

Abstract
The interplay between pathology and HIV expansion in brain tissues has not been thoroughly assessed in the HAART era. HIV-associated dementia (HAD) is marked by progressive brain infection due to recruitment and migration of macrophages in brain tissues; however, the cellular and viral events occurring prior to HAD development and death are under debate. In this study 66 brain tissues from 11 autopsies were analyzed to assess HIV-1 DNA concentration in brain tissues. In most patients without HAD, it was impossible to amplify HIV-1 from brain tissues. Amplifiable DNA was obtained from three cases of patients on HAART who died due to primary pathology other than HAD: 1) cardiovascular disease, a disease associated with HAART therapy, 2) bacterial infections, including Mycobacterium avium complex, rapid occurrence of extreme dementia and, 3) AIDS-related lymphoma with meningeal involvement. HIV-1 DNA was also amplified from multiple tissues of two HAD patients. Analysis of HIV-1 nef, gp120 and gp41 sequences showed reduced viral evolution within brain tissues for the non-HAD cases relative to patients with clinical and histological HAD. The present study is the first to show a potential correlation between HIV-1 evolutionary patterns in the brain and different neuropathologies.
Keywords: dementia, atherosclerosis, neuropathology, CNS infection, macrophages, HIV-1 nef, genetic analysis
Introduction
Before the introduction of highly active antiretroviral therapy (HAART), 20–30% of patients with late-stage acquired immunodeficiency syndrome (AIDS) developed HIV-associated dementia (HAD) (Gonzalez-Scarano and Martin-Garcia, 2005). With HAART therapy, the incidence of HAD has reportedly been cut in half, although it persists as a significant and debilitating disease (Sacktor, 2002; Sacktor et al, 2001a; Sacktor et al, 2001b). While studies concerning the evolution of HIV-1 within HAD patients are numerous, this is the first study to analyze the expansion of HIV-1 within the brain due to other neuropathologies that may develop in association with HIV-infection. Additionally, most studies concerning HIV-1 viruses in brain tissues have relied on pre-HAART patient autopsies. These earlier studies have generated some controversy over the extent of HIV-1 viral burden in the brain, signature sequences associated with central nervous system (CNS) infection and the extent of compartmentalization of viruses within brain tissues (Glass et al, 1995; Johnson et al, 1996; Ohagen et al, 2003; Van Marle and Power, 2005; Wiley et al, 1999). For many years, the well-accepted theory concerning HIV-1 expansion in the brain has suggested that HIV-1 invasion of the CNS occurs early in infection, followed by a slow evolution of HIV-1 until the onset of late-stage disease. In the last few years, however, an increasing body of research has explored alternate routes to HIV-1 brain invasion (Alexaki et al, 2008; Fischer-Smith et al, 2008; Fischer-Smith and Rappaport, 2005; Kramer-Hammerle et al, 2005).
The causes of death for HIV-infected individuals are numerous and have changed since the introduction of HAART. While a handful of AIDS-defining illnesses still persist (e.g., wasting syndrome, non-Hodgkin’s lymphoma, cytomegalovirus disease, Pneumocystis carinii infection, atypical mycobacteria infection, brain toxoplasmosis, Kaposi’s sarcoma, tuberculosis, dementia) other non-AIDS-defining illnesses are also common (e.g., hepatitis-C infection, non-AIDS-defining lymphomas, cardiovascular disease, liver dysfunction). In the United States, the life-span of HIV-infected individuals has increased along with the relative proportion of deaths attributed to non-AIDS defining diseases (Palella et al, 2006). The degree to which both AIDS and non-AIDS defining illnesses impact viral burden and expansion in the brain is unknown.
In the present study, neuropathology reports were compiled from eleven patients who died from various complications of HIV-1 and a quantitative polymerase chain reaction (qPCR) was used to identify the presence or absence of HIV-1 DNA in brain tissues collected at autopsy (Table 1). Five of the patients had amplifiable amounts of HIV DNA within brain tissues. 3.3kb HIV-1 sequences from a panel of their brain and body tissues were cloned and sequenced. Genetic analyses were used to investigate potential differences in HIV-1 evolutionary patterns among different patients.
Table 1.
Summary of cases examined
| ACSR Number-Lab ID | HAD | Degree of Neuropathology1 | Other Primary Pathology2 | HAART3 |
|---|---|---|---|---|
| 3007238-NV | None | none | yes | |
| 3007235-KA | None | 1 | yes | |
| 3007241-FL | None | 1 | yes | |
| 3007239-YZ | None | 1 | yes | |
| 3007240-OK | None | 1 | yes | |
| 3007243-DE | None | 1 | yes | |
| 3007234-AZ | None | 3 | CVD | yes |
| 3007242-BW | Progressive | 2 | Meningeal Lymphoma | yes |
| 3007233-DY | Acute | 1 | MAC | yes |
| 3007244-GA | Progressive | 5 | Pulmonary Edema | yes |
| 3006990-CX | Progressive | 5 | no | |
The Degree of Neuropathology is estimated on a scale of 1 to 5, with 1 being slight edema and/or vacuolization or other similar minimal tissue damage and 5 being profound macrophage infiltration or other severe abnormalities. In the CVD patient, the primary neuropathology observed was atherosclerosis while in the lymphoma patient the primary neuropathology was leptomeningeal lymphoma.
All patients eventually died with multiple pathologies common to end-stage AIDS; noted in this column are the primary non-neurological causes of death of the patients, as shown in table 2, detectable HIV in most brain tissues.
HAART therapy was frequently stopped in the weeks or days prior to death.
Methods
Tissue collection
Frozen autopsy tissues from patients and accompanying pathology records were obtained from the University of California at San Francisco AIDS and Cancer Resource (ACSR) (url: http://acsr.ucsf.edu). Some of the cases were obtained through the ACSR but provided by the San Diego National NeuroAIDS Tissue Consortium, NNTC (http://www.hivbrainbanks.org). The tissues were obtained from patients after appropriate consent and a de-identification procedure was applied. Clinical histories were handled similarly in a de-identified manner. Patient designations used throughout this study were generated randomly as shorthand used by technicians who performed the studies so as not to have any correlation to patient information. The ACSR is recognized by the Office of Biorepositories and Biospecimen Research at the National Institutes of Health as being HIPAA compliant and in accordance with the ethical standards of the Declaration of Helsinki. Additionally, all material was obtained under approval from the UCSF committee on human research.
Characterization of patient’s specimens for qPCR
Frozen specimens from 13 patient’s multi-site postmortem autopsies who died due to various complications of AIDS were obtained (Table 1). Standard pathologic definitions of tissue-based disease were confirmed by pathologic review prior to initiation of DNA extraction for quantitative HIV-1 DNA analyses.
