

Abstract
The retinoblastoma (Rb) tumor suppressor is a key regulator of cell cycle checkpoints but also protects against cell death induced by stresses such as DNA damage and death receptor ligation. We report here that cell death of Rb-deficient cells exposed to key genotoxic agents was associated with increased expression of S phase–specific E2F target genes and cell death consistently occurred in the S phase of the cell cycle. Cell cycle arrest induced by serum starvation prevented S phase entry, attenuated DNA damage, and promoted survival, suggesting that Rb-null cells die due to a failure to prevent S phase entry. DNA damage–induced death of Rb-null cells was associated with nucleotide depletion, higher activity of poly-ADP-ribose-polymerase (Parp), and cell death that was primarily necrotic. Knockdown of Parp-1 or chemical inhibition of Parp activity prevented nucleotide depletion and restored the viability of Rb-deficient cells to wild-type levels. Furthermore, chemical inhibition of Parp activity in vivo attenuated the cytotoxic effects of cisplatin against Rb-deficient tumors, arguing that Parp inhibitors should not be used therapeutically in combination with genotoxic drugs against tumors that are inactivated for the Rb tumor suppressor.
Introduction
Loss of the retinoblastoma (Rb) tumor suppressor sensitizes cells to the cytotoxic effects of DNA-damaging agents used as cancer chemotherapeutic agents in the clinic (1-4). However, the mechanistic basis of genotoxic drug sensitivity induced by Rb loss is not understood.
Two models have been proposed to explain the activity of pRb in protecting against cell death (5). One model proposes that pRb protects against death indirectly by inducing cell cycle arrest, whereas the other identifies a more direct role for pRb in the transcriptional repression of cell death genes, although neither model precludes the other (5). Work from mouse models and overexpression studies with viral oncoproteins identify E2Fs as the key targets of pRb in preventing cell death (5, 6). However, this does not resolve whether pRb is acting directly to repress death genes or indirectly by blocking the cell cycle as E2Fs have been shown to regulate both cell cycle genes (7, 8) and cell death genes such as Apaf-1, caspases, p73, and Bim (9-12).
To distinguish between the role of pRb in promoting survival through the induction of cell cycle arrest, as opposed to direct repression of cell death genes, we compared how wild-type and Rb-null mouse embryonic fibroblasts (MEF) responded to genotoxic agents in terms of cell cycle, E2F target gene expression, levels of DNA damage, and nucleotide depletion. We show that loss of pRb resulted in a failure to undergo cell cycle arrest, increased DNA damage, elevated poly-(ADP-ribose)-polymerase (Parp) activity, and nucleotide depletion compared with wild-type cells and led to necrotic cell death. Furthermore, we show that inhibiting Parp activity protected Rb-null MEFs against DNA damage–induced necrosis. For the first time, this work identifies elevated Parp-1 activity as a key factor in determining the sensitivity of Rb-deficient cells to death induced by DNA damage, and consequently, has implications for the use of PARP inhibitors in cancer therapy.
Results
DNA Damage–Induced Cell Death of Rb-Null MEFs Is Prevented by Serum Starvation
To determine why loss of the Rb tumor suppressor sensitized cells to death induced by genotoxic agents, we used primary Rb-null MEFs that have previously been shown to undergo cell death following treatment with a variety of chemotherapeutic agents (1-3). Consistent with previous work, we showed that Rb-null MEFs were more sensitive to killing induced by cisplatin compared with wild-type MEFs at the same passage number (Fig. 1A), and that the sensitivity to cisplatin was dose-dependent (Fig. 1B). Furthermore, we observed that Rb-null MEFs were also more sensitive to killing by two other chemotherapeutic drugs, i.e., etoposide and hydroxyurea (Fig. 1C). In support of a role for pRb in protecting MEFs against cell death induced by genotoxic agents, pRb is dephosphorylated 16 hours following treatment of wild-type MEFs with cisplatin (Fig. 1D, lane 6), and activation of pRb in this manner was associated with the induction of cellular senescence of wild-type MEFs by 48 hours following treatment, as measured by staining for senescence-associated β-galactosidase (Fig. 1E).
