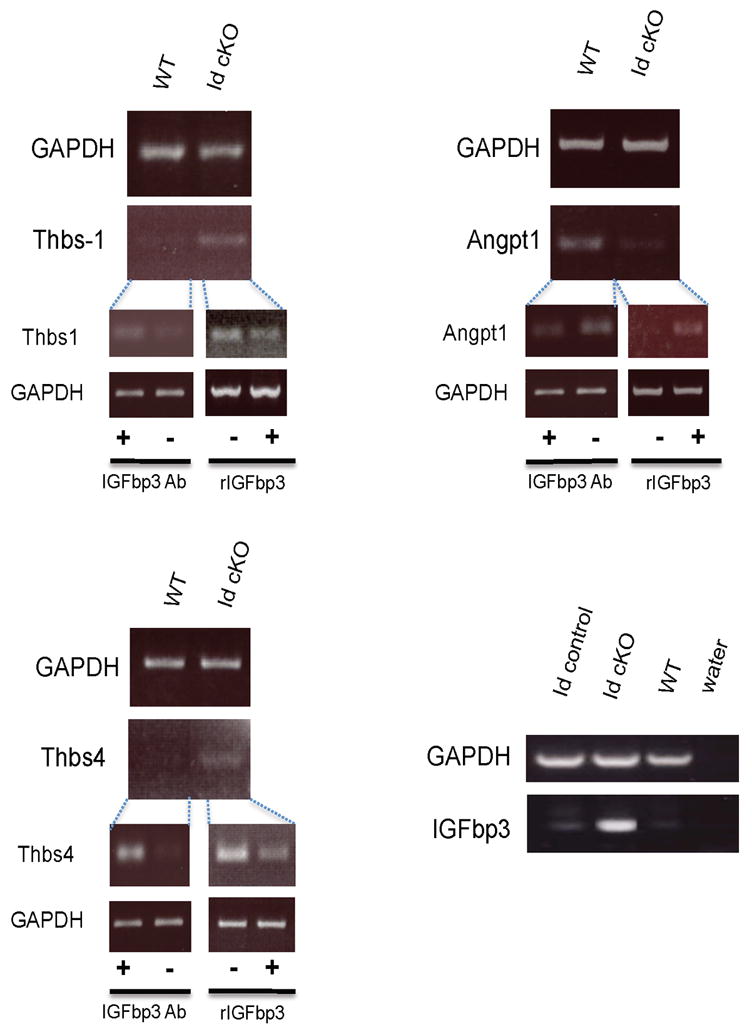
Dysregulation of vascular, fibrotic and hypertrophic markers in Id cKO hearts is validated by semi qRT-PCR analysis. Abbreviations: Thbs: thrombospondin; Angpt: angiopoietin; rIGFbp3: recombinant mouse IGFbp3, Ab: antibody.
Fibrotic tissue was apparent in areas surrounding large vessels (n=14)(Fig 3N, compare with 3M), but their endothelial lining appeared normal (n=14)(data not shown). Fibrosis was not observed in the epimyocardium (n=14)(data not shown). This vascular-initiated pattern of fibrosis differed from that of another model in which the defect is triggered by myocardial apoptosis (n=5)(Fig 3O)(Yamamoto et al., 2003). The latter is exemplified by overexpression of the mammalian sterile 20 like-kinase 1 (Mst1) gene in the cardiac myocytes (Yamamoto et al., 2003). As opposed to the Id cKO hearts (Fig. 3N), fibrosis in Mst1 Tg hearts was evenly distributed throughout the intercellular (interstitial) space of cardiac myocytes (n=5)(Fig 3O). The phenotype observed (a disorganized myocardium next to the endocardium) resembles that of the partially rescued Id dKO neonates with maternal injection of IGF1 (Fraidenraich et al., 2004).
Monthly echocardiographic studies revealed that the left ventricular ejection fraction (LVEF) or fractional shortening (FS) of Id cKO mice was reduced (n=56)(Table I). The onset of heart dysfunction was at P90 (n=8)(data not shown). In the most severely affected Id cKO mice, LVEF was below 50% (Table I) (Id cKO in Fig 3J, EF: 21% and FS: 7.9%, compare with WT in Fig 3I, EF: 64.7% and FS: 29.4%). Young pups (n=11)(P30 and P60) did not show low EF or FS readings (data not shown). This suggests that the cardiac enlargement observed in young pups is compensatory for an intrinsic myocardial dysfunction. No significant difference in heart rate was observed at any stage (Table I and data not shown). Control hearts from Tie2Cre+Id1F/−Id3+/− mice or other mice with higher Id copy number than Id cKO mice did not display enlargement, fibrosis, endocardial compromise or LVEF reduction (Fig 3B and data not shown). Thus, P180 Id cKO mice develop a vascular-initiated cardiac phenotype.
Analysis of gene expression profiles
We studied the molecular consequences of conditional ablation of Id genes in the P180 heart (Fig. 4). Gene expression profiles were compared in clustering analysis. The effect of genetic modification in Id gene (Id1 KO, Id3 KO and Id1/3 cKO) was compared with the WT control heart. The sample clustering with relative gene expression levels revealed that the Id cKO heart has distinct pattern from the heart lacking either Id1 or Id3 (Fig 4A). We next evaluated the association of gene sets over the regulated genes using Gene Ontology (GO) database. The significant GO terms (biological processes and cellular component) were summarized in heatmap with statistical scores (Fig 4B). Id cKO showed strong regulation in cell adhesion (GO:0007155), actin filament-based process (GO:0030029), vascular development (GO:0001944) and oxidation-reduction (GO:0055114). Those pathways are implicated in pathological cardiac remodeling(Lehman and Kelly, 2002; Ruwhof and van der Laarse, 2000). Id cKO also showed strong regulation in extracellular matrix (GO:0031012) and extracellular region part (GO:0044421). The significant GO terms are listed in STable I.
Figure 4. Microarray analysis of Id cKO hearts.