Quantitative PCR
For all patient samples, qPCR was used to measure and identify the presence of HIV-1 DNA in six brain tissues (meninges, grey matter, white matter, temporal cortex, cerebrum and basal ganglia). Viral genomic DNA was prepared (QIAamp DNA Mini Kit, QIAGEN company, Valencia, CA) and quantified by optical density (OD260, NanoDrop® ND-1000 UV-Vis Spectrophotometer, NanoDrop Technologies, Wilmington, Delaware). HIV-1 copy number/genomic equivalent within each tissue was calculated by qPCR using primers SK 38 (5 ′-ATAATCCACCTATCCCAGTAGGAGAAAT-3′) and SK 39 (5 ′-TTTGGTCCTTGTCTTATGTCCAGAATGC-3′) yielding an 110bp fragment of the gag genome. The single-copy human c-jun gene was used as a genomic equivalent standard for quantification purposes. The amplification protocol comprised one cycle of denaturation at 95 °C for 10 minutes, followed 60 cycles of 95 °C for 15 seconds, 60 °C for 5 seconds, and 72 °C for 12 seconds (Roche’s FastStart DNA Master SYBR Green 1 Kit, Roche Applied Science, Indianapolis, IN). 5000 cells/1 copy HIV-1 was used as a cut-off, with all values less than 5000 cells/1 HIV-1 copy considered HIV-1 positive (Table 2). This sensitivity limitation was due to the small volume of material used for PCR amplification in the Roche Light Cycler system utilized in the current study and positive specimens would therefore have relatively abundant copies of HIV-1 available for evolutionary studies.
Table 2.
HIV-1 positivity in brains of AIDS patients by quantitative PCR
| Patient group | MG | GM | WM | TC | CE | BG | HIV-1 Positive rate in brain |
|---|---|---|---|---|---|---|---|
| Non-demented | 16.7% | ||||||
| 3007238-NV | ND | ND | ND | ND | ND | ND | |
| 3007235-KA | + | ND | ND | ND | ND | ND | |
| 3007241-FL | ND | ND | ND | ND | ND | ND | |
| 3007239-YZ | ND | + | ND | ND | + | ND | |
| 3007240-OK | ND | ND | ND | ND | ND | + | |
| 3007243-DE | ND | ND | ND | + | ND | + | |
| Atherosclerosis | 83% | ||||||
| 3007234-AZ | + | + | ND | + | + | + | |
| Meningeal Lymphoma | 100% | ||||||
| 3007242-BW | + | + | + | + | + | + | |
| MAC/HAD | 83% | ||||||
| 3007233-DY | + | + | + | + | ND | + | |
| HAD | 89% | ||||||
| 3007244-GA | + | + | + | + | + | ND | |
| 3006990-CX | + | + | + | + | + | ||
MG = Meninges, GM = Frontal Lobe Grey Matter, WM = Frontal Lobe White Matter: TC = Temporal Cortex: CE = Cerebellum, BG = Basal Ganglia. ND = not detectable, + = detectable HIV (less than 5000 cells/1 HIV copy), dashed lines = data not available.
Case studies and tissue staining
Based on qPCR results, five case studies and their available brain and body tissues were chosen for DNA extraction and sequencing of partial HIV-1 genomes. Most tissues used for sequencing had several parallel fixed brain tissues available for immunohistochemical staining. These tissues were stained with anti-CD68 and with anti-HIVp24. All antibodies were obtained from Dako Inc. and were used as previously described (Lamers et al, 2009) following manufacturer’s instructions. CD68 and p24 staining were evaluated subjectively on a scale of 0 to 3, where 0 equals no positivity and 3 equals a highly positive result.
Medical and autopsy reports provided additional information for each case study as follows.
Subject AZ had been on HAART for five years. The subject had severe CVD and HCV liver disease leading to a gastrointestinal bleed and death. The patient stopped taking HAART one week prior to death. The brain was normal except for widespread atherosclerosis. Importantly, this particular patient could have suffered from virus-independent artherosclerosis due to age (>50 years old).
Subject DY had been on HAART for six years. During treatment, the patient had reoccurring episodes of MAC infection. In the week prior to death, the patient became profoundly demented. At autopsy, his brain did not show the typical histology of patients with HAD; the tissue was essentially normal with no inflammation of the meninges, cortex or basal ganglia. MNGCs and slight macrophage infiltration into the white matter was consistent with a low level HIV-1 encephalitis.
Subject BW had been on HAART for four years prior to death. The subject had disseminated lymphoma with significant leptomeningeal lymphoma. The basal ganglia, frontal and temporal lobes were populated with multinucleated giant cells, consistent with HIV-1 encephalitis.
Subject GA had been on HAART for four years. Along with myriad of complications due to HIV-1 infection, it is noted in medical reports that the patient suffered from progressive dementia in the months prior to death. There was perivascular inflammation throughout his brain consistent with HAD along with macrophages in the perivascular infiltrates.
Subject CX had a two-year history of AIDS dementia prior to death and before the availability of HAART. One month before death, he became more severely demented and cachectic, with seizures and cardiac arrest noted as the cause of death. Throughout the brain grey and white matter there was marked astrocytosis, microgliosis, and macrophage infiltration. The frontal cortex contained MNGCs and the meninges were fibrotic.
HIV-1 PCR, cloning, and data management was performed as previously described (Lamers et al, 2009).
Detection of recombination sequences
The gp120, nef and gp41 fragments were isolated from the amplified 3.3kb fragment using HIVbase@ software (Lamers et al, 2004). For each protein, recombinant sequences were identified in each tissue as previously described (Lamers et al, 2009) using an algorithm coupling split-decomposition networks with PHI-test for recombination (Salemi and Goodenow, 2008). All identified recombinant sequences were removed prior to distance or phylogenetic analysis. A linear regression analysis was performed to compare the number of recombinant sequences for each patient vs. the length of the domain using the PAST statistical package (Hammer et al, 2001).
Viral tropism
Prediction of viral tropism was performed using the geno2pheno program (www.geno2pheno.org). This program is useful in the identification of either CCR5 or CXCR4 tropic viruses, but usually will classify dual-tropic viruses as CXCR4 isolates. Subject DY was the only case in our analysis to receive a CXCR4 classification using geno2pheno; therefore, a subsequent analysis using a sensitive neural network approach was utilized to distinguish if the subject’s sequences clustered more highly with dual-tropic or CXCR4 sequences (Lamers et al, 2008).
Distance analysis
Divergence in each protein along with standard error was calculated for sequences derived from each patient’s brain tissues using the Jukes-Cantor method and the maximum likelihood composite method with 1000 bootstrap replicates in MEGA 4.0 (Tamura et al, 2007).
