

Abstract
Disturbances in GABAA receptor trafficking contribute to several neurological and psychiatric disorders by altering inhibitory neurotransmission. Identifying mechanisms that regulate GABAA receptor trafficking could lead to better understanding of disease pathogenesis and treatment. Here, we show that protein kinase Cε (PKCε) regulates the N-ethylmaleimide-sensitive factor (NSF), an ATPase critical for membrane fusion events, and thereby promotes the trafficking of GABAA receptors. Activation of PKCε decreased cell surface expression of GABAA receptors and attenuated GABAA currents. Activated PKCε associated with NSF, phosphorylated NSF at serine 460 and threonine 461, and increased NSF ATPase activity, which was required for GABAA receptor downregulation. These findings identify new roles for NSF and PKCε in regulating synaptic inhibition through downregulation of GABAA receptors. Reducing NSF activity by inhibiting PKCε could help restore synaptic inhibition in disease states in which it is impaired.
Introduction
GABAA receptors are heteropentameric ligand-gated ion channels that mediate most fast inhibitory neurotransmission (Olsen and Sieghart, 2008). Impaired GABAA receptor signaling contributes to several neuropsychiatric conditions including epilepsy (Goodkin et al., 2008), schizophrenia (Charych et al., 2009), anxiety and depression (Kalueff and Nutt, 2007; Charych et al., 2009), substance abuse (Kumar et al., 2004), and pain (Knabl et al., 2008). GABAA receptors have therefore been important targets for drug development, although current drugs can produce limiting side effects including sedation, amnesia, and dependence. Understanding mechanisms that regulate GABAA receptor trafficking could increase knowledge about the pathophysiology of these disease states and provide clues for developing alternative drugs that modulate inhibitory neurotransmission with fewer side effects.
Tumor-promoting phorbol esters decrease cell surface GABAA receptors in several preparations (Leidenheimer et al., 1992; Leidenheimer and Chapell, 1997; Chapell et al., 1998; Connolly et al., 1999; Cinar and Barnes, 2001; Balduzzi et al., 2002; Meier et al., 2003; Herring et al., 2005). This effect is presumed to be caused by activation of protein kinase C (PKC) (Kittler and Moss, 2003; Leidenheimer, 2008). PKC is not a single entity but a family of nine kinases with several splice variants that transduce signals involving lipid second messengers (Song and Messing, 2005). Phorbol esters activate seven isozymes (α, β, γ, δ, ε, η, and θ), each with specialized functions that are generally not redundant. In addition to PKCs, phorbol esters activate several other proteins such as protein kinase D, chimerins, RasGRP, and Munc-13 (Brose and Rosenmund, 2002). Thus, it is not certain that phorbol esters regulate GABAA receptor trafficking by activating a specific PKC isozyme or another phorbol ester-responsive protein.
PKC substrates that mediate GABAA receptor downregulation are also unknown. Although several phosphorylation sites have been identified on intracellular loops of GABAA γ2 and β subunits, alanine substitutions do not prevent receptor downregulation by phorbol esters (Chapell et al., 1998; Connolly et al., 1999). Other possible substrates include proteins associated with GABAA receptors (Chen and Olsen, 2007). One candidate is the N-ethylmaleimide-sensitive factor (NSF) (Matveeva et al., 2001), an ATPase that regulates membrane fusion events (Zhao et al., 2007) and is important for stability of AMPA receptors at excitatory synapses (Hanley et al., 2002). When overexpressed, NSF can decrease cell surface levels of GABAA receptors (Goto et al., 2005). NSF also interacts with GABARAP (GABAA receptor-associated protein), and this interaction is proposed to modulate GABAA receptor trafficking (Kittler et al., 2001; Leil et al., 2004; Marsden et al., 2007). However, the hypothesis that endogenous NSF truly regulates GABAA receptor trafficking remains to be tested.
We found that mice lacking PKCε (Prkce−/− mice) are hypersensitive to drugs that increase GABAA receptor activation, including benzodiazepines, ethanol, neurosteroids, and barbiturates (Hodge et al., 1999, 2002). Because diminished PKC-stimulated trafficking of GABAA receptors could contribute to this phenotype, we hypothesized that PKCε regulates GABAA receptor trafficking. Here, we demonstrate that PKCε decreases the number of GABAA receptors at the cell surface by binding, phosphorylating, and activating NSF.
Materials and Methods
Animal care.
Prkce−/− mice were generated by homologous recombination in J1 embryonic stem cells (Khasar et al., 1999). Male and female Prkce+/− mice were maintained on inbred 129S4 and C57BL/6J backgrounds and crossed to produce Prkce+/− C57BL/6J × 129S4 F1 hybrid mice for breeding. These mice were intercrossed to generate F2 hybrid Prkce+/+ and Prkce−/− littermates for experiments. Mice were genotyped by PCR of tail DNA. All animals used were males between 2 and 4 months of age at the time of experimentation. All procedures were conducted in accordance with institutional Institutional Animal Care and Use Committee policies.
Reagents.
Peptides were synthesized by AnaSpec. An affinity-purified, polyclonal rabbit anti-phospho-NSF (S460) antibody (1:50) was generated against the phosphopeptide 455RHIKA[pS]TKVEV465 by ProSci. The ATP analog-specific kinase inhibitor 1-naphthyl-4-amino-5-(4-methylphenyl)-7-(t-butyl)pyrazolo[3,4-d]-pyrimidine (1Na-PP1) was synthesized as described previously (Bishop et al., 1999). The MyrPalm-CFP expression plasmid was kindly provided by A. Newton (University of California, San Diego, La Jolla, CA). 6,7-Dinitroquinoxaline-2,3(1H,4H)-dione (DNQX) and d(−)-2-amino-5-phosphonopentanoic acid (d-APV) were purchased from Tocris Bioscience. Other reagents were from Sigma-Aldrich or from the sources listed below.
Cell culture.
HEK293 cells stably expressing rat α1β2γ2S GABAA receptors and the human analog-specific PKCε mutant (AS-PKCε, PKCε M486A) were generated as described previously (Qi et al., 2007). Cells were maintained in minimal essential medium with 10% fetal bovine serum, 0.2 mm l-glutamine, 200 μg/ml Geneticin (G418), 100 μg/ml streptomycin, and 100 U/ml penicillin at 37°C in a humidified atmosphere of 5% CO2 and 95% air. Primary cultures of hippocampal neurons were prepared from postnatal day 1 (P1) to P3 mice and cultured at 105 cells per 200 mm2 in Neurobasal A medium containing B27 and Glutamax (Invitrogen) at 37°C in a humidified atmosphere of 6% CO2 and 94% air. One-half of the medium was changed the next day and every 7 d thereafter for up to 3 weeks.
Cell surface biotinylation.
HEKGR-AS-PKCε cells were grown on 100 mm poly-l-ornithine (100 μg/ml in 15 mm borate buffer)-coated tissue culture dishes to 80–90% confluence. Cells were rinsed twice at 37°C with buffer containing 145 mm NaCl, 5 mm KCl, 10 mm glucose, 1 mm MgCl2, and 1 mm CaCl2 (adjusted with Tris base to pH 7.5), and incubated at 37°C with drugs in 10 ml of the same buffer. Cells were then rinsed with ice-cold PBS and cell surface proteins biotinylated using 1.5 mg/ml sulfo-NHS biotin (Pierce) as described previously (Qian et al., 1997). Biotinylated proteins were recovered from protein lysates on Immunopure Immobilized Monomeric Avidin beads (Pierce) and separated by SDS-PAGE on duplicate NuPAGE 4–12% Bis-Tris gels (Invitrogen). Proteins in one gel were transferred to nitrocellulose and subjected to Western blot analysis with rabbit anti-GABAA γ2 antibody (1:1000; Alpha Diagnostics International) and HRP-conjugated goat anti-rabbit IgG (1:1000; Millipore Bioscience Research Reagents). Immunoreactive bands were visualized using enhanced chemiluminescence (Pierce) and quantified by scanning densitometry with ImageJ (http://rsbweb.nih.gov/ij/). To control for recovery of biotinylated proteins, we stained the duplicate gel with Coomassie blue and quantified 45–55 kDa proteins by scanning densitometry, and then used these results to normalize Western blot results for each corresponding biotinylated sample. Cell lysates were examined by Western blot analysis to detect levels of total γ2 subunits. Values for γ2 immunoreactivity were normalized for GAPDH immunoreactivity detected in the same samples using rabbit anti-GAPDH antibody (1:2000; Abcam).
Cell electrophysiology.
HEK293 cells were plated on 35 mm plastic dishes coated with poly-d-lysine. Conventional whole-cell recordings were made with recording electrodes (5–8 MΩ) filled with the following (in mm): 145 N-methyl-d-glucamine (NMDG)-Cl, 2 MgCl2, 0.1 CaCl2, 5 EGTA, 10 HEPES, and 2 Mg2+-ATP, pH 7.3, adjusted with HCl. Cells were continuously perfused with an external solution (in mm: 145 NaCl, 3 KCl, 1.5 CaCl2, 1 MgCl2, 10 HEPES, and 10 d-glucose, pH 7.4, adjusted with NaOH). Cells were voltage clamped at −75 mV and whole-cell current was elicited by increasing concentrations of GABA that were fast applied for 5 s. There was a 1 min washout period between each GABA application with 1Na-PP1 or vehicle present in the washout solution. All recordings were obtained at room temperature. Data were expressed as current density by dividing whole-cell current by whole-cell capacitance and averaged to generate sigmoid dose–response curves for each treatment condition. For mouse hippocampal neurons (6–12 d in vitro), recording electrodes (5–8 MΩ) contained the following (in mm): 145 NMDG-Cl, 1 MgCl2, 5 HEPES, 4 Mg2+-ATP, 2 Na+-ATP, adjusted to pH 7.3 with HCl. The external control solution used to continuously perfuse the neurons contained the following (in mm): 142 NaCl, 3 KCl, 1.5 CaCl2, 1 MgCl2, 10 HEPES, 10 d-glucose, adjusted to pH 7.4 with NaOH. The GABA dose–response relationship was found to be similar for Prkce+/+ and Prkce−/− neurons, and a sub-EC50 concentration of 10 μm was chosen for current stability experiments during which GABA was applied for 5 s every 2.5 min for up to 40 min. All recordings were made at 33°C with cells voltage clamped at −75 mV. The ψεRACK peptide (HDAPIGYD; 5 μm) (Chen et al., 2001) was added to the pipette solution to activate PKCε. GABA and 1Na-PP1 were applied by local perfusion, and whole-cell currents were filtered at 2 kHz and digitized at 5 kHz. Where used, the final concentration of DMSO applied to neurons was <0.1% (v/v), which had no effect on GABAA receptor currents.