(A) Comparison of gene expression profiles in Id KO mice and mice with IGFbp3 treatment. Columns are comparisons and rows are genes. Comparisons are Id1 KO vs. WT, Id3 KO vs. WT, Id cKO vs. WT, and IGFbp3 treated Id cKO vs. Id cKO. The log2(Ratio) values were clustered and presented in a heatmap, according to the color scale shown at the bottom. The heatmap contains 4,378 transcripts with fold change >=1.5 in at least one comparison. Clustering was carried out by hierarchical clustering using Pearson correlation. (B) Significant GO terms associated with regulated genes shown in (A). Top, Biological Process (BP); Bottom, Cellular Component (CC). GO terms were analyzed by hypergeometric test. P-values were converted to −log10(P-value) for terms more associated with upregulated genes or log10(P-value) for terms more associated with downregulated ones, and are shown in a heatmap according to the color scale shown at the bottom. The top 20 and 10 GO terms are shown for BP and CC, respectively. Id1 KO: full absence of Id1 in Id1−/−Id3+/−Tie2Cre+; Id3 KO: full absence of Id3 in Id1F/−Id3−/−. See Supplemental Table I for the complete list of GO terms.
Vascular, fibrotic and hypertrophic markers are dysregulated in the adult Id cKO hearts
Microarray analysis of the extracellular matrix revealed that over 10 collagen genes were upregulated relative to WT, including types I (ID:12842) and III (ID:12825) (STable II). Thrombospondin1 (Thbs1, ID:21825), a potent anti-angiogenic secreted factor (Folkman, 1995; Good et al., 1990), which is repressed by Id1 (Volpert et al., 2002), was up-regulated in the Id cKO hearts relative to WT hearts (STable II and Fig 5). Thbs4 (ID: 21828) was also upregulated (STable II and Fig 5). Several Thbs-carrying domain proteins, like a disintegrin-like and metallopeptidase with thrombospondin (Adamts4, 9 and 1, IDs: 240913, 101401 and 11504 respectively) proteins (Brocker et al., 2009; Guo et al.), were also up-regulated (STable II). Adamts proteins play key roles in extracellular matrix remodeling, including degradation of collagen fibers (Guo et al.). Angiopoietin-1 (Angpt1, ID: 11600), a secreted factor produced by myocytes that signals to the endocardium through the Tie2 receptor and whose absence during development leads to trabeculation defects (Suri et al., 1996), was down-regulated (STable II, and Fig 5) in the Id cKO hearts. As in the phenotype of Id dKO embryos, lack of Angpt1 causes embryonic lethality at midgestation and a thin myocardium (Suri et al., 1996). Thus, important paracrine signals of the cardiac vasculature are dysregulated in the Id cKO adult hearts. In the Id dKO embryos, IGF1 (ID: 16000) and IGFbp4 (ID: 16010) were down- and up-regulated respectively (Fraidenraich et al., 2004). However, IGF1 and IGFbp4 were unchanged in the Id cKO adult hearts. Instead, IGFbp-3, 5 and 7 (IDs: 16009, 16011 and 29817 respectively) were upregulated (STable II, Fig 5 and data not shown). In P180 WT hearts, IGFbp3 was detected in a few small-caliber (nonmyocardial) cells that appose larger cardiac myocytes (Fig 6A and C). IGFbp3 was also detected in the endothelial lining of larger vessels (Fig 6A inset). Concordant with upregulation of IGFbp3 (STable II and Fig 5), IGFbp3 was enhanced in the P180 Id cKO hearts (Fig 6B, B inset and D), to become detectable throughout the extracellular interstitium (Fig 6B and D). Therefore, IGFbp3 is expressed primarily in the nonmyocardial compartment, including endothelial cells, and its expression is enhanced in the P180 Id cKO heart.
Figure 6. IGFbp3 is enhanced in Id cKO hearts.
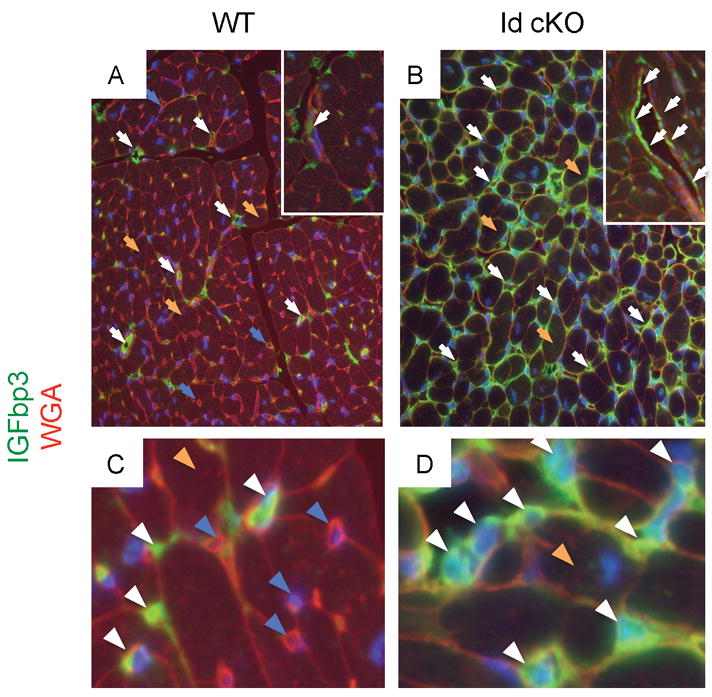
IGFbp3 protein is detected primarily in the nonmyocardial compartment of WT, including endothelial cells (A, A inset and C). Note enhancement of IGFbp3 protein expression in the Id cKO (B, B inset and D). Abbreviations: WGA: wheat germ agglutinin. White arrowheads (A–D): IGFbp3-positive small caliber nonmyocardial cells; blue arrowheads (A, C): IGFbp3-negative nonmyocardial cells; orange arrowheads (A–D): cardiac myocytes. Magnification: A and B: 200X; A and B insets and C and D: 400X.