Consensus V3 amino acid sequences and charges
Consensus amino acid V3 brain sequences were constructed with the program Seqpublish at the HIV-1 database at Los Alamos website (http://www.hiv.lanl.gov/). In Figure 1, capital letters in the alignment are those positions where ≥95% identity was observed between all sequences. A lower case letter indicates an identity between ≥50% and 95%. A question mark indicates that <50% similarity existed at the position. Overall charges for consensus sequences were calculated by adding positive charges (K and R) and subtracting negative charges (D and E).
Figure 1. V3 loop consensus sequences.
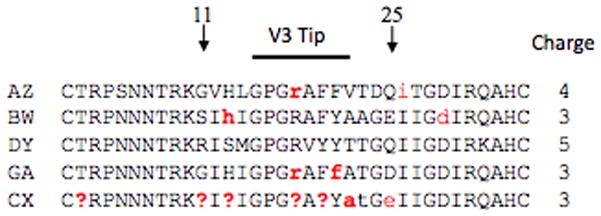
The 35 consensus amino acids that span the V3 loop domain along with the overall charge of the domain are shown for each patient. Conserved positions, those that occur at a level of 95% or more, are shown in black and variable positions are shown in red. Lower case letters indicate a position that is conserved at a level of 51–94%, whereas a question mark indicates a position where no majority is observed.
Phylogenetic analysis
Sequences were codon aligned using MEGA 4.0 and manipulated by hand when necessary to maximize the alignment. Phylogenies for gp120, gp41 and nef were inferred using a Bayesian framework assuming a relaxed molecular clock and a constant size population prior using the Markov Chain Monte Carlo (MCMC) method in the BEAST software package version 1.4.8 (http://evolve.zoo.ox.ac.uk/beast/). This method allows for a full Bayesian reconstruction of evolutionary parameters taking into account phylogenetic uncertainty. Each aligned data set was partitioned in 1st+2nd and 3rd codon positions and the parameters of the nucleotide substitution (HKY+Γ+I) were estimated independently for each partition. Because all samples were obtained from the same time point, an evolutionary rate of 1.0 was assumed. The Markov Chain was run for 30,000,000 generations with sampling every 3000th generation. Convergence of the MCMC was assessed by calculating the effective sampling size (ESS) of the combined runs. All parameter estimates showed significant ESS (>250). The maximum a posterior clade credibility tree (MAP tree) was obtained for each patient after a 50% burn-in with target tree branch lengths using the program TreeAnnotator (http://beast.bio.ed.ac.uk/TreeAnnotator). Estimates of the median root height (scaled in substitutions per site) averaged over all trees in the posterior distribution (less a 50% burn-in) were obtained for each patient and for the clade containing the first appearance of brain sequences. Phylogenies were visualized using Figtree (http://tree.bio.ed.ac.uk/software/figtree/).
Genbank Accession numbers for the sequences analyzed in this study are GQ868779-GQ869380.
Results
Quantitative PCR
HIV-1 was detected via qPCR in only 6 out of 35 tissues examined from the patients with minor neuropathic changes in autopsy tissues and no clinical signs of dementia at the time of death. Two of these patients had a positive result in two out of five brain tissues, while the rest had one or less tissues with a positive qPCR result (Table 1). In the individual with atherosclerosis, 4 out of 5 tissues exhibited a positive QPCR. A similar result was found in the patient with MAC co-infection while the patient with leptomeningeal lymphoma had a positive qPCR for all tissues examined. In the two patients with clinical HAD prior to death with extensive signs of macrophage-associated inflammation of brain tissues, 100% of the tissues sampled were positive by qPCR. The consistent finding in the survey was that a positive qPCR was related to cases with histological damage seen within brain tissues, except in the case of patient DY.
CD68 and P24 staining of frontal lobe
Of the five subjects studied, only one showed complete absence of CD68+ macrophages in frontal lobe white matter and minimal active viral replication: the patient with CVD. In the next three cases shown in Table 3, a moderate number of CD68+ macrophages were observed, while a high degree of replicating virus was found in the patient with lymphoma (BW), a moderate degree in the patient with MAC (DY) and a slight degree in the patient with dementia on HAART (GA). The non-HAART dementia patient (CX) showed both very high CD68 and P24 staining, indicating that a large number of resident macrophages were present and producing viruses.
Table 3. CD 68 and P24 staining results for frontal lobe white matter.
Subjective results are listed on a scale of 0–3, with 0 meaning no positivity and 3 associated with the highest level of staining.
| Subject | CD68 | P24 |
|---|---|---|
| AZ | 0 | 1 |
| BW | 1–2 | 3 |
| DY | 1–2 | 2 |
| GA | 1–2 | 0–1 |
| CX | 3 | 3 |
Consensus V3 amino acid sequences, charges and viral tropism
The consensus amino acid sequence for each patient (Figure 1) showed that most variable amino acid positions occurred within the V3 tip. Positions 11 and 25, which are often positively charged amino acids in CXCR4 viruses (Cho et al, 1998; Milich et al, 1993), were usually negative except in the case of position 11 for patient DY. Patient DY was also interesting in that there were no variations in the 35 amino acids that comprise the V3 loop. The overall charge of all V3 loop amino acid sequences for most patients was consistent with CCR5 viruses, except in the case of patient DY, who showed a consensus V3 loop charge of +5. A neural network approach to determining viral tropism indicated with high probability that the viruses in DY’s brain were dual-tropic.
Sample diversity and recombination rates
Pair-wise nucleotide diversity and the proportion of recombinant sequences within each patient’s brain sequences are shown in Figure 2 for three different HIV-1 proteins (gp120, gp41 and nef). The two HAD patients had four- to five-fold increases in overall diversity compared to that of the CVD patient. In fact, the diversity in all three HIV-1 proteins in the brain for the CVD patient was negligible; however, it is important to note that fewer brain nucleotide sequences were available to analyze for patient AZ, which could account for the reduced diversity in the data set. A linear regression analysis comparing gp41, gp120 and nef length vs. number of recombinants in each region was performed in order to investigate whether recombination occurred at similar rates in different HIV-1 proteins from different individuals. Both HAD patients showed strong and highly significant correlation (CX, p< 0.0005, r= 0.97; GA p< 0.0005, r= 0.96). In the non-HAD patients, the correlation was significant (p), but the correlation coefficient (r) was much lower, indicating no correlation existed (p< 0.0005 and AZ r=0.14; BW, r= 0.35; DY r=0.54).
Figure 2. Nef, gp120 and gp41 genetic variation in sequences generated from brain tissues of different patients.

