

Abstract
Th2 lymphocytes deliver essential signals for induction of asthmatic airway inflammation. We previously found that airway antigen challenge induces recruitment of Gr-1+ neutrophils prior to the recruitment of Th2 cells. We examined, therefore, whether Gr-1+ cells contribute to the development of Th2-dependent airway inflammation. Systemic depletion of Gr-1+ cells using the RB6-8C5 monoclonal antibody reduced Th2 cell recruitment following intranasal antigen challenge. The levels of both matrix metalloproteinase (MMP)-9 and the tissue inhibitor of matrix metalloproteinases-1 (TIMP-1) mRNA were up-regulated in the lungs of mice 12 h after intranasal antigen challenge. Up-regulation of TIMP-1 was independent of Gr-1+ cells, whereas up-regulation of MMP-9 RNA and total gelatinolytic activity were dramatically reduced in mice depleted of Gr-1+ cells. At 24 h after challenge, total lung collagenolytic activity was also up-regulated, in a Gr-1+ cell-dependent fashion. Systemic inhibition of MMP-8 and MMP-9 reduced the airway recruitment of Th cells, resulting in significantly reduced eosinophilic inflammation. These data suggest that antigen challenge via the airway activates Gr-1+ cells and consequently MMPs to facilitate the recruitment of Th cells in the airway inflammatory response.
Keywords: asthma, airway inflammation, MMP, TIMP
Introduction
Asthma is a complex disease, characterized by airway obstruction and bronchial hyperresponsiveness (AHR). Bronchial inflammation is a crucial component of asthma pathogenesis, and accumulating data suggest that asthma is due to an aberrant Th2 immune response to commonly inhaled antigens [1]. Despite the clear role of Th2 cells in this disease, little is known about the mechanisms that govern the recruitment of Th2 cells into airways.
Although they are generally present in numbers lower than the numbers of mononuclear cells, polymorphonuclear neutrophils (PMN) also accumulate in the airways of patients with allergic and non-allergic asthma [2, 3]. After allergen challenge of asthmatic patients or mice, PMN are the first inflammatory cells to accumulate within the airways [4–6], and the numbers of PMN recovered from the lungs of patients with allergic asthma by bronchoalveolar lavage (BAL) 4 h after allergen challenge have been calculated to be approximately 90-fold higher than the numbers of PMN recovered from the lungs of healthy controls [7].
In mice, PMN are characterized by surface expression of the Gr-1 antigen [8]. Gr-1 is also expressed by plasmacytoid dendritic cells (pDCs) [9, 10], by a subset of monocytes/macrophages [11, 12], and by immature eosinophils [13]. PMN and perhaps other Gr-1+ cells can be depleted from the circulation of mice by systemic treatment with the anti-Gr-1 monoclonal antibody (mAb), RB6-8C5 [14]. Depletion of circulating Gr-1+ cells has been shown to reduce the severity of inflammatory diseases of the liver [15–17] and eye [15] by inhibition of effector T cell recruitment. Similar results were also seen in an experimental model of fungal allergic airways disease [18]. We speculate that these Gr-1+ cells might contribute to recruitment of CD4+ T cells to the airways via products that they release when they are activated in the tissues. One of the classes of biomolecules produced or induced by Gr-1+ cells that might participate in Th cell recruitment are the family of matrix metalloproteinases (MMPs). Although it is not known whether pDCs or Gr-1+ monocytes /macrophages produce biologically significant quantities of MMPs, PMN have been shown to produce high levels of collagenases and gelatinases including MMP-8 (neutrophil collagenase) and MMP-9 (gelatinase B) [19]. While expression of MMP-8 and MMP-9 can be regulated at the level of gene transcription [20, 21], both enzymes can also be stored within PMN granules, and can be rapidly liberated in response to pro-inflammatory stimuli [19]. A major action of these enzymes is to remodel the extracellular matrix, a process that is thought to facilitate leukocyte trafficking through endothelial barriers and into solid organs [19]. In addition, MMPs can activate chemotactic molecules by proteolytic cleavage [22]. The functions of MMPs are regulated by tissue inhibitors of metalloproteinase (TIMPs). Together, MMPs and TIMPs have broad potential to participate in cell recruitment into the tissues in which they are produced.
We have previously demonstrated that, after airway antigen challenge, adoptively transferred Th2 cells are less efficiently recruited into lungs and airways than adoptively transferred Th1 cells [23]. Using the DO11.10 mouse strain (DO) that carries a transgenic TCR specific for ovalbumin (OVA) peptide 323–339 and intranasal (i.n.) challenge of the airway with OVA, we found that the combination of antigen and endotoxin acts synergistically, recruiting both Th1 and Th2 cells into the site of antigen-induced inflammation [24]. We showed further that recruitment of adoptively transferred Th1 and Th2 cells into the airways of Toll-like receptor 4 mutant (TLR4m) mice was reduced compared to their recruitment into the airways of wild type animals, resulting in less severe airway inflammation in the TLR4m mice following airway antigen challenge. In this system, when mice received adoptive transfer of DO Th2 cells without co-transfer of DO Th1 cells, high doses of i.n. OVA (30 µl of ≥ 0.03% w/v, designated ‘high dose challenge’) were required to induce recruitment of the Th2 cells to the airways and to generate eosinophil associated inflammation. In contrast, when DO Th2 cells were co-transferred with DO Th1 cells, i.n challenge with as little as 30 µl of 0.003% OVA (defined as ‘low dose challenge’) resulted in recruitment of both Th1 and Th2 cells together with development of inflammation with a prominent eosinophil component.
TLR4m mice challenged with antigen via the airway also showed impaired recruitment of PMN into the lungs. PMN are usually recruited rapidly to the airways within minutes to hours after antigen challenge [7]. In contrast, most of the recruited Th cells enter the lung beginning one to two days after antigen challenge [23]. Together, these data led us to hypothesize that the initial TLR4-dependent recruitment of PMN might play an important role in preconditioning the lungs to facilitate the subsequent recruitment of Th1 and Th2 cells.
In this report, we show that Gr-1+ cells play a crucial role in Th1 cell and Th1 cell-dependent Th2 cell recruitment to the airways. MMPs produced at early time points after antigen challenge also participate importantly in this recruitment. Systemic administration of inhibitors of MMP-8 and MMP-9 reduces the recruitment of Th1 and Th2 cells to the airways after antigen challenge, resulting in decreased severity of eosinophilic inflammation. Since expression of TIMP-1 is also up-regulated after antigen challenge, and since a major function of this molecule is to inhibit the MMPs that are induced by Gr-1+ cells, we postulate that the impact of these molecules depends on the local ratios of their expression in key lung microenvironments.
Results
Systemic depletion of Gr-1+ cells results in reduced recruitment of Th1 and Th2 cells into the airways
In previous studies, we showed that PMN are recruited into the lungs and airways shortly after i.n. OVA challenge and approximately 1 d before adoptively transferred OVA-specific DO Th cells enter the tissue [24]. This recruitment of PMN was dependent on normal function of TLR4. Because TLR4-deficient mice showed both failure of neutrophil recruitment and reduced recruitment of adoptively transferred DO Th cells, we hypothesized that PMN might play an immunoregulatory role in facilitating the T cell recruitment that is induced following airway antigen challenge. To test this hypothesis, we used systemic administration of an anti-Gr-1 mAb to deplete PMN and possibly other Gr-1+ cells from the circulation prior to airway antigen challenge. We injected either the depleting anti-Gr-1 or a control mAb i.v. into recipient mice 1 day before adoptive transfer of in vitro differentiated DO Th1 and Th2 cells. A second injection of anti-Gr-1 or control mAb was administered i.v. when mice received the first i.n. OVA challenge, extending the period of neutrophil depletion through at least the second day after challenge (data not shown). The i.n. challenge was with the low dose of 0.003% OVA that we have shown induces Th1 cell-dependent Th2 cell recruitment [24]. The challenge was repeated 6 h later. To assess the extent of OVA-induced airway inflammation, we directly measured the numbers of Th1 and Th2 cells recruited into the airways using flow cytometry of BAL cells. Intranasal OVA challenge of mice following adoptive transfer of DO Th1 and Th2 cells resulted in the recruitment of both Th1 and Th2 cells to the airways (Fig. 1A). Treatment with anti-Gr-1 but not the control mAb resulted in a dramatic reduction of the numbers of Th1 and Th2 cells recovered in the BAL of OVA-challenged mice.
Figure 1. Reduced recruitment of Th1 and Th2 cells into the airways of mice depleted of Gr-1+ cells.
