

Since its introduction in 1953 by Child and associate,3 the experimental procedure of portacaval transposition has received intensive study in many laboratories. Such investigations have shown that portacaval transposition can be performed in dogs with relatively small risk,8, 32 that animals so altered can live for long periods of time in good health, and that hepatic function tests are not deranged,3, 8, 29, 31 except for abnormalities of response to oral ammonia loads.29 Although gastric hyperacidity has been shown to be a consequence of the operation,4, 5, 20, 25, 30 this has not proved to be the cause of long-term morbidity in dogs.
In this communication a previously unreported result of portacaval transposition in dogs will be described—that of hepatic de-glycogenation. The observation that a substantial reduction in hepatic glycogen content follows portacaval transposition in dogs prompted the use of this operative procedure for the treatment of an 8-year-old child with hepatic glycogen storage disease and portal cirrhosis.
METHODS
Animal studies
A total of 45 mongrel dogs weighing 7.0 to 22.7 kilograms were used. Seventeen animals, employed in hepatic glycogen studies, underwent portacaval transposition by means of a rnodification32 of Child’s3 method. Thirteen of these 17 dogs also had ligation of all venous tributaries to the ileocaval system from the inguinal ligaments to the diaphragm, with the single exception of the renal veins, in order to prevent the uncontrolled development of systemic venous collaterals. Since the results of hepatic deglycogenation were similar in those groups which did and those which did not have this additional procedure, the results from all 17 experiments were ultimately pooled for analysis. Four additional control dogs had a sham operation in which the same dissection was carried out. The portal vein was then occluded for 10 minutes and the inferior vena cava for 20 minutes, times comparable to those required for the performance of the actual transposition in the test group.
After a 20 hour fast, open liver biopsies were obtained under sodium pentobarbital anesthesia at the time of, and 36 to 75 days after, transposition or sham operation. The liver tissue was immediately frozen on dry ice and analyzed in Denver for glycogen content. The method of Bloom and his associates1 was used to separate the trichloroacetic acid (TCA) soluble from the TCA insoluble fractions. Quantitative determination of these glycogen fractions was then carried out by the anthrone method of Seifter and his associate.26 Results were expressed as milligrams of glycogen per gram of wet liver tissue.
After a period of training, oral glucose tolerance tests were performed in the unanesthetized state 2 days before the pre- and posttransposition biopsies in all 17 dogs of the test group. The glucose dose of 0.5 Gm. per kilogram was given in solution by gastric intubation. Plasma glucose was measured with an autoanalyzer. Before transposition, the hyperglycemic response to oral glucose was measured only in the femoral vein. Postoperatively, simultaneous specimens were obtained from an artery, the femoral vein, and the distal portal vein via a multiple-catheter sampling technique.31
In another group of 24 animals weighing 7.0 to 21.5 kilograms, transposition and ligation of ileocaval venous collaterals were carried out. Four to 12 weeks later, the effect of constant intravenous glucagon infusion (15 mcg. in 45 minutes) was measured, comparing the effect of transfemoral (intraportal) administration with that observed with the systemic (via a foreleg vein) route. The tests with fore- and hind-leg vein glucagon infusion were performed one week apart in randomized order.
Human studies
Detailed analysis of the biochemical defect in the patient with glycogen storage disease was carried out before and 8½ months after portacaval transposition, following a 7 hour fast. Biopsies of the liver and the rectus muscle were immediately frozen on dry ice and subsequently analyzed in St. Louis for glycogen content and enzymatic activity. Glycogen was isolated from alkaline digests of the tissues and the structures of the polysaccharide preparations were determined by differential enzymatic degradation with phosphorylase and amylo-1,6-glucosidase.18
Homogenates of the liver and muscle were assayed for glucose-6-phosphatase,13 liver phosphorylase, muscle phosphorylase, α-glucosidase,11 UDPG glycogen synthetase and amylo-1,6-glucosidase. Amylo-1,6-glucosidase was determined with the widely used method of Hers,9 as well as with a recently described technique.14, 15
In addition, red cell glycogen content was measured.27 Serum lipids were determined in the following fractions: cholesterol and esters, fatty acids, nonesterified fatty acids, phospholipids, and neutral fats. Urine steroids and catechol levels were determined (by Dr. Dalton Jenkins). Quick one-stage prothrombin, specific prothrombin and proconvertin, accelerator globulin, and Stuart assay were performed. Numerous standard liver function tests were measured and the responses to oral glucose, intravenous glucagon, and various gastric stimulants are described below. The size of the liver and of the spleen were determined before and after operation with a liver scan technique employing radioactive gold.
RESULTS
Effect of transposition upon hepatic glycogen content
Fourteen of the 17 dogs studied had reduction in hepatic glycogen content 45 to 75 days after transposition. The total glycogen decreased an average of 51 percent (Fig. 1). The principal loss was in the TCA soluble glycogen, the mean decrease being 70 per cent. TCA insoluble glycogen decreased an average of only 11 percent. The changes in total and TCA soluble glycogen were significant at the 2 percent (p < .02) and 1 percent (p < .01) levels, respectively. The change in TCA insoluble glycogen was not significant.
Fig. 1.