Each two-letter code on the x-axis indicates the subject ID. Subject pathology is shown above the graph (CVD = cardiovascular disease, ARL = AIDS-related lymphoma, HAD = HIV-1 associated dementia, MAC = Mycobacterium avium complex). Colored bars represent percent nucleotide diversity within brain sequences in specific gene regions according to the color legend to the right of the graph. Standard error bars are also given. The number along each colored bar indicates the percentage of recombinant sequences detected in that gene region. The number of sequences used in each calculation is shown in parentheses below the subject ID.
Phylogenetic analysis
Similar to pair-wise distance analysis, Bayesian estimates of the root height for nef, gp41 and gp120 DNA sequences indicated increased evolution within both HAD patient’s sequence populations when compared to sequences from the other subjects (nef phylogenies shown in Figure 3). The median height of the root averaged over all trees in the posterior distribution, scaled in substitutions/site, is shown under the phylogenies and was smallest for subject AZ at 0.0182, followed by 0.0286 for patient DY, 0.0445 for patient BW, 0.0481 for patient CX and 0.0656 for patient GA. A similar pattern was seen in the median root height for clades containing the majority of brain sequences (AZ=0.004, DY=0.013, BW=0.0337, CX=0.045 and GA= 0.043). Interestingly, all patients except BW, whose primary brain pathology was meningeal lymphoma, showed some mixing of brain and non-brain sequences. Subject AZ had a single monophyletic clade containing all brain sequences and 12 non-brain sequences. Subject BW was the only subject with a single highly supported monophyletic clade containing only brain sequences. Subject’s DY phylogeny was similar to subject AZ’s in that a majority of the brain sequences were found in a single monophyletic clade. The two HAD subjects, CX and GA, had a more evolved sequence population including several highly supported clades and sub-clades containing sequences from brain tissues.
Figure 3. Bayesian phylogenetic analysis of nef proteins from sequences derived from lymphoid and non-lymphoid tissues.

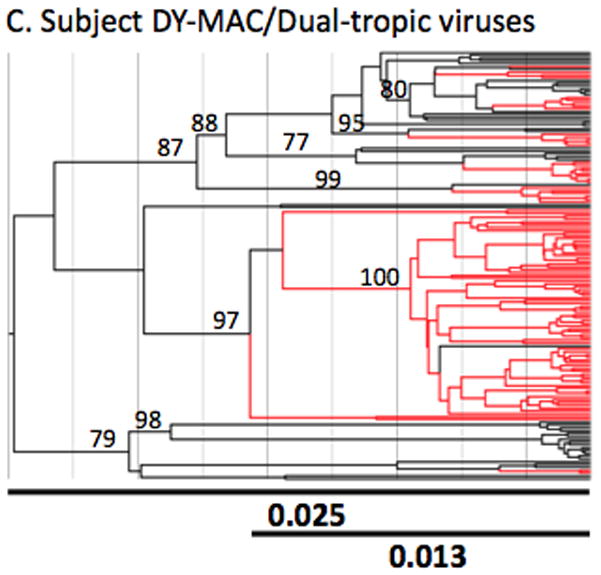

The Bayesian MAP trees were inferred as detailed in the methods section. The subject name appears above each analysis (Panels A-E). Tropism for patient DY was not determined experimentally and was implied by computational tests and viral behavior. Branches containing sequences generated from non-lymphoid (brain) tissues are shown in red whereas those generated from lymphoid tissues are shown in black. Branch lengths are drawn in scale proportional to nucleotide substitutions per site. The two lines at the bottom of each tree represents the length of the entire tree (top line) and the length of the tree up to the earliest brain clade (bottom line) in units of nucleotide substitutions per site (bold numbers). Each number along an internal branch represents the posterior probability for that branch. Only posterior probabilities >75% are indicated.
Discussion
In this study qPCR was utilized to identify HIV-1 DNA within 66 brain tissues from 13 autopsies of HIV-infected individuals with and without clinical and histological indications of HAD. These results indicated that most tissues from patients without HAD contained little amplifiable HIV-1 DNA, whereas two patients with both clinical and histological HAD contained amplifiable HIV-1 DNA in all brain tissues analyzed. Three other patients also contained positive qPCR results in brain tissues, each with a different pathology: CVD, MAC co-infection, and non-Hodgkins AIDS-related lymphoma.
Histological staining of frontal lobe tissues for HIV-1 showed varied degrees of replicating and resident macrophages among patients with differing pathologies. In patient AZ, the CD68+ staining was negative; these results suggest that brain viruses found in patient AZ’s frontal lobe had either not been present long enough to activate brain macrophages or that those viruses that had entered the frontal lobe had migrated from an atherosclerotic plaque, a rich pathogenic macrophage process. Either interpretation is consistent with late viral entry into brain tissues due to atherosclerosis. The next three cases (BW, DY, GA) were interesting in that they all showed a moderate degree of HIV-1 infected macrophages and varying degrees of replicating virus. It is also noteworthy that patient DY staining results indicated the existence of a population of activated macrophages; however, neuropathology reports describe the brain as grossly normal and unremarkable. As in patient AZ, this may indicate that the infected cell population within the frontal lobe was relatively new. The fact that subjects BW, DY and GA all show less positive CD68 and P24 results than patient CX may be due to the effect of HAART. Certainly it is well reported that manifestations of HIV-associated CNS involvement have generally become less severe and more manageable in patients on HAART.
A variety of sequence analysis highlighted differences in viral evolution between HAD and non-HAD patients. Recombination rates were more prevalent in both HAD patients, indicating that there were more viruses replicating within super-infected macrophages in brain tissues. This observation was previously noted in gp120 DNA sequences from damaged tissues (Lamers et al, 2009). The positive correlation seen in the HAD patients between protein length and the number of recombinants may indicate less selective constraints among proteins, random generation of recombinant events across the HIV-1 genome, or that most sequences were derived from viruses that were sequestered within a macrophage reservoir for a much longer period of time. The fact that a correlation between domain length and number of recombinants was not present in the three non-HAD individuals may indicate selective constraints on particular proteins evolving under particular conditions, such as populations of viruses that had recently been exposed to a more complex immune environment. It should be noted that the small number of variables in this regression analysis (three data points) makes it impossible to say with certainty that either conclusion is correct, still, if it is true that the viruses from these patients entered the brain at significantly different times, the results provide another piece of evidence that indicates different evolutionary genetic patterns in lymphoid rather than non-lymphoid tissues. A full genome study would help confirm these results. Patients BW and DY showed a statistical increase in diversity in gp120 over gp41, while both HAD patients demonstrated a significant increase in diversity of gp41 over gp120 sequences. This difference may be due to different selective pressures on the surface glycoprotein (gp120) and the transmembrane/fusion glycoprotein (gp41) during different disease pathologies.
