

Abstract
In the search for a selective delta-opioid receptor agonist, (-)-(1R,5R,9R)-5,9-dimethyl-2’-hydroxy-2-(6-hydroxyhexyl)-6,7-benzomorphan hydrochloride ((-)-NIH 11082) and the (+)-enantiomer were synthesized and tested. (-)-NIH 11082 displayed antinociceptive activity in the paraphenylquinone test (PPQ test) in male ICR mice [ED50 = 1.9 (0.7 – 5.3) mg/kg, s.c.] and showed little, if any, activity in the tail-flick and hot-plate assays. The (+)-enantiomer was essentially inactive indicating stereoselectivity. Opioid receptor subtype characterization studies indicated that naltrindole, a delta-opioid receptor antagonist, was potent versus the ED80 of (-)-NIH 11082 in the PPQ test [AD50 = 0.75 (0.26 – 2.20) mg/kg, s.c]. beta-Funaltrexamine and nor-binaltorphimine, selective mu- and kappa-receptor antagonists, respectively, were inactive versus the ED80 of (-)-NIH 11082. In rats with inflammation-induced pain, (-)-NIH 11082 produced antihyperalgesic effects that were attenuated by naltrindole. In morphine-dependent rhesus monkeys of both sexes, (-)-NIH 11082 neither substituted for morphine nor exacerbated withdrawal signs in the dose range of 4.0 to 32.0 mg/kg, s.c. Neither convulsions nor other overt behavioral signs were observed in any of the species tested. The results indicate that (-)-NIH 11082 has delta-opioid receptor properties.
Keywords: Selective delta opioid; (-)-(1R,5R,9R)-5,9-Dimethyl-2’-hydroxy-2-(6-hydroxyhexyl)-6,7-benzomorphan hydrochloride ((-)-NIH 11082); antinociception; antihyperalgesia; mouse; rat; rhesus monkey
1. Introduction
Preclinical studies with delta opiates have implicated their use in a wide spectrum of conditions involving analgesic (Cowan et al., 1985, Jiang et al., 1990), opiate craving (Brandt et al., 2001), tolerance and dependence (Abdelhamid et al., 1991), depression (Baamonde et al., 1992, Broom et al., 2002), diarrhea (Broccado and Improta, 1992), neuropathic and inflammatory pain (Fraser et al.2000, Mika et al., 2001), potentiation of mu-opioid agonist analgesia (Porreca et al., 1992), respiration (Cheng et al.,1993) immune function (Mazumder et al.,1993) and Parkinson’s disease(Hudzik et al., 2000). This impressive list of possible therapeutic applications has intensified the search for delta opioids. Nonpeptidic delta-opioid-receptor agonists that cross the blood-brain barrier such as (±)-4-[(alpha-R*)-alpha-[(2S*,5R*)-4-allyl-2,5-dimethyl-1-piperazinyl]-3-hydroxbenzyl]-N,N-diethylbenzamide (BW-373U86) (Chang et al., 1993) and (+)-4-[(alphaR)-alpha-((2S,5R)-4-allyl-2,5-dimethyl-1-piperazinyl)-3-methoxybenzyl}N,N-diethylbenzamide (SNC-80), were synthesized and studied (Calderon et al., 1994). Unfortunately, seizures and/or convulsions limited their therapeutic potential.
Previous studies in our laboratory with (-)-(1R,5R,9R)-5,9-dimethyl-2’-hydroxy-2-(methyl through decyl)-6,7-benzomorphans revealed a transition of activity (May et al.,1994). Antinociceptive acitivy in the mouse went from agonist ((-)-methyl) to agonist antagonist ((-)-ethyl, (-)-propyl and (-)-butyl) and back to agonist ((-)-pentyl, (-)-hexyl, -(-)-heptyl, and (-)-octyl). The (-)-nonyl and (-)- decyl homologs were inactive. Furthermore, in vitro binding studies revealed different rank order potencies at mu-, kappa- and delta-opioid receptors. Conceivably, these results could reflect the interaction of these compounds with specific amino acids in the transmembrane helices of the opioid receptors. We speculated that other N-substituted (-)-benzomorphans might possess selective delta-opioid activity. Because known delta-opioid receptor agonists were active only in the PPQ test in our laboratory, our strategy was simple. We prepared and selected, for further study, compounds that were potently active in the PPQ test in mice and whose activity was selectively antagonized by naltrindole and were devoid of undesirable effects associated with known delta-opioids. In order to demonstrate that the observed results were not peculiar to mice additional studies in rats and monkeys using other models were conducted.
2. Materials and Methods
All procedures involving animals were carried out in accordance with the Guide for the Care and Use of Laboratory Animals (National Research Council, 1996) and were approved by the Institutional Animal Care and Use Committee of Virginia Commonwealth University. The animal facilities are certified by the American Association of Laboratory Animal Care.
2.1 Antinociceptive Studies in mice
For the antinociceptive studies, ICR (Institute of Cancer Research) male outbred mice (Harlan Sprague-Dawley, Inc., Indianapolis, IN) were used. The mice weighed 20–30 g each and were housed on a 12/12 h light/dark cycle, in groups of five, in polycarbonate cages. They had free access to food and water. Each animal was tested only once. The number of mice per treatment regimen was 6 to 8. When applicable, MPE ED 50s and 80s and AD 50s were calculated by using computerized probit analysis (Bliss, 1967), otherwise the data was expressed as % change.