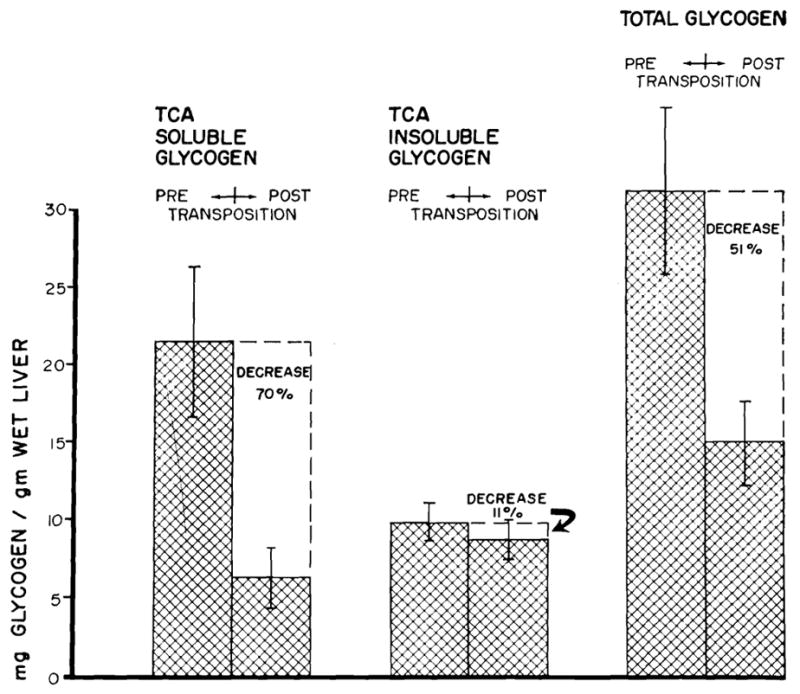
Mean changes in liver glycogen fractions in 17 dogs following transposition. Vertical lines represent ±1 standard error.
Effect of sham operation upon hepatic glycogen content
The 4 control dogs had a mean total hepatic glycogen content of 30.6 mg. per gram wet liver tissue; TCA soluble glycogen, of 20.9 mg. per gram; and TCA insoluble glycogen, of 9.6 mg. per gram. These figures were almost identical to those recorded in the test group. Thirty-six days after the sham operation, total hepatic glycogen content was increased in all 4 animals. The mean TCA soluble fraction was 50.9 mg. per gram, the TCA insoluble fraction was 10 mg. per gram, and the total glycogen was 60.9 mg. per gram. The increase in TCA soluble glycogen is probably due to the better diet available to the animals in the laboratory than had been their usual fare. The increased glycogen content observed in the control group lends increased significance to the de- creases noted in the test animals, which were obtained from the same sources and treated in the same way.
Effect of portacaval transposition upon glucose tolerance test
The effect upon peripheral venous blood of the same oral glucose load before and after operation is depicted in Fig. 2. The magnitude of the hyperglycemic response was almost halved after operation, and the duration of the rises in blood sugar was somewhat lessened. The differences were of borderline significance (p < 0.1). After transposition, the postprandial hyperglycemia was considerably greater in the splanchnic venous drainage than in the peripheral arteries or femoral vein.
Fig. 2.
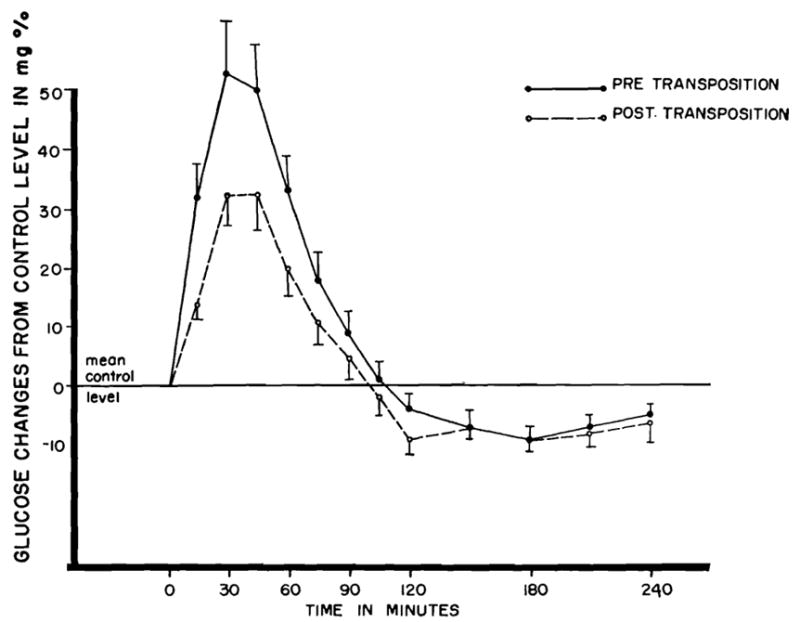
Venous plasma glucose changes pre- and posttransposition following the intraastric administration of 0.5 Gm. glucose per kilogram body weight. Means from 17 dogs. Vertical lines represent ±1 standard error.
Effect of glucagon infusion in dogs with portacaval transposition
With portal infusion (via the femoral vein) the hyperglycemic response was greater (Fig. 3) than with glucagon infusion into a foreleg vein. The difference in effect by the two routes of administration was significant at the 1 percent level (p < .01).
Fig. 3.