Phylogenetic trees for both HAD subjects contained multiple highly supported sub-clades containing sequences from brain consistent with a longer period of infection within brain tissues during HAD development. This finding is also supported by the larger median root height of the posterior distribution of sequences in CX and GA. On the other hand, the primary brain clades for patients DY and AZ contained no supported sub-clades. The phylogeny for subject BW differed from the other four in that brain and non-brain sequences each branched in highly supported monophyletic clades. However, patient BW was similar to the HAD patients in that the tree showed well-supported sub-clades within brain sequences. Various mechanisms can contribute to the degree of compartmentalization of HIV-1 populations seen in a phylogenetic analysis, such as the high rate of mutation of HIV-1 in vivo, differences in selective pressures imposed by the immune system, differences in local concentrations of antiviral drugs, cellular tropism, drug resistance, and level of pathogenesis (Zarate et al, 2007). The lack of compartmentalization in subject AZ and DY is not surprising if the viruses that populated the brain were due to arterial leakage near death. The compartmentalization of subject BW brain viruses may reflect the nature of lyphomagenesis in patients with HIV-1 infection; BW’s brain may have been populated near death with an aggressive strain of HIV-1 closely associated with metastatic tissues. It is also possible that some of the sequences represented archival proviruses from latently infected cells. Yet, the substantial genetic heterogeneity observed, coupled with p24 positive staining discussed in table 3 indicate that at least some viruses are replicating within productively infected brain cells. Overall, discerning which sequences reflect latent or productive infection does not impact the principal finding that different viral evolutionary patterns can be observed in patients with different pathology.
Williams and Hickey (Williams and Hickey, 2002) proposed a popular model to describe the development of HAD: during the early phase of the disease, the immune system still controls the infection in brain macrophages; later the immune system becomes unable to prevent the emergence of productively infected cells as they continue to amass and damage surrounding tissues. The continuous accumulation in the brain of activated macrophages initiates a self-amplifying cycle that eventually leads to the development of HAD. The model explains latency, extensive brain damage observed at autopsy and the separate evolution of brain from body viral isolates (Salemi et al, 2005; Williams and Hickey, 2002). Later, Fischer-Smith et al. (Fischer-Smith et al, 2008; Fischer-Smith and Rappaport, 2005) described another model for HIV-1 infection of the brain; they proposed that brain tissues may also become infected during late-stage disease via the expansion of more virulent viruses, uncontrolled viral replication and alterations in the myeloid differentiation pathway that results in an activated monocyte subset capable of CNS tissue invasion. In vitro experiments have implicated direct infection of brain astrocytes by HIV-1 as contributing to HAD development (Zheng et al, 1999). Changes in cell environment, like the elevation in the level of cytokines such as TNF-α and IL-1β, might also activate virus production within CNS tissues (Gorry et al, 2005; Kramer-Hammerle et al, 2005).
HIV-associated dementia generally develops in late-stage HIV-1 disease (CD4 cells/μL<50). The onset is usually subtle, developing over weeks or months. At autopsy, the brains from patients with clinically identified HAD commonly show marked astrocytosis, microgliosis and a numerical increase of resident macrophages actively replicating virus. The macrophage infiltrate is most prominent in the white matter of the frontal lobe, although usually no area of the central nervous system is spared (Williams and Hickey, 2002). The general theory for the development of HAD is that infected macrophages carry HIV-1 into brain tissues where the infection evolves primarily in microglia and macrophages (McGrath, 1997; Williams et al, 2001). Macrophages bind viruses that are specific for the CCR5 and CCR3 chemokine receptors (Alkhatib et al, 1996; Deng et al, 1996). Once infected, macrophages can produce a variety of proinflammatory cellular neurotoxins, including tumor necrosis factor-alpha, cytokines, interleukins, chemokines, nitric oxide, and excitatory amino acids (Dou et al, 2004). Some studies have suggested that the damage caused by macrophages within tissues may be more correlated to their release of inflammatory mediators in the CNS than to the actual viral load in the brain (Hult et al, 2008; Williams et al, 2008).
Two of the HAD patients studied (GA and CX), one on HAART and the other not on therapy, presented with a slower-progressing clinical dementia and showed classic histological signs of HAD at the time of death. Phylogenetic analysis, recombination, distance and charge analysis in these patients all support the theory of Williams and Hickey as well as earlier observations from our group (Salemi et al, 2005), wherein the brain is infected early, sequences in the brain evolve separate from the body, and a self-amplifying macrophage infection ensues.
Another patient, AZ, did not develop HAD; however, the patient did have a severe case of CVD. CVD is the most common non-AIDS-defining illness leading to death during HAART. It is uncertain if this is a result of long-term survival during HAART therapy or an indirect toxic effect of the therapy (Friis-Moller et al, 2003; Holmberg et al, 2002; Sudano et al, 2006). What is clear is that HIV-infected patients in the HAART era are at a significantly increased risk for myocardial infarction (Friis-Moller et al, 2003), endothelial dysfunction (Stein et al, 2001), osteonecrosis (Monier et al, 2000), coronary artery calcification (Meng et al, 2002), and atherosclerosis (Hsue et al, 2004). During the evolution of atherosclerosis, fatty lipids are deposited within arteries. Macrophages called in to consume the lipids are transformed into activated fat containing vacuoles called foam cells. The sites of damaged endothelium attract more macrophages to the site of lipid deposition and eventually form an early atherosclerotic site, also called a fatty streak. Fatty streaks cause vascular breakdown, bleeding and a self-propagating inflammatory response to injury (Linton and Fazio, 2003). In certain ways, this process is similar to what has been described during the perivascular macrophage inflammatory response associated with HAD (Salemi et al, 2005; Williams et al, 2001; Williams and Hickey, 2002); however, the CVD process differs in that the perivascular HIV-1 infected macrophages in CVD are associated with arteries, rather than central nervous system veins involved in HAD. Patient AZ exhibited little diversity in sequence populations between brain and body isolates and little brain tissue damage other than widespread atherosclerosis. Phylogenetic, distance, and recombination analysis confirm a late-stage brain infection likely due to arterial leakage from atherosclerosis.