2. 1.1 Paraphenylquinone Abdominal Stretching Test (PPQ test)
The mice were injected s.c. with doses of (+)- or (-)- NIH 11082 or its and 10 min later received an intraperitoneal injection of a freshly prepared solution of paraphenylquinone (2 mg/ml/kg) as previously described by Pearl et al., (1966) and later modified (Aceto et al., 1997). Mice were then placed in cages in groups of three each. The total number of stretches for the groups during each 1-min period was then counted at 10 and 15 min postinjection. A stretch was characterized by an elongation of the body, development of tensions in the abdominal muscles and extension of the hind limbs. The antinociceptive response was expressed as the percentage inhibition of the paraphenyquinone-induced stretching response and was calculated as [(1 − (total number of stretches in the medicated mice)/total number of stretches in the control mice)] × 100.
Selective opioid receptor-antagonist subtype tests were conducted against the s.c. 80% effective dose (ED80) of (-)- NIH11082. Routes of administration and pretreatment times for the selective opioid receptor antagonists were: beta-funaltrexamine (beta-FNA) intracerebroventricular (i.c.v), 4 hr; nor-binaltorphimine (nor-BNI) s.c., 2 h; and naltrindole, s.c., 30 min. Antagonist receptor doses and pretreatment times were based on previous studies (Aceto et al., 1996, 1997).
Intracerebroventricular injections were conducted as described by Pedigo et al., (1975). Mice were lightly anesthetized with ether and an incision was made in the scalp so that the bregma was exposed. Injections were performed using a 26-gauge needle fitted with a sleeve of polyethylene 20 tubing to control the depth of the injection. An injection volume of 5 l was administered at a site 2 mm lateral to the bregma, at a depth of 2 mm.
2.1. 2 Tail-flick Test
Antinociception was assessed by the tail-flick method described by D’Amour and Smith, (1941), as modified by Dewey and co investigators, (1970). Briefly, the mouse’s tail was placed in a groove, which contained a slit under which was located a photoelectric cell. When the heat source of noxious stimulus was turned on, the heat focused in the tail, and the animal responded by flicking its tail out of the groove. Light thus passed through the slit and activated the photocell which, in turn stopped the recording timer. A control response (2–4 s) was determined for each mouse. To minimize tissue damage a maximum latency of 10 s was imposed. Antinociceptive response was calculated as percent maximal possible effect (%MPE), where %MPE = [(test −control)/ (10 − control)] × 100. The mice were tested 20 min after the s.c. administration of NIH 11082 or its (+)-enantiomer. In the TF versus morphine study, the percent antagonism was calculated as [1 − (antagonist + agonist MPE/ (agonist MPE 80)] × 100.
2.1. 3 Hot Plate Test
The method used was a modification of that described by Eddy and Leimbach (Eddy and Leimbach, 1953) and later modified (Atwell and Jacobson, (1978). Mice were placed into a 10-cm wide glass cylinder on a hot plate (Thermojust Apparatus, Richmond, VA) maintained at 56.5°C. Control latencies were determined for each mouse. Only mice that gave control response latencies in the range of 6 to 10 s on both trials served as subjects. The reaction time was scored when the animal jumped or licked its paws. Activity was scored as positive if the mouse jumped, licked or shook its paws at least 5 s beyond its control latency. Cut-off time was 15 s. Percent activity for each dose tested was calculated as (total number of mice scored as positive) / (total number tested) × 100.
2. 2 Testing of Arthritic Rats
For the arthritic tests, twenty male Lewis rats approximately 60 days of age were purchased from Charles Rivers Laboratories (Raleigh, NC). Rats were individually housed, had free access to food and water and were maintained on 12/12 h light/dark cycle.
The procedure was described previously (Cook and Nickerson, 2005) Briefly, on Day 1 animals were injected intradermally in the base of the tail with 0.1 ml of 5.0 mg/ml Complete Freund’s Adjuvant (heat-killed Mycobacterium, Difco Laboratories, Detroit, MI) or 0.1 ml of vehicle (mineral oil).
Nociceptive tests were conducted on days 19 and 21 post vehicle/ Complete Freund’s Adjuvant administration. Nociceptive sensitivity was assessed by determining thresholds in response to mechanical pressure applied to the hindpaws of the rats. Rats were lightly restrained in a towel and a mechanical stimulus was applied with an analgesia meter (Ugo Basile, Varese, Italy). A 500 g cut-off was imposed on all tests. The % antihyperalgesic effect was also determined based upon the following equation: % Antihyperalgesia = [(observed - Complete Freund’s Adjuvant baseline)/(vehicle baseline - Complete Freund’s Adjuvant baseline)] × 100. Differences in the antihyperalgesic effects of 30 and 60 mg/kg (-)-NIH 11082 over 120 min were determined using a repeated measures ANOVA.
For the effects of (-)-NIH 11082 alone and in combination with naltrindole, mean paw-pressure thresholds were determined at each time point and compared to the mean non-drug baseline paw pressure threshold using a repeated measures ANOVA followed by a Dunnett’s post-hoc test. The area under the time-course curve was calculated for the effects of (-)-NIH 11082 alone and for the effects of (-)-NIH 11082 in combination with naltrindole. A paired T-test was used to determine significance.
2.3 Morphine-Dependent Rhesus Monkeys
Male and female rhesus monkeys (Macaca mulatta) were purchased from the Caribbean Primate Research Center (PR). As experimental subjects they weighted 2.5 to 7.5 kg. They were housed in pens in socially compatible groups of 4 or 5. The monkeys received 3 mg/kg, s.c. of morphine sulfate every 6 h. All the animals had received morphine for at least 3 months and were maximally dependent on morphine (Deneau, 1956). Additional details were reported by Aceto and coworkers (1977).