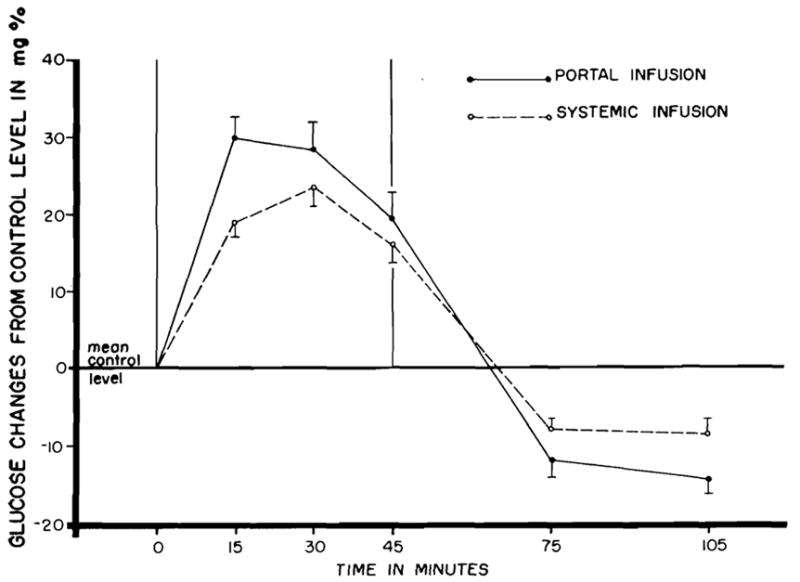
Arterial plasma glucose changes during and following 45 minute infusions of 15 mcg. of crystalline glucagon by two different routes. Mean changes from 24 dogs. Vertical lines represent ±1 standard error.
CLASSIFICATION O F GLYCOGENOSIS
Glycogen storage diseases are hereditary disorders of carbohydrate metabolism which have been classified on the basis of demonstrated enzymatic deficiencies7, 11, 12 Type I glycogenosis is characterized by a deficiency of glucose-6-phosphatase6; Type II by a lack of α-glucosidase11; Type III by a deficiency of amylo-1,6-glucosidase17; Type IV by a presumed deficiency of the branching enzyme16, 28; Type V by a lack of muscle phosphorylase21, 23; and Type VI of unknown etiology, but primarily affecting the liver. The case described below involved an amylo-1,6-glucosidase deficiency.
CASE REPORT
The patient, an 8½-year-old girl, was born on Feb. 13, 1955, the product of a normal pregnancy. An older brother is healthy. The child’s maternal grandmother died of cirrhosis of the liver at the age of 44. During the first few months of life the child had abdominal swelling, irritability, and constipation. The diagnosis of glycogen storage disease was made from an open liver biopsy at the age of 7 months. During the first 8 years of her life pronounced hepatomegaly was present, the liver being palpable from 5 to 7 cm. below the right costal margin. Splenomegaly became increasingly prominent. At the ages of 1, 2, and 5 years, the patient had relatively severe convulsive episodes lasting from 1 to 2 hours, which were relieved on each occasion with an intravenous glucose infusion. Blood sugars were not determined at these times. She was maintained on a high protein, high carbohydrate diet with multiple nocturnal feedings. From the age of 3, she suffered from frequent nose bleeds and easy bruisability. At the age of 6, she had a relatively severe hematemesis, the cause for which was not determined. Her principal complaint before admission to Colorado General Hospital on Sept. 20, 1963, was enlargement of the abdomen that made ambulation difficult.
Physical examination
A large, upper abdominal mass was palpable 7 cm. below the right costal margin and 9 cm. below the left costal margin. There were a number of scattered ecchymoses on both legs, thighs, and over the lower abdomen. The child was of short stature.
Laboratory examination
Hematocrit was 30 percent, white count 4,000 per mm.,3 and platelet count 88,000. Urinalysis was normal. Thymol turbidity was 4.0 and serum bilirubin 1.8 mg. percent, with 0.5 mg. percent conjugated. Alkaline phosphatase was 6.7 Bodansky units and SGOT was 220 SF units. The quick prothrombin time ranged between 40 and 50 percent, with a slight depression of proconvertin (Factor VII), which ranged from 39 to 54 percent, and depression of the Stuart factor, which ranged from 45 to 60 percent. Thromboelastograms showed a slight delay of clotting. The patient’s euglobulin lysis time was 50 minutes (normal, 2 to 4 hours). Total plasma proteins were 6.7 Gm. percent, with a 1.23 A/G ratio. Serum cholesterol and esters, fatty acids, phospholipids and neutral fat were all normal. Nonesterified fatty acids were 1,695 μEq. per liter (normal, 315 to 1,200).
Glucose and glucagon tests were performed after an 8 to 10 hour fast. The preoperative glucose tolerance test was flattened, with a rise from a fasting level of 69 mg. percent to a maximum 138 mg. percent in 30 minutes, and a return to control values by 3 hours. There was no response whatever to intravenous administration of 20 mcg. per kilogram of glucagon. Red cell glycogen was 400 mcg. per gram Hgb. (normal, 22 to 109 mcg. per gram Hgb.). Seven fasting blood sugar evaluations were made; the lowest value recorded was 55 mg. percent and the highest was 87 mg. percent. Urinary steroids and catecholamines were normal.
Chest x-ray and electrocardiogram were normal. The liver scan and other x-ray studies showed a greatly enlarged liver as well as splenomegaly. Esophageal varices or duodenal ulcer could not be seen with a gastrointestinal series. An intravenous pyelogram showed slightly enlarged kidneys.