FIGURE 1.

Rb-null MEFs are sensitized to cell death induced by genotoxic agents. A. Phase contrast photography of untreated and cisplatin-treated (24 h) wild-type and Rb-null MEFs showing cell death of Rb-null MEFs (white arrows, bottom right) treated with cisplatin but much fewer dying cells evident in cisplatin-treated wild-type MEF cultures at this time point. Cells with altered morphology indicative of imminent cell death are evident in cisplatin-treated Rb-null cultures (black arrows, bottom right). B. The percentages of dying cells in cultures of wild-type and Rb-null MEFs growing exponentially for 24 h in increasing concentrations of cisplatin (16, 25, and 50 μmol/L) were determined by flow cytometric analysis of propidium iodide uptake in three separate experiments. C. Flow cytometric analysis of propidium iodide uptake by wild-type and Rb-null MEFs left untreated or treated with 16 μmol/L of cisplatin, 20 μmol/L of etoposide, or 2 mmol/L of hydroxyurea for 24 h. D. Western blot analysis of pRb in wild-type MEFs treated with 16 μmol/L of cisplatin for 0, 1, 2, 4, 8, 16, and 24 h. E. Staining for SA-β-galactosidase activity in wild-type MEFs treated with 16 μmol/L of cisplatin for 24 h.
To determine whether the sensitivity of Rb-null MEFs to cisplatin-induced death was associated with increased expression of known E2F-regulated cell death genes, we carried out microarray analyses of gene expression changes in wild-type and Rb-null MEFs before and after treatment with 16 μmol/L of cisplatin (Table 1)4 and validated the expression of various known E2F target genes by real-time reverse transcription-PCR (Fig. 2A). Although we did not observe significant increases in the expression of Apaf-1, caspase-3, or p73 (known E2F target genes implicated in apoptosis) in Rb-null MEFs compared with wild-type MEFs, either before or 24 hours after drug treatment, we did observe elevated expression of genes encoding regulators of DNA replication and S phase progression. Notably, Cdc6, dhfr, cyclin E2, and p49 primase were expressed at elevated levels in Rb-null MEFs compared with wild-type MEFs, both before and after cisplatin treatment (Table 1; Fig. 2A). These results indicated that cisplatin-induced cell death of Rb-null MEFs was associated with the deregulation of E2F-regulated cell cycle genes (and DNA replication genes in particular).
TABLE 1.
The Complete Data Set of Gene Expression Changes in Wild-type and Rb-Null MEFs Before and After Treatment with 16 μmol/L of Cisplatin
| E2F target genes | Rb-null/wild-type |
|||||
|---|---|---|---|---|---|---|
| Untreated |
Cisplatin-treated |
|||||
| Values | Fold change | P | Values | Fold change | P | |
| Cell cycle | ||||||
| Cdc6 | 357.1/62.5 | 5.710 | 0.005 | 1,013.0/293.5 | 3.450 | 0.009 |
| Orc-1 | 1,887.9/364.9 | 5.170 | <0.001 | 1,339.3/483.1 | 2.770 | <0.001 |
| dhfr | 337.6/108.6 | 3.110 | 0.010 | 819.2/226.1 | 3.620 | <0.001 |
| DNA pol-α2 | 429.2/161.1 | 2.660 | 0.002 | 604.2/268.9 | 2.250 | <0.001 |
| Cyclin E2 | 611.9/103.5 | 5.910 | <0.001 | 1,632.2/433.8 | 3.760 | <0.001 |
| Cyclin A2 | 1,455.8/501.1 | 2.900 | <0.001 | 1,161.9/808.3 | 1.440 | <0.001 |
| Geminin | 777.9/222.6 | 3.490 | <0.001 | 1,150.7/485.6 | 2.370 | <0.001 |
| Primase p49 | 522.2/92.5 | 5.650 | <0.001 | 1,194.8/281.4 | 4.250 | <0.001 |
| RPA-2 | 763.1/269.4 | 2.830 | <0.001 | 1,782.6/598.8 | 2.980 | <0.001 |
| Cell death | ||||||
| Apaf-1 | 687.3/459.7 | 1.490 | 0.001 | 720.4/742.9 | 0.970 | 0.711 |
| p73 | 31.0/42.1 | 0.740 | 0.189 | 30.3/41.2 | 0.730 | 0.199 |
| Caspase-3 | 1,096.7/705.2 | 1.560 | <0.001 | 2,314.3/1,735.4 | 1.330 | <0.001 |
NOTE: Values for expression levels in untreated Rb-null MEFs compared with untreated wild-type MEFs are shown for representative E2F target genes involved in regulating DNA replication or cell death. The fold change in values is shown and the significance of the change represented by P value. P < 0.01 is considered significant. Changes in expression levels of these same target genes in response to cisplatin treatment of Rb-null and wild-type MEFs is shown at the right side of the table. As determined by microarray analysis (available at http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE6206).