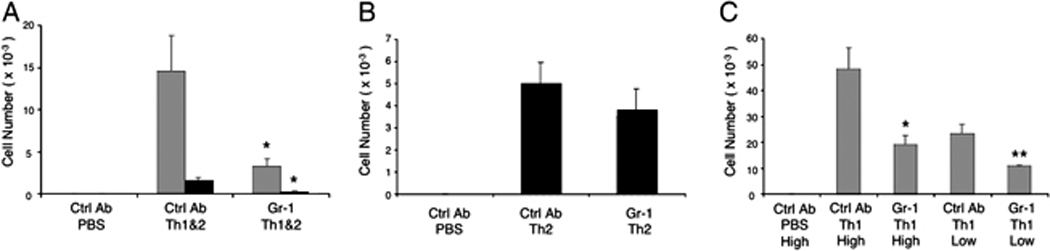
(A) Mice received Th1 and Th2 cells or PBS i.v. were challenged i.n. with the low dose of OVA as described in Materials and Methods. A group of mice were treated with an anti-Gri-1 mAb one day before the transfer of in vitro cultured Th cells and at the same time of the 1st OVA challenge, while other groups of mice received an isotype control mAb as indicated. Three days after Ag challenge DO Th1 cells (gray bars) and Th2 cells (black bars) were recovered from the BAL and enumerated by flow cytometry. (B) Th2 cells were counted in BAL of mice that received Th2 cells only prior to challenge i.n. with the high dose of OVA. (C) Numbers of Th1 cells in the airways were determined 3 days after high or low dose OVA challenge of mice that received Th1 cells only. Data show mean ± SEM (n = 5 mice per group; *p<0.05 and **p<0.01, vs. groups treated with control mAb). Similar results were obtained in at least 3 replicate experiments.
Using this experimental system, we could assess whether Gr-1+ cells contributed selectively to the recruitment of Th1 cells, Th2 cells, or both. Using a high dose (0.03%) of i.n. OVA, recruitment of Th2 cells could be elicited without a requirement for co-recruitment of Th1 cells. We, therefore, transferred DO Th2 cells alone into naïve mice and, after treatment with either anti-Gr-1 or a control mAb, we challenged with the high dose of i.n. OVA. Interestingly, treatment with anti-Gr-1 mAb did not cause a significant reduction in the numbers of Th2 cells recruited after this high-dose OVA challenge (Fig. 1B). As shown previously, only very low numbers of Th2 cells were recovered from the airways of mice that received Th2 cells and that were challenged with either the low dose of OVA or with PBS, regardless of whether they were treated with anti-Gr-1 mAb or not (data not shown).
Our finding that airway Th2 cell recruitment was insensitive to depletion of Gr-1+ cells might have implied that Th2 cells do not rely on signals from Gr-1+ cells for their recruitment to the airways. Alternatively, our data might have indicated that a requirement for signals from Gr-1+ cells could be overcome by administration of high doses of the challenge antigen. To test the impact of high antigen challenge doses, we performed a similar experiment after adoptive transfer of DO Th1 cells without Th2 cells (Fig. 1C). In this experiment, the recruitment of Th1 cells was significantly reduced following depletion of Gr-1+ cells, regardless of whether a low dose or a high dose of antigen was used (Fig. 1C).
It is well recognized that in addition to PMN, eosinophils, plasmacytoid dendritic cells and a subset of monocytes/macrophages also express the Gr-1 surface antigen. Although it has been established that systemic treatment with the RB6-8C5 mAb very effectively depletes both PMN and eosinophils [25], it has not known whether treatment with this antibody depletes the Gr-1+ monocyte/macrophage subset or plasmacytoid dendritic cells. To address this question, we injected either a control mAb or RB6-8C5 i.v. and measured the numbers of Gr-1+ cells in these subsets in the lung 1d after the Ab treatment (Supplementary Figs. 1 and 2). Compared to mice treated with control Ab, mice that had received RB6-8C5 contained very low numbers of PMN (Gr-1hi, CD11b+) and Gr-1+ monocytes/macrophages (Gr-1int, CD11b+) in the lung (Supplementary Fig. 1). Notably, the numbers of plasmacytoid dendritic cells (CD11b−, Gr-1lo, CD11cint, Ly6C+, B220+) (Supplementary Fig. 1) or Th1 (TCRβ+, KJ1-26+, IFN-γ+) cells (Supplementary Fig. 2) were not altered by treatment with RB6-8C5. Eosinophils are not present in detectable numbers in the lungs or airways of either control mice or OVA-sensitized and challenged mice until between the 2nd and 5th day after challenge after CD4+ T cells have been recruited to the lung tissue, thus there are no alterations in eosinophil numbers during this early portion of the response induced by airway antigen challenge. We conclude from these experiments that PMN and possibly Gr-1+ monocytes/macrophages provide signals that help to induce recruitment of Th1 cells, without having a direct effect on recruitment of Th2 cells.
Airway antigen challenge induces up-regulation of lung MMP-9 mRNA in a Gr-1+-cell dependent fashion
MMPs and TIMPs have been shown to affect Th cell recruitment in vitro [26] and were consequently of particular interest as potential effector molecules in the Gr-1+ cell-dependent Th1 and Th2 cell recruitment that we have observed. In order to investigate whether MMPs and TIMPs play a critical role in this model, we first measured their mRNA expression levels in lungs 12 and 24 h after OVA challenge. We chose these time points because airway neutrophil inflammation peaked at 12 h and had largely resolved by 24 h after the antigen was administered and because the majority of Th cells were not recruited to the airways until 2 days after antigen challenge. Lungs from groups of mice were harvested, and RNAs from these tissues were analyzed by RNase protection assay (RPA). At 12 h after the challenge, compared to unchallenged mice, the expression of TIMP-1 was increased in groups of mice challenged i.n. with OVA (Fig. 2A). This over-expression of TIMP-1 mRNA was maintained at 24 h after the challenge (Fig. 2B). In contrast, MMP-9 was up-regulated in lungs 12 h after OVA challenge but had returned to steady state baseline levels at 24 h after challenge (Fig. 2B). Over this time course, antigen challenge elicited no detectable change in expression of MMP-8 mRNA (data not shown). Treatment of mice with anti-Gr-1 mAb ablated the enhanced MMP-9 expression seen at 12 h after challenge but had no effect on baseline MMP-9 expression. In contrast, treatment with anti-Gr-1 did not alter either baseline or stimulated expression of TIMP-1. The intensities of the MMP-9 (Fig. 2C) and TIMP-1 (Fig. 2D) bands at the 12 h time point were quantified using a PhosphoImager. These analyses confirmed that MMP-9 expression was increased substantially after antigen challenge, but not in the lungs of animals that had been treated with anti-Gr-1 mAb (Fig. 2C). In contrast, expression levels of TIMP-1 mRNA were up-regulated equally after antigen challenge in mice treated with control and anti-Gr-1 mAb (Fig. 2D). In order to investigate the potential cellular sources of MMP-8 and MMP-9, we purified PMN (CD11b+ Gr-1hi cells), the Gr-1+ subset of macrophage/monocytes (CD11b+ Gr-1int/lo), resident macrophages plus dendritic cells (CD11b− CD11c+), and other cells (CD11b− CD11c−) by flow sorting from the pool of cells recovered from collagenase-digested lungs of OVA-sensitized and challenged mice. We analyzed MMP-8 and MMP-9 mRNA expression in these purified cell populations using real time RT-PCR (Supplementary Fig 3). Using this sensitive approach, we detected expression of MMP-8 and MMP-9 only in the purified CD11b+ Gr-1hi PMN fraction. With the caveat that this approach would not detect gene expression restricted to stromal cell types that are not released from the tissue by collagenase/DNase digestion, these data indicate that Gr-1+ PMN are responsible for the normal up-regulation of MMP-9 mRNA that is observed after airway antigen challenge. In contrast to the expression of MMP-9 mRNA, the expression of TIMP-1 mRNA was up-regulated in antigen-challenged lungs in a Gr-1+ cell-independent fashion and MMP-8 mRNA expression was not detectably altered after antigen challenge.
Figure 2. Effect of Gr-1+ cell depletion on antigen-induced lung MMP-9 mRNA expression.