There were 4 or 5 monkeys to a group. Each test was initiated by the s.c. injection of (-)-NIH 11082, morphine control or vehicle into the monkeys of a group that had not received morphine for 14 to 15 h and that showed definite signs of withdrawal (Aceto et al., (1997). Each animal was randomly chosen to receive one of the following treatments: (a) a dose of (-)-NIH 11082; (b) morphine control (3 mg/kg); or (c) vehicle control 1 ml/kg sterile water. Withdrawal signs were scored absent or present once during each of 5 consecutive 30 min- observation periods. Withdrawal signs included slowing of movement, drowsiness (sitting with eyes closed and lethargic or being indifferent to surroundings), fighting, vocalizing, rigidity of abdominal muscles, vocalization during palpations, restlessness (pacing), tremorousness, coughing, retching, vomiting, wet-dog shakes, and masturbation. A minimal 2-week recuperation period was allowed between tests. Test data were grouped according to time, dose and drug and analyzed using the nonparametric Kruskal-Wallis ANOVA test followed by post hoc comparisons using the Mann-Whitney test. Data were analyzed using statistical software (StatView 512+ Brainpower, Inc, Agoura Hills, CA). The significance level for all studies was set at P<0.05.
2.4 Synthesis of the stereoisomers of NIH 11082
For the synthesis of (-)-NIH 11082, a mixture of 0.4 g of (-)-(1R,5R,9R)-5-9-dimethyl-2’-hydroxy-6,-7-benzomorphan; 0.36 g of 6-bromo-1-hexanol, 0.5g of KHC0, 5 ml of tetrahydrofuran and 1 ml of dimethylfornamide was heated under reflux for 5 hours. The tetrahydrofuran was evaporated in vacuo and the residue treated with 5-10 ml of water and about 7 ml of ethyl acetate. The water layer was extracted with another 5–7 ml of ethyl acetate. The combined extracts were dried with about 3 g of magnesium sulfate and evaporated in vacuo to give 0.75 of the ethereal solution with 2 ml of 1M HCl gave a semisolid. After 1-5 days at 0°C, the ether was decanted from semisolid hexorphan HCl. Addition of 10-15 ml of acetone gave after warming, stirring and overnight cooling at 0°C, 0.46 g of hexorphan HCl, m.p. 190-195°C. NIH 11082 was dissolved in 1-2 ml of warm methanol and the solution diluted to 15-20 ml with acetone. Evaporation on the hot-plate to the appearance of crystals (to 4-5 ml) and overnight cooling at 15-20°C gave 0.4 g of TLC-pure hexorphan·HCl (m.p. 196-7°C. Analysis: calculated for C20H32ClN02: C, 67.87; H, 9.12; N., 3.96; Found: C, 68.08; H, 9.05; N, 3.96. The chemical structure is shown in Figure 1.
Figure 1.
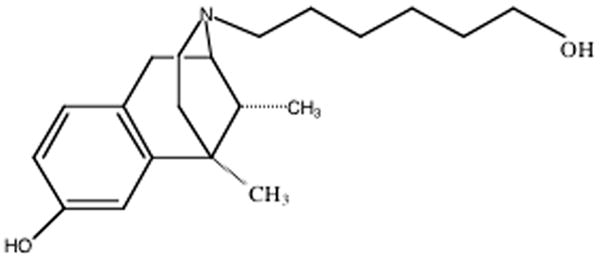
Chemical structure of (-)-(1R,5R,9R)-5,9-Dimethyl-2’-hydroxy-2-(6-hydroxyhexyl)-6,7-benzomorphan hydrochloride ((-)-NIH 11082)
The (+)-(1S,5S,9S) enantiomer was similarly prepared from (+)-(1S,5S,9S)-5,9dimethyl-2’hydroxy-6,7-benzomorphan. It melted at 195-7°C. Analysis calculated for C20H32ClN02: C, 67.87; H, 9.12; N., 3.96. Found: C, 68.03; H, 9.07; N,3.90.
2.5 Chemicals and Solutions
In the mouse studies, NIH 11082 and its (+)-enantiomer were dissolved in 10% hydroxypropyl-beta-cyclodextrin (Cargill, Cedar Rapid, IA) in distilled water. This solution was filtered using a 0.2 micron filter. Then appropriate dilutions were made using the same vehicle. Morphine sulfate and paraphenylquinone were purchased from Mallinckrodt and Sigma, respectively (St Louis, MO). beta-Funaltrexamine hydrochloride (beta-FNA), nor-binaltorphimine hydrochloride (nor-BNI) and naltrindole hydrochloride were obtained from the National Institute on Drug Abuse. (-)- and (+)-5-9-Dimethyl-2’-hydroxy-6,-7-benzomorphan was supplied by F.I. Carroll, RTI International Research, Triangle Park, NC. 6-Bromo-1-hexanol was purchased from Sigma-Aldrich, St Louis, MO. All drugs were dissolved in sterile water for injection, unless otherwise indicated, and administered as the salt.
3. Results
The results of the antinociception studies with (-)- and (+)- NIH 11082 are presented in Table 1. (-)-NIH 11082 is potently active on the PPQ test (ED50 = 1.9 (0.7 – 5.3, mg/kg/s.c.) and displayed no activity in the tail-flick and hot plate tests at doses of 1, 10 and 30 mg/kg, s.c.. It lacked opioid antagonist properties when tested versus the ED80 of morphine in the tail flick test. Because (+)-NIH 11082 was essentially inactive on the antinociceptive tests, opiate subtype studies were not conducted with this enantiomer. As summarized in Table 2, naltrindole, the delta opioid receptor antagonist was effective versus the ED80 of (-)-NIH 11082 in the PPQ test (AD50 = 0.75 (0.26 – 2.20 mg/kg, s.c.). beta-FNA the mu-opioid receptor antagonist was weakly active producing only 26% antagonism at 30 g/brain and nor BNI was completely inactive at doses of 1, 10 and 30 mg/kg, s.c.