Microarray analysis of markers of hypertrophy revealed that atrial and brain natriuretic precursor peptides (Nppa or ANF and Nppb or BNF, IDs: 230899 and 18158), were upregulated relative to WT (STable II, and data not shown)(Gardner, 2003). The developmental Myh7 gene (β-MHC, ID: 140781) was also upregulated while the adult Myh6 (α-MHC, 17888) was unchanged, suggesting an isozyme shift of MHC expression (STable II)(Rajabi et al., 2007; van Rooij et al., 2007).
As expected, Id1 and Id3 (ID: 15901 and 15903 respectively) were both downregulated relative to WT (STable II). Loss of Id1 in the Id cKO heart was further confirmed by semi qRT-PCR and Western blot (data not shown). Id4 (ID: 15904), which is not expressed and may not play a role in the heart (Jen et al., 1996; Jen et al., 1997), was unchanged (STable II). However, Id2 (ID: 15902), whose pattern of expression and function overlaps with those of Id1 and Id3 (Jen et al., 1996; Jen et al., 1997), was slightly upregulated (STable II), suggesting a modest compensatory mechanism between members of the Id family. As expected, hearts from Tie2Cre+Id1−/−Id3+/− and Id1F/+Id3−/− mice, which contain higher Id levels than those from Id cKO mice, did not reveal severe changes in gene expression profiles (STable II). The gene expression studies suggest that a panel of vascular, fibrotic and hypertrophic markers is altered in the hearts of Id cKO mice.
Reversion of vascular and fibrotic, but not hypertrophic markers in IGFbp3-treated Id cKO heart explants
Because Id and IGFbp3 are expressed in the endothelial compartment, Id represses IGFbp3, and the IGF axis plays a role in the rescue of Id KO embryos, we determined if IGFbp3 plays a role on the reversion of gene expression profiles in Id cKOs. To this end, we incubated transverse sections of P180 Id cKO hearts in the presence and absence of mouse recombinant IGFbp3. In this condition, the potential interactions between the multiple layers (myocardium – epicardium – endocardium - endothelium) remain intact. We found that IGFbp3 treatment reversed expression of genes dysregulated in Id cKO (Fig 4A, IGFbp3 treated vs not treated in Id cKO). Notably, an opposite regulation was apparent in genes that belong to the extracellular matrix and extracellular region part (GO:0031012 and GO:0044421 respectively, compare Id1/3 cKO with IGFbp3 treated) (Fig 4B).
IGFbp3 treatment reversed expression profiles of specific members of the vascular and fibrotic pathways (STable II). The anti-angiogenic Thbs4 and Thbs1 were down-regulated by IGFbp3 addition (STable II, and Fig 5). This suggests that this potent, Id-dependent, anti-angiogenic Thbs family can be normalized by IGFbp3 administration. Conversely, expression of the pro-angiogenic Angpt1 was up-regulated by IGFbp3 addition (STable II and Fig 5). Thus, IGFbp3 promotes expression of pro-angiogenic markers and represses expression of anti-angiogenic markers in Id cKO hearts. Regarding key components of the fibrotic tissue, a direct effect on specific expression of 2 collagen genes was observed (type I and type XV, IDs: 12842 and 12819 respectively, STable II and data not shown). Despite the fact that Thbs1/4 were affected, none of the Adamts (4, 9 and 1) proteins, which contain a Thbs domain, were changed by IGFbp3 addition (STable II). Surprisingly, however, no direct effect of IGFbp3 was observed on the regulation of hypertrophic markers, like ANF, BNF or βMHC (STable II). Id2 expression was not affected by IGFbp3 administration (STable II). Similarly, no effect on IGFbp 3, 5 and 7 was observed. However, IGFbp4 was down-regulated (STable II), suggesting a compensatory cross-talk among members of the IGFbp family. We also incubated transverse sections of WT hearts in the presence of an antibody reactive with IGFbp3. The treatment resulted in Thbs4/1 upregulation and Angpt1 downregulation (Fig 5). As expected, this outcome in WT with IGFbp3 antibody (dysregulation) was opposite to that in Id cKO with recombinant IGFbp3 (reversion). The experiments suggest that addition of recombinant IGFbp3 in Id-deficient hearts is corrective, and that a block of IGFbp3 mimics the effect produced by Id-deficiency.
Vascular compromise takes place outside the heart
Because the vascular-specific recombination of Id1 was not restricted to the heart, we determined if endothelial ablation of Id1 also led to angiogenic defects outside of the heart. To this end, P60 Id cKO (n=6) or WT (n=6) mice were subjected to excisional skin wounding (n=12, 2 wounds per mouse), and the wound healing response was studied over a period of 8 days (Fig. 7). Wound closure was markedly delayed in the Id cKO relative to WT (Fig. 7A–D insets). Accordingly, histopathological examination revealed a defective angiogenesis (Fig. 7B, D, F, compare with 7A, C, E). Overall, granulation tissue formation was less dense at the Id cKO border zone of the wound (relative to WT) (Fig. 7B, D, compare with 7A, C). At day 6, a burst of small, Id-positive endothelial capillaries, accompanied by extensive granulation, emerged from the uninjured skin towards the injured skin at the border zone, underneath a region of new epithelium (Fig. 7A). However, in the wounds of the Id cKO mice, granulation was markedly less dense (Fig. 7B). Consistent with almost complete loss of wound healing activity (Fig. 7B inset), capillary development was sparse (Fig. 7B). By day 8, the wounds were largely healed in WT mice (Fig. 7C inset), but the wounds in the Id cKO mice were still unhealed (Fig. 7D inset). At the histological level, less cellularity or hypoplasticity was apparent in the Id cKO mice. (Fig. 7D, compare with 7C). Strikingly, a network of large caliber and distended (Id-negative) vessels was prevalent in the Id cKO wound (Fig. 7D, F, H). Thus, this experiment supports the notion that the Id cKO mice have a compromised angiogenic response to external injury.
Figure 7. Angiogenesis is compromised in Id cKO excisional skin wounds.