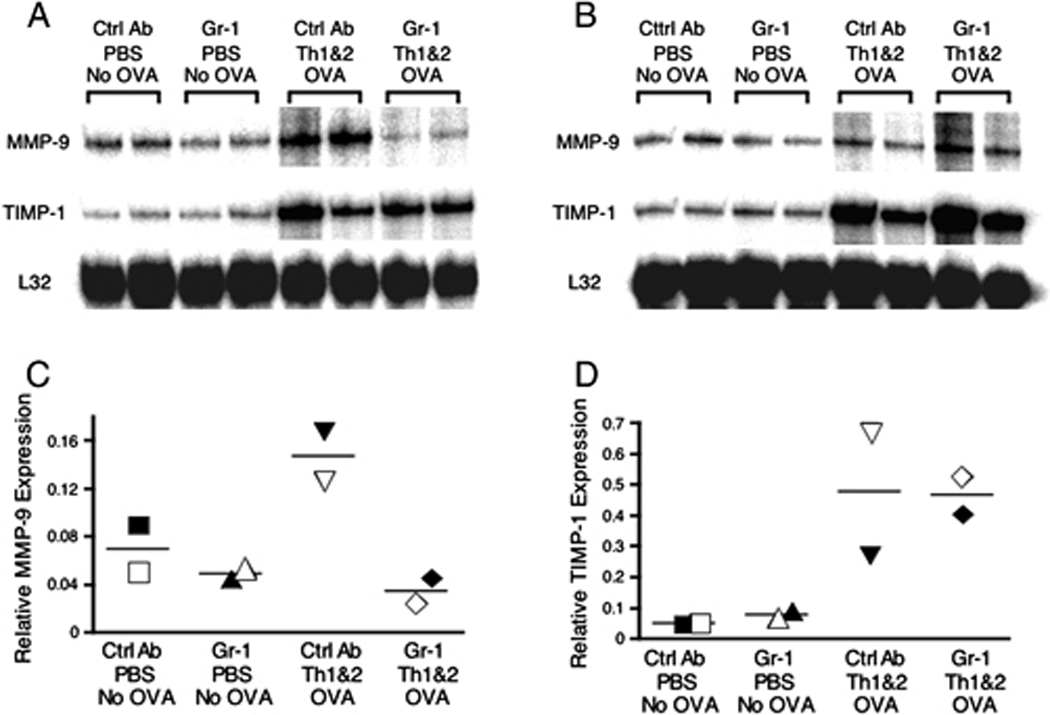
Lung RNA was extracted 12 h (panel A) or 24 h (panel B) after challenge, and the relative expression levels of MMP-9 and TIMP-1 were measured using RPA. To quantify the relative abundance of MMP-9 (C) and TIMP-1 (D) mRNA, the gel shown in panel A was analyzed using a PhosphoImager (GE Healthcare, Piscataway, NJ, USA). Each data point was normalized for gel loading based on the expression of the ribosomal RNA L32. Data shown are representative of 2 independent experiments.
Intranasal OVA challenge up-regulates lung gelatinase and collagenase activities in a Gr-1+ cell-dependent fashion
It is well established that many MMPs must be activated after secretion in order to express their protease function [27]. In order to assess the functional significance of the MMP-8 and MMP-9 gene expression that we observed at the RNA level, we also tested, therefore, whether gelatinase or collagenase activities were present in control or OVA-challenged lungs. We felt it was particularly important to assess gelatinolytic and collagenase activities in situ because the expression of TIMP-1 mRNA was up-regulated after antigen challenge, and the functional proteolytic activity was expected to be determined locally by the combined levels of the proteases themselves and of the endogenous protease inhibitors. In order to focus on the role of Gr-1+ cells as modulators of proteolytic activities, we employed in situ zymography to assess the overall activities of gelatinase and collagenase in the OVA-challenged lungs of mice that had been treated with or without the depleting anti-Gr-1 mAb. We tested lungs 12 and 24 h after antigen challenge. Airway OVA challenge resulted in little change in total gelatinolytic activity (Fig. 3A and 3B). Since OVA challenge induced increased expression of both MMP-9 and TIMP-1 (Fig. 2A), these data suggested that antigen challenge resulted locally in balanced changes in both the gelatinolytic proteinase and the endogenous proteinase inhibitors, maintaining unaltered total gelatinolytic function. In contrast, gelatinolytic activities were reduced at both 12 and 24 h after antigen challenge in the lungs of mice treated with anti-Gr-1 mAb. This was consistent with the OVA-induced up-regulation of TIMP-1 mRNA expression together with no change in the expression of MMP-9 mRNA in mice treated with anti-Gr-1 mAb (Fig. 2).
Figure 3. Reduced gelatinolytic and collagenolytic activities in the lungs of mice depleted of Gr-1+ cells.
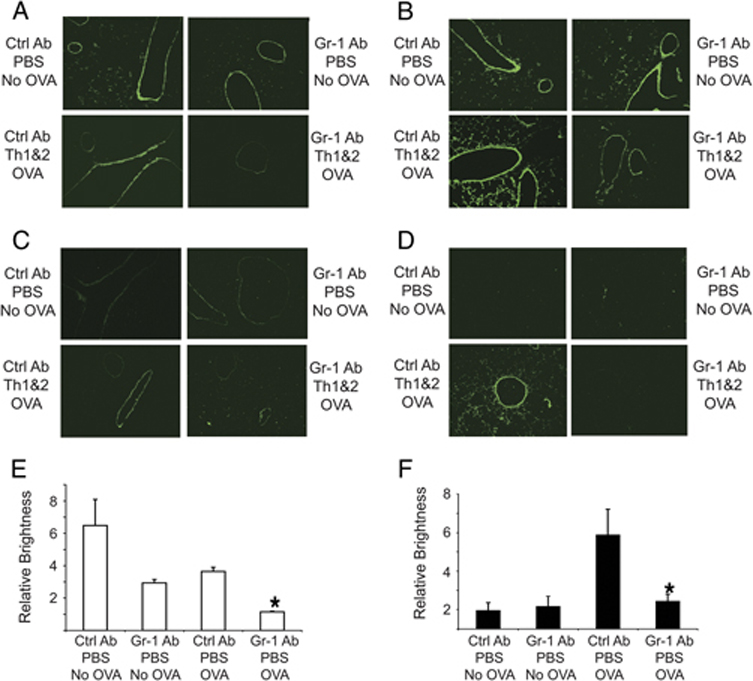
Mice received DO Th1 + Th2 cells followed by anti-Gr-1 or isotype control mAb and were challenged with the low dose of OVA. 12 h (A, C) or 24 h (B, D) after antigen challenge, lung sections were overlaid with fluorescently labeled DQ gelatin (A, B) or DQ collagen type I (C, D). (Magnification 10X) The relative intensities of gelatinolytic (E) or collagenolytic (F) activities in the lungs of mice (3 mice per data point) 24 h after PBS or OVA challenge (B and D) were quantified using the ImageJ software. Data represent means ± SEM, (*p<0.05, vs. groups treated with control mAb). Data shown are representative of 3 independent experiments.
In contrast to the results using in situ zymography with gelatin, in situ analysis of collagenolytic activity showed only very low levels of activity at baseline or at 12 h after OVA challenge, with no detectable effect of treatment with anti-Gr-1 mAb (Fig. 3C); however, at 24 h after OVA challenge, there was a substantial induction of collagenolytic activity that appeared to be absolutely dependent on Gr-1+ cells (Fig. 3D). Since analysis of total lung RNA had showed no increase in MMP-8 transcripts at either 12 or 24 h after airway challenge, the upregulation of lung collagenolytic activity seen here could represent release of preformed MMP-8 rather than de novo biosynthesis of this proteinase. Similar data were obtained from the mice that were challenged with OVA but that had not been treated with adoptive transfer of transgenic Th cells (data not shown). We quantified the overall intensity of gelatinase or collagenase activities from the in situ zymograms performed 24 h after antigen challenge by analyzing positive pixels using the ImageJ software package (Figs. 3E and 3F). These data demonstrated that there were substantial reductions of gelatinolytic activity in the lung of OVA-sensitized and challenged mice that had been treated with anti-Gr-1 mAb (Fig. 3E). Quantitative analysis of gelatinolytic activity in lung sections prepared 12 h after Ag challenge showed similar results (data not shown). In contrast, collagenolytic activities observed by in situ zymography in the lungs of OVA-sensitized and challenged mice analyzed 12 h after challenge did not show significant differences compared to unchallenged mice (data not shown); however, ImageJ quantitation demonstrated a several-fold increase in collagenase activity in the lungs of mice treated with the control Ab at 24 h after the Ag challenge (Fig. 3F). This increase was largely prevented by treatment with the anti-Gr-1 antibody. Altogether, these results indicate that Gr-1+ cells, not Th cells, contribute importantly to the maintenance of lung gelatinolytic activity and to the upregulation of lung collagenolytic activity after airway antigen challenge.
Down-regulation of MMP-9 activity in the lungs of OVA-challenged mice treated with anti-Gr-1 mAb
In order to determine which of the two mouse gelatinolytic MMPs (MMP-2 or MMP-9) [19] was responsible for the gelatinase activities detected by in situ zymography in lungs of OVA-challenged mice (Fig. 3A and 3B), we performed gelatinolytic PAGE-zymography. At 12 h after OVA challenge, MMP-9 activity was modestly increased in homogenates of lungs from challenged mice that had received adoptively transferred DO Th1 and Th2 cells, and was dramatically reduced in lungs of mice that had been treated with anti-Gr-1 mAb before airway antigen challenge (Fig. 4A). MMP-2 activity was not changed in the lungs of mice analyzed under any of these test conditions. By quantifying the intensity of each zymographic band and plotting the normalized values (Fig. 4B), we observed that antigen challenge caused an increase in total MMP-9 activity, but that removal of Gr-1+ cells ablated not only the challenge-induced increase in MMP-9 activity, but also the baseline activity that was observed in unchallenged mice. We also analyzed MMP-2 and MMP-9 activity in homogenates of lungs harvested 24 h after antigen challenge, and found results indistinguishable from those observed at 12 hours after challenge (data not shown). Similar results were obtained in OVA-challenged mice that had not been treated with adoptive transfer of transgenic Th cells, indicating that the decreased activity of MMP-9 was independent of Th cell (data not shown). Altogether, these data led us to conclude that, in mice depleted of Gr-1+ cells, MMP-9 activity was selectively reduced shortly after airway antigen challenge.