A fourth patient, DY, suffered for several years with reoccurring MAC infection, one of the most common bacterial opportunists. Dual infection in HIV-1 patients with other opportunistic infections increases viral replication in both macrophages and CD4+ T-cells (Wahl et al, 2000; Wahl et al, 1998; Wahl et al, 1999), especially in late-stage disease when the CD4+ T-cells are depleted and HIV-1 continues to replicate within macrophage populations. Wahl et al. (Wahl et al, 1999) found that MAC up-regulates TNF-α expression, which subsequently enhances HIV-1 replication within macrophages. Other studies have also observed this finding (Denis and Ghadirian, 1994; Havlir et al, 2001; MacArthur et al, 2000). Additionally, patient DY likely harbored dual-tropic viruses. Infection of an individual with dual-tropic HIV-1 has been linked to drug resistance, rapid progression to AIDS and hastened death. Although brain autopsy specimens were normal, this patient developed a severely aggressive dementia in the days prior to death. This was the only patient observed with no apparent histology abnormalities in which amplifiable HIV-1 DNA was present. Phylogenetic, recombination, distance analysis and primary sequence analysis all suggest the rapid expansion of an aggressive form of virus that evolved near the time of death. One plausible explanation for this patient’s pathology could be the Fisher-Smith Hypothesis; however, another interesting hypothesis was presented in 1999 concerning the infection of astrocytes with HIV-1 (Zheng et al, 1999). This study showed through in vitro experiments that brain astrocytes are capable of binding viruses via the CXCR4 co-receptor. Once infected, astrocytes cause the release of a variety of pro-inflammatory cytokines that could cause neuronal dysfunction (Zheng et al, 1999). Astrocyte infection is associated with moderate to severe dementia (Ranki et al, 1995). By combining genetic analysis, pathology, histology and theories concerning HIV-1 infection of the CNS, an interesting hypothesis for the rapid development of HAD minus brain tissue damage in this patient can be constructed, 1) co-infection with MAC super-activated macrophages that were producing dual-tropic viruses, 2) dual tropic viruses could enter the brain via macrophages, 3) because of the ability to utilize both the CCR5 and CXCR4 co-receptors, these viruses could bind both macrophages and astrocytes directly, causing a rapid and amplified production of cytotoxic and neurotoxic proteins from several cell-types and 4) this amplified cascade of events would disrupt neuronal signaling faster and could cause observed dementia before an accumulation of macrophages within brain tissues.
The fifth patient in the study, patient BW, had developed NH-ARL in the weeks prior to death that had metastasized into the CNS where it formed a neoplasm in the meninges. Since the development of HAART, the occurrence of NH-ARL has dropped approximately 50%, but it still occurs at a rate higher than in the normal population. NH-ARL in HIV-1 infected patients is diffusely aggressive and almost always fatal. In addition to common symptoms of lymphoma, HIV-1 infected patients with CNS lymphoma may present with seizures, headache, altered mental status, or other focal neurological deficits. Although symptoms of dementia may be present, the subsequent histological examination will be different than seen in patients with clinical HAD, showing less overall macrophage infiltrate into deep brain tissues. Patient BW was interesting in comparison to patients DY and AZ in that the individual’s brain viruses were highly compartmentalized when compared body sequences, which could indicate the spread of a potentially later-stage, lymphoma-specific virus that travelled within metastatic cells to the brain meninges where it spread to other brain tissues. A previous study showed that macrophages containing HIV-1 taken from a human NH-ARL tumor induced a tumor in a SCID model that containing related HIV, thus showing that HIV-1 infected macrophages were implicated in tumorogenesis in ARL (Zenger et al, 2002). As ARL is an explosive disease process, this could also explain BW’s high rate of viral production as indicated by p24 staining.
Theories on how and when HIV-1 enters CNS tissues have been presented for many years. In the pre-HAART era, HIV-1 infection progressed faster and it is likely that fewer mechanisms for viral CNS entry existed. Additionally, studies on the prevalence of HAD within different subtypes of HIV-1 in similar populations points to the fact that a genetic determinant for CNS invasion of HIV-1 exists (Sacktor et al., 2009). In developed countries where HAART therapy is common, greatly reduced cases of HAD have been noted; however, the evolution of different mechanisms for the invasion of HIV-1 into brain tissues may be occurring and may further complicate the debate over how and when HIV-1 evolves within the CNS. Although the number of case studies examined here are small, the number of sequences examined are large and the results from the analysis presented support several mechanisms of HIV-1 viral entry into CNS tissues, including some that are associated with non-AIDS defining illnesses. The study highlights the fact that HIV-1 infection in the brain is likely specific to the host and to the host’s developing pathologies and physical reaction to HAART therapy.
Acknowledgments
The authors would like to thank the National Institutes of Health and the many individuals at the AIDS and Cancer Specimen Resource that assisted in the careful preparation of study materials. Additionally the authors would like to thank Sara Granier, Tulio de Oliveira, Leanne Huysentruyt, John P. Lamers and Elizer Masliah for assistance in sequence management, manuscript editing and sample collection. The project was primarily funded through NIH R01 MH073510-01 and NIH R01 NS063897-01A2. Additional support was provided to S.L. Lamers through SBIR grant DMI-0349669. Additional support was provided to M.S. McGrath through the West Coast AIDS and Cancer Specimen Resource Consortium U01 CA066529-12. Additional support for M. Salemi was provided by grants AI065265 and HD32259.