Id cKO mice display defective angiogenesis, associated with compromised granulation tissue formation by 6 days (A and B). Accordingly, the process of wound closure is delayed (A and B insets). Distended, enlarged vasculature and negligible Id1 expression are apparent in the still unhealed wounds of Id cKO mice by 8 days (C–H, C and D insets). White arrowheads (A and B): small capillaries; white and yellow arrowheads (C–F): small and large capillaries respectively; white arrowheads (G–H): Id1-positive cells adjacent to E–F respectively. Dotted black circles (A–D insets): perimeter of the excisional skin wounds. Magnification: A and B: 400X; C–H: 200X; Scale bars (A–D, insets): 5 mm.
Discussion
In this study we ablated Id1 and Id3 in the endocardium and endothelium, starting at mid-gestation development. The embryos escaped mid-gestation lethality, but developed perinatal and adult cardiac phenotypes. Thus, the heart of the conditional KO mice appears to be sensitive to loss of Id genes in the vasculature.
Commonalities and differences between the neonatal and the adult cardiac phenotypes
The cardiac phenotype, starting at late gestation, presents varying features throughout development and into adulthood. At birth, the dual phenotype of neonatal death and survival resembles that of IGF1 KO mice (Liu et al., 1993). The neonatal cardiac phenotype (enlargement, VSDs, trabeculation defects) also resembles that of Id dKO pups partially rescued by maternal injection of recombinant IGF1(Fraidenraich et al., 2004). Some features of the neonatal phenotype (enlargement, endocardial/endothelial defects) are also apparent in the adult heart, some (VSDs and trabeculation) do not exist and others (vascular fibrosis and functional deficits) emerge in the adult. It is likely that absence of VSDs in the adult heart, rather than reflecting corrective activities overtime, indicates a milder phenotype of the fraction of adult mice that survives postnatally.
Vascular compromise outside of the heart
The vascular phenotype is not confined to the heart, as recombination takes place in the endothelial network globally (Kisanuki et al., 2001). However, not all endothelial cells and/or apposing tissues are equally affected. For example, the Id cKO placentas are devoid of Id1, yet they do not display an obvious morphological phenotype. Interestingly, a process that requires a de novo generation of vasculature or neo-angiogenesis (i.e. healing of a skin injury) appears to be highly sensitive to Id levels, as the appearance of the vasculature in Id cKO wounded mice is delayed and aberrant. Interestingly, despite being two different processes, both the heart and the healing wound lead to adjacent areas with low cellularity. Notably, the presence of a network of distended vessels in the latter process resembles the phenotype of the coronary vasculature of Id cKO neonates and the brain vasculature of Id dKO embryos, which die of brain hemorrhage (Lyden et al., 1999). Because Tie2 is also expressed in the myeloid lineage at low levels, additional indirect cardiac effects caused by ablation of Id1 in this compartment cannot be formally ruled out. Although the pregnant females undergo similar vascular processes in the reproductive system, the Id cKO females produce normal litters when crossed with Id control males. However, the Id cKO females have to be paired for prolonged periods of time (in some cases for 4 months) to accomplish successful pregnancies (unpublished data). Whether the compromised/delayed capacity of the Id cKO females to become pregnant is attributed to neoangiogenesis defects, it remains to be determined.
Thrombospondin-1 and -4 are altered in Id cKO mice
Few effectors of the Id pathway have been identified thus far (Bai et al., 2007; Ciarrocchi et al., 2007; Di et al., 2006; Fraidenraich et al., 2004; Lee et al., 2009; Lin et al.; Lyden et al., 1999; Volpert et al., 2002). This is partly because the Id genes and their effectors are linked via non-cell autonomous pathways, which involve the production of secreted factors (IGF1 for example). Thus, Id genes and their effectors are not necessarily expressed in the same tissues, making the identification of the latter factors cumbersome. One established effector of the Id pathway is Thbs1, a potent anti-angiogenic factor (Folkman, 1995), which is repressed by Id1 (Volpert et al., 2002). The Id-dependent regulation of Thbs1 is indirect, as expression of Id and Thbs1 does not overlap (Volpert et al., 2002). In addition, repression of Thbs1 by Id1 is not mediated by canonical E-box DNA consensus binding sites in the Thbs1 promoter region (Volpert et al., 2002), supporting the notion that the regulation is indirect. Although Thbs1 is expressed outside of the endothelium, its ultimate function is to inhibit angiogenesis (Folkman, 1995). Thus, Thbs1 feeds back to the endothelium, where Id1 is normally expressed. In the absence of Id signals, Thbs1 is upregulated, resulting in inhibition of angiogenesis (Volpert et al., 2002). In addition to Thbs1, we identified another member of the Thbs family that responds to Id: Thbs4, which is also upregulated in the Id cKO hearts.
Angiopoietin-1 is altered in Id cKO mice
Thbs is not the sole vascular target of Id. Angpt1, an essential regulator of vascular remodeling (Davis et al., 1996; Folkman and D’Amore, 1996; Suri et al., 1996), is downregulated in the Id cKO hearts. Interestingly, like in the Id dKO embryos, loss of Angpt1 causes an embryonic lethal phenotype, which is characterized by disruption of the trabeculation network and by a thin myocardial wall (Suri et al., 1996). Embryos deficient for Angpt1 exhibit abnormal vascular architecture where the principal defect is a failure to recruit smooth muscle and pericyte precursors in the heart (Suri et al., 1996). Furthermore, a point mutation in the human Tie2 gene (the receptor of the ligand Angpt1) may lead to aberrant Tie2 function and Angpt1 downregulation, and may play a role in the etiology of spontaneous venous malformations (Vikkula et al., 1996). Notably, the vasculature of the Tie2 mutant patients is distended (as in the case of the vasculature of the skin-wounded Id cKO mice), due to a reduction of surrounding smooth muscle vascular cells (Vikkula et al., 1996). However, despite a striking similarity of roles of the Angpt1 and Id pathways, no genetic link between these two genes has been previously reported.