Figure 4. Depletion of Gr-1+ cells ablates detectable MMP-9 activity in lungs of challenged mice.
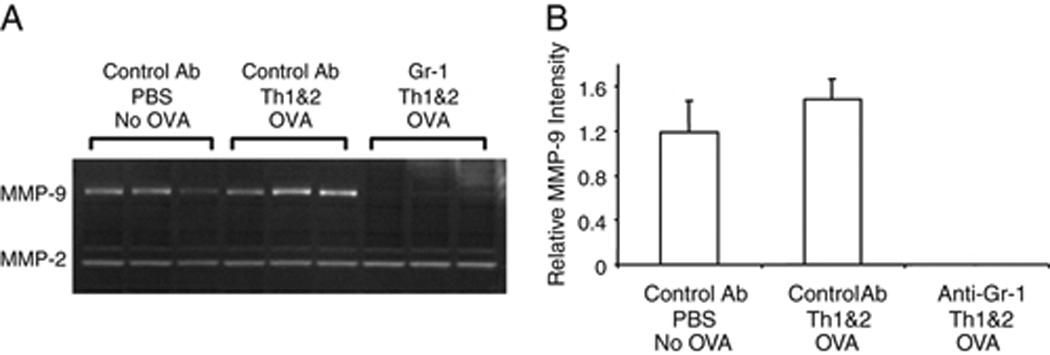
Mice were treated with anti-Gr-1 or control mAb followed by DO Th1 and Th2 cells and challenged with the low dose of OVA. (A) 12 h later, proteins were extracted from lungs and analyzed by PAGE-zymography to identify the activities of MMP-2 and MMP-9. (B) Gelatinase activities in the PAGE zymograms were quantified using a Kodak Image Analysis workstation. The signals for MMP-9 activity from the lungs of mice that were treated with the anti-Gr-1 mAb were below the detection limit; therefore, the observed differences compared to the signals in mice that received the control mAb were highly significant. Data shown are representative of three independent experiments, each with 2 animals per group.
Given that our prior RPA showed that MMP-9 gene expression was maintained at a low level in antigen challenged mice that had been pre-treated with anti-Gr-1 mAb, we hypothesize that the absence of detectable MMP-9 gelatinolytic activity in lung homogenates of mice treated in this fashion indicates that a TIMP acts to inhibit the low levels of MMP-9 that are expressed. TIMP-1 is probably responsible for this effect because mRNA expression levels of the other members of the TIMP family were not regulated after OVA challenge (data not shown). Interestingly, under the same conditions that lead to extinction of MMP-9 activity, the gelatinolytic activity that was due to MMP-2 was not detectably suppressed (Fig. 4A). These data suggest that MMP-2 and MMP-9 are differentially available to inhibition by TIMP-1 in the lung.
MMP-9 is primarily localized near endothelial cells in the lung
Since the gelatinolytic activities mediated by MMP-9 and by MMP-2 differed in their abilities to be inhibited by TIMPs, we next analyzed the tissue localization of MMP-9 in the lungs of mice that had received adoptive transfer of DO Th1 and Th2 cells prior to OVA challenge. We compared total gelatinolytic activity (Fig. 5A) and MMP-9 immunoreactivity (Fig. 5B) in serial sections prepared from lungs harvested 12 h after airway antigen challenge. The highest levels of gelatinolytic activity co-localized with the endothelial cell layers of medium to large sized blood vessels in the lungs and also with the epithelial cell layers of the conducting airways. There were also low levels of gelatinase activity in the parenchymal areas of the lungs. In contrast, immunofluorescent staining with an anti-MMP-9 mAb showed that MMP-9 protein co-localized primarily with the endothelial cell layers that showed gelatinolytic activity. MMP-9 immunoreactivity was also present apparently on cells that were dispersed throughout the lung parenchyma. Interestingly, MMP-9 immunoreactivity was not found associated with the epithelial cells of conducting airways, suggesting that the gelatinolytic activity present in these areas might indicate selective expression of MMP-2 in this tissue compartment. We have not, however, been able to test this hypothesis directly because we have not identified any anti-murine MMP-2 antibodies suitable for immunofluorescent or immunohistochemical staining. Also of interest, the MMP-9 that was localized in the lung parenchyma showed little gelatinolytic activity when analyzed by in situ zymography (Fig. 5A). This suggests that the MMP-9 that is located in the lung parenchyma either has not been proteolytically activated or is inhibited locally by TIMPs. Altogether, these data demonstrate that MMP-9 whose activity is regulated following airway antigen challenge is expressed at or near the endothelial cell layers in the lungs, a location that would enable it to play an important role in the recruitment of leukocytes from the circulation to the lung parenchyma and airways.
Figure 5. MMP-9 immunoreactive material and gelatinase activity co-localize in endothelial cell layers of airway-associated blood vessels.
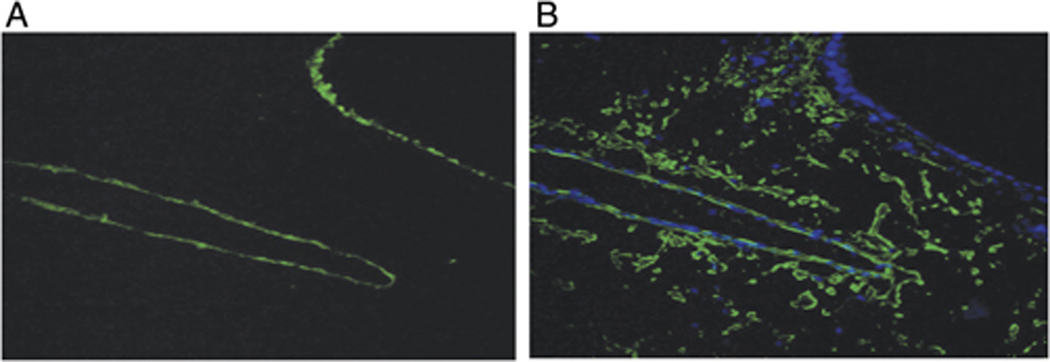
In vitro differentiated DO Th1 and Th2 cells were transferred into naïve mice and were followed by i.n. OVA challenge. 12 h after antigen challenge, lungs were frozen in OCT and serial sections were prepared. (A) One section was analyzed for gelatinolytic activity by in situ zymography. (B) A serial section was fixed, stained for MMP-9 (green), counterstained with Hoechst 33342 (blue), and analyzed using epifluorescence microscopy (Magnification 40X). Results are representative of three independent experiments.
Treatment with inhibitors of MMP-8 and MMP-9 reduces recruitment of Th1 and Th2 cells to the airways, also reducing eosinophilic inflammation
These studies have established that Gr-1+ cells contribute importantly to the recruitment of adoptively transferred Th1 and Th2 cells to the lungs, and that Gr-1+ cells participate in the local up-regulation of expression of MMP-9 gelatinolytic activity and MMP-8 collagenolytic activity. To assess whether these changes in metalloproteinase activity are physiologically significant, we used systemic inhibitors to test the roles of MMP-8 and MMP-9 in the development of Th cell-driven airway inflammation after i.n. antigen challenge. Among the three collagenases (MMP-1, MMP-8, and MMP-13) that are expressed in mice, we focused for this experiment on MMP-8 because PMN are the major source of MMP-8, and because PMN store and can secrete large amount of this enzyme. For these experiments, mice were treated first with adoptive transfer of DO Th1 and Th2 cells, and then 30 minutes before the first i.n. antigen challenge, the mice were treated with inhibitors of MMP-8 and MMP-9 administered by i.p. injection. Additional doses of inhibitors were injected daily until the airway inflammatory cells were analyzed 72 h after the antigen challenge. Due to their chemical characteristics and the nature of the solvent required to dissolve them for injection, we were not able to administer sufficiently high doses of these inhibitors to achieve full suppression of MMP activity without observing nonspecific effects (see Methods). All mice that received Th cells and that were challenged with i.n. OVA developed eosinophilic airway inflammation; however, treatment with inhibitors of MMP-8 and MMP-9 reduced the quantity of airway inflammation as determined by both total leukocyte counts and by analysis of the numbers of airway eosinophils (Fig. 6). The reduced inflammation in mice treated with inhibitors of MMP-8 and MMP-9 in vivo was associated with reduced recruitment of Th1 and Th2 cells (Fig. 6). Additional studies using the inhibitors individually showed no significant reduction in airway inflammatory cells after airway antigen challenge (data not shown). These data supported the conclusion that MMP-8 and MMP-9 together play a crucial role in recruiting Th1 and Th2 cells into the airways.