FIGURE 2.
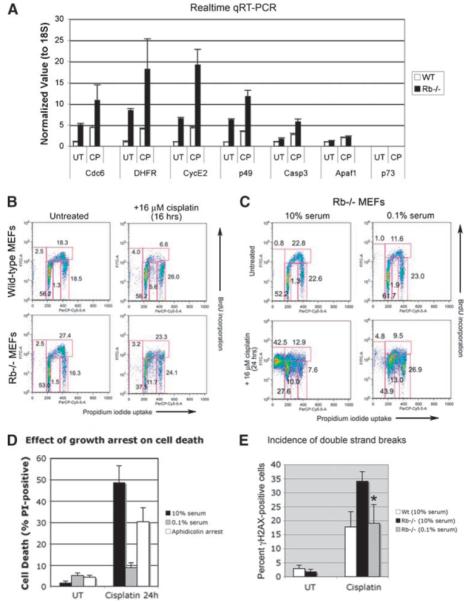
Growth arrest induced by serum starvation protects against cell death in S phase. A. Real-time PCR quantification of the expression of representative E2F target genes identified by microarray analysis as being deregulated in MEFs by loss of pRb. B. Flow cytometric analysis of BrdUrd incorporation and DNA content (propidium iodide uptake by fixed cells) of wild-type and Rb-null MEFs treated for 16 h with 16 μmol/L of cisplatin, compared with untreated control cultures, as a measure of cell cycle phase distribution. C. Flow cytometric analysis of BrdUrd incorporation and DNA content (propidium iodide uptake by fixed cells) of untreated and 16 μmol/L of cisplatin-treated Rb-null MEFs at 24 h, in the presence of 10% or 0.1% serum, as a measure of cell cycle phase distribution. D. Histogram of three separate experiments illustrating the relative protective effect of serum deprivation or pretreatment with aphidicolin for 48 h against cell death of cisplatin-treated Rb-null MEFs compared with cisplatin-treated Rb-null MEFs indicates a statistically significant difference in the levels of cell death (P < 0.05). E. Histogram of three separate experiments illustrating the protective effect of serum deprivation against DNA double-strand breaks (measured by immunolabeling for γH2AX) in cisplatin-treated Rb-null MEFs indicates a statistically significant difference in γH2AX staining in cells grown in 0.1% serum compared with 10% serum (*, P < 0.05).
To determine whether growth arrest failure explained the death of cisplatin-treated Rb-null MEFs, we compared the effect of cisplatin on cell cycle checkpoint control in wild-type and Rb-null MEFs. Prior to cisplatin treatment, Rb-null MEFs showed an increased proportion of cells in S phase (27.4%) compared with wild-type MEFs (18.3%; Fig. 2B). Wild-type MEFs showed a marked decrease in the number of cells in S phase by 16 hours (6.6%) following treatment with 16 μmol/L of cisplatin (Fig. 2B). By contrast, Rb-null MEFs failed to show any significant decrease in the percentage of S phase cells by 16 hours (23.3%) following cisplatin treatment (Fig. 2B). Furthermore, whereas wild-type MEFs accumulated in G1 (56.2%) and G2-M (26.0%) by 16 hours post-cisplatin treatment, Rb-null MEFs showed a marked decrease in G1 cells (37.5%) relative to untreated Rb-null MEFs (53.6%), although the G2 checkpoint seemed intact in Rb-null MEFs (24.1% compared with 16.3% in untreated cells). In contrast to cisplatin-treated wild-type MEFs or untreated Rb-null MEFs, we noted an increased number of Rb-null MEFs with a 2N-4N DNA content (11.7%) that was bromodeoxyuridine (BrdUrd)-negative or low, indicating that Rb-null MEFs were failing to progress through S phase (Fig. 2B).