Table 1.
Antinociceptive profile of activity of the (-)- and (+)-NIH 11082
| Antinociceptive Test | ED50 (mg/kg, s.c.) / 95% C L or % change | |
|---|---|---|
| (-)- NIH 11082 | (+)-NIH 11082 | |
| Paraphenylquinone | 1.9 (0.7 – 5.3) | 35% at 10 and 28 % at 30 |
| Hot Plate | Inactive at 1, 10 and 20% at 30 | Inactive at 10 and 30 |
| Tail Flick | Inactive at 1, 10 and 30 | Inactive at 1, 10 and 30 |
| Tail Flick vs Morphine Sulfate ED80 | Inactive at 1,10 and 30 | Inactive at 1 10 and 30 |
Table 2.
Antagonist activity of selective opioid receptor antagonists versus the ED80 of (-)-NIH11082 in the PPQ test in mice.
| Selective Opioid Receptor Antagonist | (-)-NIH 11082 ED80 ,s.c. |
|---|---|
| Beta-FNA i.c.v. | 3% at 1, 9% at 10 and 26% at 30 υg/brain |
| nor-BNI, s.c. | Inactive at 1, 10 and 30 |
| Naltrindole, s.c. | 0.75 (0.26 – 2.20) |
(-)-NIH 11082 produced long lasting antihyperalgesic effects in Complete Freund’s Adjuvant-treated rats (Figure 2). A dose of 30 mg/kg (-)-NIH11082 produced a maximal effect of approximately 50% at 60 min post administration. A dose of 60 mg/kg (-)-NIH 11082 produced approximately an 80% effect from 30-120 min post administration and falling to a 34% effect by 210 min. Comparison of the time-course from 30-120 min indicated a main effect of dose (F(1,42)=25.59, P=0.0002). Tests of simple effects indicated that 60 mg/kg (-)-(-)-NIH11082 produced greater % antihyperalgesia than 30 mg/kg (-)-NIH11082 at 30, 90 and 120 min.
Figure 2.
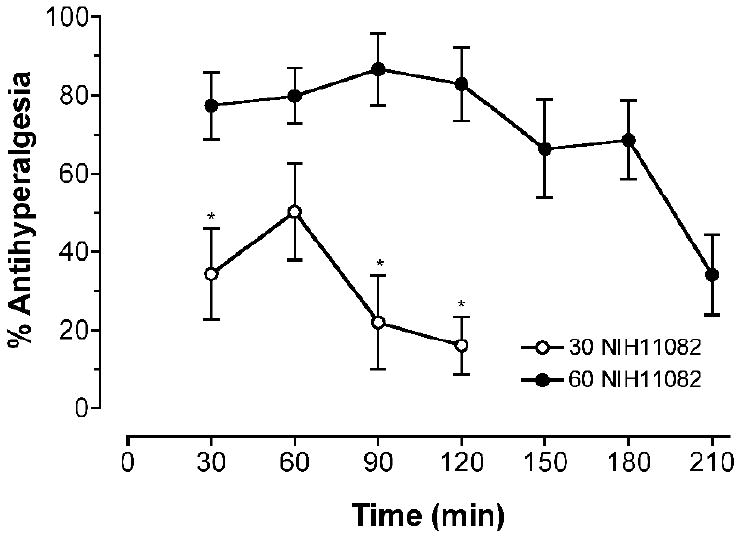
Antihyperalgesic effects of 30 and 60 mg/kg (-)-NIH11082 in Complete Freund’s Adjuvant-treated rats. Baseline paw pressure thresholds in the rats treated with 30 mg/kg were 70 g and 100% antihyperalgesia was equal to 158 g (Baselines significantly different, t=7.41, P<0.0001). Baseline paw pressure thresholds in the rats treated with 60 mg/kg were 55 g and 100% antihyperalgesia was equal to 125 g (Baselines significantly different, t=12.67, P<0.0001). * indicates a significant difference between the doses.
The effects of 60 mg/kg (-)-NIH 11082 alone and in combination with 10 mg/kg naltrindole are shown in Figure 3. For 60 mg/kg (-)-NIH11082 alone there was a main effect of time (F(7,56)=12.37, P<0.0001) on paw pressure thresholds such that thresholds were significantly greater than baseline from 90-180 min. When combined with naltrindole, there was a main effect of time (F(7,56)=12.37, P<0.0001) on paw pressure thresholds such that thresholds were significantly greater than baseline from 60-210 min. Naltrindole attenuated increases in thresholds based upon area under the curve analysis (t=3.49, P=0.008) (Figure 3 inset).
Figure 3.