Figure 6. Pharmacologic inhibition of MMP-8 and MMP-9 decreases eosinophilic airway inflammation.
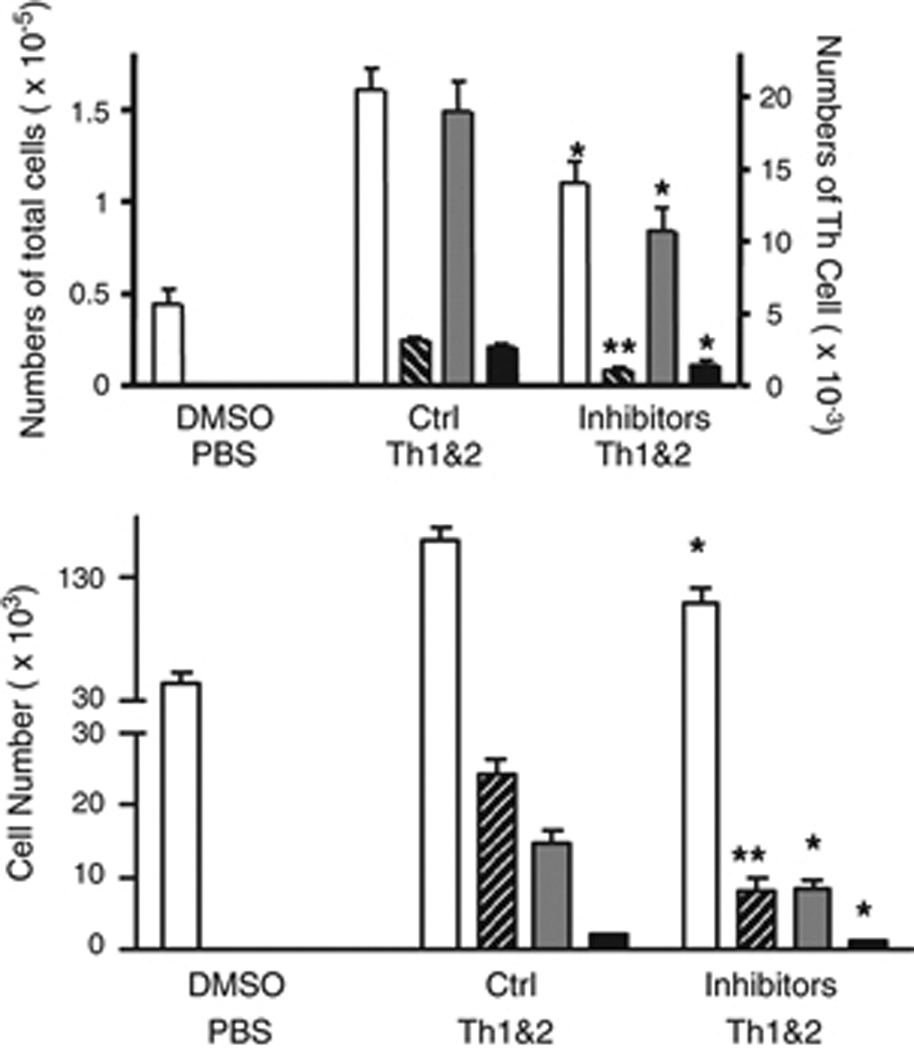
Mice received DO Th1 and Th2 cells followed by i.n. airway challenge with the low dose of OVA (0.003%). Inhibitors of MMP-8 and MMP-9 or control inhibitors were administered daily, beginning 30 minutes before the airway challenge. Three days later, total BAL cells (open bars), BAL eosinophils (cross-hatched bars), DO Th1 (gray bars) and Th2 cells (black bars) were enumerated (mean ± SEM,; * and **: p<0.05 and p<0.01, compared to groups (5 mice per group) treated with inactive control inhibitors). Similar results were obtained in 4 additional experiments.
Discussion
The experiments reported here demonstrate that Gr-1+ cells provide crucial signals for the recruitment of Th1 and Th2 cells into the airways following intranasal antigen challenge. Our data show further that MMPs and TIMP-1 are key mediators supporting this recruitment. When mice received an airway antigen challenge under conditions that lead to mobilization of Gr-1+ cells to the lungs, total lung MMP-9 and TIMP1 mRNAs were up-regulated 12 h after challenge, and MMP-9 gelatinolytic activity was also modestly increased, indicating that the increase in MMP-9 expression was greater than the increase in TIMP-1 expression. In contrast, when mice were treated with a depleting anti-Gr-1 mAb prior to airway OVA challenge, the expression of MMP-9 mRNA remained at baseline levels 12 and 24 h after challenge indicating that under normal conditions Gr-1+ cells are needed to provide signals that lead to activation of MMP-9 expression. In contrast, the expression of TIMP-1 mRNA remained increased indicating that the antigen-induced increase of TIMP-1 was via a mechanisms independent of Gr-1+ cells. In the context of the unstimulated MMP-9 mRNA expression, the fraction of the gelatinase activity that was due to MMP-9 was drastically decreased in mice that had been depleted of Gr-1+ cells. This was probably due to inhibition of lung MMP-9 by locally increased production of TIMP-1. In contrast, collagenase activity as visualized by in situ zymography was increased substantially in an absolutely Gr-1+ cell-dependent fashion in the lungs of mice challenged with antigen, but without an increase in the levels of MMP-8 mRNA. This suggests that the primary regulation of MMP-8-induced collagenase activity is either at the level of secretion of intracellular stores of the enzyme, or at the level of activation of the secreted proenzyme and that this process is dependent on the presence of Gr-1+ cells in the lungs. Finally, the increased expression of collagenase and gelatinase activity for the expression of allergic airway inflammation was established because inhibition of MMP-8 and MMP-9 activities in vivo reduced airway recruitment of both Th1 and Th2 cells, and consequently recruitment of other inflammatory cells.
With the caveat that pDCs and small subsets of monocytes/macrophages also express Gr-1 and can be either depleted (Supplemental Fig. 1) or functionally impaired in mice treated systemically with anti-Gr-1 antibody [28, 29], PMN remain the most likely cell type to facilitate Th cell recruitment into airways via this MMP-dependent mechanism because: i) pDCs were not depleted in the lungs of mice treated with the RB6-8C5 mAb; ii) PMN have been shown to produce large amounts of MMP-8 and MMP-9 [19]; iii) in the lungs of OVA sensitized and challenged mice, the mRNAs of MMP-8 and MMP-9 were predominantly found in PMN; iv) the time course of MMP-9 mRNA expression correlates with the time course of PMN migration into the airways, peaking at 12 h and returning to baseline by 24 h after antigen challenge [5]; v) PMN from the BAL of asthmatic patients have previously been shown to produce MMP-9 [30]; and vi) local recruitment of PMN has previously been shown to change the phenotype of endothelial cells at sites of inflammatory cell recruitment [31, 32].
Gr-1+ cells and the MMPs that they induce or express themselves have been shown to play key roles in other mouse models of inflammatory diseases. First, Lee and her coworkers showed that depletion of Gr-1+ cells using the same anti-Gr-1 mAb used here resulted in reduced expression of MMP-9 and reduced neo-angiogenesis in the corneas of mice infected with herpes simplex virus [15]. These authors suggested that PMN were the major sources of MMP-9 because these leukocytes had prominently infiltrated the cornea shortly after virus infection. The roles of Gr-1+ cells and MMPs in the recruitment of CD4+ T cells and other leukocytes were not, however, investigated in this ocular virus infection model. In another study investigating inflammation by hepatitis B virus (HBV), expression of HBV protein in the liver led to the recruitment of adoptively transferred HBV-specific CD8+ T cells, as well as antigen nonspecific CD4+ and CD8+ T cells [16, 17]. In this experimental model, systemic depletion of Gr-1+ cells did not affect the recruitment of HBV-specific CD8+ T cells, but did lead to a significant reduction in the recruitment of antigen nonspecific CD4+ and CD8+ T cells, as well as other leukocytes. This resulted in less tissue damage in the HBV protein-expressing liver. Our study differs from the above studies in that the systemic depletion of Gr-1+ cells in our model resulted in reduced recruitment of antigen-specific CD4+ Th1 and Th2 cells into the airways. Possible explanations for the differences between our study and those previously published include the following: 1) the mechanisms governing the recruitment of OVA-specific CD4+ Th2 cells and virus-specific CD8+ T cells may differ; 2) differences between the microenvironments of the lung and the liver may result in altered lymphocyte homing behavior in these two organs; 3) the expression of TIMP-1 (not measured in the liver inflammation model) may vary importantly in the liver compared to the lung and may be differentially regulated following airway antigen challenge compared to over the course of a viral infection; and 4) the phenotypes of the transgenic anti-OVA CD4+ T cells and the anti-HBV CD8+ T cells in terms of the factors that govern their tissue recruitment may differ.