By 24 hours following cisplatin treatment, there were increased numbers of Rb-null MEFs with sub-G1 DNA content (42.5%; Fig. 2C). At the same time, there was a marked reduction in the number of Rb-null MEFs in G1 (27.6%) and G2-M (7.6%). Similar to observations at 16 hours following cisplatin treatment (Fig. 2B), we also noted an increase in the number of BrdUrd-negative/low cells with a 2N-4N DNA content (10.0%), indicating that cells were continuing to enter S phase but failing to progress through to G2-M. These results suggest that Rb-null MEFs undergo cell death following cisplatin treatment due to a failure to undergo cell cycle arrest.
To further assess whether cisplatin-induced cell death of Rb-null MEFs was the consequence of failed cell cycle arrest, we examined the effect of inducing growth arrest for the sensitivity of Rb-null MEFs to cisplatin. Although Rb-null MEFs were refractory to G1 arrest induced by roscovitine and transforming growth factor-β1 (data not shown), they are sensitive to growth arrest induced by serum starvation (13, 14) in a p107/p130-dependent manner (13, 14) and to growth arrest induced by pretreatment with aphidicolin that inhibits DNA polymerase (3). We noted that serum starvation caused Rb-null MEFs to accumulate in G1 (61.7%) and reduced numbers in S phase (11.6%; Fig. 2C). Importantly, we observed that serum starvation for 48 hours protected Rb-null MEFs from cell death induced by 24 hours of treatment with cisplatin (Fig. 2D). Aphidicolin treatment also protected Rb-null MEFs from cell death induced by cisplatin but not as effectively as serum starvation (Fig. 2D).
Although cisplatin-induced DNA adduct formation is similar between wild-type and Rb-null MEFs (3, 15), we observed increased levels of DNA double-strand breaks, as measured by intracellular staining for γH2AX (Fig. 2E), in cisplatin-treated Rb-null MEFs compared with wild-type controls. We observed that cisplatin induced an almost 3-fold higher level of γH2AX-positive double-strand breaks in Rb-null MEFs relative to wild-type MEFs (Fig. 2E). Furthermore, serum starvation markedly reduced γH2AX staining in serum-starved Rb-null MEFs compared with cycling Rb-null MEFs following cisplatin treatment (Fig. 2E), indicating that serum starvation and/or cell cycle arrest prevented double-strand break formation in cisplatin-treated Rb-null MEFs.
Cisplatin Induces Necrotic Cell Death of Rb-Null MEFs
Previous work has implicated pRb in protecting against apoptotic cell death (1, 16, 17), but other work has shown that Bax−/−Bak−/− cells undergo necrosis in response to alkylating agents (18). To determine the type of cell death induced in Rb-null MEFs by cisplatin treatment and other DNA-damaging agents, we examined different features of apoptotic versus necrotic cell death. Caspase inhibition failed to protect Rb-null MEFs from cisplatin-induced cell death (Fig. 3A and C), although it did protect Rb-null MEFs from death induced by Fas treatment (Fig. 3B), indicating that cisplatin-induced death was caspase-independent. Indeed, caspase inhibition seemed to increase the cell death of cisplatin-treated MEFs. Furthermore, electron microscopic analysis of the ultrastructure of Rb-null MEFs treated with cisplatin for 24 hours (Fig. 3D), revealed numerous signs of necrotic cell death, including loss of nuclear membrane integrity (Fig. 3D, red arrow), cytoplasmic vacuolation (Fig. 3D, black arrow), and loss of plasma membrane integrity. Additionally, the level of tumor necrosis factor-α (TNF-α) produced by macrophages cultured in medium conditioned by cisplatin-treated Rb-null MEFs was higher than that secreted in response to medium conditioned by cisplatin-treated wild-type MEFs or untreated Rb-null MEFs (Fig. 3E), indicative of increased release of proinflammatory molecules by cisplatin-treated Rb-null cells, such as that which occurs during necrosis. HMG-B1 is one such proinflammatory molecule released from the nucleus of necrotic cells (19), and we detected increased release of nuclear HMG-B1 from cisplatin-treated Rb-null MEFs but not in similarly treated wild-type MEFs (Fig. 3F), consistent with Rb-null MEFs dying by necrosis in response to cisplatin. Similar results were observed in the treatment of Rb-null MEFs with 20 μmol/L of etoposide or 2 mmol/L of hydroxyurea (data not shown), indicating that the effect of cisplatin on the type of cell death induced in Rb-null MEFs was not unique.