Slice electrophysiology.
Male mice (P45–P65) were anesthetized with halothane and then decapitated. Their brains were rapidly removed and put in ice-cold artificial CSF (ACSF) containing the following (in mm): 124 NaCl, 26 NaHCO3, 2 KCl, 2 CaCl2, 2 MgCl2, 1.25 KH2PO4, 10 glucose, equilibrated with 95% O2 and 5% CO2. Horizontal hippocampal sections (300 μm) were cut with a vibratome in ice-cold ACSF. Slices were left to recover at 30°C for 30 min before transfer to a recoding chamber at room temperature. Slices were continuously superfused with ACSF at 2 ml/min at room temperature. Whole-cell recordings of CA1 hippocampal pyramidal neurons were obtained with electrodes that had a resistance of 5–8 MΩ when filled with pipette solution containing the following (in mm): 145 K-gluconate, 2 MgCl2, 10 HEPES, 0.5 EGTA, 2 Mg-ATP, Na-GTP, and 5 phosphocreatine, pH 7.3, adjusted with KOH; osmolarity, 270–280 mOsm. Cells were held under voltage clamp at −45 mV, and responses were low-pass filtered at 2 kHz, and digitized at 10 kHz. Synaptic responses were evoked by monophasic current pulses (200 μs; 30 μA; 4 Hz) delivered by a pair of monopolar tungsten electrodes placed in CA1 stratum radiatum. Responses mediated by GABAA receptors were pharmacologically isolated by adding 10 μm DNQX and 30 μm d-APV to the ACSF to block glutamate receptors. Addition of bicuculline abolished these currents indicating that they were generated by GABAA receptors. Recordings began immediately after establishing the whole-cell configuration. Only recordings that showed a <30% change in series resistance over the 20–40 min recording period were analyzed.
Immunoprecipitation.
HEKGR-AS-PKCε cells were cultured as described above for biotinylation studies. Cells were collected in buffer containing 50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 5 mm EDTA, 5 mm EGTA, 1% NP-40, and Complete protease inhibitor mixture (Roche Applied Science) and lysed by freezing and thawing. Lysates were centrifuged at 20,817 × g for 15 min at 4°C. Whole forebrains from 2- to 4-month-old mice were homogenized using 15 strokes of a Teflon-glass homogenizer in ice-cold homogenization buffer containing 25 mm HEPES, pH 7.4, 150 mm NaCl, 1% Triton X-100, 1 mm EDTA, 0.5 mm ATP, Complete protease inhibitor mixture (Roche Applied Science), phosphatase inhibitor mixture 1 (Sigma-Aldrich), and 1 mm PMSF. The homogenate was centrifuged at 10,000 × g for 10 min at 4°C. Supernatants (500 μg protein in 1 ml buffer) were incubated overnight at 4°C with rabbit anti-NSF antibody (5 μg/ml; Millipore Biotechnology) and protein A-agarose (50 μl; Roche Applied Science) and separated on NuPAGE 4–12% Bis-Tris gels (Invitrogen) in MOPS (4-morpholinepropanesulfonic acid) running buffer. Proteins were examined by Western blot analysis with goat anti-PKCε (1:1000; Santa Cruz Biotechnology), mouse anti-NSF antibody (1:1000; BD Biosciences), or anti-GABAA receptor γ2 subunit (1:500; PhosphoSolutions). Phosphorylation of NSF at S460 was detected using polyclonal rabbit anti-NSF (S460) antibody (diluted 1:50). Immunoreactivity was detected using appropriate HRP-conjugated anti-IgG secondary antibodies (1:1000; Jackson ImmunoResearch) followed by enhanced chemiluminescence (Pierce).
Pull-down assay.
FLAG-tagged human PKCε (in pcDNA3 from A. Toker, Harvard University, Cambridge, MA) was purified from COS-7 cells using the FLAG system (Sigma-Aldrich) (Qi et al., 2007). FLAG-human PKCε was immobilized on anti-FLAG antibody M2 conjugated to agarose (50 μl; Sigma-Aldrich), and then incubated for 2 h at 4°C with 0, 2.5, or 5 μg of recombinant His6-NSF in binding buffer (10 mm Tris-HCl, pH 7.5, 300 mm NaCl, 2 mm EDTA, 0.5 mm ATP). After centrifugation at 20,817 × g for 5 min, beads were washed three times with binding buffer. Bound proteins were separated in NuPAGE Bis-Tris gels (Invitrogen) and detected by Western blot analysis with monoclonal anti-NSF antibody (1:1000; BD Biosciences) and polyclonal anti-PKCε antibody SN134 (1:1000) (Qi et al., 2007). Immunoreactivity was quantified by scanning densitometry using ImageJ, and results for NSF immunoreactivity were normalized to PKCε immunoreactivity.
Gel overlay assay.
For overlay assays, NSF (0.5 μg) was run on NuPAGE Bis-Tris gels (Invitrogen) and transferred to nitrocellulose Hybond C Extra membranes (GE Healthcare). Membranes were incubated in blocking buffer containing TBS-Tween 20 and 2% nonfat milk for 1 h at room temperature and incubated overnight at 4°C with 10 nm recombinant PKCε (Invitrogen) in overlay buffer containing 50 mm Tris-HCl, pH 7.5, 10 mm β-mercaptoethanol, 200 mm NaCl, 1 mm EGTA, 0.1% BSA, 1% polyethylene glycol, 0.3 μg/μl l-α-phosphatidylserine, 62 μg/ml 1,2-diolein, and 0.03% Triton X-100. After being washed for 10 min three times in TBS-Tween 20 containing 1% nonfat milk, the membranes were incubated at room temperature for 4 h with anti-PKCε antibody SN134 (1:1000) in TBS-Tween 20 containing 1% nonfat milk. Membranes were then washed for 10 min with TBS-Tween 20 four times, and PKCε bound to membranes was detected by HRP-conjugated anti-rabbit IgG and enhanced chemiluminescence. The amount of mutant NSF loaded in each lane was detected by anti-NSF antibody (1:5000; BD Biosciences), HRP-conjugated donkey anti-mouse IgG antibodies (1:1000; Jackson ImmunoResearch), and enhanced chemiluminescence.
Immunofluorescence microscopy.
HEKGR-AS-PKCε cells were plated on poly-d-lysine (BD Biosciences)-coated chamber slides at 5 × 103 cells per 200 mm2 and grown overnight in minimal essential medium with 10% fetal bovine serum, 0.2 mm l-α-glutamine, 200 μg/ml G418, 100 μg/ml streptomycin, and 100 U/ml penicillin at 37°C in a humidified atmosphere of 5% CO2 and 95% air. To identify plasma membrane, we transfected these cells with MyrPalm-CFP, which encodes cyan fluorescent protein fused to the myristoylation and palmitoylation sequence of Lyn kinase (Violin et al., 2003), using the SuperFect Transfection Reagent (QIAGEN). Cells were used for experiments 24 h later. After drug treatment, cells were fixed for 15 min with 4% paraformaldehyde in PBS, permeabilized for 5 min with 0.1% Triton X-100 in PBS, and blocked for 1 h in 10% normal donkey serum (NDS) and 0.2% BSA in PBS. Samples were incubated overnight at 4°C with the following primary antibodies diluted in PBS containing 2% NDS and 0.2% BSA: chicken anti-GFP (green fluorescent protein) (1:100; Invitrogen), mouse anti-NSF (1:200; BD Biosciences), and goat anti-PKCε (1:100; Santa Cruz Biotechnology). After three washes with PBS, cells were incubated with donkey DyLight 649-conjugated anti-goat, DyLight 549-conjugated anti-mouse, and DyLight 488-conjugated anti-chicken IgY secondary antibodies (1:200; Jackson ImmunoResearch).
Mouse hippocampal neurons were plated on poly-d-lysine (BD Biosciences)-coated chamber slides at 105 cells per 200 mm2 for 14–21 d before treatment with drugs. To detect GABAA receptors, live cultures were incubated with rabbit anti-GABAA receptor γ2 subunit antibody (1:100; Alomone Labs) diluted in Ringer's solution containing 0.5 μm tetrodotoxin for 90 min at room temperature. After three 10 min washes in Ringer's solution, neurons were fixed with methanol for 10 min at −20°C, washed with PBS, and incubated for 90 min at room temperature with mouse anti-gephyrin (1:200; Synaptic Systems) in PBS containing 10% NDS. To detect NSF and gephyrin, methanol-fixed cultures were incubated with rabbit anti-NSF (1:200; Millipore Biotechnology) and mouse anti-gephyrin antibodies. After three washes with PBS, cells were incubated with DyLight 549-conjugated anti-rabbit and DyLight 488-conjugated anti-mouse IgG secondary antibodies (1:200; Jackson ImmunoResearch).
Immunofluoresence was detected using a Zeiss LSM 510 confocal microscope (Carl Zeiss MicroImaging) with a Plan Apochromat 63×/1.40 numerical aperture oil-immersion objective. Quantification of colocalization was performed using ImageJ (Abramoff et al., 2004) with the PSC colocalization plug-in (French et al., 2008). For studies using HEK293 cells, at least 26 cells were analyzed for each treatment condition. For experiments with neurons, neurites were analyzed in at least seven images (1024 × 1024 pixels; 7 pixels/μm) for each treatment condition. Results are presented as Spearman's rank correlation coefficients. Unlike the Pearson's product–moment correlation coefficient, which measures the strength of the linear relationship between two signals, the Spearman's coefficient can detect linear and nonlinear relationships (French et al., 2008). The analysis gives values from −1 (a complete negative correlation) to +1 (a complete positive correlation), with zero representing no correlation. Nontransfected HEK293 cells and gephyrin-negative neurites were excluded from the analyses. Results were analyzed using Mann–Whitney tests and differences between median values were considered significant where p < 0.05.