On Sept. 30, 1963, muscle and liver biopsies were obtained. Histologically, the liver tissue appeared to have a high glycogen content, and there was moderately severe portal cirrhosis (Fig. 4,A). The hepatic glycogen content was elevated (10.1 percent). The muscle glycogen content was normal (1.6 percent). Amylo-1,6-glucosidase activity was absent in the liver homogenates, whereas a significant degree of activity was demonstrable in the muscle. Glucose-6-phosphatase, muscle phosphorylase, UDPG glycogen synthetase, and α-glucosidase were not abnormal. Liver phosphorylase was slightly depressed.* These data established the diagnosis of Type III,- B glycogen storage disease (Table I).
Fig. 4.

Biopsies 2 weeks before and 8½ months after portacaval transposition. Note the foamy appearance of the glycogen-laden hepatocytes in both specimens, as well as the evidence of cirrhosis. A, Preoperative, B, postoperative. (Hematoxylin and eosin. Original magnification ×32.) An increase in mononuclear cells in the periportal areas is present in the second specimen.
Table I.
Enzymatic activities of homogenates prepared from liver and muscle biopsies of the patient treated with portacaual transposition (activities in μmoles substrate utilized per minute per gram tissue)
| Liver |
Muscle |
|||
|---|---|---|---|---|
| October, 1963 | July, 1964 | October, 1963 | July, 1964 | |
| Glucose-6-phosphatase | 1.9 | 2.1 (2.0–5.3)* | ||
| α-glucosidase | 1.11 (0.7–2.4) | .06 (0.03–0.10) | ||
| Amylo- l,6-glucosidase | 0 | 0 (0.04–0.18) | .042 | .047 (0.03–0.07) |
| Phosphorylase | 17.4 | 10.3 (16–33) | 98 | 84 (45–123) |
| UDPG glycogen synthetase | 1.1 | 3.0 (1.4–3.5) | 1.8 | 2.7 (1.5–3.9) |
| Glycoeen content | 10.1 per cent | 9.7 per cent | 1.6 per cent | 1.2 per cent |
The figures in parentheses are the normal range.
Portacaval transposition
The child underwent portacaval transposition and total-body hypothermia of 30° C. A thoracoabdominal incision was made (Fig. 5). Upon entering the abdomen, numerous serpiginous venous collaterals were seen on the abdominal wall, in the retroperitoneal space, and in the portal triad. The portal vein was dissected free from the pancreas to just below its division in the liver hilum and the inferior vena cava was cleaned off from the renal veins to the liver. Both vessels were then grasped with Potts clamps as far proximally and distally as possible and the vessels were transected (Fig. 5,B). I t had been planned to use external bypass from the inferior to the superior vena cava during the time of vascular occlusion, but application of the clamps on both vessels did not cause any fall in blood pressure or rise in pulse and this step was there- fore omitted. Anastomosis of the distal portal vein to the proximal vena cava was performed first (Fig. 5,C) and anastomosis of the distal vena cava to the proximal was performed next (Fig. 5,D). There were no technical difficulties nor any undue tension on the reconstructed vessels.
Fig. 5.
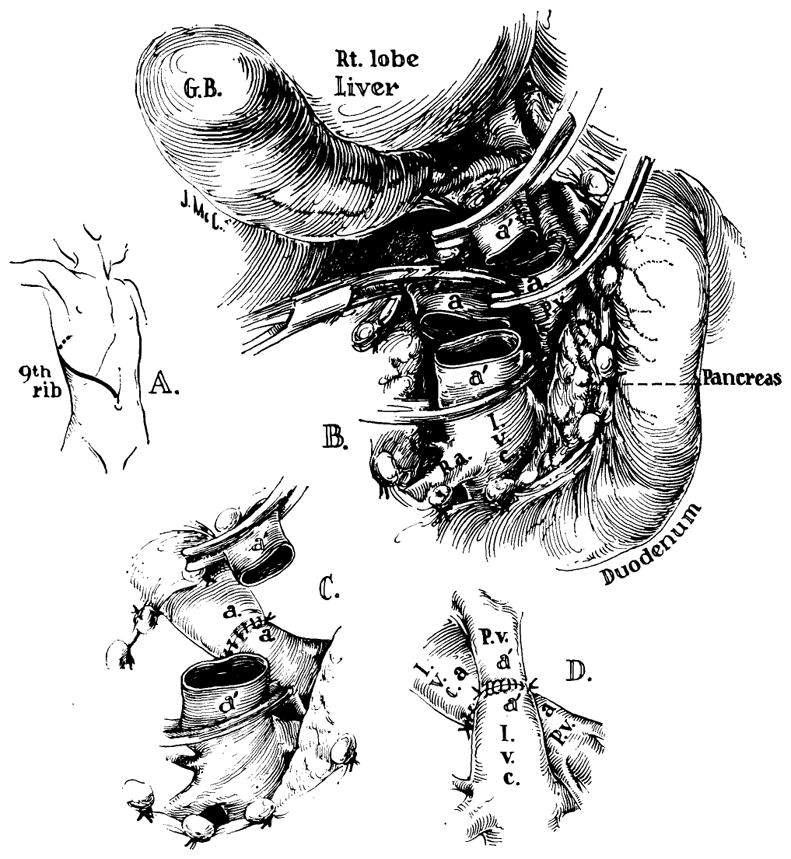
Technique of portacaval transposition. The operation was done under hypothermia. A, Incision; B, dissection completed and vessels transected; C, portacaval anastomosis performed first; D, the second anastomosis completed.