FIGURE 3.
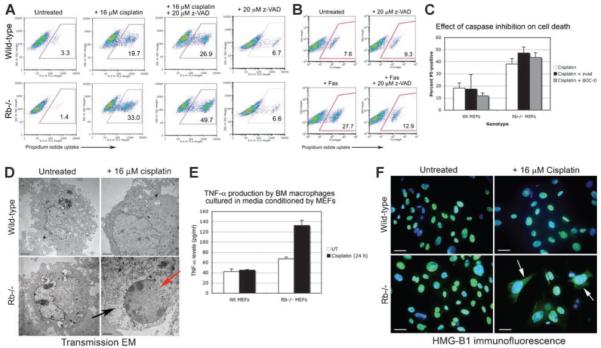
Rb-null MEFs die by necrosis in response to cisplatin, etoposide, and hydroxyurea. A. Flow cytometric analysis of propidium iodide uptake from wild-type and Rb-null MEFs to determine the effect of caspase inhibition on the incidence of cell death of untreated MEFs or of MEFs exposed to 16 μmol/L of cisplatin for 24 h. B. Flow cytometric analysis of cell death induced by Fas treatment of Rb-null MEFs in the presence or absence of 20 μmol/L of z-VAD as a positive control for results in A. C. Histogram of three separate experiments illustrating the failure of two different caspase inhibitors (z-VAD, 100 μmol/L; or Boc-D-fmk, 20 μmol/L) to protect against cell death of cisplatin-treated Rb-null MEFs. D. Electron micrographs of untreated and cisplatin-treated wild-type and Rb-null MEFs showing increased cell size and enlarged nucleus in cisplatin-treated wild-type MEFs (top right) consistent with a senescent phenotype and loss of nuclear membrane integrity (red arrow, bottom right) and cytoplasmic vacuolation (black arrow, bottom right) in Rb MEFs consistent with the incidence of necrosis. E. TNF-α production by primary bone marrow macrophages cultured in medium conditioned by untreated or cisplatin-treated wild-type or Rb-null MEFs. F. Immunofluorescent staining of wild-type and Rb-null MEFs, untreated (left) or treated with 16 μmol/L of cisplatin for 24 h (right) to detect the nuclear release of HMG-B1 to the cytoplasm as a measure of necrosis.