IGFbp3 corrects gene expression profiles
Although expression of Thbs1 is regulated by serum, and there are serum response elements in the promoter region of Thbs1(Framson and Bornstein, 1993), the non-cell autonomous link between Id and Thbs is not fully delineated. Here we identified IGFbp3, a downstream member of the Id pathway, as a potential mediator of Id/Thbs interaction (Cubbage et al., 1990; Jogie-Brahim et al., 2009). IGFbp3 is a circulating factor (Jogie-Brahim et al., 2009) present in failing hearts (Pucci et al., 2009). IGFbp3 is detected in the nonmyocardial compartment, including the endothelium, and found to spread throughout the extracellular region of the myocardium in the Id cKO. Transgenic overexpression of IGFbp3 causes cardiomegaly (Murphy et al., 1995) and external administration of recombinant IGFbp3 (or a blocking IGFbp3 antibody) results in Thbs normalization (or dysregulation). Interestingly, IGFbp3 targets other families of angiogenic factors, as Angpt1 levels can also be reversed (or dysregulated) by IGFbp3 protein (or IGFbp3 antibody) treatment. Thus, IGFbp3 overproduction in the Id cKO hearts may be cardioprotective, as it may ameliorate the phenotype of the Id cKO hearts by reversing gene expression profiles of Id-dependent, vascular genes. Importantly, external administration of IGFbp3 may aid in the rescue of the Id cKO cardiac phenotype.
In the Id cKO mice, it is not clear if the deposition of collagen fibers is secondary to the appearance of low cellularity regions. Our data suggest however, that IGFbp3 protein has a direct effect on the expression (down-regulation) of specific members of the collagen family. Members of a family with collagen-degrading activity (Adamts) (Brocker et al., 2009; Guo et al.) are also upregulated in the Id cKO hearts. These proteins bear Thbs motifs. However, IGFbp3 administration does not significantly change expression of Adamts. This observation reinforces the notion that the potential beneficial effect of IGFbp3 on reducing the formation of fibrotic tissue may come from inhibition of expression and not activation of degradation of collagen.
It is possible that Id1 represses IGFbp3 in the endothelium, as both proteins are detected there. However, we do not know if Id1 and IGFbp3 are expressed in the same subset of the endothelium. Whether the endothelial regulation is direct or indirect, it remains to be determined. Because IGFbp3 is enhanced in the Id cKO and spreads throughout the myocardial interstitium, we cannot rule out that IGFbp3 is also produced by other cell types, like cardiac fibroblasts or myocytes. Future studies will help determine if there is a paracrine mechanism of Id1/IGFbp3 regulation.
IGFbp3 action: IGF1-dependent or IGF1–independent?
The fact that IGF1 and IGFbp3 play different roles in the Id deficient heart at different stages raises the question of whether the role of IGFbp3 in adult heart disease is dependent or independent of IGF1 signaling. In other words, does IGFbp3 act via a canonical mode of action, by binding and inhibiting the function of IGF1? (Granata et al., 2007; Jogie-Brahim et al., 2009; Rajah et al., 1997). Our results suggest that the direct effect of IGFbp3 on angiogenic and collagen gene expression profiles in the Id cKO heart explants does not appear to implicate IGF1. In addition, IGF1 (embryonic) and IGFbp3 (adult) appear to have different targets. While IGFbp3 targets the vasculature and fibrotic tissue, IGF1 targets the myocardium (IGF1 corrects proliferation deficits of the myocardium but fails to correct vascular and structural defects in IGF1-treated Id dKO embryos). Nevertheless, we cannot discount a potential canonical effect of IGFbp3 in processes that do not implicate reversion of gene expression profiles.
Is the IGF axis a non-cell autonomous link between Id and its effectors?
The perinatal and the adult phenotypes are not similar. The common denominator is the defect in the endocardium/endothelium, which is the main source of Id. IGF1 is reduced in Id dKO embryos but unchanged in the adult Id cKO heart. IGFbp4 is enhanced in Id dKO embryos but unchanged in the adult Id cKO heart. Conversely, IGFbp3 is unchanged in the postnatal heart, but enhanced in the adult Id cKO heart. Whether loss of endocardial/endothelial-Id impacts on stage-specific non-cell autonomous links (members of the IGF axis), which in turn impact on different effectors, it remains to be determined. Future experiments involving administration or transgenic overexpression of IGF members will help elucidate their roles as potential non-cell autonomous links in the Id pathway.
Conclusion
The results obtained in this study suggest that loss of developmental Id in the endothelium of Id cKO mice results in postnatal vascular-associated cardiac phenotypes. The heart becomes sensitive to the vascular compromise at two postnatal stages, one at late development and another in the adult. The latter defects are accompanied by dysregulation of vascular, fibrotic and hypertrophic pathways, along with the production of IGFbp3. Excess of IGFbp3 in turn reverses vascular and fibrotic, but not hypertrophic gene expression profiles in heart explants. Blocking IGFbp3 promotes dysregulation, thus mimicking the effect produced by Id loss. Recently, a polymorphism in the Id3 gene has been associated with vascular intima-media thickness in humans (Doran et al.). Therefore, the elucidation of the roles of Id genes in the developmental vasculature and their consequences in the postnatal heart will have tremendous implications for human congenital and adult fibrotic cardiomyopathy.
Supplementary Material
(A–D) MF20 and CD31 demarcate myocardium and endocardium/endothelium respectively. Magnification: 40X.
Size and morphology of placentas of Id cKO embryos appear normal (A, B and A, B, insets). Accordingly, the endothelial network of both embryonic and maternal components of the placentas appear normal (C, E, G and H), despite that no Id1 is detected in either the embryonic or maternal components (D and F, I and J, adjacent sections to C, E, G and H respectively). Red and yellow arrowheads (A, B, insets and C–F): embryonic and maternal components of the placentas respectively; white arrowheads (G, H): endothelial network; orange arrowhead (I): Id positive cells in adjacent section to (G). Abbreviations: WT embryo (WT mother): placentas obtained from E17.5 WT embryos harbored by WT mothers; Id cKO embryo (Id cKO mother): placentas obtained from E17.5 Id cKO embryos harbored by Id cKO mothers. Magnification: A, B: 100X; A, B, insets: 10X; C–F: 20X; G–J: 100X.