Response to portacaval transposition
There was rapid recovery from the operative procedure and the patient was discharged on the ninth postoperative day. Swelling of the lower extremities was not observed. Venograms 3 months after operation demonstrated patency of the anastomosis (Fig. 6). There was no disturbance of renal function, all postoperative blood urea nitrogen values and creatinine clearances (Ccr) being normal. A number of studies were performed 3 weeks, and 3, 8½, and 11 months postoperatively.
Fig. 6.
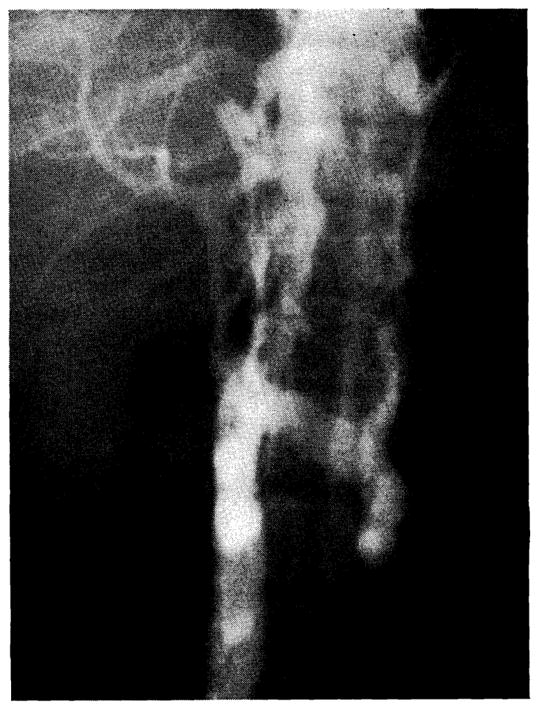
Inferior venocavagram performed 3 months after clinical portacaval transposition. The portacaval anastomosis is indicated (arrow). Note that some dye reaches the liver, but a large amount of the vena caval blood passes through paravertebral collaterals.
The abnormalities in liver function present before operation have been relatively unchanged. Postoperative serum bilirubins, prothrombin times, SGOT, thymol turbidity, plasma proteins, serum lipids, glucose tolerance, and glucagon tolerance have all remained essentially unchanged. Urine steroids and catecholamines remained within normal limits.
Evidence of hypersplenism persisted postoperatively, the white count ranging from 2,600 to 8,200, and platelet counts from 40,000 to 180,000. Hematocrit has increased from 27 to 37 percent.
Alterations in gastric secretion
A comparison of basal, peptone-induced, and histamine-induced gastric secretion are shown in Table II before and after transposition. Although there appeared to be a n increased response to peptone broth stimulation, this was not a marked change and was unattended by alterations in basal secretion. The acid concentration after maximal histamine stimulation appeared to be elevated, although this rise was accompanied by low-volume secretion so that the total acid produced was essentially unchanged. The long-term effect of transposition on gastric secretion will probably not be dissimilar to that which results from portacaval anastomosis. Postoperative gastrointestinal series was normal on Sept. 9, 1964.
Table II.
Effect of human portacaval transposition upon gastric secretion
| Gastric secretion test | Volume (ml.) | HCl (mEq./L.) | Amount HCl per hour (mEq.) |
|---|---|---|---|
| One hour basal | 45 | 21 | 0.95 |
| 31 | 36 | 1.12 | |
| 34 | 51 | 1.7 | |
| 21 | 23 | 0.5 | |
| 29 | 18 | 0.5 | |
| Peptone broth stimulation (1 hour) | 48 | 43 | 2.1 |
| 29 | 35 | 1.02 | |
| 30 | 81 | 2.4 | |
| 26 | 48 | 1.25 | |
| 52 | 76 | 4.0 | |
| Maximum 30 minute histamine response* (15 to 45 minutes after histamine) | 48 | 59 | 5.6 |
| 22 | 75.5 | 3.32 | |
| 20 | 104 | 4.2 | |
| 22 | 78 | 3.4 | |
| 25 | 100 | 5.0 |
Note: Results are listed chronologically within each test according to the following sequence: Oct. 9, 1963 (Preoperative); Nov. 8, 1963; Jan. 10, 1964; July 1, 1964; and Sept. 9, 1964.
Dose: 0.04 mg. per kilogram body weight of histamine acid phosphate S.Q. 100 mg. Benadryl was given intramuscularly 30 minutes before histamine. Note that histamine volumes are for 30 minutes, but that HCl output is expressed per hour.
Analysis of postoperative liver biopsy
On July, 2, 1964, laparotomy was performed for liver biopsy. Histologic study of the section revealed somewhat more cirrhosis than was seen a t the time of the first biopsy (Fig. 4,B), with more cellular infiltration in the periportal areas. Biochemical analysis for glycogen content and structure and for enzyme content were unchanged.
Growth and development
The child, who weighed 65 pounds prior to transposition, weighed 74.2 pounds 11 months later. Her height, which was 49 inches, increased to 53½ inches during this time (Fig. 7). On the Harvard growth chart these changes constituted a weight jump from the sixtieth to the seventy-fifth percentile, and a change from the tenth to the fiftieth percentile in height. Her abdominal girth was markedly reduced, due primarily to a reduction in spleen size. By physical examination and at re-exploration after 7 months, diminution in liver size was also noted, but this could not be objectively documented with a liver scan. The night feedings which she had received during most of her life were discontinued without harmful sequelae. She receives a regular diet. Fifteen postoperative fasting plasma glucoses have been obtained and these have ranged from 62 to 75. She has had 3 or 4 upper respiratory infections in the past year, all controlled with the appropriate antibiotic therapy.