In vitro kinase assay. In vitro phosphorylation of NSF or GST- γ2S by PKCε was performed using 0.07 μm FLAG-tagged human PKCε purified from COS-7 cells (Qi et al., 2007) in 10 μl of kinase buffer containing 20 mm HEPES, pH 7.4, 0.1 mm EGTA, 0.03% Triton X-100, 10 mm MgCl2, 0.48 μg/μl l-α-phosphatidylserine (Avanti Polar Lipids), 1 μm phorbol 12-myristate, 13-acetate (PMA), and 0.5 mm ATP. The kinase reaction was initiated at 37°C by adding 10.8 pmol of recombinant NSF or 18.8 pmol of GST- γ2S and 10 μCi of [γ-32P]ATP. At different time points, 10 μl aliquots of the reaction mixture were removed and added to 2.5 μl of 5× SDS buffer to stop the reaction. Proteins were separated on NuPAGE Bis-Tris gels (Invitrogen) and stained using SimplyBlue (Invitrogen). Phosphorylated NSF was detected by phosphorimaging. The specific radioactivity per mole of [γ-32P]ATP was determined by counting diluted aliquots of the stock [γ-32P]ATP solution to obtain a conversion value for calculating the molar ratio of 32P to NSF. The stoichiometry of phosphate incorporation into NSF was expressed as moles of incorporated γ-32P per moles of NSF.
Purification of NSF.
Recombinant His-tagged Chinese hamster ovary NSF cDNA in the vector pQE9 was kindly provided by Dr. Sidney Whiteheart (University of Kentucky, Lexington, KY). Recombinant His-NSF and its mutants were isolated from Escherichia coli M15 by chromatography on nickel-agarose columns (QIAGEN). To preserve the enzymatic activity of NSF, 0.5 mm ATP were added to the lysis buffer and 0.5 mm ADP were added to the washing and elution buffers. Bacterial cultures were grown at 37°C in LB broth containing 100 μg/ml ampicillin and 25 μg/ml kanamycin until the OD600 reached 0.6, and then induced with 1 mm IPTG (isopropyl β-d-thiogalactopyranoside) for 4 h. The cells were collected and resuspended in 25 ml of lysis buffer containing 20 mm HEPES, pH 8.0, 50 mm NaCl, 0.5 mm ATP, 5 mm MgCl2, 5 mm β-mercaptoethanol, and 1 mm PMSF. Extracts were solubilized by sonication. The unsolubilized material was removed by centrifugation at 200,000 × g for 30 min at 4°C. After affinity binding of the supernatant to nickel-agarose columns, the columns were washed once with lysis buffer containing 10 mm imidazole and once with lysis buffer containing 0.5 mm ADP and 20 mm imidazole. The proteins were eluted using 100 mm HEPES at pH 8.0, 300 mm NaCl, 10% glycerol, 0.5 mm ADP, and 300 mm imidazole buffer. After concentration in Centricon K50 columns, the proteins were dialyzed against buffer containing 20 mm HEPES at pH 7.4, 100 mm NaCl, 10% glycerol, 1 mm MgCl2, 0.5 mm ADP, and 1 mm β-mercaptoethanol and stored at −80°C. The concentration of recombinant NSF was determined by comparison with bovine serum albumin standards on NuPAGE Bis-Tris gels (Invitrogen). The purity of the final proteins was >95%, as assessed by Coomassie blue staining.
Mass spectrometry.
NSF proteins were phosphorylated in vitro by PKCε for 3 h as described above. The reaction mixtures were fractionated by SDS-PAGE and stained with Colloidal Blue (Invitrogen). Gel slices containing NSF were digested with trypsin or AspN protease. Samples from the digests were analyzed by nano-liquid chromatography–mass spectrometry/mass spectrometry (nano-LC-MS/MS) using a LC-Packings HPLC (Dionex) coupled to a QStar XL mass spectrometer (Applied Biosystems). Peptides were first desalted on a 300 μm × 5 mm PepMap C18 trap column with 0.1% formic acid in HPLC-grade water at a flow rate of 20 μl/min. After being desalted for 6 min, peptides were backflushed onto a LC Packings 75 μm × 15 cm C18 nano column (3 μm; 100 A) at a flow rate of 200 nl/min. Peptides were eluted with a 30 min gradient of 3–40% acetonitrile in 0.1% formic acid. Mass ranges for the MS survey scan and MS/MS were m/z 300–1800 and m/z 50–1800, respectively. The scan times for MS and MS/MS were 1.0 and 2.0 s, respectively. The top three, multiply charged ions with MS peak intensity >30 counts/scan were chosen for MS/MS fragmentation with a precursor ion dynamic exclusion of 60 s. Data were searched against a home-built database that included the NSF sequence. Four modifications were included in the database search as follows: carbamidomethyl (C), oxidation (M), phospho (ST), and phospho (Y).
NSF peptide phosphorylation.
The following peptides (based on amino acids 452–468 of NSF) were dissolved in ddH2O to prepare 1 mm stocks: native NSF (AMNRHIKASTKVEVDME), S460A mutant (AMNRHIKAATKVEVDME), T461A mutant (AMNRHIKASAKVEVDME), and S460A/T461A double mutant (AMNRHIKAAAKVEVDME). Human recombinant PKCε (Invitrogen) was diluted in 0.1 mg/ml BSA, 0.05% Triton X-100 to 4 ng/μl. Phosphorylation was performed with 9.5 nm PKCε and 100 μm NSF peptide in 25 μl of kinase buffer containing 20 mm Tris-HCl, pH 7.5, 10 mm MgCl2, 1 μm PMA, 0.48 mg/ml l-α-phosphatidylserine, 0.25 mm EGTA, 0.1 mg/ml BSA, 2.5 μm ATP, and 0.5 μCi of [32γ-P]ATP. After 90 min incubation at 27°C, the reaction was terminated by adding 12.5 μl of 7.5 m guanidine hydrochloride. Eight microliters of reaction mixture were spotted onto a streptavidin-coated SAM Biotin Capture membrane (Promega). The membrane was washed once with 2 m NaCl for 30 s, three times with 2 m NaCl for 2 min, four times with 2 m NaCl in 1% H3PO4 for 2 min, and twice with double-distilled H2O for 30 s. The membrane was air dried at 27°C for 60 min, and radioactivity remaining on the membrane was measured by liquid scintillation counting.
ATPase assay.
NSF (600 ng) was phosphorylated in vitro for 30 min using 200 ng of PKCε as described above but in the presence of 1 mm ATP without [γ-32P]ATP. ATPase assays were then performed in 30 μl of 25 mm Tris-HCl, pH 9.0, 100 mm KCl, 0.65 mm β-mercaptoethanol, 2 mm MgCl2, 0.5 mm ATP, and 10% glycerol for 1 h at 37°C. The reaction was initiated by the addition of 10 μCi of [γ-32P]ATP. An aliquot (1 μl) of the reaction mixture was separated on cellulose PEI thin-layer plates (J. T. Baker) in 0.8 m acetic acid and 0.8 m LiCl and analyzed using a Typhoon 9410 scanner (GE Healthcare). The ATPase activity of NSF was calculated as the percentage of released inorganic 32P using the following formula: ATPase activity = 32P/(32P + [γ-32P]ATP).
Cell surface ELISA.
Hippocampal neurons were grown on four-well poly-d-lysine-coated slides. After drug treatment at 37°C in medium, slides were placed on ice and washed twice in ice-cold PBS, fixed for 10 min in ice-cold PBS containing 3% paraformaldehyde, and washed three times with PBS. Neurons were then incubated with rabbit anti-GABAA γ2 (extracellular) antibody (1:250; Alomone Labs), followed anti-rabbit IgG (H+L) (1:5000; Jackson ImmunoResearch) at room temperature for 1 h, washed six times with ice-cold PBS, and incubated with 0.5 ml per well of 3,3′,5,5′-tetramethylbenzidine (Sigma-Aldrich) for 1 h at room temperature. The supernatant (100 μl) was transferred to a 96-well plate, and absorbance was measured at 655 nm. Results were expressed as a percentage of absorbance detected in neurons treated with vehicle alone.
Statistical analysis.
Quantitative data were expressed as mean ± SEM values and analyzed using Prism 5.0 (GraphPad Software). Data were tested for normality by the Kolmogorov–Smirnov test and then for differences between means by t tests or one-way ANOVA with Newman–Keuls post hoc tests. Data that were not normally distributed were analyzed by Mann–Whitney tests, or by Kruskal–Wallis and post hoc Dunn's multiple-comparison tests. Dose–response curves were analyzed by nonlinear curve fitting. Values of p < 0.05 were considered to be statistically significant.
Results
PKCε regulates the cell surface level of GABAA receptors
To examine the role of PKCε in GABAA receptor trafficking, we investigated phorbol ester-stimulated downregulation of GABAA receptors. We used HEKGR-AS-PKCε cells, which stably express α1β2γ2 GABAA receptors and an ATP analogue-specific mutant of PKCε (AS-PKCε) that can be selectively inhibited by the AS-kinase inhibitor 1Na-PP1 at concentrations that do not affect native kinases (Qi et al., 2007). We treated these cells with the PKC activator PMA, with or without 1Na-PP1, and measured the level of GABAA γ2 subunits at the cell surface by Western blot analysis of biotinylated cell surface proteins (Fig. 1A). PMA (30 nm) reduced the cell surface level of γ2 subunits by 43%, whereas 1 μm 1Na-PP1 increased it by 33% and antagonized the effect of PMA. These treatments did not alter the total level of γ2 subunits in the cell lysate (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
Figure 1.