Significance score was calculated as described in Methods. If a GO term showed significance in any biological setting (significance score > 2, hypergeometric p-value < 0.01), it was included. The GO terms were sorted by maximum absolute score of each row. Id1 KO: full absence of Id1 in Id1−/−Id3+/−Tie2Cre+; Id3 KO: full absence of Id3 in Id1F/−Id3−/−.
Comparisons are Id1 KO vs. WT, Id3 KO vs. WT, Id cKO vs. WT, and IGFbp3 treated Id cKO vs. Id cKO. Both log2(Ratio) and Z score are shown. Z score is standardized log2(Ratio). Affymetrix Transcript ID, NCBI Gene ID, and Gene Symbol and description are shown. NA indicates that expression is not detectable on the microarray. Id1 KO: full absence of Id1 in Id1−/−Id3+/−Tie2Cre+; Id3 KO: full absence of Id3 in Id1F/−Id3−/−; IGFbp3: IGFbp3-treated Id1/3 cKO.
Acknowledgments
We thank Dr. R. Benezra (Memorial Sloan Kettering Cancer Center) for providing Id mice, Dr. J. Sadoshima (UMDNJ) for providing Mst1 mice, Dr. L Wu (UMDNJ) for advisement with placental experiments and Dr. P Soteropoulos at the Center for Applied Genomics at UMDNJ-NJMS for microarray analysis.
Sources of funding
D.F is funded by the AHA (GIA-0755909T) and the NIH (HL076568 and HL094905). S.J.L. is funded by the NIH (GM068636). B.T is funded by the NIH (HL098802)
Footnotes
Disclosures
none
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature cited
- Bai G, Sheng N, Xie Z, Bian W, Yokota Y, Benezra R, Kageyama R, Guillemot F, Jing N. Id sustains Hes1 expression to inhibit precocious neurogenesis by releasing negative autoregulation of Hes1. Dev Cell. 2007;13:283–97. doi: 10.1016/j.devcel.2007.05.014. [DOI] [PubMed] [Google Scholar]
- Brocker CN, Vasiliou V, Nebert DW. Evolutionary divergence and functions of the ADAM and ADAMTS gene families. Hum Genomics. 2009;4:43–55. doi: 10.1186/1479-7364-4-1-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien KR, Moretti A, Laugwitz KL. Development. ES cells to the rescue. Science. 2004;306:239–40. doi: 10.1126/science.1104769. [DOI] [PubMed] [Google Scholar]
- Chien KR, Olson EN. Converging pathways and principles in heart development and disease: CV@CSH. Cell. 2002;110:153–62. doi: 10.1016/s0092-8674(02)00834-6. [DOI] [PubMed] [Google Scholar]
- Ciarrocchi A, Jankovic V, Shaked Y, Nolan DJ, Mittal V, Kerbel RS, Nimer SD, Benezra R. Id1 restrains p21 expression to control endothelial progenitor cell formation. PLoS One. 2007;2:e1338. doi: 10.1371/journal.pone.0001338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cubbage ML, Suwanichkul A, Powell DR. Insulin-like growth factor binding protein-3. Organization of the human chromosomal gene and demonstration of promoter activity. J Biol Chem. 1990;265:12642–9. [PubMed] [Google Scholar]
- Davis S, Aldrich TH, Jones PF, Acheson A, Compton DL, Jain V, Ryan TE, Bruno J, Radziejewski C, Maisonpierre PC, Yancopoulos GD. Isolation of angiopoietin-1, a ligand for the TIE2 receptor, by secretion-trap expression cloning. Cell. 1996;87:1161–9. doi: 10.1016/s0092-8674(00)81812-7. [DOI] [PubMed] [Google Scholar]
- Di K, Ling MT, Tsao SW, Wong YC, Wang X. Id-1 modulates senescence and TGF-beta1 sensitivity in prostate epithelial cells. Biol Cell. 2006;98:523–33. doi: 10.1042/BC20060026. [DOI] [PubMed] [Google Scholar]
- Doran AC, Lehtinen AB, Meller N, Lipinski MJ, Slayton RP, Oldham SN, Skaflen MD, Yeboah J, Rich SS, Bowden DW, McNamara CA. Id3 is a novel atheroprotective factor containing a functionally significant single-nucleotide polymorphism associated with intima-media thickness in humans. Circ Res. 106:1303–11. doi: 10.1161/CIRCRESAHA.109.210294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folkman J. Angiogenesis inhibitors generated by tumors. Mol Med. 1995;1:120–2. [PMC free article] [PubMed] [Google Scholar]
- Folkman J, D’Amore PA. Blood vessel formation: what is its molecular basis? Cell. 1996;87:1153–5. doi: 10.1016/s0092-8674(00)81810-3. [DOI] [PubMed] [Google Scholar]
- Fraidenraich D, Benezra R. Embryonic stem cells prevent developmental cardiac defects in mice. Nat Clin Pract Cardiovasc Med. 2006;3(Suppl 1):S14–7. doi: 10.1038/ncpcardio0402. [DOI] [PubMed] [Google Scholar]
- Fraidenraich D, Stillwell E, Romero E, Wilkes D, Manova K, Basson CT, Benezra R. Rescue of cardiac defects in id knockout embryos by injection of embryonic stem cells. Science. 2004;306:247–52. doi: 10.1126/science.1102612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Framson P, Bornstein P. A serum response element and a binding site for NF-Y mediate the serum response of the human thrombospondin 1 gene. J Biol Chem. 1993;268:4989–96. [PubMed] [Google Scholar]
- Gardner DG. Natriuretic peptides: markers or modulators of cardiac hypertrophy? Trends Endocrinol Metab. 2003;14:411–6. doi: 10.1016/s1043-2760(03)00113-9. [DOI] [PubMed] [Google Scholar]