Fig. 7.
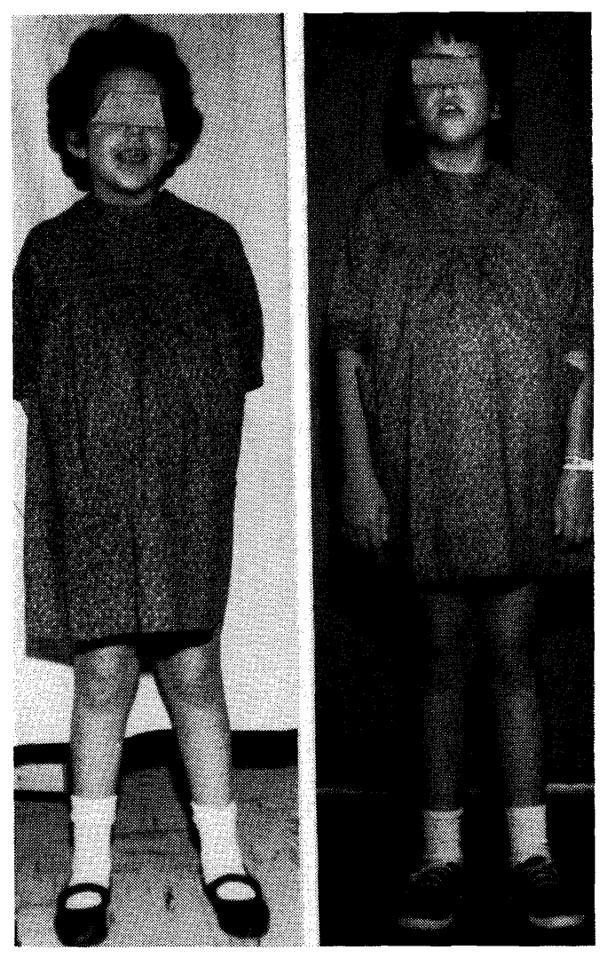
Photographs of patient who underwent portacaval transposition taken one week before (A) and 11 months after (B) operation. An idea of the gain in height can be obtained, since the child is wearing the same dress for both photographs
DISCUSSION
While it has been said by Silen and his associates29 that the liver of the dog with Eck fistula has a reduced glycogen content, the same authors did not believe that a similar deglycogenation occurred after portacaval transposition. Failure to observe this effect may have been due to differences in methodology, since these authors studied autopsy tissue by means of a histochemical determination which does not differentiate TCA soluble from TCA insoluble glycogen. Failure to freeze tissue immediately after biopsy is known to result in rapid changes of TCA soluble fraction.24 With rapid freezing of the biopsy tissue and quantitative biochemical analysis, a substantial reduction in hepatic glycogen was demonstrated in the present study, the principal loss being in the TCA soluble fraction.
It is noteworthy that the same dogs which exhibited a loss of hepatic glycogen also had a change in the glucose tolerance test after transposition. The hyperglycemic response to an oral glucose load was of shorter duration and reduced intensity. This is in contrast to the finding after Eck fistula, in which a diabetic curve is encountered.22
Despite the aforementioned changes in carbohydrate metabolism, the studies with glucagon infusion demonstrate the capacity of the liver for glycogenolysis. Very small doses of glucagon (15 mcg. total) delivered by constant infusion over a 45 minute period caused substantial hyperglycemic responses, either with hindlimb or forelimb infusion. The effect was greater with hindlimb infusion, in which at least part of the glucagon passed directly into the liver, a finding which is in accord with earlier studies.2 It is noteworthy that the child who received 10 to 30 times as much glucagon per weight over a much shorter period of time did not respond with elevations in blood sugar.
In the patient reported, the presence of portal cirrhosis, portal hypertension, and hypersplenism constituted indications for decompression of the splanchnic bed. That portal decompression can sometimes benefit patients with abnormal hepatic deposition of metabolites was demonstrated by Imparato,19 who performed an end-to-side portacaval anastomosis on a 10-year-old child with Gaucher’s disease. The patient was proved by biopsy to have the abnormal depositions of cerebroside in the skeleton, testicle, spleen, mesenteric lymph nodes, and liver. The indication for operation was the presence of massive ascites, which was promptly relieved and which had not recurred 2½ years later. It is of interest that a reduction of hepatomegaly was incidentally observed postoperatively. Unfortunately, metabolic studies were not recorded.
The present case, which required portal decompression, also provided an unusual opportunity to see whether changes in carbohydrate metabolism similar to those observed in dogs with transposition would occur in a patient with glycogen storage disease, recognizing that the enzyme deficiency in the child would diminish the degree of response. Thus the inherent capacity for glycogenolysis in the child would not be altered by the operation, but the liver would be removed from the immediate portal hyperglycemia following meals, and more glucose would be made available for peripheral utilization. The possible value of portal blood flow diversion for this purpose was suggested by Field.7
Working with this hypothesis, the only suitable type of case would be that of the patient with an enzyme defect confined to the liver, in which case the liver would be placed in a more advantageous position, in that its reduced rate of glycogenolysis could be brought into closer conformity to the rate of glycogen storage. If enzyme defects are not present in other tissue, the result will not then be accelerated deposition of glycogen in the heart and muscles and kidneys. The child reported in the present study fulfilled these criteria. The only enzyme deficiency was amylo-1,6-glucosidase and the abnormal glycogen depots were confined to the liver.