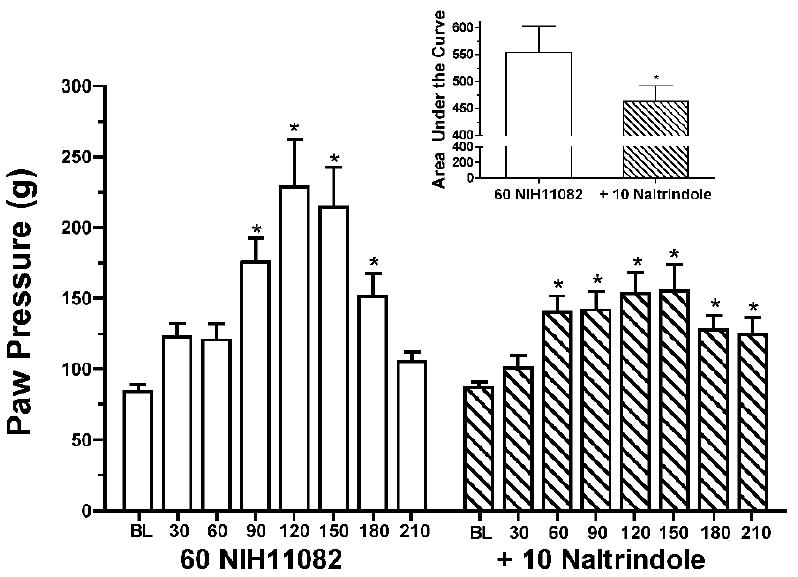
Effects of 60 mg/kg (-)-NIH11082 alone and in combination with 10 mg/kg naltrindole on paw pressure thresholds in Complete Freund’s Adjuvant-treated rats. Data above baseline represent baseline thresholds prior to drug administration. * indicates a significant increase in thresholds relative to baseline. Inset: Area under the time-course curve for 60 mg/kg (-)-NIH11082 alone and in combination with 10 mg/kg naltrindole. * indicates that naltrindole significantly attenuated the effects of(-)- NIH11082.
In morphine-dependent monkeys at doses of 4, 8, 16 and 32 mg/kg, s.c., (-)-NIH 11082 neither substituted for morphine nor exacerbated withdrawal. Of course, in the morphine control group, full suppression of withdrawal signs was evident and the saline controls remained in withdrawal for the duration of the study (2 1/2 h). The results of the Kruskal-Wallis analysis of variance revealed significant overall differences among treatments at each 30-min time point: (30 min- H=10.991, P=.0516); (60 min- H=13.715, P=.0175); (90 min- H=14.144, P=.0147); (120 min- H=13.680, P=.0178) and (150 min-H=14.585, P=.0123).
These differences were due primarily to the fact that the morphine-treated group completely suppressed withdrawal signs. Post hoc analysis revealed no significant differences between the vehicle-treated group and any and all of the (-)-NIH 11082 doses. In other words, the vehicle– and all (-)-NIH 11082-treated monkeys neither substituted for morphine nor exacerbated withdrawal. Finally, no convulsions or unusual behaviors were noted.
4. Discussion
(-)-NIH 11082 and SNC80 have similar pharmacological profiles of activity in our laboratory in the mouse and rhesus monkey. In studies reported earlier, SNC80, given s.c., was active in the PPQ test (ED50 = 3.8 (1.6 – 9.3 mg/kg, s.c.). The s.c. AD50 for naltrindole versus the ED80 of SNC in the PPQ test was 5.48 (2.97 – 10.11 mg/kg). In addition, SNC80 showed little, if any, activity on the tail-flick, hot plate and tail-flick versus morphine assays (Aceto et al., 1996). Gallantine and Meert (2005) reported that SNC80 produced dose-dependent antinociception in the acetic acid writhing test and in the warm-water tail-withdrawal test in rats. Our findings contrast with those demonstrating that SNC80 produced tail-flick antinociception following i.c.v. administration in rats (Fraser et al., 2000) and produced hotplate and warm-water tail-withdrawal antinociception in mice (Bilsky et al., 1995). However, we did not study (-)- NIH 11082 or SNC80 in the rat, by these routes of administration or using those assays.
(-)-NIH 11082-induced antinociception was blocked by the selective delta-opioid receptor antagonist naltrindole indicating that SNC80 and (-)-NIH 11082 may share similar mechanisms of action. The selective mu- and kappa-opioid receptor antagonists, beta-FNA and nor-BNI were inactive in this respect. SNC80-induced antinociception was reported to be antagonized by naltrindole but not beta-FNA or nor-BNI. (Bilsky, et al., 1995).
The nonpeptidic delta-receptor agonists BW373U86 and SNC80 were shown to produce naltrindole-reversible convulsions in mice (Comer et al. 1993, Broom et al. 2000). In our hands, SNC80 produced short-lived convulsions when given intravenously at 1 and 10 mg/kg. (unpublished data). Convulsions were not observed with either SNC80 or (-)-NIH 11082 in the dose range of 1 to 30 mg/kg, s.c. By the subcutaneous and intraperitoneal routes of administration, Bilsky and co workers (1995) noted brief convulsions with SNC80 at a relatively high dose of 100 mg/kg.
In Complete Freund’s Adjuvant-treated rats with mechanical hyperalgesia, the antihyperalgesic effects of (-)-NIH 11082 were attenuated by the delta opioid antagonist naltrindole. These results are similar to those obtained following i.c.v. administration of the delta agonist SNC80 (Fraser et al., 2000) and following direct injection of the delta peptide [D-Ala2,Glu4] deltorphin in the rostral ventromedial medulla (Hurley and Hammond, 2000) in Complete Freund’s Adjuvant-treated rats experiencing thermal hyperalgesia. In addition to the involvement of supraspinal receptors in mediating delta opioid-induced hyperalgesia, delta-opioid receptors localized in the inflamed paw may be involved (Stein et al., 1989).
(-)-NIH 11082 neither substituted for morphine nor exacerbated withdrawal in morphine-dependent monkeys. Similar results were reported previously with SNC80 in morphine-dependent monkeys (Aceto et al., 1996). These results provide additional evidence that (-)-NIH 11082 is devoid of mu-opioid receptor activity. Finally, no convulsions or other unusual behaviors were noted with either compound when given subcutaneously.