PKCε regulates cell surface levels of GABAA receptors. A, HEKGR-AS-PKCε cells were incubated for 1 h with 0.03% DMSO vehicle (con) (n = 10), 1 μm 1Na-PP1 (n = 10), 30 nm PMA (n = 5), or 1Na-PP1 plus PMA (n = 4). The top panel is a representative Western blot of biotinylated proteins detected using anti-γ2-subunit antibodies. *p < 0.05 compared with untreated control (100%) by one-sample t tests. B, GABA-stimulated currents recorded from HEKGR-AS-PKCε cells pretreated for 1 h with 0.03% DMSO vehicle (control; n = 16) or 3 μm 1Na-PP1 (n = 14). 1Na-PP1 increased the GABA current density from 38.8 ± 3.8 to 54.7 ± 4.4 pA/pF (p = 0.0086) without altering the EC50 for GABA, which was 13.8 μm (log EC50 = −4.86 ± 0.25) in control and 8.7 μm (log EC50 = −5.06 ± 0.23) in 1Na-PP1-treated cells (p = 0.554). C, GABA-gated currents recorded every 2.5 min for 30 min from cultured Prkce+/+ hippocampal neurons treated with buffer (control; n = 6) or with the PKCε activator 5 μm ψεRACK (n = 10), and from Prkce−/− neurons treated with ψεRACK (n = 6). D, Normalized amplitudes at each time point were expressed relative to current measured at time 0. Compared with vehicle-treated Prkce+/+ cells (control; n = 4) treatment of Prkce+/+ neurons with ψεRACK (n = 5) decreased current amplitude, whereas in Prkce−/− neurons (n = 4) ψεRACK had no effect (Ftime (12,120) = 11.59, p < 0.0001; Fcondition (2,120) = 20.05, p = 0.0003; Ftime by condition (24,120) = 6.47, p < 0.0001). *p < 0.05 versus control by Bonferroni's post hoc test. E, Representative eIPSC traces recorded from hippocampal pyramidal neurons in acute brain slices treated with buffer (control), 5 μm ψεRACK, or 5 μm scrambled ψεRACK peptide. F, Normalized amplitudes of eIPSCs averaged every 3 min and expressed relative to current measured immediately after establishing whole-cell patch configuration at time = 0. Compared with control cells (n = 5), treatment with ψεRACK (n = 5) decreased eIPSC amplitude over the 30 min test period, whereas scrambled ψεRACK (n = 4) had no effect (Ftime (12,132) = 6.85, p < 0.0001; Ftreatment (2,132) = 23.41, p = 0.0001; Ftime by treatment (24,132) = 5.73, p < 0.0001). *p < 0.05 versus control by Bonferroni's post hoc test. Error bars indicate SEM.
These findings suggested that PKCε regulates the level of GABAA receptors at the cell surface. To investigate this hypothesis further, we measured GABA-stimulated currents in HEKGR-AS-PKCε cells. Treatment with 3 μm 1Na-PP1 increased the GABA current density by 41% without altering the EC50 for GABA (Fig. 1B). This increase in GABA efficacy was similar in magnitude to the increase in cell surface γ2 subunits induced by 1Na-PP1 (Fig. 1A), indicating that PKCε regulates GABA efficacy by altering the number of functional GABAA receptors at the cell surface.
We next investigated whether PKCε also regulates GABAA receptors in neurons by studying mouse hippocampal neurons from P1–P3 wild-type and Prkce−/− mice cultured for 6–12 d in vitro. Treatment with 5 μm ψεRACK, a specific PKCε activator (Chen et al., 2001) administered via the recording pipette, reduced GABA-stimulated currents in wild-type neurons but had no effect on Prkce−/− neurons (Fig. 1C,D). In wild-type brain slices from P45–P65 mice, 5 μm ψεRACK administered in the recording pipette reduced electrically evoked, GABA-mediated inhibitory postsynaptic currents in hippocampal pyramidal neurons, whereas 5 μm of a scrambled peptide had no effect (Fig. 1E,F). These results demonstrate that PKCε downregulates native GABAA receptors in hippocampal neurons, which leads to decreased GABA-mediated inhibitory synaptic transmission.
PKCε associates with NSF and GABAA γ2 subunits
We next investigated mechanisms by which PKCε regulates the number of GABAA receptors at the cell surface. Since PKC phosphorylation of GABAA receptor subunits does not regulate receptor trafficking (Chapell et al., 1998; Connolly et al., 1999), we considered as possible substrates proteins that associate with GABAA receptors and are known to regulate trafficking of other receptors. We focused attention on NSF since it regulates the trafficking of AMPA receptors (Collingridge et al., 2004). We found that NSF coimmunoprecipitated with both PKCε and GABAA γ2 subunits in lysates of HEKGR-AS-PKCε cells (Fig. 2A). Incubation with 1 μm PMA for 45 min increased the amount of coimmunoprecipitated PKCε by ∼60%, whereas 1Na-PP1 decreased it by 60% and prevented the effect of PMA (F(3,8) = 16.4; p = 0.0009) (Fig. 2A). In contrast, the amount of GABAA γ2 that coimmunoprecipitated with NSF was not altered by either treatment. The abundance of NSF, PKCε, and γ2 in the lysates was also not altered by these treatments (supplemental Fig. 2A, available at www.jneurosci.org as supplemental material).
Figure 2.

PKCε interacts directly with NSF in a complex containing GABAA receptor γ2 subunits. A, The left panel is a representative experiment showing proteins immunoprecipitated from HEKGR-AS-PKCε cell lysates with anti-NSF antibody and detected by Western blot analysis with anti-NSF, anti-PKCε, or anti-γ2-subunit antibodies. Data are quantified in the right panel, showing that, after 45 min of treatment, 1 μm 1Na-PP1 decreased and 1 μm PMA increased the amount of PKCε coimmunoprecipitated with NSF [n = 3; *p < 0.05 compared with control (con) and †p < 0.05 compared with PMA by Newman–Keuls post hoc test]. B, Representative Western blots with anti-NSF, anti-PKCε, and anti-γ2-subunit antibodies of brain lysates from Prkce+/+ and Prkce−/− mice after immunoprecipitation with anti-NSF antibody. C, The top panel shows representative Western blots from a pull-down assay using recombinant NSF and purified FLAG-PKCε (2 μg) immobilized on anti-FLAG antibody-conjugated agarose. The amount of NSF pulled down with immobilized PKCε increased linearly with the amount of NSF added to the assay (bottom panel; n = 4). D, Representative overlay assay showing Ponceau S-stained-NSF and GST immobilized on a nitrocellulose membrane after SDS-PAGE (PONCEAU S; left) and PKCε immunoreactivity detected using an anti-PKCε antibody (PKCε overlay; right) after incubation of the membrane with recombinant PKCε. This experiment were repeated three times with similar results. Error bars indicate SEM.
To test whether PKCε, NSF, and GABAA receptors also form a protein complex in the brain, we prepared whole-brain lysates from Prkce+/+ and Prkce−/− mice. The abundance of NSF and GABAA γ2 subunits was similar in Prkce+/+ and Prkce−/− lysates (supplemental Fig. 2B, available at www.jneurosci.org as supplemental material). NSF could be coimmunoprecipitated with PKCε and GABAA γ2 subunits in lysates from Prkce+/+ mice (Fig. 2B). The amount of GABAA γ2 coimmunoprecipitated with NSF from Prkce−/− brain lysates was 97 ± 3% of the amount coimmunoprecipitated from Prkce+/+ lysates (n = 3 for each genotype; p = 0.45), indicating that the association between NSF and GABAA γ2 subunits occurs independently of PKCε. Reciprocal immunoprecipitations using antibodies against PKCε or GABAA γ2 confirmed their association with NSF in HEKGR-AS-PKCε cell and Prkce+/+ brain lysates (supplemental Fig. 3, available at www.jneurosci.org as supplemental material).
NSF and PKCε may interact directly or require another protein to bind in a complex. To investigate these possibilities, we first performed a pull-down assay using FLAG epitope-tagged PKCε immobilized on agarose beads and His-tagged NSF (Fig. 2C). There was a linear correlation between the amount of NSF immunoreactivity recovered from the PKCε-agarose beads and the amount of His-tagged NSF that was added to the assay (rp = 0.995; p = 0.0204; n = 4). We also performed a gel overlay assay using NSF immobilized on nitrocellulose membranes (Fig. 2D). Recombinant PKCε bound selectively to immobilized NSF but not to immobilized GST (Fig. 2D). These findings indicate that NSF and PKCε can bind directly to each other.
Activation of PKCε causes NSF to translocate to the plasma membrane
Since phorbol esters cause translocation of PKC to the plasma membrane (Song and Messing, 2005) and our results indicated that NSF interacts with activated PKCε, we predicted that PMA would also cause NSF to translocate to the plasma membrane. We examined this possibility by performing immunofluorescence studies in intact HEKGR-AS-PKCε cells using MyrPalm-CFP as a membrane marker (Violin et al., 2003). Treatment with PMA (1 μm) for 60 min increased the colocalization of NSF with MyrPalm-CFP, but addition of 1 μm 1Na-PP1 for 15 min before PMA blocked colocalization of NSF and MyrPalm-CFP (Fig. 3A,B) (p < 0.0001, Kruskal–Wallis test). PMA also increased the colocalization of PKCε and MyrPalm-CFP, and this effect was reduced by pretreatment with 1Na-PP1 (Fig. 3A,C) (p < 0.0001, Kruskal–Wallis test). These results indicate that phorbol esters induce a translocation of NSF to the plasma membrane that depends on PKCε activity.
Figure 3.