After portacaval transposition the child appeared to have been considerably benefited, with a marked decrease in the abdominal girth, a reduction in the size of the upper abdominal masses, a spurt in growth, and a marked increase in physical activity. While the hypersplenism is still present, there has been improvement, the most marked change being in the platelet count.
It is possible that most or all of these effects were the consequence of the portal decompression, although the accelerated growth rate is not easily explained on this basis. Almost certainly the reduction in splenomegaly was due to the relief of portal hypertension. The biochemical analyses performed before operation were essentially unchanged afterward, the enzyme concentration and glycogen content of both liver and muscle being essentially the same. While a reduction in hepatomegaly did occur, it was not of great magnitude. It is possible that such a change in liver size would not be reflected in the tissue analyses, since these were expressed in units per wet weight. Thus, although the concentrations of enzyme and glycogen might remain the same, this would not preclude an over-all decrease in the total amount of glycogen stored in the liver.
In the absence of more decisive evidence, however, it must be conceded that the outcome may not have been different from that which would have occurred with a standard portacaval shunt. If this is so, the child incurred the slight increase in operative risk which would attend the performance of a second vascular anastomosis. Balancing this added risk is the theoretical benefit which might derive from the possibility that the total hepatic blood flow was less severely reduced than would result from a portacaval shunt.
The ultimate prognosis in this case is not known. Both before and after the operation, there has been evidence of serious and continued liver disease. The combination of portal cirrhosis with Type IIIB glycogen storage disease is extremely uncommon and may be fortuitous. Liver function has been stable since the time of operation. Histologically, there appears to have been some progression of the cirrhotic process and the rapidity with which this develops will be of far greater significance in limiting survival than the metabolic defect.
SUMMARY
An investigation was conducted of the influence of portacaval transposition upon carbohydrate metabolism in 45 dogs. In 17 dogs, hepatic glycogen content was measured before and from 45 to 75 days after transposition. A reduction in glycogen content, principally in the TCA soluble fraction, was noted in 14 animals. The mean loss of total glycogen was 51 percent, and the mean loss of TCA soluble glycogen was 70 percent. In control animals hepatic deglycogenation did not occur. Despite the reduction in hepatic glycogen content, the animals were capable of glucagon-induced glycogenolysis using very small test doses. After transposition, a greater response to intraportal injection was noted as compared to that obtained with systemic venous infusions.
Other alterations in carbohydrate metabolism were also measured. These included a reduction in the duration and magnitude of the hyperglycemic response to oral glucose loads. The profile of glycemic response under these conditions was studied, and demonstrated to be greatest in the portal vein, least in the peripheral venous blood, and of intermediate magnitude in the peripheral arteries.
Based upon the hepatic deglycogenating effect of portacaval transposition in dogs, this operation was used for the treatment of an 8½-year-old child with glycogen storage disease and concomitant portal cirrhosis. The portacaval transposition was performed in preference to a standard portacaval shunt. The enzyme defect in the patient was extensively studied before and after transposition. Prior to surgery, she was demonstrated to have Type IIIB glycogenosis (amylo-1,6-glucosidase deficiency confined to the liver). Eight and one half months after operation, the quantities of glycogen in liver and muscle and the enzyme activities showed no significant alteration.
The clinical response to portacaval transposition was gratifying. There has been a decrease in the hepatosplenomegaly, rapid growth, a diminution in the pre-existing hypersplenism, and a considerable increase in the child’s physical activity. Most of these benefits are ascribable to the effective portal decompressive procedure. Whether any metabolic benefit derived from the portacaval transposition is problematical.
Acknowledgments
Aided by United States Public Health Service Grants AM 06283. A 6344, HE 07735, AM 07772, FR 00051, and GM 04761.
Footnotes
Full details of the analytic methods and interpretation of results will be provided upon request to Dr. Barbara Illingworth, Department of Biological Chemistry, Washington University School of Medicine, St. Louis.