Also, in our laboratory, (-)-NIH 11082 exhibited antidepressant-like activity in a mouse tail-suspension test that was antagonized by naltrindole. It was also devoid of locomotor stimulant properties. (manuscript submitted for publication). Although SNC80 has locomotor stimulant properties, it was shown shown that the antidepressant effect is still evident after the locomotor stimulant effects have dissipated (Broom et al., 2002).
Unlike SNC80 where a good correlation between in vitro and in vivo activity was reported, in selective binding and [35S]GTPgS assays on (-)-NIH 11082, Traynor and co workers (2005) reported that (-)-NIH 11082 had high binding affinity at mu- and kappa-opioid receptors and less affinity at delta-opioid receptors. The results of the [35S]GTPgS assays were as follows: maximal agonist activity at mu-opioid receptors was 50.5 ± 6.7% with an EC50 = 303 ± 57 nM; maximal agonist activity at kappa-opioid receptors was 21.7 ± 4.1% with an EC50 = 1346 ± 514 nM and maximal agonist activity at delta-opioid receptors was 9.3 ±.4 % with an EC50 of 555 ± 149 nM. Traynor and his coinvestigators (2005) concluded that (-)-NIH 11082 was a partial agonist at mu- and kappa-opioid receptors with low potency. NIH 11082 had almost no efficacy at delta-opioid receptors.
Presumably, these data explained why (-)-NIH 11082 was active only on the PPQ test and not in antinociceptive tests involving heat in spite of its high binding affinity (Traynor et al., 2005). Other explanations are possible. For example, Alves and coworkers (2004) demonstrated that different classes of delta-opioid ligands (peptide, nonpeptide, antagonist, inverse agonist and partial agonist) all induced different receptor conformational states in the human delta-opioid receptor. (-)-NIH 11082 may interact allosterically producing a conformational change that can still be modulated by naltrindole. Other possibilities suggest themselves. For example, heterodimerization of mu-and delta-opioid receptors has been proposed as a mechanism for enhancing morphine analgesia (Gomes et al., 2004). Also, (-)-NIH11082 may be operating through the delta-opioid endogenous system. It may even interact with other receptors that impinge on delta-opioid neurons and trigger activity in intracellular pathways. Obviously, other studies are required to characterize the mechanism of action of (-)-NIH 11082.
In summary, in vivo, (-)-NIH 11082 has pharmacological properties associated with delta-opioid receptor agonists. Its actions are stereoselective and opioid subtype specific for the delta-opioid receptor. Remarkably, of the 50 analogs and homologs that were synthesized, (-)-NIH 11082 was the only compound in the series whose activity in the PPQ test was selectively antagonized by naltrindole. This unique structural specificity may provide further insights regarding molecular events involving delta opioids.
Acknowledgments
The authors wish to thank Barbara Kipps and Larry D. Hughes, for their expert technical assistance during the execution of these studies. Some of the data were presented at the 64th and 67th Annual Scientific Meetings, The Committee on Problems of Drug Dependence. Supported by National Institute on Drug Abuse contracts DA 5-8059, DA 8-8088, DA 05274 and DA 3-8823.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abdelhamid EE, Sultana M, Portoghese PS, Takemori AE. Selective blockage of delta opioid receptors prevents the development of morphine tolerance and dependence in mice. J Pharmacol Exp Ther. 1991;258:299–303. [PubMed] [Google Scholar]
- Aceto MD, Bowman ER, Harris LS, May EL. Dependence studies of new compounds in the rhesus monkey, rat and mouse. In: Harris LS, editor. NIDA Research Monograph; Problems of Drug Dependence, 1996: Proceedings of the 58th Annual Scientific Meeting, of the Committee on Problems of Drug Dependence, Inc; Rockville, MD: U. S. Department of Health and Human Services; 1997. pp. 338–395. [Google Scholar]
- Aceto MD, Harris LS, Bowman ER. Etorphines: mu-opioid receptor selective antinociception and low physical dependence capacity. Eur J Pharmacol. 1997;338:215–223. doi: 10.1016/s0014-2999(97)81924-3. [DOI] [PubMed] [Google Scholar]
- Aceto MD, Bowman ER, Harris LS, May EL. Dependence studies of new compounds in the rhesus monkey, rat and mouse. In: Harris LS, editor. NIDA Research Monograph; Problems of Drug Dependence, 1995: Proceedings of the 57th Annual Scientific Meeting, The Committee on Problems of Drug Dependence, Inc; Rockville, MD: U.S. Department of Health and Human Services; 1996. pp. 408–468. [Google Scholar]
- Aceto MD, Flora RE, Harris LS. The effects of naloxone and nalorphine during the development of morphine dependence in rhesus monkeys. Pharmacol. 1977;15:1–9. doi: 10.1159/000136657. [DOI] [PubMed] [Google Scholar]