References
- Alexaki A, Liu Y, Wigdahl B. Cellular reservoirs of HIV-1 and their role in viral persistence. 2008;6:388–400. doi: 10.2174/157016208785861195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkhatib G, Combadiere C, Broder CC, Feng Y, Kennedy PE, Murphy PM, Berger EA. CC CKR5: a RANTES, MIP-1alpha, MIP-1beta receptor as a fusion cofactor for macrophage-tropic HIV-1. Science. 1996;272:1955–8. doi: 10.1126/science.272.5270.1955. [DOI] [PubMed] [Google Scholar]
- Cho MW, Lee MK, Carney MC, Berson JF, Doms RW, Martin MA. Identification of determinants on a dualtropic human immunodeficiency virus type 1 envelope glycoprotein that confer usage of CXCR4. 1998;72:2509–15. doi: 10.1128/jvi.72.3.2509-2515.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng H, Liu R, Ellmeier W, Choe S, Unutmaz D, Burkhart M, Di Marzio P, Marmon S, Sutton RE, Hill CM, Davis CB, Peiper SC, Schall TJ, Littman DR, Landau NR. Identification of a major co-receptor for primary isolates of HIV-1. 1996;381:661–6. doi: 10.1038/381661a0. [DOI] [PubMed] [Google Scholar]
- Denis M, Ghadirian E. Mycobacterium avium infection in HIV-1-infected subjects increases monokine secretion and is associated with enhanced viral load and diminished immune response to viral antigens. 1994;97:76–82. doi: 10.1111/j.1365-2249.1994.tb06582.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou H, Kingsley JD, Mosley RL, Gelbard HA, Gendelman HE. Neuroprotective strategies for HIV-1 associated dementia. Neurotoxicity Research. 2004;6:503–21. doi: 10.1007/BF03033447. [DOI] [PubMed] [Google Scholar]
- Fischer-Smith T, Bell C, Croul S, Lewis M, Rappaport J. Monocyte/macrophage trafficking in acquired immunodeficiency syndrome encephalitis: lessons from human and nonhuman primate studies. 2008;14:318–26. doi: 10.1080/13550280802132857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer-Smith T, Rappaport J. Evolving paradigms in the pathogenesis of HIV-1-associated dementia. Expert Reviews in Molecular Medicine. 2005;7:1–26. doi: 10.1017/S1462399405010239. [DOI] [PubMed] [Google Scholar]
- Friis-Moller N, Sabin CA, Weber R, d’Arminio Monforte A, El-Sadr WM, Reiss P, Thiebaut R, Morfeldt L, De Wit S, Pradier C, Calvo G, Law MG, Kirk O, Phillips AN, Lundgren JD Group DCoAEoA-HDDS. Combination antiretroviral therapy and the risk of myocardial infarction. 2003;350:1993–2003. doi: 10.1056/NEJMoa030218. [DOI] [PubMed] [Google Scholar]
- Glass JD, Fedor H, Wesselingh SL, McArthur JC. Immunocytochemical quantitation of human immunodeficiency virus in the brain: correlations with dementia. 1995;38:755–62. doi: 10.1002/ana.410380510. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Scarano F, Martin-Garcia J. The neuropathogenesis of AIDS. 2005;5:69–81. doi: 10.1038/nri1527. [DOI] [PubMed] [Google Scholar]
- Gorry PR, Churchill M, Crowe SM, Cunningham AL, Gabuzda D. Pathogenesis of macrophage tropic HIV-1. 2005;3:53–60. doi: 10.2174/1570162052772951. [DOI] [PubMed] [Google Scholar]
- Hammer O, Harper DAT, Ryan PD. Palaeontologia Electronica. Coquina Press; 2001. PAST: paleontological statistics software for education and data analysis; p. 9. [Google Scholar]
- Havlir DV, Torriani FJ, Schrier RD, Huang JY, Lederman MM, Chervenak KA, Boom WH. Serum interleukin-6 (IL-6), IL-10, tumor necrosis factor (TNF) alpha, soluble type II TNF receptor, and transforming growth factor beta levels in human immunodeficiency virus type 1-infected individuals with Mycobacterium avium complex disease. 2001;39:298–303. doi: 10.1128/JCM.39.1.298-303.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmberg S, Moorman A, Williamson J, Tong T, Ward D, Wood K, Greenberg A, Janssen R. Protease inhibitors and cardiovascular outcomes in patients with HIV-1-1. 2002;360:1747–8. doi: 10.1016/S0140-6736(02)11672-2. [DOI] [PubMed] [Google Scholar]
- Hsue PY, Lo JC, Franklin A, Bolger AF, Martin JN, Deeks SG, Waters DD. Progression of atherosclerosis as assessed by carotid intima-media thickness in patients with HIV infection. Circulation. 2004;109:1603–8. doi: 10.1161/01.CIR.0000124480.32233.8A. [DOI] [PubMed] [Google Scholar]
- Hult B, Chana G, Masliah E, Everall I. Neurobiology of HIV. International Review of Psychiatry. 2008;20:3–13. doi: 10.1080/09540260701862086. [DOI] [PubMed] [Google Scholar]
- Johnson RT, Glass JD, McArthur JC, Chesebro BW. Quantitation of human immunodeficiency virus in brains of demented and nondemented patients with acquired immunodeficiency syndrome. 1996;39:392–5. doi: 10.1002/ana.410390319. [DOI] [PubMed] [Google Scholar]
- Kramer-Hammerle S, Rothenaigner I, Wolff H, Bell JE, Brack-Werner R. Cells of the central nervous system as targets and reservoirs of the human immunodeficiency virus. 2005;111:194–213. doi: 10.1016/j.virusres.2005.04.009. [DOI] [PubMed] [Google Scholar]
- Lamers S, Beason S, Dunlap L, Compton R, Salemi M. HIVbase: a PC/Windows-based software offering storage and querying power for locally held HIV-1 genetic, experimental and clinical data. 2004;20:436. doi: 10.1093/bioinformatics/btg445. [DOI] [PubMed] [Google Scholar]
- Lamers SL, Salemi M, Galligan DC, De Oliveira T, Fogel GB, Granier SC, Zhao L, Brown JN, Morris A, Masliah E, McGrath MS. Extensive HIV-1 intra-host recombination is common in tissues with abnormal histopathology. PLoS ONE. 2009;4:e5065. doi: 10.1371/journal.pone.0005065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamers SL, Salemi M, McGrath MS, Fogel GB. Predicting HIV R5, X4 and dual tropic phenotype using evolved neural networks. IEEE/ACM Transactions In Computational Biology and Bioinformatics. 2008;5:291–300. doi: 10.1109/TCBB.2007.1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linton MF, Fazio S. Macrophages, inflammation, and atherosclerosis. 2003;27:S35–40. doi: 10.1038/sj.ijo.0802498. [DOI] [PubMed] [Google Scholar]
- MacArthur RD, Lederman MM, Benson CA, Chernoff MC, MacGregor RR, Spritzler J, Mahon LF, Yen-Lieberman B, Purvis S. Effects of Mycobacterium avium complex-infection treatment on cytokine expression in human immunodeficiency virus-infected persons: results of AIDS clinical trials group protocol 853. The Journal of Infectious Diseases. 2000;181:1486–90. doi: 10.1086/315370. [DOI] [PubMed] [Google Scholar]
- McGrath MS. T-cells and macrophages in HIV disease. Clinical Reviews In Allergy and Immunology. 1997;14:359–366. doi: 10.1007/BF02771752. [DOI] [PubMed] [Google Scholar]
- Meng Q, Lima JA, Lai H, Vlahov D, Celentano DD, Strathdee SA, Nelson KE, Wu KC, Chen S, Tong W, Lai S. Coronary artery calcification, atherogenic lipid changes, and increased erythrocyte volume in black injection drug users infected with human immunodeficiency virus-1 treated with protease inhibitors. American Heart Journal. 2002;144:642–8. doi: 10.1067/mhj.2002.125009. [DOI] [PubMed] [Google Scholar]