Membrane translocation of NSF in response to activation of PKCε. A, HEKGR-AS-PKCε cells were transfected with MyrPalm-CFP and incubated for 60 min with control vehicle (0.1% DMSO; Con), 1 μm 1Na-PP1, or 1 μm PMA, or with 1Na-PP1 for 15 min before addition of PMA for another 60 min. CFP (green), NSF (red), and PKCε (blue) immunoreactivity were detected by confocal microscopy. Merged images indicate colocalization of CFP and NSF (yellow) or CFP and PKCε (cyan). PMA (1 μm) induced translocation of NSF and PKCε to cell membrane. B, C, The colocalization between these proteins was quantified and expressed as Spearman's rank correlation coefficients. The results indicate that 1 μm PMA increased the colocalization of both NSF (n = 31–57 cells) and PKCε (n = 26–38 cells) with CFP. *p < 0.05 compared with other conditions (Dunn's multiple-comparisons test). Scale bar, 10 μm. Error bars indicate SEM.
We next investigated the association of NSF and GABAA receptors with gephyrin, which is involved in the synaptic localization of GABAA receptors (Essrich et al., 1998). We examined neurites of primary hippocampal neurons cultured from P1–P3 mice for 14–21 d in vitro and found that 200 nm PMA increased the colocalization of NSF and gephyrin (p < 0.0001), increasing the Spearman's coefficient from 0.467 ± 0.008 in cells treated with the 0.02% DMSO vehicle (n = 14) to 0.538 ± 0.010 in cells treated with PMA (n = 14) (Fig. 4A). In contrast, PMA reduced colocalization of gephyrin and GABAA γ2 subunits (p = 0.0012), decreasing the Spearman's coefficient from 0.547 ± 0.017 in DMSO-treated cultures (n = 7) to 0.374 ± 0.030 in PMA-treated cultures (n = 15) (Fig. 4B). These results suggest that PKC activation recruits NSF to inhibitory synapses while downregulating the number of GABAA receptors.
Figure 4.
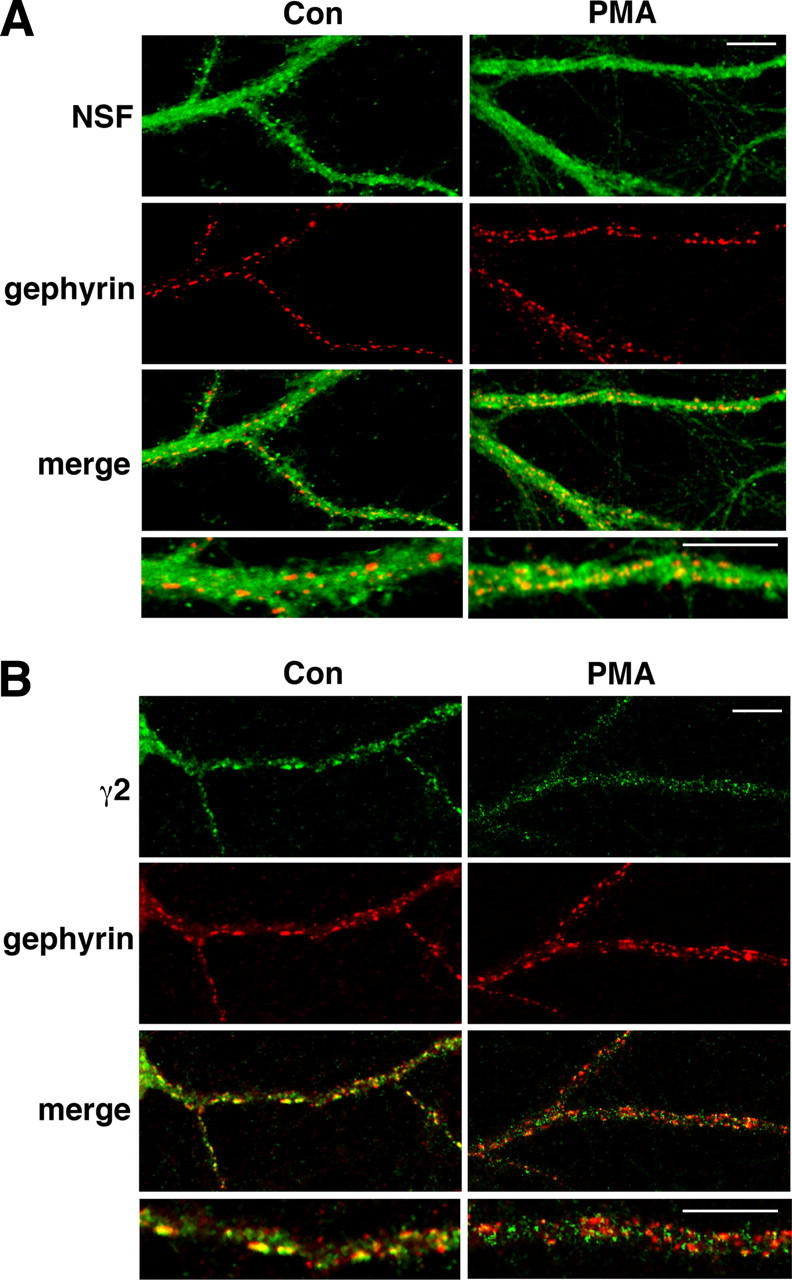
PKC activation alters the colocalization of NSF and GABAA receptors with gephyrin. A, PMA increases the colocalization of NSF with gephyrin in cultured hippocampal neurons. Neurons were incubated with the control vehicle (0.02% DMSO; n = 14 images) or 200 nm PMA (n = 14). NSF (green) and gephyrin (red) were detected by confocal microscopy. Representative neurites in a 250 × 500 μm region are shown. Merged images show colocalization of NSF with gephyrin in yellow. B, Compared with control (0.02% DMSO; n = 7 images), 200 nm PMA (n = 15) decreased the colocalization of GABAA γ2 subunits with gephyrin. Merged images show colocalization of γ2 subunits (green) and gephyrin (red) in yellow. Higher magnification merged images are shown in the bottom panels. Scale bars, 10 μm.
PKCε phosphorylation enhances the ATPase activity of NSF
Since PKCε and NSF interact, we next tested whether NSF is a PKCε substrate. In vitro kinase assays showed that PKCε phosphorylates NSF at a rate similar to the major intracellular loop of the GABAA γ2S subunit (GST-γ2S), which contains a PKCε phosphorylation site at S327 (Fig. 5A) (Qi et al., 2007). PKCε phosphorylated NSF to a maximal stoichiometry of 0.917 ± 0.069 (n = 3). Nano-LC-MS/MS of in vitro phosphorylated and trypsin-digested NSF identified a single phosphorylated peptide that contains one serine and one threonine residue (underlined) (ASTKVEVDMEK; amino acids 459–469) (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). To test whether PKCε phosphorylates either site, we generated NSF peptides containing S460A, T461A, or both mutations. PKCε phosphorylated an NSF peptide containing the native sequence, but showed reduced ability to phosphorylate peptides containing either mutation (F(3,8) = 85.54; p < 0.0001) (Fig. 5B). These results indicate that PKCε phosphorylates NSF at both S460 and T461 in vitro.
Figure 5.
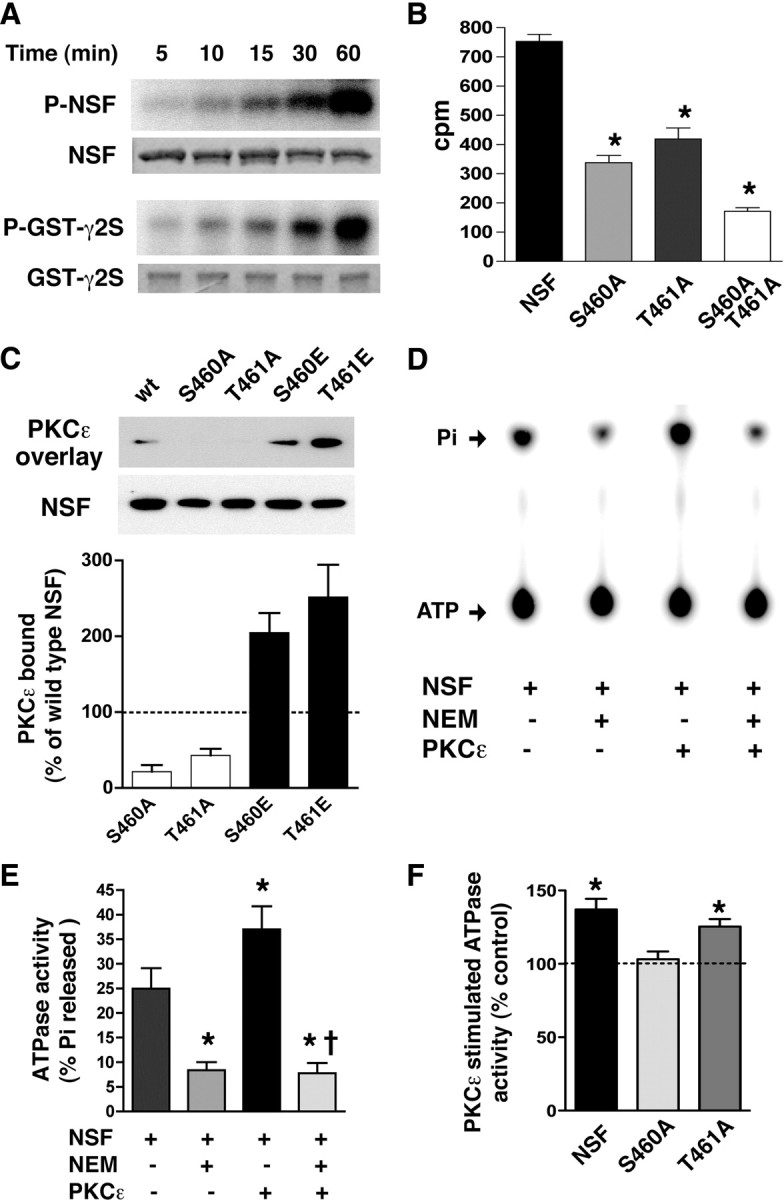
PKCε phosphorylates NSF at S460 and regulates NSF ATPase activity. A, Representative autoradiograph of phosphorylated NSF (top panel) and a scanned image of a Coomassie blue-stained gel (bottom panel). B, In vitro phosphorylation of NSF peptides by PKCε is reduced by alanine mutations at S460 and T461 (n = 3 each). *p < 0.05 compared with native NSF by Newman–Keuls post hoc tests. C, Representative overlay assay showing NSF mutants immobilized on a nitrocellulose membrane after SDS-PAGE with PKCε immunoreactivity detected using an anti-PKCε antibody (PKCε overlay) after incubation of the membrane with recombinant PKCε. PKCε binding to alanine mutants (S460A, T461A) was reduced while binding to glutamate mutants (S460E, T461E) was increased (p < 0.05, one-sample t tests; n = 5). D, Representative autoradiograph of a thin-layer chromatogram showing increased NSF ATPase activity after PKCε phosphorylation of NSF. Radiolabeled ATP and released inorganic phosphate (Pi) are indicated by arrows. E, PKCε phosphorylation increased NSF ATPase activity and NEM blocked both baseline and PKCε-stimulated activity (n = 3). *p < 0.05 compared with NSF and †p < 0.05 compared with NSF+PKCε but not NSF+NEM by Newman–Keuls post hoc tests. F, The S460A NSF mutation prevented enhancement of ATPase activity by PKCε (n = 3). *p < 0.05 compared with the corresponding untreated control (100%) by one-sample t tests. Error bars indicate SEM.