- Good DJ, Polverini PJ, Rastinejad F, Le Beau MM, Lemons RS, Frazier WA, Bouck NP. A tumor suppressor-dependent inhibitor of angiogenesis is immunologically and functionally indistinguishable from a fragment of thrombospondin. Proc Natl Acad Sci U S A. 1990;87:6624–8. doi: 10.1073/pnas.87.17.6624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granata R, Trovato L, Lupia E, Sala G, Settanni F, Camussi G, Ghidoni R, Ghigo E. Insulin-like growth factor binding protein-3 induces angiogenesis through IGF-I- and SphK1-dependent mechanisms. J Thromb Haemost. 2007;5:835–45. doi: 10.1111/j.1538-7836.2007.02431.x. [DOI] [PubMed] [Google Scholar]
- Guo C, Wang Y, Liang H, Zhang J. ADAMTS-1 contributes to the antifibrotic effect of captopril by accelerating the degradation of type I collagen in chronic viral myocarditis. Eur J Pharmacol. 2010;629:104–10. doi: 10.1016/j.ejphar.2009.12.009. [DOI] [PubMed] [Google Scholar]
- Jen Y, Manova K, Benezra R. Expression patterns of Id1, Id2, and Id3 are highly related but distinct from that of Id4 during mouse embryogenesis. Dev Dyn. 1996;207:235–52. doi: 10.1002/(SICI)1097-0177(199611)207:3<235::AID-AJA1>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Jen Y, Manova K, Benezra R. Each member of the Id gene family exhibits a unique expression pattern in mouse gastrulation and neurogenesis. Dev Dyn. 1997;208:92–106. doi: 10.1002/(SICI)1097-0177(199701)208:1<92::AID-AJA9>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- Jogie-Brahim S, Feldman D, Oh Y. Unraveling insulin-like growth factor binding protein-3 actions in human disease. Endocr Rev. 2009;30:417–37. doi: 10.1210/er.2008-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastner P, Mark M, Chambon P. Nonsteroid nuclear receptors: what are genetic studies telling us about their role in real life? Cell. 1995;83:859–69. doi: 10.1016/0092-8674(95)90202-3. [DOI] [PubMed] [Google Scholar]
- Kisanuki YY, Hammer RE, Miyazaki J, Williams SC, Richardson JA, Yanagisawa M. Tie2-Cre transgenic mice: a new model for endothelial cell-lineage analysis in vivo. Dev Biol. 2001;230:230–42. doi: 10.1006/dbio.2000.0106. [DOI] [PubMed] [Google Scholar]
- Lee JY, Kang MB, Jang SH, Qian T, Kim HJ, Kim CH, Kim Y, Kong G. Id-1 activates Akt-mediated Wnt signaling and p27(Kip1) phosphorylation through PTEN inhibition. Oncogene. 2009;28:824–31. doi: 10.1038/onc.2008.451. [DOI] [PubMed] [Google Scholar]
- Lehman JJ, Kelly DP. Transcriptional activation of energy metabolic switches in the developing and hypertrophied heart. Clin Exp Pharmacol Physiol. 2002;29:339–45. doi: 10.1046/j.1440-1681.2002.03655.x. [DOI] [PubMed] [Google Scholar]
- Lin J, Guan Z, Wang C, Feng L, Zheng Y, Caicedo E, Bearth E, Peng JR, Gaffney P, Ondrey FG. Inhibitor of differentiation 1 contributes to head and neck squamous cell carcinoma survival via the NF-kappaB/survivin and phosphoinositide 3-kinase/Akt signaling pathways. Clin Cancer Res. 16:77–87. doi: 10.1158/1078-0432.CCR-08-2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JP, Baker J, Perkins AS, Robertson EJ, Efstratiadis A. Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF receptor (Igf1r) Cell. 1993;75:59–72. [PubMed] [Google Scholar]
- Lyden D, Young AZ, Zagzag D, Yan W, Gerald W, O’Reilly R, Bader BL, Hynes RO, Zhuang Y, Manova K, Benezra R. Id1 and Id3 are required for neurogenesis, angiogenesis and vascularization of tumour xenografts. Nature. 1999;401:670–7. doi: 10.1038/44334. [DOI] [PubMed] [Google Scholar]
- Murphy LJ, Rajkumar K, Molnar P. Phenotypic manifestations of insulin-like growth factor binding protein-1 (IGFBP-1) and IGFBP-3 overexpression in transgenic mice. Prog Growth Factor Res. 1995;6:425–32. doi: 10.1016/0955-2235(95)00026-7. [DOI] [PubMed] [Google Scholar]
- Nam HS, Benezra R. High levels of Id1 expression define B1 type adult neural stem cells. Cell Stem Cell. 2009;5:515–26. doi: 10.1016/j.stem.2009.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter PS, Brady JE, Yan L, Chen W, Engelhardt S, Wang Y, Sadoshima J, Vatner SF, Vatner DE. Inhibition of p38 alpha MAPK rescues cardiomyopathy induced by overexpressed beta 2-adrenergic receptor, but not beta 1-adrenergic receptor. J Clin Invest. 2007;117:1335–43. doi: 10.1172/JCI29576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pucci A, Zanini C, Granata R, Ghignone R, Iavarone A, Broglio F, Sorrentino P, Bergamasco L, Rinaldi M, Ghigo E. Myocardial insulin-like growth factor-1 and insulin-like growth factor binding protein-3 gene expression in failing hearts harvested from patients undergoing cardiac transplantation. J Heart Lung Transplant. 2009;28:402–5. doi: 10.1016/j.healun.2008.12.022. [DOI] [PubMed] [Google Scholar]
- Rajabi M, Kassiotis C, Razeghi P, Taegtmeyer H. Return to the fetal gene program protects the stressed heart: a strong hypothesis. Heart Fail Rev. 2007;12:331–43. doi: 10.1007/s10741-007-9034-1. [DOI] [PubMed] [Google Scholar]