- Milich L, Margolin B, Swanstrom R. V3 loop of the human immunodeficiency virus tye 1 Env protein: interpreting sequence variability. 1993;67:5623–34. doi: 10.1128/jvi.67.9.5623-5634.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monier P, McKown K, Bronze MS. Osteonecrosis complicating highly active antiretroviral therapy in patients infected with human immunodeficiency virus. 2000;31:1488–92. doi: 10.1086/317503. [DOI] [PubMed] [Google Scholar]
- Ohagen A, Devitt A, Kunstman K, Gorry P, Rose P, Korber B, Taylor J, Levy R, Murphy R, Wolinsky S, Gabuzda D. Genetic and functional analysis of full-length human immunodeficiency virus type 1 env genes derived from brain and blood of patients with AIDS. 2003;77:12336–12345. doi: 10.1128/JVI.77.22.12336-12345.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palella FJJ, Baker RK, Moorman AC, Chmiel JS, Wood KC, Brooks JT, Holmberg SD. Mortality in the highly active antiretroviral therapy era: changing causes of death and disease in the HIV outpatient study. Journal of Acquired Immune Deficiency Syndromes. 2006;43:27–34. doi: 10.1097/01.qai.0000233310.90484.16. [DOI] [PubMed] [Google Scholar]
- Ranki A, Nyberg M, Ovod V, Haltia M, Elovaara I, Raininko R, Haapasalo H, Krohn K. Abundant expression of HIV Nef and Rev proteins in brain astrocytes in vivo is associated with dementia. 1995;9:1001–8. doi: 10.1097/00002030-199509000-00004. [DOI] [PubMed] [Google Scholar]
- Sacktor N. The epidemiology of human immunodeficiency virus-associated neurological disease in the era of highly active antiretroviral therapy. 2002;8:115–21. doi: 10.1080/13550280290101094. [DOI] [PubMed] [Google Scholar]
- Sacktor N, Lyles RH, Skolasky R, Kleeberger C, Selnes OA, Miller EN, Becker JT, Cohen B, McArthur JC, Study MAC. HIV-associated neurologic disease incidence changes: Multicenter AIDS Cohort Study, 1990–1998. 2001a;56:257–60. doi: 10.1212/wnl.56.2.257. [DOI] [PubMed] [Google Scholar]
- Sacktor N, Tarwater PM, Skolasky RL, McArthur JC, Selnes OA, Becker J, Miller EN (MACS) MfACS. CSF antiretroviral drug penetrance and the treatment of HIV-associated psychomotor slowing. 2001b;57:542–4. doi: 10.1212/wnl.57.3.542. [DOI] [PubMed] [Google Scholar]
- Salemi M, Goodenow MM. An exploratory algorithm to identify intra-host recombinant viral sequences. 2008;49:618–28. doi: 10.1016/j.ympev.2008.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salemi M, Lamers S, Yu S, de Oliveira T, Fitch W, McGrath M. Phylodynamic analysis of human immunodeficiency virus type 1 in distinct brain compartments provides a model for the neuropathogenesis of AIDS. 2005;79:11343–11352. doi: 10.1128/JVI.79.17.11343-11352.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein JH, Klein MA, Bellehumeur JL, McBride PE, Wiebe DA, Otvos JD, Sosman JM. Use of human immunodeficiency virus-1 protease inhibitors is associated with atherogenic lipoprotein changes and endothelial dysfunction. Circulation. 2001;104:257–62. doi: 10.1161/01.cir.104.3.257. [DOI] [PubMed] [Google Scholar]
- Sudano I, Spieker LE, Noll G, Corti R, Weber R, Luscher TF. Cardiovascular disease in HIV infection. American Heart Journal. 2006;151:1147–55. doi: 10.1016/j.ahj.2005.07.030. [DOI] [PubMed] [Google Scholar]
- Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular evolutionary genetics analysis (MEGA) software version 4.0. 2007;24:1596–9. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- Van Marle G, Power C. Human immunodeficiency virus type 1 genetic diversity in the nervous system: evolutionary epiphenomenon or disease determinant? 2005;11:107–28. doi: 10.1080/13550280590922838. [DOI] [PubMed] [Google Scholar]
- Wahl SM, Greenwell-Wild T, Hale-Donze H, Moutsopoulos N, Orenstein JM. Permissive factors for HIV-1 infection of macrophages. 2000;68:303–10. [PubMed] [Google Scholar]
- Wahl SM, Greenwell-Wild T, Peng G, Hale-Donze H, Doherty TM, Mizel D, Orenstein JM. Mycobacterium avium complex augments macrophage HIV-1 production and increases CCR5 expression. 1998;95:12574–9. doi: 10.1073/pnas.95.21.12574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl SM, Greenwell-Wild T, Peng G, Hale-Donze H, Orenstein JM. Co-infection with opportunistic pathogens promotes human immunodeficiency virus type 1 infection in macrophages. The Journal of Infectious Diseases. 1999;179:S457–60. doi: 10.1086/314814. [DOI] [PubMed] [Google Scholar]
- Wiley CA, Achim CL, Christopherson C, Kidane Y, Kwok S, Masliah E, Mellors J, Radhakrishnan L, Wang G, Soontornniyomkij V. HIV mediates a productive infection of the brain. 1999;13:2055–9. doi: 10.1097/00002030-199910220-00007. [DOI] [PubMed] [Google Scholar]
- Williams K, Corey S, Westmoreland S, Pauley D, Knight H, Debakker C, Alvarez X, Lackner A. Perivascular macrophages are the primary cell type productively infected by simian immunodeficiency virus in the brains of macaques: implications for the neuropathogenesis of AIDS. The Journal of Experimental Medicine. 2001;193:905–916. doi: 10.1084/jem.193.8.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams KC, Hickey WF. Central nervous system damage, monocytes and macrophages, and neurological disorders in AIDS. 2002;25:537–62. doi: 10.1146/annurev.neuro.25.112701.142822. [DOI] [PubMed] [Google Scholar]
- Williams R, Dhillon NK, Hegde ST, Yao H, Peng F, Callen S, Chebloune Y, Davis RL, Buch SJ. Proinflammatory cytokines and HIV-1 synergistically enhance CXCL10 expression in human astrocytes. 2008:0894–1491. doi: 10.1002/glia.20801. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarate S, Kosakovsky S, Shapshak P, Frost S. Comparative study of methods for detecting sequence compartmentalization in human immunodeficiency virus type 1. 2007;81:6643–6651. doi: 10.1128/JVI.02268-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenger E, Abbey NW, Weinstein MD, Kapp L, Reis J, Gofman I, Millward C, Gascon R, Elbaggari A, Herndier BG, McGrath MS. Injection of human primary effusion lymphoma cells or associated macrophages into severe combined immunodeficient mice causes murine lymphomas. 2002;62:5536–42. [PubMed] [Google Scholar]
- Zheng J, Thylin MR, Ghorpade A, Xiong H, Persidsky Y, Cotter R, Niemann D, Che M, Zeng YC, Gelbard HA, Shepard RB, Swartz JM, Gendelman HE. Intracellular CXCR4 signaling, neuronal apoptosis and neuropathogenic mechanisms of HIV-1-associated dementia. 1999;98:185–200. doi: 10.1016/s0165-5728(99)00049-1. [DOI] [PubMed] [Google Scholar]