To determine whether PKCε phosphorylation of NSF regulates NSF function, we first examined the effect of phosphorylation on the interaction of NSF with PKCε using gel overlay assays. Mutation of S460 or T461 to alanine disrupted binding to PKCε (Fig. 5C). Conversely, mimicking phosphorylation by glutamic acid substitution at these residues facilitated binding to PKCε. The results indicate that phosphorylation of NSF at S460 and T461 is necessary for its interaction with PKCε.
We next investigated whether PKCε phosphorylation regulates the enzymatic activity of NSF. Phosphorylation of NSF by PKCε increased NSF ATPase activity by 50% (F(3,8) = 17.34; p = 0.0007) (Fig. 5D,E). To ensure that this increase in activity, measured as hydrolysis of ATP, was attributable to NSF and not to PKCε, we treated samples with the NSF inhibitor N-ethylmaleimide (NEM). This treatment abolished nearly all ATPase activity, indicating that the ATPase activity measured in this assay was attributable to NSF. To determine whether S460 and T461 are both necessary for regulation of NSF by PKCε, we examined NSF variants containing these mutations and found that only the S460A mutation prevented enhancement of NSF ATPase activity by PKCε (Fig. 5F). These results indicate that PKCε phosphorylation of NSF at S460 increases the catalytic activity of NSF.
Since these in vitro studies showed that S460 is involved not only in enhancing NSF ATPase activity but also in the interaction of NSF with PKCε, we investigated whether PKCε phosphorylates S460 in vivo. We used an affinity-purified, phosphospecific antibody raised against a peptide containing phospho-S460. This anti-phospho-NSF (S460) antibody detected recombinant NSF phosphorylated by PKCε in vitro, but not the nonphosphorylated protein (Fig. 6A). Anti-phospho-NSF (S460) immunoreactivity was decreased by 35% in Prkce−/− brain lysates compared with wild-type tissue (Fig. 6B,C), consistent with the prediction that PKCε directly phosphorylates brain NSF at S460 in vivo.
Figure 6.

PKCε phosphorylates NSF in vivo. A, An affinity-purified rabbit anti-phospho-NSF (S460) antibody was generated that detects recombinant NSF phosphorylated by PKCε (P), but not unphosphorylated NSF (N). The antibody was not immunoreactive against the NSF S460A mutant before or after incubation with PKCε in a kinase reaction. B, Representative Western blot showing that the anti-phospho-NSF (S460) antibody detected less immunoreactivity in brain lysates from Prkce−/− mice than in lysates from wild-type littermates. C, The ratio of P-NSF (S460) to NSF immunoreactivity was reduced in samples from Prkce−/− (n = 4) compared with Prkce+/+ (n = 4) mice (*p < 0.05, two-tailed t test). Error bars indicate SEM.
NSF regulates the level of GABAA receptors at the cell surface
To determine whether NSF ATPase activity regulates the cell surface level of GABAA receptors, we used a peptide inhibitor of NSF (NSFi) (Morrell et al., 2005), which consists of an NSF oligomerization domain attached to a protein transduction domain from the human immunodeficiency virus transactivator of transcription (TAT) protein. NSFi penetrates cells and blocks NSF ATPase activity (Morrell et al., 2005). We hypothesized that, if NSF acts downstream of PKCε, then inhibiting NSF function should increase cell surface levels of GABAA receptors and prevent PKCε-induced downregulation of GABAA receptors. We first examined HEKGR-AS-PKCε cells and found that treatment for 1 h with 30 nm PMA reduced cell surface GABAA γ2 immunoreactivity by 35% (Fig. 7A), as expected (Fig. 1A). Treatment for 75 min with 1 μm NSFi increased cell surface GABAA γ2 by 52% and preincubation with 1 μm NSFi for 15 min before addition of 30 nm PMA prevented the effect of PMA (Fig. 7A). A control TAT peptide had no effect compared with the DMSO (0.02%) vehicle-treated control. PMA and NSFi did not alter the level of total γ2 immunoreactivity in cell lysates (supplemental Fig. 5A, available at www.jneurosci.org as supplemental material).
Figure 7.

NSF ATPase activity regulates cell surface levels of γ2 subunits and evoked GABAA currents. A, HEKGR-AS-PKCε cells were incubated with the DMSO vehicle as a control (n = 13), 1 μm TAT (n = 11), TAT plus 30 nm PMA (n = 10), 1 μm NSFi (n = 8), or NSFi plus PMA (n = 7). Biotinylated cell surface proteins were compared by Western blot analysis with anti-γ2 subunit antibodies. NSFi increased the level of cell surface GABAA γ2 subunits and blocked PMA-induced downregulation. *p < 0.05 compared with control vehicle (100%) by one-sample t tests. B, Hippocampal neurons were treated with buffer alone as control (n = 18), or with 1 μm each of scrambled (scr) ψεRACK (n = 6), scrambled (scr) NSFi (n = 6), ψεRACK (n = 12), NSFi (n = 12), or NSFi plus ψεRACK (n = 12). Cell surface GABAA γ2 subunits were determined by ELISA using an antibody against extracellular domains of γ2 subunits. The ψεRACK peptide decreased γ2 immunoreactivity at the cell surface, whereas NSFi increased it and blocked the effect of ψεRACK. *p < 0.0003 compared with control (100%) by one-sample t tests. C, D, Amplitudes of eIPSCs were averaged every 3 min and expressed relative to current measured in the first 3 min immediately after establishing whole-cell patch configuration. Peptides were administered in the patch pipette. Treatment with 5 μm ψεRACK (n = 6) decreased the eIPSC amplitude, whereas 2 μm NSFi (n = 7) increased it and prevented the effect of ψεRACK (NSFi plus ψεRACK; n = 5). Results in D are mean values from the last 6 min of recording (24–30 min after break in). *p < 0.05 compared with NSFi plus ψεRACK by Newman–Keuls test. Error bars indicate SEM.
We next determined whether NSFi similarly regulates GABAA receptors in neurons. For these studies, we used an ELISA that allowed us to detect γ2 subunits using fewer cells than required by the biotinylation assay. Treatment of hippocampal neurons from Prkce+/+ mice with 1 μm ψεRACK for 1 h decreased cell surface γ2 subunits by 26%, whereas treatment for 75 min with 1 μm NSFi increased them by 60% and preincubation with 1 μm NSFi for 15 min before addition of 1 μm ψεRACK completely prevented downregulation by ψεRACK (Fig. 7B). Control scrambled versions of both peptides had no effect compared with untreated control cells. ψεRACK and NSFi did not alter the level of total γ2 immunoreactivity in cell lysates (supplemental Fig. 5B, available at www.jneurosci.org as supplemental material).
Based on these findings, we predicted that inhibiting NSF would increase GABAA currents. To test this prediction, we examined eIPSCs in hippocampal slices (Fig. 7C,D) while administering 2 μm NSFi, 5 μm ψεRACK, or both peptides in the internal recording solution. We found that ψεRACK decreased evoked GABAA currents in CA1 neurons by ∼62%, whereas NSFi increased them by 98% and inhibited the effect of ψεRACK (F(2,17) = 11.75; p = 0.0009). To ensure that NSFi acted postsynaptically, we administered it together with ψεRACK through the patch pipette. As a result, NSFi was less effective in reversing the effect of ψεRACK in hippocampal slices than in neuronal cultures (Fig. 7B), since neurons in culture could be preincubated with NSFi for 15 min before addition of ψεRACK to the medium. These results in hippocampal slices demonstrate that inhibiting NSF increases GABAA currents at inhibitory synapses on hippocampal CA1 neurons, and are consistent with NSF acting downstream of PKCε in mediating downregulation of GABAA receptors.
Discussion
Our results identify a PKCε–NSF signaling pathway that regulates inhibitory synapses by promoting GABAA receptor downregulation. We found that PKCε phosphorylates NSF in its catalytic domain, thereby increasing NSF ATPase activity. Several upstream activators known to regulate GABAA receptor trafficking by a PKC-dependent mechanism could activate this signaling pathway. Examples include brain-derived neurotrophic factor in mouse superior colliculus (Henneberger et al., 2002) and rat hippocampal neurons (Brünig et al., 2001), serotonin in rat prefrontal cortical pyramidal neurons (Feng et al., 2001), and acetylcholine in rat superior cervical ganglion neurons (Brandon et al., 2002). Thus, PKCε and NSF may be critical for downregulation of inhibitory neurotransmission induced by several neurotransmitters and growth factors.
Altered NSF-dependent regulation of GABAA receptor trafficking could contribute to several phenotypes in Prkce−/− mice. These mice show decreased anxiety-like behavior and suppression of the hypothalamic–pituitary–adrenal axis, both of which can be reversed by low doses of the GABAA receptor antagonist bicuculline (Hodge et al., 2002), indicating that GABAA receptor signaling is enhanced in Prkce−/− mice. Reduced NSF-stimulated GABAA receptor trafficking may explain the increased endogenous GABA tone in these mice and their supersensitivity to several different classes of GABAA receptor agonists (Hodge et al., 1999, 2002).