- Rajah R, Valentinis B, Cohen P. Insulin-like growth factor (IGF)-binding protein-3 induces apoptosis and mediates the effects of transforming growth factor-beta1 on programmed cell death through a p53- and IGF-independent mechanism. J Biol Chem. 1997;272:12181–8. doi: 10.1074/jbc.272.18.12181. [DOI] [PubMed] [Google Scholar]
- Ruwhof C, van der Laarse A. Mechanical stress-induced cardiac hypertrophy: mechanisms and signal transduction pathways. Cardiovasc Res. 2000;47:23–37. doi: 10.1016/s0008-6363(00)00076-6. [DOI] [PubMed] [Google Scholar]
- Ruzinova MB, Benezra R. Id proteins in development, cell cycle and cancer. Trends Cell Biol. 2003;13:410–8. doi: 10.1016/s0962-8924(03)00147-8. [DOI] [PubMed] [Google Scholar]
- Schneider JS, Vitale JM, Terzic A, Fraidenraich D. Blastocyst Injection of Embryonic Stem Cells: A Simple Approach to Unveil Mechanisms of Corrections in Mouse Models of Human Disease. Stem Cell Rev Rep. 2009 doi: 10.1007/s12015-009-9089-6. [DOI] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–1. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Sucov HM, Dyson E, Gumeringer CL, Price J, Chien KR, Evans RM. RXR alpha mutant mice establish a genetic basis for vitamin A signaling in heart morphogenesis. Genes Dev. 1994;8:1007–18. doi: 10.1101/gad.8.9.1007. [DOI] [PubMed] [Google Scholar]
- Suri C, Jones PF, Patan S, Bartunkova S, Maisonpierre PC, Davis S, Sato TN, Yancopoulos GD. Requisite role of angiopoietin-1, a ligand for the TIE2 receptor, during embryonic angiogenesis. Cell. 1996;87:1171–80. doi: 10.1016/s0092-8674(00)81813-9. [DOI] [PubMed] [Google Scholar]
- van Rooij E, Sutherland LB, Qi X, Richardson JA, Hill J, Olson EN. Control of stress-dependent cardiac growth and gene expression by a microRNA. Science. 2007;316:575–9. doi: 10.1126/science.1139089. [DOI] [PubMed] [Google Scholar]
- Vikkula M, Boon LM, Carraway KL, 3rd, Calvert JT, Diamonti AJ, Goumnerov B, Pasyk KA, Marchuk DA, Warman ML, Cantley LC, Mulliken JB, Olsen BR. Vascular dysmorphogenesis caused by an activating mutation in the receptor tyrosine kinase TIE2. Cell. 1996;87:1181–90. doi: 10.1016/s0092-8674(00)81814-0. [DOI] [PubMed] [Google Scholar]
- Volpert OV, Pili R, Sikder HA, Nelius T, Zaichuk T, Morris C, Shiflett CB, Devlin MK, Conant K, Alani RM. Id1 regulates angiogenesis through transcriptional repression of thrombospondin-1. Cancer Cell. 2002;2:473–83. doi: 10.1016/s1535-6108(02)00209-x. [DOI] [PubMed] [Google Scholar]
- Yamamoto S, Yang G, Zablocki D, Liu J, Hong C, Kim SJ, Soler S, Odashima M, Thaisz J, Yehia G, Molina CA, Yatani A, Vatner DE, Vatner SF, Sadoshima J. Activation of Mst1 causes dilated cardiomyopathy by stimulating apoptosis without compensatory ventricular myocyte hypertrophy. J Clin Invest. 2003;111:1463–74. doi: 10.1172/JCI17459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida K, Taga T, Saito M, Suematsu S, Kumanogoh A, Tanaka T, Fujiwara H, Hirata M, Yamagami T, Nakahata T, Hirabayashi T, Yoneda Y, Tanaka K, Wang WZ, Mori C, Shiota K, Yoshida N, Kishimoto T. Targeted disruption of gp130, a common signal transducer for the interleukin 6 family of cytokines, leads to myocardial and hematological disorders. Proc Natl Acad Sci U S A. 1996;93:407–11. doi: 10.1073/pnas.93.1.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A–D) MF20 and CD31 demarcate myocardium and endocardium/endothelium respectively. Magnification: 40X.
Size and morphology of placentas of Id cKO embryos appear normal (A, B and A, B, insets). Accordingly, the endothelial network of both embryonic and maternal components of the placentas appear normal (C, E, G and H), despite that no Id1 is detected in either the embryonic or maternal components (D and F, I and J, adjacent sections to C, E, G and H respectively). Red and yellow arrowheads (A, B, insets and C–F): embryonic and maternal components of the placentas respectively; white arrowheads (G, H): endothelial network; orange arrowhead (I): Id positive cells in adjacent section to (G). Abbreviations: WT embryo (WT mother): placentas obtained from E17.5 WT embryos harbored by WT mothers; Id cKO embryo (Id cKO mother): placentas obtained from E17.5 Id cKO embryos harbored by Id cKO mothers. Magnification: A, B: 100X; A, B, insets: 10X; C–F: 20X; G–J: 100X.
Significance score was calculated as described in Methods. If a GO term showed significance in any biological setting (significance score > 2, hypergeometric p-value < 0.01), it was included. The GO terms were sorted by maximum absolute score of each row. Id1 KO: full absence of Id1 in Id1−/−Id3+/−Tie2Cre+; Id3 KO: full absence of Id3 in Id1F/−Id3−/−.
Comparisons are Id1 KO vs. WT, Id3 KO vs. WT, Id cKO vs. WT, and IGFbp3 treated Id cKO vs. Id cKO. Both log2(Ratio) and Z score are shown. Z score is standardized log2(Ratio). Affymetrix Transcript ID, NCBI Gene ID, and Gene Symbol and description are shown. NA indicates that expression is not detectable on the microarray. Id1 KO: full absence of Id1 in Id1−/−Id3+/−Tie2Cre+; Id3 KO: full absence of Id3 in Id1F/−Id3−/−; IGFbp3: IGFbp3-treated Id1/3 cKO.