- Alves ID, Cowell SM, Salamon Z, Devanathan S, Tollin G, Hruby VJ. Different structural states of the proteolipid membrane are produced by ligand binding to the human delta-opioid receptor as shown by plasmon-waveguide resonance spectroscopy. Mol Pharmacol. 2004;65:1248–1257. doi: 10.1124/mol.65.5.1248. [DOI] [PubMed] [Google Scholar]
- Atwell L, Jacobson AE. The search for less harmful analgesics. Lab Animal. 1978;7:42–47. [Google Scholar]
- Baamonde A, Daugé V, Ruiz-Gayo M, Fulga IG, Turcaud S, Foumié Roques BP, Zaluski MC, Roques BP. Antidepressant-type effects of endogenous enkephalins protected by systemic RB 101 are mediated by opioid delta and dopamine D1 receptor stimulation. Eur J Pharmacol. 1992;216:157–166. doi: 10.1016/0014-2999(92)90356-9. [DOI] [PubMed] [Google Scholar]
- Bilsky EJ, Calderon SN, Wang T, Berstein RN, Davis P, Hruby VJ, McNutt RW, Rothman RB, Rice KC, Porreca F. SNC 80, a selective nonpeptidic and systemically active opioid delta agonist. J Pharmacol Exp Ther. 1995;273:359–366. [PubMed] [Google Scholar]
- Bliss CI. Statistics in Biology. McGraw-Hill; New York: 1967. p. 439. [Google Scholar]
- Brandt MR, Furness MS, Rice KC, Fischer BD, Negus SS. Studies of tolerance and dependence with the delta-opioid agonist SNC80 in rhesus monkeys responding under a schedule of food presentation. J Pharmacol Exp Ther. 2001;299:629–637. [PubMed] [Google Scholar]
- Broccardo M, Improta G. Antidiarrheal and colonic antipropulsive effects of spinal and supraspinal administration of the natural delta opioid receptor agonist, [D-Ala2]deltorphin II, in the rat. Eur J Pharmacol. 1992;218:69–72. doi: 10.1016/0014-2999(92)90148-w. [DOI] [PubMed] [Google Scholar]
- Broom DC, Jutkiewicz EM, Folk JE, Traynor JR, Rice KC, Woods JH. Convulsant activity of a non-peptidic delta-opioid receptor agonist is not required for its antidepressant-like effects in Sprague-Dawley rats. Psychopharmacology. 2002;164:42–48. doi: 10.1007/s00213-002-1179-y. [DOI] [PubMed] [Google Scholar]
- Calderon SN, Rothman RB, Porreca F, Flippen-Anderson JL, McNutt RW, Xu H, Smith LE, Bilsky EJ, Davis P, Rice KC. Probes for narcotic receptor mediated phenomena. 19. Synthesis of (+)-4-[(alpha R)-alpha-((2S,5R)-4-allyl-2,5-dimethyl-1-piperazinyl)-3- methoxybenzyl]-N,N-diethylbenzamide (SNC 80): a highly selective, nonpeptide delta opioid receptor agonist. J Med Chem. 1994;37:2125–128. doi: 10.1021/jm00040a002. [DOI] [PubMed] [Google Scholar]
- Chang KJ, Rigdon GC, Howard JL, McNutt RW. A novel, potent and selective nonpeptidic delta opioid receptor agonist BW373U86. J Pharmacol Exp Ther. 1993;267:852–857. [PubMed] [Google Scholar]
- Cheng PY, Wu D, Decena J, Soong Y, McCabe S, Szeto HH. Opioid-induced stimulation of fetal respiratory activity by [D-Ala2]deltorphin I. Eur J Pharmacol. 1993;230:85–88. doi: 10.1016/0014-2999(93)90413-c. [DOI] [PubMed] [Google Scholar]
- Cook CD, Nickerson MD. Nociceptive sensitivity and opioid antinociception and antihyperalgesia in Freund’s Adjuvant-induced arthritic male and female rats. J Pharmacol Exp Ther. 2005;313:449–559. doi: 10.1124/jpet.104.077792. [DOI] [PubMed] [Google Scholar]
- Comer SD, Hoenicke EM, Sable AI, McNutt RW, Chang KJ, De Costa BR, Mosberg HI, Woods JH. Convulsive effects of systemically administration of the delta opioid agonist BW373U86 in mice. J Pharmacol Exp Ther. 1993;267:888–895. [PubMed] [Google Scholar]
- Cowan A, Zhu XZ, Mosberg HI, Omnaas JR, Porreca F. Direct dependence studies in rats with agents selective for different types of opioid receptor. J Pharmacol Exp Ther. 1988;246:950–955. [PubMed] [Google Scholar]
- D’Amour FE, Smith DL. A method for determining loss of pain sensation. J Pharmacol Exp Ther. 1941;72:74–79. [Google Scholar]
- Deneau GA. Dissertation. University Microfilms International; Ann Arbor Michigan, U.S.A: 1956. An analysis of the factors influencing the development of physical dependence to narcotic analgesics in the rhesus monkey with methods for predicting physical dependence in man. [Google Scholar]
- Dewey WL, Harris LS, Howes JF, Nuite JA. The effects of various neurohormonal regulators on the activity of morphine and the narcotic antagonists in the tail-flick and phenylquinone tests. J Pharmacol Exp Ther. 1970;175:435–442. [PubMed] [Google Scholar]
- Eddy NB, Leimbach D. Synthetic analgesics. II. Dithienylbutenyl- and dithienylbutylamines. J Pharmacol Exp Ther. 1953;107:385–393. [PubMed] [Google Scholar]
- Evans CJ, Keith DE, Jr, Morrison H, Mangendzo K, Edwards RH. Cloning of a delta opioid receptor by functional expression. Science. 1992;258:1952–1955. doi: 10.1126/science.1335167. [DOI] [PubMed] [Google Scholar]