Phosphorylation of NSF by PKCε not only increased NSF ATPase activity but also regulated the subcellular localization of NSF. Unlike most substrate–kinase interactions, which are transient, activation of PKCε stimulated the formation of an NSF–PKCε complex that translocated to the plasma membrane. PKCε-induced translocation of NSF could be important for regulating GABAA receptor density at the cell surface since, when translocated to the cell membrane, NSF is better able to encounter cell surface GABAA receptors and regulate their trafficking.
Previously, it was thought that PKC does not contribute to constitutive GABAA receptor trafficking since nonselective PKC inhibitors do not alter the density of cell surface GABAA receptors (Chapell et al., 1998; Connolly et al., 1999). However, our finding that PKCε inhibition increases cell surface γ2 subunits and GABA efficacy indicates that PKCε regulates constitutive receptor trafficking and that another PKC isozyme may exert an opposing effect. Inhibition of NSF also increased cell surface levels of γ2 subunits in addition to preventing PKCε-stimulated receptor downregulation. Together, these results reveal important roles for NSF and PKCε in both constitutive and stimulated GABAA receptor trafficking.
NSF is comprised of three domains, an N-terminal domain and two homologous ATP binding domains, D1 and D2 (Zhao et al., 2007). The N-terminal domain interacts with soluble NSF attachment proteins (SNAPs) and soluble NSF attachment protein receptors (SNAREs). The D1 domain hydrolyzes ATP and the D2 domain mediates NSF homohexamerization. To understand why phosphorylation of the D1 domain at S460 but not T461 enhances the catalytic activity of NSF, we modeled the D1 domain of NSF using the crystal structure of the homologous D2 domain (PDB number 1D2N) (Fig. 8) (Lenzen et al., 1998). Results using structure-based sequence alignment (http://www.cgl.ucsf.edu/chimera) showed that S460 and T461 lie in the middle of the α9 helix. Both residues are solvent accessible, but only S460 is in direct contact (∼3.7 Å) with D425 and E428, two acidic residues in the α7 helix (Fig. 8). Phosphorylation at S460 on the α9 helix adds a negatively charged phosphate predicted to repel these nearby acidic residues in the α7 helix. This repulsion could alter the conformation of adjacent α6 and α8 helices, which directly bind ATP, and thereby enhance the catalytic function of NSF when PKCε is activated. In contrast, the side chain of T461 projects outward, away from other NSF residues. Thus, phosphorylation of T461 is unlikely to alter the conformation of NSF, but instead could regulate its interaction with other molecules such as PKCε.
Figure 8.

Model of the NSF D1 domain showing that S460 within the α9 helix (orange-red) lies in close proximity (∼3.7 Å) to D425 and E428 on the α7 helix (gold). The side chains of S460, D425, E428, and T461 are shown in blue, and the oxygen atoms on the side chains are shown in red.
In contrast to our findings, Marsden et al. (2007) previously reported that NMDA receptor activation stimulates exocytosis of GABAA receptors that is dependent on calcium/calmodulin-dependent kinase II (CaMKII) and NSF. Using hippocampal neurons cultured from P0 rats, they found that NMDA increases the association of NSF with GABAA β2/3 subunits, and that a peptide inhibitor of NSF blocks NMDA-induced increases in GABAA receptors at the cell surface. There are technical differences between their study and ours that could explain why our findings are different. These include choice of species and use of different peptides with different mechanisms of action. The peptide we used inhibits homo-hexamerization of NSF and abolishes NSF ATPase activity (Morrell et al., 2005). The peptide inhibitor used by Marsden and colleagues mimics the NSF binding site of α- and β-SNAPs and is designed to prevent the binding of NSF to SNAPs rather than inhibit NSF activity (Lledo et al., 1998). Moreover, the kinases that we studied, PKCε versus CaMKII, are different, which raises the intriguing possibility that these kinases determine whether NSF upregulates or downregulates synaptic GABAA receptors. In vitro NSF appears to be a substrate of CaMKII (Hirling and Scheller, 1996), but how this kinase regulates NSF function is not yet known.
Long-term depression of AMPA currents occurs through NMDA-stimulated removal of AMPA receptors from postsynaptic membranes (Hanley, 2008). This process requires the interaction of GluA2 subunits with the protein interacting with C-kinase-1 (PICK1). NSF binds to GluA2 subunits and acts as a disassembling chaperone using energy derived from ATP hydrolysis to disassociate AMPA receptors from PICK1, thereby inhibiting GluA2 endocytosis and stabilizing AMPA receptors at synapses (Hanley et al., 2002). As shown by our current findings, PKCε-phosphorylated NSF instead promotes downregulation of GABAA receptors. Although the effects of NSF on these two types of receptors are quite different, they are additive in producing a net enhancement of excitatory neurotransmission. Thus, reducing PKCε and NSF activity could prove beneficial for resetting the balance of excitation and inhibition in disorders associated with neuronal hyperexcitability. Our findings suggest that inhibitors of PKCε and NSF could provide an approach for achieving this goal.
Footnotes
This work was supported by United States Public Health Service Grant AA013588 and funds provided by the State of California for medical research on alcohol and substance abuse through University of California, San Francisco (UCSF) (R.O.M.). We thank S. Whiteheart (University of Kentucky) for the NSF cDNA construct, P. Sung and P. Chi (Yale University) for advice on the ATPase activity assay, Y. Li (University of Texas Southwestern) for mass spectrometry, C. Johnston (UCSF) for statistical advice, and A. Newton (University of California, San Diego) for the MyrPalm-CFP expression plasmid.
References
- Abramoff MD, Magelhaes PJ, Ram SJ. Image processing with ImageJ. Biophotonics Int. 2004;11:36–42. [Google Scholar]
- Balduzzi R, Cupello A, Robello M. Modulation of the expression of GABA(A) receptors in rat cerebellar granule cells by protein tyrosine kinases and protein kinase C. Biochim Biophys Acta. 2002;1564:263–270. doi: 10.1016/s0005-2736(02)00460-1. [DOI] [PubMed] [Google Scholar]
- Bishop AC, Kung C-Y, Shah K, Witucki L, Shokat KM, Liu Y. Generation of monospecific nanomolar tyrosine kinase inhibitors via a chemical genetic approach. J Am Chem Soc. 1999;121:627–631. [Google Scholar]
- Brandon NJ, Jovanovic JN, Smart TG, Moss SJ. Receptor for activated C kinase-1 facilitates protein kinase C-dependent phosphorylation and functional modulation of GABAA receptors with the activation of G-protein-coupled receptors. J Neurosci. 2002;22:6353–6361. doi: 10.1523/JNEUROSCI.22-15-06353.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brose N, Rosenmund C. Move over protein kinase C, you've got company: alternative cellular effectors of diacylglycerol and phorbol esters. J Cell Sci. 2002;115:4399–4411. doi: 10.1242/jcs.00122. [DOI] [PubMed] [Google Scholar]
- Brünig I, Penschuck S, Berninger B, Benson J, Fritschy JM. BDNF reduces miniature inhibitory postsynaptic currents by rapid downregulation of GABAA receptor surface expression. Eur J Neurosci. 2001;13:1320–1328. doi: 10.1046/j.0953-816x.2001.01506.x. [DOI] [PubMed] [Google Scholar]
- Chapell R, Bueno OF, Alvarez-Hernandez X, Robinson LC, Leidenheimer NJ. Activation of protein kinase C induces gamma-aminobutyric acid type A receptor internalization in Xenopus oocytes. J Biol Chem. 1998;273:32595–32601. doi: 10.1074/jbc.273.49.32595. [DOI] [PubMed] [Google Scholar]
- Charych EI, Liu F, Moss SJ, Brandon NJ. GABAA receptors and their associated proteins: implications in the etiology and treatment of schizophrenia and related disorders. Neuropharmacology. 2009;57:481–495. doi: 10.1016/j.neuropharm.2009.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Hahn H, Wu G, Chen CH, Liron T, Schechtman D, Cavallaro G, Banci L, Guo Y, Bolli R, Dorn GW, 2nd, Mochly-Rosen D. Opposing cardioprotective actions and parallel hypertrophic effects of delta PKC and epsilon PKC. Proc Natl Acad Sci U S A. 2001;98:11114–11119. doi: 10.1073/pnas.191369098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZW, Olsen RW. GABAA receptor associated proteins: a key factor regulating GABAA receptor function. J Neurochem. 2007;100:279–294. doi: 10.1111/j.1471-4159.2006.04206.x. [DOI] [PubMed] [Google Scholar]
- Cinar H, Barnes EM., Jr Clathrin-independent endocytosis of GABAA receptors in HEK 293 cells. Biochemistry. 2001;40:14030–14036. doi: 10.1021/bi011025t. [DOI] [PubMed] [Google Scholar]
- Collingridge GL, Isaac JT, Wang YT. Receptor trafficking and synaptic plasticity. Nat Rev Neurosci. 2004;5:952–962. doi: 10.1038/nrn1556. [DOI] [PubMed] [Google Scholar]