Not definitively addressed in any of these studies is the question of which cell type(s) produce the physiologically relevant MMPs. Although Gr-1+ cells, especially PMN, can produce large amounts of MMPs, further studies will be necessary to address whether in vivo PMN themselves produce the functionally significant MMPs, or rather stimulate other cells, such as endothelial or epithelial cells, to produce these important enzymes. Our studies using in situ zymography show that gelatinase and collagenase activity are localized to the areas of blood vessels and medium-sized airways even prior to antigen challenge. This suggests either that, in the lung, cells that are not leukocytes express significant quantities of MMPs, or that leukocyte-derived MMPs are retained in association with these structures after they have been secreted from the originally producing cells.
Studies by Vermaelen and colleagues [32] and by McMillan and colleagues [33] have examined the role of MMP-9 in the development of airway inflammation using MMP-9-deficient mice. Interestingly, these two reports yielded substantially different data. In Vermaelen’s study, a protocol that induces asthmatic airway inflammation in wild type mice yielded reduced airway inflammation in MMP-9−/− animals. Their data also suggested that, in the absence of MMP-9, airway dendritic cells were not efficiently replenished in the course of an ongoing antigen-driven airway inflammatory response, and that this was mechanistically involved in the reduced airway inflammation observed in the MMP-9-deficient animals. In contrast, McMillan and colleagues found that MMP-9−/− mice developed more severe inflammation than wild type mice following airway sensitization and challenge [33]. Since these two groups used different protocols to sensitize and challenge the mice, it is difficult to dissect the mechanisms underlying the differences that were observed.
In another experimental model of asthma in mice [34], systemic treatment with MMP inhibitors prior to and following airway antigen challenge reduced both the quantity of recruited airway inflammatory cells and the extent of AHR, supporting the participation of MMPs in the inflammatory pathophysiology. Lee and colleagues demonstrated that treatment with doxycyclin (a broadly active MMP inhibitor) and other synthetic MMP inhibitors reduced airway inflammation in a murine model of toluene diisocyanate-induced asthma [35]. Neither of these two studies, however, addressed the recruitment of Th cells. Lastly, Park and colleagues, using an experimental model of fungal allergic airway disease, found an association between lung MMP-9 activity, airway neutrophil recruitment, and airway reactivity [18].
Many studies have detected MMPs in the airways of human subjects with asthma. Important insights have come from more recent studies that examined the ratio of the activities of the MMPs and the TIMPs, and correlated these ratios with disease pathogenesis. It has been generally accepted that increased ratios of MMP-9 to TIMP-1 in BAL [4, 30], serum [36], or sputum [37, 38] are indicators of severe asthma. Consistent with this, treatment of asthmatic patients with glucocorticoids both reduces expression of MMP-9 and increases expression of TIMP-1 [37]; however, the extent to which these changes are required for the expression of human asthma remains undefined.
Our studies demonstrate an important action of Gr-1+ cells, most likely PMN, as mediators of key signals for the recruitment of Th cells to the airways, thereby supporting the development of asthmatic airway inflammation. The products that mediate this Gr-1+ cell effect includes MMPs, with the ratio between MMP-9 and/or MMP-8 and TIMP-1 appearing to play a crucial role in the Th cell recruitment process. Our findings support a model in which innate activation of the airway causes the recruitment of PMN and perhaps other Gr-1+ cells to the airways, and these cells, using an MMP-dependent mechanism, condition the lung microenvironment for subsequent recruitment of Th1 and Th2 cells. Recruitment of Th cells ultimately supports the full expression of eosinophil-predominant asthmatic airway inflammation. This model suggests that both Gr-1+ cells and MMPs will be fruitful targets for development of new anti-inflammatory asthma therapies.
Materials and Methods
Mice
BALB/c mice (6–12 wks old) were from Jackson Laboratory, Bar Harbor, ME, USA. DO mice [39] and Thy1.1+ DO mice were gifts of Kenneth Murphy (Washington University, St. Louis, MO, USA) and Osami Kanagawa (RIKEN, Yokoyama, Japan), respectively. All mice were kept in microisolator cages in the specific pathogen-free Animal Resources Program facility at the University of Alabama at Birmingham (UAB). All experiments were approved by the Institutional Animal Care and Use Committee at UAB.
Adoptive transfer and challenge
OVA-specific Th1 and Th2 cells were generated by two rounds of in vitro culture of DO spleen cells with irradiated syngeneic spleen antigen presenting cells, OVA, and cytokines as described [40]. At d 7 of the second round of culture, approximately 50% of the CD4+ cells in the Th1 cell culture were IFN-γ+IL-4− and 40% of the cells in the Th2 cell culture were IFN-γ−IL-4+ based on intracellular staining with anti-cytokine antibodies after culture for 6 h with PMA (10 ng/ml), ionomycin (1 µM), and monensin (2 µM). Seven days after the second stimulation, 107 Th1 and/or Th2 cells were injected i.v. into naïve recipient mice. The following d, under light anesthesia with Isoflurane (Schering-Plough Animal Health Corp., Union, NJ, USA), mice were challenged twice, 6 h apart with OVA (grade V, Sigma, St. Louis, MO, USA). Intranasal challenges were with 0.03% (designated “high dose”) or 0.003% (designated “low dose”) OVA in 30 µl of PBS. We have shown previously that DO cells differentiated in this fashion maintain their polarized Th1 and Th2 phenotype after adoptive transfer and airway antigen challenge [23]. This conclusion was based on brief stimulation in vitro of cells recovered from BAL with PMA and ionomycin prior to analysis by intracellular cytokine staining. In the current study, we have used the KJ1-26 T cell receptor clonotypic antigen to mark the transgenic Th cells and the allotypic Thy1.1 and Thy1.2 antigens to distinguish Th1 and Th2 cells recovered in the BAL.
Analysis of airway and lung inflammation
Three d after challenge, mice were sacrificed by lethal injection; airway cells were collected by BAL; and finally airway leukocytes were analyzed by flow cytometry and by staining of cytospin preparations [24]. In some experiments, the left upper lobe was tied off after lavage and frozen at −80°C for either RNA or protein analysis. The rest of the lungs were inflated with a 1:1 mixture of Optimal Cutting Temperature compound (OCT, Sakura Finetek, Torrance, CA, USA) and PBS, and frozen for analysis by in situ zymography.
Treatment in vivo with neutralizing antibodies and protease inhibitors
Hybridomas producing a rat IgG2b mAb specific for mouse Gr-1 (clone RB6-8C5)[41] and a rat IgG2b isotype control mAb were provided by Emil Unanue (Washington University) and John Kearney (UAB), respectively. Purified anti-Gr-1 or the isotype control mAb (100 µg in 50 µl of PBS) was given i.v. the day before Th cells were transferred, and again at the time of the first OVA challenge. Following this protocol, a greater than 98% depletion of blood PMN was achieved, and no detectable PMN or eosinophils were seen in BAL samples throughout the time course of these experiments. Inhibitors of MMP-8 (IC50=4nM) and MMP-9 (IC50=5nM), and an inactive molecule, chemically related to MMP-8 as a control were purchased from Calbiochem (La Jolla, CA, USA) and injected i.p. (2 mg/kg of MMP-8 and control inhibitor, 7.65 mg/kg of MMP-9 inhibitor) in 50 µl of DMSO once a day starting 30 min before the first antigen challenge.
RNase Protection Assay
Following BAL, total lung RNAs were extracted using the RNeasy kit (Qiagen). Probes for MMP-9, TIMP-1, and L32 were kindly provided by Dr. Iain L. Campbell (Univ. of Sydney, Sydney, Australia) [42]. Levels of MMP-9, TIMP-1, and L32 RNA were determined using the RiboQuant RPA kit (BD Biosciences).
In situ zymography
In situ gelatinase and type I collagenase zymography were performed as previously described [16, 43]. Frozen sections (8 µm) of lungs were overlaid with assay solution (50 mM Tris-HCl, 5 mM CaCl2, 9.2 mM NaN3, pH 7.5) containing either 40 µg/ml FITC-labeled DQ gelatin or 40 µg/ml FITC-labeled DQ collagen type I (Invitrogen, Eugene, OR, USA). Sections were incubated at 37°C for 18 h and examined by fluorescence microscopy.