- Fraser GL, Gaudreau GA, Clarke PB, Menard DP, Perkins MN. Antihyperalgesic effects of delta opioid agonists in a rat model of chronic inflammation. Br J Pharmacol. 2000;129:1668–1672. doi: 10.1038/sj.bjp.0703248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallantine EL, Meert TF. A comparison of the antinociceptive and adverse effects of the mu-opioid agonist morphine and the delta-opioid agonist SNC80. Basic Clin Pharmacol Toxicol. 2005;97:39–51. doi: 10.1111/j.1742-7843.2005.pto_97107.x. [DOI] [PubMed] [Google Scholar]
- Gomes I, Jordan BA, Gupta A, Trapaidze N, Nagy V, Devi LA. Heterodimerization of mu and delta opioid receptors: a role in synergy. J Neurosci. 2000;20:RC110. doi: 10.1523/JNEUROSCI.20-22-j0007.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handa BK, Land AC, Lord JA. Analogues of beta-LPH61-64 possessing selective agonist activity at mu-opioid receptors. Eur J Pharmacol. 1981;70:531–540. doi: 10.1016/0014-2999(81)90364-2. [DOI] [PubMed] [Google Scholar]
- Hudzik TJ, Howell A, Payza K, Cross AJ. Antiparkinsonism potential of delta-opioid receptor agonists. Eur J Pharmacol. 2000;396:101–107. doi: 10.1016/s0014-2999(00)00209-0. [DOI] [PubMed] [Google Scholar]
- Hurley RW, Hammond DL. The analgesic effects of supraspinal mu and delta opioid receptor agonists are potentiated during persistent inflammation. J Neurosci. 2000;20:1249–1259. doi: 10.1523/JNEUROSCI.20-03-01249.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Q, Takemori AE, Sultana M, Portoghese PS, Bowen WD, Mosberg HI, Porreca F. Differential antagonism of opioid delta antinociception by [D-Ala2,Leu5,Cys6]enkephalin and naltrindole 5’-isothiocyanate: evidence for delta receptor subtypes. J Pharmacol Exp Ther. 1991;257:1069–1075. [PubMed] [Google Scholar]
- May EL, Aceto MD, Bowman ER, Bentley C, Martin BR, Harris LS, Medzihradsky F, Mattson MV, Jacobson AE. Antipodal alpha-N-(methyl through decyl)-N-normetazocines (5,9alpha-Dimethyl-2’-hydroxy-6,7-benzomorphans): In vitro and in vivo properties. J Med Chem. 1994;37:3408–3418. doi: 10.1021/jm00046a026. [DOI] [PubMed] [Google Scholar]
- Mika J, Przewlocki R, Przewlocka B. The role of delta-opioid receptor subtypes in neuropathic pain. Eur J Pharmacol. 2001;415:31–37. doi: 10.1016/s0014-2999(01)00814-7. [DOI] [PubMed] [Google Scholar]
- Mosberg HI, Hurst R, Hruby BJ. Bis penicillamine enkephalins possess highly improved specificity towards delta opioid receptors. Proc Natl Acad Sci U S A. 1983;80:5871–5874. doi: 10.1073/pnas.80.19.5871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazumder S, Nath I, Dhar MM. Immunomodulation of human T cell responses with receptor selective enkephalins. Immunol Lett. 1993;35:33–38. doi: 10.1016/0165-2478(93)90144-q. [DOI] [PubMed] [Google Scholar]
- Meng F, Xie GX, Thompson RC. Cloning and pharmacological characterization of the rat kappa opioid receptor. Proc Natl Acad Sci U S A. 1993;90:9954–9958. doi: 10.1073/pnas.90.21.9954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearl J, Harris LS. Inhibition of writhing by narcotic antagonists. J Pharmacol Exp Ther. 1966;154:319–324. [PubMed] [Google Scholar]
- Pedigo NW, Dewey WL, Harris LS. Determination and characterization of the antinociceptive activity of intraventricularly administered acetylcholine in mice. J Pharmacol Exp Ther. 1975;193:845–852. [PubMed] [Google Scholar]
- Porreca F, Takemori AE, Sultana M, Portoghese PS, Bowen WD, Mosberg HI. Modulation of mu-mediated antinociception in the mouse involves opioid delta-2 receptors. J Pharmacol Exp Ther. 1992;263:147–152. [PubMed] [Google Scholar]
- Portoghese PS, Sultana M, Nagase H, Takemori AE. A highly selective delta 1-opioid receptor antagonist: 7-benzylidenenaltrexone. Eur J Pharmacol. 1992;218:195–196. doi: 10.1016/0014-2999(92)90167-3. [DOI] [PubMed] [Google Scholar]
- Portoghese PS, Nagase H, Maloney Huss KE, Lin CE, Takemori AE. Role of spacer and address components in peptidomimetic delta opioid receptor antagonists related to naltrindole. J Med Chem. 1991;34:1715–1720. doi: 10.1021/jm00109a027. [DOI] [PubMed] [Google Scholar]
- Stein C, Millan MJ, Shippenberg TS, Peter K, Herz A. Peripheral opioid receptors mediating antinociception in inflammation. Evidence for involvement of mu, delta and kappa receptors. J Pharmacol Exp Ther. 1989;248:1269–1275. [PubMed] [Google Scholar]
- Thompson RC, Mansour RA, Ludens JH. Cloning and pharmacological characterization of a rat mu opioid receptor. Neuron. 1993;11:903–913. doi: 10.1016/0896-6273(93)90120-g. [DOI] [PubMed] [Google Scholar]
- Traynor JR, Fantegrossi W, Woods JH. Evaluation of Compounds for Opioid Activity. In: Dewey WL, editor. NIDA Research Monograph; Problems of Drug Dependence, 2004: Proceedings of the 66th Annual Scientific Meeting, The Committee on Problems of Drug Dependence, Inc; Rockville, MD: U.S Department of Health and Human Services; 2005. pp. 131–159. [Google Scholar]