- Connolly CN, Kittler JT, Thomas P, Uren JM, Brandon NJ, Smart TG, Moss SJ. Cell surface stability of gamma-aminobutyric acid type A receptors. Dependence on protein kinase C activity and subunit composition. J Biol Chem. 1999;274:36565–36572. doi: 10.1074/jbc.274.51.36565. [DOI] [PubMed] [Google Scholar]
- Essrich C, Lorez M, Benson JA, Fritschy JM, Lüscher B. Postsynaptic clustering of major GABAA receptor subtypes requires the gamma 2 subunit and gephyrin. Nat Neurosci. 1998;1:563–571. doi: 10.1038/2798. [DOI] [PubMed] [Google Scholar]
- Feng J, Cai X, Zhao J, Yan Z. Serotonin receptors modulate GABAA receptor channels through activation of anchored protein kinase C in prefrontal cortical neurons. J Neurosci. 2001;21:6502–6511. doi: 10.1523/JNEUROSCI.21-17-06502.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French AP, Mills S, Swarup R, Bennett MJ, Pridmore TP. Colocalization of fluorescent markers in confocal microscope images of plant cells. Nat Protoc. 2008;3:619–628. doi: 10.1038/nprot.2008.31. [DOI] [PubMed] [Google Scholar]
- Goodkin HP, Joshi S, Mtchedlishvili Z, Brar J, Kapur J. Subunit-specific trafficking of GABAA receptors during status epilepticus. J Neurosci. 2008;28:2527–2538. doi: 10.1523/JNEUROSCI.3426-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto H, Terunuma M, Kanematsu T, Misumi Y, Moss SJ, Hirata M. Direct interaction of N-ethylmaleimide-sensitive factor with GABAA receptor beta subunits. Mol Cell Neurosci. 2005;30:197–206. doi: 10.1016/j.mcn.2005.07.006. [DOI] [PubMed] [Google Scholar]
- Hanley JG. PICK1: a multi-talented modulator of AMPA receptor trafficking. Pharmacol Ther. 2008;118:152–160. doi: 10.1016/j.pharmthera.2008.02.002. [DOI] [PubMed] [Google Scholar]
- Hanley JG, Khatri L, Hanson PI, Ziff EB. NSF ATPase and alpha-/beta-SNAPs disassemble the AMPA receptor-PICK1 complex. Neuron. 2002;34:53–67. doi: 10.1016/s0896-6273(02)00638-4. [DOI] [PubMed] [Google Scholar]
- Henneberger C, Juttner R, Rothe T, Grantyn R. Postsynaptic action of BDNF on GABAergic synaptic transmission in the superficial layers of the mouse superior colliculus. J Neurophys. 2002;88:595–603. doi: 10.1152/jn.2002.88.2.595. [DOI] [PubMed] [Google Scholar]
- Herring D, Huang R, Singh M, Dillon GH, Leidenheimer NJ. PKC modulation of GABAA receptor endocytosis and function is inhibited by mutation of a dileucine motif within the receptor beta 2 subunit. Neuropharmacology. 2005;48:181–194. doi: 10.1016/j.neuropharm.2004.09.015. [DOI] [PubMed] [Google Scholar]
- Hirling H, Scheller RH. Phosphorylation of synaptic vesicle proteins: modulation of the alpha SNAP interaction with the core complex. Proc Natl Acad Sci U S A. 1996;93:11945–11949. doi: 10.1073/pnas.93.21.11945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodge CW, Mehmert KK, Kelley SP, McMahon T, Haywood A, Olive MF, Wang D, Sanchez-Perez AM, Messing RO. Supersensitivity to allosteric GABAA receptor modulators and alcohol in mice lacking PKCepsilon. Nat Neurosci. 1999;2:997–1002. doi: 10.1038/14795. [DOI] [PubMed] [Google Scholar]
- Hodge CW, Raber J, McMahon T, Walter H, Sanchez-Perez AM, Olive MF, Mehmert K, Morrow AL, Messing RO. Decreased anxiety-like behavior, reduced stress hormones, and neurosteroid supersensitivity in mice lacking protein kinase Cε. J Clin Invest. 2002;110:1003–1010. doi: 10.1172/JCI15903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalueff AV, Nutt DJ. Role of GABA in anxiety and depression. Depress Anxiety. 2007;24:495–517. doi: 10.1002/da.20262. [DOI] [PubMed] [Google Scholar]
- Khasar SG, Lin YH, Martin A, Dadgar J, McMahon T, Wang D, Hundle B, Aley KO, Isenberg W, McCarter G, Green PG, Hodge CW, Levine JD, Messing RO. A novel nociceptor signaling pathway revealed in protein kinase C epsilon mutant mice. Neuron. 1999;24:253–260. doi: 10.1016/s0896-6273(00)80837-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittler JT, Moss SJ. Modulation of GABAA receptor activity by phosphorylation and receptor trafficking: implications for the efficacy of synaptic inhibition. Curr Opin Neurobiol. 2003;13:341–347. doi: 10.1016/s0959-4388(03)00064-3. [DOI] [PubMed] [Google Scholar]
- Kittler JT, Rostaing P, Schiavo G, Fritschy JM, Olsen R, Triller A, Moss SJ. The subcellular distribution of GABARAP and its ability to interact with NSF suggest a role for this protein in the intracellular transport of GABA(A) receptors. Mol Cell Neurosci. 2001;18:13–25. doi: 10.1006/mcne.2001.1005. [DOI] [PubMed] [Google Scholar]
- Knabl J, Witschi R, Hösl K, Reinold H, Zeilhofer UB, Ahmadi S, Brockhaus J, Sergejeva M, Hess A, Brune K, Fritschy JM, Rudolph U, Möhler H, Zeilhofer HU. Reversal of pathological pain through specific spinal GABAA receptor subtypes. Nature. 2008;451:330–334. doi: 10.1038/nature06493. [DOI] [PubMed] [Google Scholar]
- Kumar S, Fleming RL, Morrow AL. Ethanol regulation of gamma-aminobutyric acid A receptors: genomic and nongenomic mechanisms. Pharmacol Ther. 2004;101:211–226. doi: 10.1016/j.pharmthera.2003.12.001. [DOI] [PubMed] [Google Scholar]
- Leidenheimer NJ. Regulation of excitation by GABAA receptor internalization. Results Probl Cell Differ. 2008;44:1–28. doi: 10.1007/400_2007_039. [DOI] [PubMed] [Google Scholar]
- Leidenheimer NJ, Chapell R. Effects of PKC activation and receptor desensitization on neurosteroid modulation of GABAA receptors. Brain Res Mol Brain Res. 1997;52:173–181. doi: 10.1016/s0169-328x(97)00255-6. [DOI] [PubMed] [Google Scholar]
- Leidenheimer NJ, McQuilkin SJ, Hahner LD, Whiting P, Harris RA. Activation of protein kinase C selectively inhibits the gamma-aminobutyric acid A receptor: role of desensitization. Mol Pharmacol. 1992;41:1116–1123. [PubMed] [Google Scholar]
- Leil TA, Chen ZW, Chang CS, Olsen RW. GABAA receptor-associated protein traffics GABAA receptors to the plasma membrane in neurons. J Neurosci. 2004;24:11429–11438. doi: 10.1523/JNEUROSCI.3355-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenzen CU, Steinmann D, Whiteheart SW, Weis WI. Crystal structure of the hexamerization domain of N-ethylmaleimide-sensitive fusion protein. Cell. 1998;94:525–536. doi: 10.1016/s0092-8674(00)81593-7. [DOI] [PubMed] [Google Scholar]
- Lledo PM, Zhang X, Südhof TC, Malenka RC, Nicoll RA. Postsynaptic membrane fusion and long-term potentiation. Science. 1998;279:399–403. doi: 10.1126/science.279.5349.399. [DOI] [PubMed] [Google Scholar]
- Marsden KC, Beattie JB, Friedenthal J, Carroll RC. NMDA receptor activation potentiates inhibitory transmission through GABA receptor-associated protein-dependent exocytosis of GABAA receptors. J Neurosci. 2007;27:14326–14337. doi: 10.1523/JNEUROSCI.4433-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matveeva EA, Whiteheart SW, Vanaman TC, Slevin JT. Phosphorylation of the N-ethylmaleimide-sensitive factor is associated with depolarization-dependent neurotransmitter release from synaptosomes. J Biol Chem. 2001;276:12174–12181. doi: 10.1074/jbc.M007394200. [DOI] [PubMed] [Google Scholar]
- Meier J, Akyeli J, Kirischuk S, Grantyn R. GABAA receptor activity and PKC control inhibitory synaptogenesis in CNS tissue slices. Mol Cell Neurosci. 2003;23:600–613. doi: 10.1016/s1044-7431(03)00079-4. [DOI] [PubMed] [Google Scholar]
- Morrell CN, Matsushita K, Lowenstein CJ. A novel inhibitor of N-ethylmaleimide-sensitive factor decreases leukocyte trafficking and peritonitis. J Pharmacol Exp Ther. 2005;314:155–161. doi: 10.1124/jpet.104.082529. [DOI] [PubMed] [Google Scholar]
- Olsen RW, Sieghart W. International Union of Pharmacology. LXX. Subtypes of gamma-aminobutyric acid(A) receptors: classification on the basis of subunit composition, pharmacology, and function. Update. Pharmacol Rev. 2008;60:243–260. doi: 10.1124/pr.108.00505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi ZH, Song M, Wallace MJ, Wang D, Newton PM, McMahon T, Chou WH, Zhang C, Shokat KM, Messing RO. Protein kinase C epsilon regulates gamma-aminobutyrate type A receptor sensitivity to ethanol and benzodiazepines through phosphorylation of gamma2 subunits. J Biol Chem. 2007;282:33052–33063. doi: 10.1074/jbc.M707233200. [DOI] [PubMed] [Google Scholar]
- Qian Y, Galli A, Ramamoorthy S, Risso S, DeFelice LJ, Blakely RD. Protein kinase C activation regulates human serotonin transporters in HEK-293 cells via altered cell surface expression. J Neurosci. 1997;17:45–57. doi: 10.1523/JNEUROSCI.17-01-00045.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song M, Messing RO. Protein kinase C regulation of GABAA receptors. Cell Mol Life Sci. 2005;62:119–127. doi: 10.1007/s00018-004-4339-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Violin JD, Zhang J, Tsien RY, Newton AC. A genetically encoded fluorescent reporter reveals oscillatory phosphorylation by protein kinase C. J Cell Biol. 2003;161:899–909. doi: 10.1083/jcb.200302125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Slevin JT, Whiteheart SW. Cellular functions of NSF: not just SNAPs and SNAREs. FEBS Lett. 2007;581:2140–2149. doi: 10.1016/j.febslet.2007.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]