Gelatin PAGE-zymography
Gelatinolytic activity was determined as described by Heo and coworkers [44]. Frozen lungs were homogenized in 1.0% Triton X-100 (Sigma-Aldrich) in PBS. The protein concentration of each sample was measured with the Dc protein assay kit (Bio-Rad, Hercules, CA, USA) using OVA as a standard. Total lung lysates were mixed with sample buffer (80 mM Tris-HCl, pH 6.8, 4% SDS, 25% Glycerol, 0.01% Bromophenol Blue) and loaded into a 10% SDS-polyacrylamide gel containing 1 mg/ml gelatin (Bio-Rad). After electrophoresis, gels were soaked in renaturation buffer (2.5% Triton X-100) for 1 h at room temperature, and then incubated in 50 mM Tris-HCl, pH 7.5, 200 mM NaCl, 5 mM CaCl2, 0.02% Brij-35 (Bio-RAD) at 37°C overnight. After incubation, gels were stained with Coomassie Brilliant Blue R-250 for 1 h and destained. Zones of gelatinolytic activity were detected as clear bands on a dark blue background. Molecular weight markers as well as individually electrophoresed samples of purified recombinant MMP-2 or MMP-9 (R&D Systems, Minneapolis, MN, USA) were used to estimate the molecular weights of gelatinolytic fractions.
Immunofluorescent Staining
Tissue sections (8 µm thick) were cut from OCT-embedded frozen lungs, then fixed with 0.1M citric acid and permeabilized with 1% Triton X-100. The endogenous peroxidases were blocked by incubation with 1% H2O2 for 15 min prior to reaction of sections with 10 µg/ml of polyclonal rabbit anti-mouse MMP-9 IgG (Chemicon, Temecula, CA, USA) or nonspecific rabbit IgG (Southern Biotech, Birmingham, AL, USA). Subsequently, the sections were washed and incubated with goat anti-rabbit IgG conjugated to HRP (Southern Biotech), and developed with the tyramide signal amplification kit (Perkin Elmer, Boston, MA, USA). Sections were counterstained with Hoechst 33342 (Invitrogen).
Statistical analyses
Statistical analyses were performed by two-tailed equal variant Student’s t test. A p value <0.05 was considered statistically significant.
Supplementary Material
Acknowledgement
We thank Joanne Murphy-Ullrich, Lisa M. Schwiebert, Daniel C. Bullard and Thuc vy Le for general discussion, Namasivayam Ambalavanan for suggestions regarding methods for analysis of MMPs, Trenton Schoeb for advice regarding histology, and Kathy May and Tea Hyun Kim for technical assistance. This work was supported in part by NIH grant HL073907 (D.D.C).
Nonstandard abbreviations used
- AHR
airway hyperresponsiveness
- APC
allophycocyanin
- BAL
bronchoalveolar lavage
- DO
DO11.10 mice
- HBV
hepatitis B virus
- mAb
monoclonal antibody
- pDC
plasmacytoid dendritic cells
- PMN
polymorphonuclear neutrophil
- RPA
RNase protection assay
- TIMP-1
tissue inhibitor of matrix metalloproteinases-1
- TLR4m
Toll-like receptor 4 mutant
- UAB
University of Alabama at Birmingham
References
- 1.Liu AH. Endotoxin exposure in allergy and asthma: reconciling a paradox. J. Allergy Clin. Immunol. 2002;109:379–392. doi: 10.1067/mai.2002.122157. [DOI] [PubMed] [Google Scholar]
- 2.Bradley BL, Azzawi M, Jacobson M, Assoufi B, Collins JV, Irani AM, Schwartz LB, Durham SR, Jeffery PK, Kay AB. Eosinophils, T-lymphocytes, mast cells, neutrophils, and macrophages in bronchial biopsy specimens from atopic subjects with asthma: comparison with biopsy specimens from atopic subjects without asthma and normal control subjects and relationship to bronchial hyperresponsiveness. J. Allergy Clin. Immunol. 1991;88:661–674. doi: 10.1016/0091-6749(91)90160-p. [DOI] [PubMed] [Google Scholar]
- 3.Gibson PG, Simpson JL, Saltos N. Heterogeneity of airway inflammation in persistent asthma: evidence of neutrophilic inflammation and increased sputum interleukin-8. Chest. 2001;119:1329–1336. doi: 10.1378/chest.119.5.1329. [DOI] [PubMed] [Google Scholar]
- 4.Kelly EA, Busse WW, Jarjour NN. Increased matrix metalloproteinase-9 in the airway after allergen challenge. Am. J. Respir. Crit. Care Med. 2000;162:1157–1161. doi: 10.1164/ajrccm.162.3.9908016. [DOI] [PubMed] [Google Scholar]
- 5.Koh YY, Dupuis R, Pollice M, Albertine KH, Fish JE, Peters SP. Neutrophils recruited to the lungs of humans by segmental antigen challenge display a reduced chemotactic response to leukotriene B4. Am. J. Respir. Cell Mol. Biol. 1993;8:493–499. doi: 10.1165/ajrcmb/8.5.493. [DOI] [PubMed] [Google Scholar]
- 6.Lommatzsch M, Julius P, Kuepper M, Garn H, Bratke K, Irmscher S, Luttmann W, Renz H, Braun A, Virchow JC. The course of allergen-induced leukocyte infiltration in human and experimental asthma. J. Allergy Clin. Immunol. 2006;118:91–97. doi: 10.1016/j.jaci.2006.02.034. [DOI] [PubMed] [Google Scholar]
- 7.Nocker RE, Out TA, Weller FR, Mul EP, Jansen HM, van der Zee JS. Influx of neutrophils into the airway lumen at 4 h after segmental allergen challenge in asthma. Int. Arch. Allergy Immunol. 1999;119:45–53. doi: 10.1159/000024174. [DOI] [PubMed] [Google Scholar]
- 8.Lagasse E, Weissman IL. Flow cytometric identification of murine neutrophils and monocytes. J. Immunol. Methods. 1996;197:139–150. doi: 10.1016/0022-1759(96)00138-x. [DOI] [PubMed] [Google Scholar]
- 9.Dalod M, Hamilton T, Salomon R, Salazar-Mather TP, Henry SC, Hamilton JD, Biron CA. Dendritic cell responses to early murine cytomegalovirus infection: subset functional specialization and differential regulation by interferon alpha/beta. J. Exp. Med. 2003;197:885–898. doi: 10.1084/jem.20021522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nakano H, Yanagita M, Gunn MD. CD11c(+)B220(+)Gr-1(+) cells in mouse lymph nodes and spleen display characteristics of plasmacytoid dendritic cells. J. Exp. Med. 2001;194:1171–1178. doi: 10.1084/jem.194.8.1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bronte V, Apolloni E, Cabrelle A, Ronca R, Serafini P, Zamboni P, Restifo NP, Zanovello P. Identification of a CD11b(+)/Gr-1(+)/CD31(+) myeloid progenitor capable of activating or suppressing CD8(+) T cells. Blood. 2000;96:3838–3846. [PMC free article] [PubMed] [Google Scholar]
- 12.Mordue DG, Sibley LD. A novel population of Gr-1+-activated macrophages induced during acute toxoplasmosis. J. Leukoc. Biol. 2003;74:1015–1025. doi: 10.1189/jlb.0403164. [DOI] [PubMed] [Google Scholar]
- 13.Tepper RI, Coffman RL, Leder P. An eosinophil-dependent mechanism for the antitumor effect of interleukin-4. Science. 1992;257:548–551. doi: 10.1126/science.1636093. [DOI] [PubMed] [Google Scholar]
- 14.Stoppacciaro A, Melani C, Parenza M, Mastracchio A, Bassi C, Baroni C, Parmiani G, Colombo MP. Regression of an established tumor genetically modified to release granulocyte colony-stimulating factor requires granulocyte-T cell cooperation and T cell-produced interferon gamma. J. Exp. Med. 1993;178:151–161. doi: 10.1084/jem.178.1.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee S, Zheng M, Kim B, Rouse BT. Role of matrix metalloproteinase-9 in angiogenesis caused by ocular infection with herpes simplex virus. J. Clin. Invest. 2002;110:1105–1111. doi: 10.1172/JCI15755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sitia G, Isogawa M, Iannacone M, Campbell IL, Chisari FV, Guidotti LG. MMPs are required for recruitment of antigen-nonspecific mononuclear cells into the liver by CTLs. J. Clin. Invest. 2004;113:1158–1167. doi: 10.1172/JCI21087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sitia G, Isogawa M, Kakimi K, Wieland SF, Chisari FV, Guidotti LG. Depletion of neutrophils blocks the recruitment of antigen-nonspecific cells into the liver without affecting the antiviral activity of hepatitis B virus-specific cytotoxic T lymphocytes. Proc. Natl. Acad. Sci. USA. 2002;99:13717–13722. doi: 10.1073/pnas.172521999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park SJ, Wiekowski MT, Lira SA, Mehrad B. Neutrophils regulate airway responses in a model of fungal allergic airways disease. J. Immunol. 2006;176:2538–2545. doi: 10.4049/jimmunol.176.4.2538. [DOI] [PubMed] [Google Scholar]