References
- 1.Bloom WL, Lewis GT, Shumpert MA, Shen TM. Glycogen fractions of liver and muscle. J Biol Chem. 1951;188:631. [PubMed] [Google Scholar]
- 2.Bürger M. Glucagon, Fortschr. Diag u Ther. 1950;1:1. [Google Scholar]
- 3.Child CG, III, Barr D, Holswade GH, Harrison CS. Liver regeneration following portacaval transposition in dogs. Ann Surg. 1953;138:600. doi: 10.1097/00000658-195310000-00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Clarke JS, Hoffs M, ElFarra S. Effect of portacaval transposition on Heidenhain pouch secretion to various stimuli. S Forum. 1959;10:134. [PubMed] [Google Scholar]
- 5.Clarke JS, McKissock PK, Cruze K. Studies on site of origin of the agent causing hypersecretion in dogs with portacaval transposition. Surgery. 1959;46:48. [PubMed] [Google Scholar]
- 6.Cori GT, Cori CF. Glucose 6-phosphatase of the liver in glycogen storage disease. J Biol Chem. 1952;199:661. [PubMed] [Google Scholar]
- 7.Field RA. In: The metabolic basis of inherited disease. Stanbury JB, Wyngaarden JB, Frederickson DS, editors. New York: McGraw-Hill Book Company. Inc; 1960. p. 187. [Google Scholar]
- 8.Heer FW, Silvius DE, Seipel RS, Harper HA. Effect of portacaval transposition on hepatic blood flow and function in normal and cirrhotic dogs. J Surg Res. 1963;3:213. doi: 10.1016/s0022-4804(63)80059-1. [DOI] [PubMed] [Google Scholar]
- 9.Hers HG. Glycogen storage disease. Rev Internat Hepatol. 1959;9:35. [Google Scholar]
- 10.Hers HG. Amylo-1,6-glucosidase activity in tissues of children with glycogen storage disease. Biochem J. 1960;76:69. [Google Scholar]
- 11.Hers HG. α-Glucosidase deficiency in generalized glycogen storage disease (Pompe’s disease) Biochem J. 1963;86:11. doi: 10.1042/bj0860011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hers HG. Advances in metabolic disorders. I. New York: Academic Press; 1964. pp. 1–44. [Google Scholar]
- 13.Illingworth B. Glycogen storage disease. Am J Clin Nutr. 1961;9:683. [Google Scholar]
- 14.Illingworth B, Brown DH. Action of amylo-1,6-glucosidase on low molecular weight substrates and the assay of this enzyme in glycogen storage disease. Proc Nat Acad Sc. 1962;48:1619. doi: 10.1073/pnas.48.9.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Illingworth B, Brown DH. Ciba Foundation Symposium: Control of glycogen metabolism. London: J. & A. C. Churchill, Ltd; In press. [Google Scholar]
- 16.Illingworth B, Cori GT. Structure of glycogens and amylopectins. III. Normal and abnormal human glycogen. J Biol Chem. 1952;199:653. [PubMed] [Google Scholar]
- 17.Illingworth B, Cori GT, Cori CF. Amylo-1,6-glucosidase in muscle tissue in generalized glycogen storage disease. J Biol Chem. 1956;218:123. [PubMed] [Google Scholar]
- 18.Illingworth B, Larner J, Cori GT. Structure of glycogens and amylopectin. I. Enzymatic determinations of chain length. J Biol Chem. 1952;199:631. [PubMed] [Google Scholar]
- 19.Imparato AM. Gaucher’s disease with ascites, response to portacaval shunt. Ann Surg. 1960;151:431. doi: 10.1097/00000658-196003000-00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kohatsu S, Gwaltney JA, Nagano K, Dragstedt LA. Mechanism of gastric hypersecretion following portacaval transposition. Am J Physiol. 1959;196:841. doi: 10.1152/ajplegacy.1959.196.4.841. [DOI] [PubMed] [Google Scholar]
- 21.Larner J, Villar-Palasi C. Enzymes in the glycogen storage myopathy. Proc Nat Acad Sc. 1959;45:1234. doi: 10.1073/pnas.45.8.1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Loeweneck M, Wachsmuth W. Alimentary hyperglycemia in dogs with Eck fistula. Klin Wchnschr. 1930;9:396. [Google Scholar]
- 23.Mommaerts WFHM, Illingworth B, Pearson CM, Guillory RJ, Seraydarian K. A functional disorder of muscle associated with the absence of phosphorylase. Proc Nat Acad Sc. 1959;45:791. doi: 10.1073/pnas.45.6.791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Merrick AW. Encephalic glycogen differences in young and adult rats. J Physiol. 1961;158:476. doi: 10.1113/jphysiol.1961.sp006781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rex JC, Code CF, ReMine WH. Gastric secretion of acid and urinary excretion of histamine in dogs with portacaval transposition. Ann Surg. 1964;160:193. doi: 10.1097/00000658-196408000-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Seifter S, Dayton S, Novic B, Muntwyler E. Quantitative detection of glycogen fractions by anthrone method. Arch Biochem. 1950;25:191. [PubMed] [Google Scholar]
- 27.Sidbury JB, Cornblath M, Fisher J, House E. Glycogen in erythrocytes of patients with glycogen storage disease. Pediatrics. 1961;27:103. [Google Scholar]
- 28.Sidbury JB, Mason J, Burns WB, Ruebner BH. Type IV glycogenosis. Report of a case roved by characterization of glycogen and studied at necropsy. Bull Johns Hopkins Hosp. 1962;111:157. [PubMed] [Google Scholar]
- 29.Silen W, Mawdsley DL, Wenick WL, Harper HA. Studies of hepatic function in dogs with Eck fistula or portacaval transposition. Arch Surg. 1957;74:964. doi: 10.1001/archsurg.1957.01280120142017. [DOI] [PubMed] [Google Scholar]
- 30.Silen W, Eiseman B. The nature and cause of gastric hypersecretion following portacaval shunts. Surgery. 1959;46:38. [PubMed] [Google Scholar]
- 31.Starzl TE, Scanlon WA, Shoemaker WC, Thornton FH, Wendel RM, Schlachter L. Studies on hepatic blood flow and rate of BSP clearance in dogs with portacaval transposition. Surgery. 1962;52:660. [PMC free article] [PubMed] [Google Scholar]
- 32.Starzl TE, Butz GW, Munger DH. A technique for portacaval transposition (Addendum) J Surg Res. 1961;1:218. doi: 10.1016/s0022-4804(61)80046-2. [DOI] [PMC free article] [PubMed] [Google Scholar]