- 19.Sternlicht MD, Werb Z. How matrix metalloproteinases regulate cell behavior. Annu. Rev. Cell Dev. Biol. 2001;17:463–516. doi: 10.1146/annurev.cellbio.17.1.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baronas-Lowell D, Lauer-Fields JL, Borgia JA, Sferrazza GF, Al-Ghoul M, Minond D, Fields GB. Differential modulation of human melanoma cell metalloproteinase expression by alpha2beta1 integrin and CD44 triple-helical ligands derived from type IV collagen. J. Biol. Chem. 2004;279:43503–43513. doi: 10.1074/jbc.M405979200. [DOI] [PubMed] [Google Scholar]
- 21.Saleem M, Kweon MH, Johnson JJ, Adhami VM, Elcheva I, Khan N, Bin Hafeez B, Bhat KM, Sarfaraz S, Reagan-Shaw S, Spiegelman VS, Setaluri V, Mukhtar H. S100A4 accelerates tumorigenesis and invasion of human prostate cancer through the transcriptional regulation of matrix metalloproteinase 9. Proc. Natl. Acad. Sci. USA. 2006;103:14825–14830. doi: 10.1073/pnas.0606747103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kruidenier L, MacDonald TT, Collins JE, Pender SL, Sanderson IR. Myofibroblast matrix metalloproteinases activate the neutrophil chemoattractant CXCL7 from intestinal epithelial cells. Gastroenterology. 2006;130:127–136. doi: 10.1053/j.gastro.2005.09.032. [DOI] [PubMed] [Google Scholar]
- 23.Randolph DA, Carruthers CJ, Szabo SJ, Murphy KM, Chaplin DD. Modulation of airway inflammation by passive transfer of allergen-specific Th1 and Th2 cells in a mouse model of asthma. J. Immunol. 1999;162:2375–2383. [PubMed] [Google Scholar]
- 24.Jung YW, Schoeb TR, Weaver CT, Chaplin DD. Antigen and lipopolysaccharide play synergistic roles in the effector phase of airway inflammation in mice. Am. J. Pathol. 2006;168:1425–1434. doi: 10.2353/ajpath.2006.050986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Su SB, Grajewski RS, Luger D, Agarwal RK, Silver PB, Tang J, Tuo J, Chan CC, Caspi RR. Altered chemokine profile associated with exacerbated autoimmune pathology under conditions of genetic interferon-gamma deficiency. Invest. Ophthalmol. Vis. Sci. 2007;48:4616–4625. doi: 10.1167/iovs.07-0233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leppert D, Waubant E, Galardy R, Bunnett NW, Hauser SL. T cell gelatinases mediate basement membrane transmigration in vitro. J. Immunol. 1995;154:4379–4389. [PubMed] [Google Scholar]
- 27.Han YP, Nien YD, Garner WL. Tumor necrosis factor-alpha-induced proteolytic activation of pro-matrix metalloproteinase-9 by human skin is controlled by down-regulating tissue inhibitor of metalloproteinase-1 and mediated by tissue-associated chymotrypsin-like proteinase. J. Biol. Chem. 2002;277:27319–27327. doi: 10.1074/jbc.M202842200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dalod M, Salazar-Mather TP, Malmgaard L, Lewis C, Asselin-Paturel C, Briere F, Trinchieri G, Biron CA. Interferon alpha/beta and interleukin 12 responses to viral infections: pathways regulating dendritic cell cytokine expression in vivo. J. Exp. Med. 2002;195:517–528. doi: 10.1084/jem.20011672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Heer HJ, Hammad H, Soullie T, Hijdra D, Vos N, Willart MA, Hoogsteden HC, Lambrecht BN. Essential role of lung plasmacytoid dendritic cells in preventing asthmatic reactions to harmless inhaled antigen. J. Exp. Med. 2004;200:89–98. doi: 10.1084/jem.20040035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cundall M, Sun Y, Miranda C, Trudeau JB, Barnes S, Wenzel SE. Neutrophil-derived matrix metalloproteinase-9 is increased in severe asthma and poorly inhibited by glucocorticoids. J. Allergy Clin. Immunol. 2003;112:1064–1071. doi: 10.1016/j.jaci.2003.08.013. [DOI] [PubMed] [Google Scholar]
- 31.Hermant B, Bibert S, Concord E, Dublet B, Weidenhaupt M, Vernet T, Gulino-Debrac D. Identification of proteases involved in the proteolysis of vascular endothelium cadherin during neutrophil transmigration. J. Biol. Chem. 2003;278:14002–14012. doi: 10.1074/jbc.M300351200. [DOI] [PubMed] [Google Scholar]
- 32.Vermaelen KY, Cataldo D, Tournoy K, Maes T, Dhulst A, Louis R, Foidart JM, Noel A, Pauwels R. Matrix metalloproteinase-9-mediated dendritic cell recruitment into the airways is a critical step in a mouse model of asthma. J. Immunol. 2003;171:1016–1022. doi: 10.4049/jimmunol.171.2.1016. [DOI] [PubMed] [Google Scholar]
- 33.McMillan SJ, Kearley J, Campbell JD, Zhu XW, Larbi KY, Shipley JM, Senior RM, Nourshargh S, Lloyd CM. Matrix metalloproteinase-9 deficiency results in enhanced allergen-induced airway inflammation. J. Immunol. 2004;172:2586–2594. doi: 10.4049/jimmunol.172.4.2586. [DOI] [PubMed] [Google Scholar]
- 34.Kumagai K, Ohno I, Okada S, Ohkawara Y, Suzuki K, Shinya T, Nagase H, Iwata K, Shirato K. Inhibition of matrix metalloproteinases prevents allergen-induced airway inflammation in a murine model of asthma. J. Immunol. 1999;162:4212–4219. [PubMed] [Google Scholar]
- 35.Lee KS, Jin SM, Kim SS, Lee YC. Doxycycline reduces airway inflammation and hyperresponsiveness in a murine model of toluene diisocyanate-induced asthma. J. Allergy Clin. Immunol. 2004;113:902–909. doi: 10.1016/j.jaci.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 36.Bosse M, Chakir J, Rouabhia M, Boulet LP, Audette M, Laviolette M. Serum matrix metalloproteinase-9:Tissue inhibitor of metalloproteinase-1 ratio correlates with steroid responsiveness in moderate to severe asthma. Am. J. Respir. Crit. Care Med. 1999;159:596–602. doi: 10.1164/ajrccm.159.2.9802045. [DOI] [PubMed] [Google Scholar]
- 37.Mattos W, Lim S, Russell R, Jatakanon A, Chung KF, Barnes PJ. Matrix metalloproteinase-9 expression in asthma: effect of asthma severity, allergen challenge, and inhaled corticosteroids. Chest. 2002;122:1543–1552. doi: 10.1378/chest.122.5.1543. [DOI] [PubMed] [Google Scholar]
- 38.Vignola AM, Riccobono L, Mirabella A, Profita M, Chanez P, Bellia V, Mautino G, D'Accardi P, Bousquet J, Bonsignore G. Sputum metalloproteinase-9/tissue inhibitor of metalloproteinase-1 ratio correlates with airflow obstruction in asthma and chronic bronchitis. Am. J. Respir. Crit. Care Med. 1998;158:1945–1950. doi: 10.1164/ajrccm.158.6.9803014. [DOI] [PubMed] [Google Scholar]
- 39.Murphy KM, Heimberger AB, Loh DY. Induction by antigen of intrathymic apoptosis of CD4+CD8+TCRlo thymocytes in vivo. Science. 1990;250:1720–1723. doi: 10.1126/science.2125367. [DOI] [PubMed] [Google Scholar]
- 40.Randolph DA, Stephens R, Carruthers CJ, Chaplin DD. Cooperation between Th1 and Th2 cells in a murine model of eosinophilic airway inflammation. J. Clin. Invest. 1999;104:1021–1029. doi: 10.1172/JCI7631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fleming TJ, Fleming ML, Malek TR. Selective expression of Ly-6G on myeloid lineage cells in mouse bone marrow. RB6-8C5 mAb to granulocyte-differentiation antigen (Gr-1) detects members of the Ly-6 family. J. Immunol. 1993;151:2399–2408. [PubMed] [Google Scholar]
- 42.Pagenstecher A, Stalder AK, Campbell IL. RNase protection assays for the simultaneous and semiquantitative analysis of multiple murine matrix metalloproteinase (MMP) and MMP inhibitor mRNAs. J. Immunol. Methods. 1997;206:1–9. doi: 10.1016/s0022-1759(97)00077-x. [DOI] [PubMed] [Google Scholar]
- 43.Oh LY, Larsen PH, Krekoski CA, Edwards DR, Donovan F, Werb Z, Yong VW. Matrix metalloproteinase-9/gelatinase B is required for process outgrowth by oligodendrocytes. J. Neurosci. 1999;19:8464–8475. doi: 10.1523/JNEUROSCI.19-19-08464.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Heo JH, Lucero J, Abumiya T, Koziol JA, Copeland BR, del Zoppo GJ. Matrix metalloproteinases increase very early during experimental focal cerebral ischemia. J. Cereb. Blood Flow Metab. 1999;19:624–633. doi: 10.1097/00004647-199906000-00005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.