Necrosis of Rb-Null MEFs Is Associated with Nucleotide Depletion and Elevated Parp Activity
Given that necrotic cell death is linked to energetic failure resulting from ATP depletion (18), we examined how cisplatin treatment affected ATP levels in wild-type and Rb-null MEFs. We observed that ATP was more rapidly depleted by cisplatin treatment in Rb-null MEFs compared with wild-type MEFs (Fig. 4A). ATP can be generated in the cytosol by glycolysis in the presence of NAD+ and thus we measured NAD+ levels at different time points following cisplatin treatment (Fig. 4B). We noted that NAD+ was increasingly depleted in Rb-null MEFs, such that by 24 hours, Rb-null MEFs contained less than one third of the NAD+ levels detected in untreated cells, whereas wild-type MEFs retained 78% of the NAD+ levels compared with untreated cells (Fig. 4B). We also observed that the addition of 10 mmol/L of NAD+ to the culture medium reduced the levels of cell death detected in cisplatin-treated Rb-null MEFs to that observed with cisplatin-treated wild-type MEFs (Fig. 4C). Given that serum starvation inhibited cell death, we examined how serum starvation affected NAD+ depletion in cisplatin-treated Rb-null MEFs and observed that it blocked NAD+ depletion completely (Fig. 4D). These results suggest that Rb-null MEFs are sensitized to cisplatin-induced cell death due to increased NAD+ depletion arising from growth arrest failure. NAD+ is a substrate for the Parp family of enzymes that are activated in response to DNA damage (20, 21). Given that cisplatin-induced NAD+ depletion in Rb-null MEFs is associated with increased DNA damage (Fig. 2E), that PARP-mediated NAD+ depletion induced the necrosis of Bak−/−;Bax−/− MEFs treated with alkylating agents (18), and that Parp-1 activity induced HMG-B1 release from the nucleus (22), we examined Parp expression and activity in cisplatin-treated wild-type and Rb-null MEFs. Parp-1 levels were similar between wild-type and Rb-null MEFs (Fig. 4E), although a small amount of caspase-cleaved Parp-1 (89 kDa) was observed in both wild-type and Rb-null MEFs treated with cisplatin (Fig. 4E, lanes 4 and 8), consistent with low-level caspase activity.
FIGURE 4.
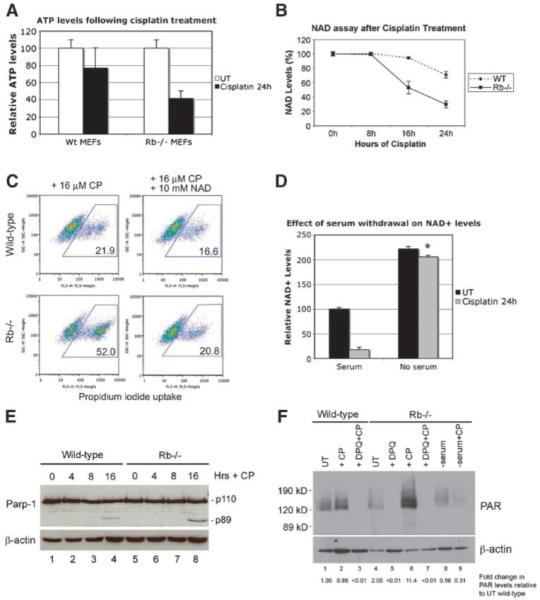
NAD depletion and PARP activity explain the necrotic death of Rb-null MEFs. A. Determination of ATP levels in wild-type and Rb-null MEFs before and after treatment with 16 μmol/L of cisplatin for 24 h. B. Determination of cytosolic NAD+ in wild-type and Rb-null MEFs before and after treatment with 16 μmol/L of cisplatin for 24 h. C. Flow cytometric analysis of propidium iodide uptake from wild-type and Rb-null MEFs to determine the effect of the addition of 10 mmol/L of NAD+ on the incidence of cell death induced by 24 h of treatment with 16 μmol/L of cisplatin. D. Determination of cytosolic NAD+ in Rb-null MEFs before and after treatment with 16 μmol/L of cisplatin for 24 h in the presence of either 10% or 0.1% serum. *, P < 0.001, statistically significant recovery in NAD+ levels due to growth in 0.1% serum compared with growth in 10% serum. E. Western blot analysis of Parp-1 expression in wild-type and Rb-null MEFs before and after treatment with 16 μmol/L of cisplatin for 16 h. F. Western blot analysis of levels of PARsylated proteins in wild-type (lanes 1-3) and Rb-null MEFs (lanes 4-9), before (lanes 1 and 4) and after cisplatin treatment (lanes 2 and 6), and as a function of pretreatment (30 min) with DPQ (lanes 3, 5, and 7) or prior serum starvation for 48 h (lanes 8 and 9). Densitometry for PAR levels was normalized to levels of μ-actin and expressed as a fold change, as indicated.
When we measured the levels of poly-ADP-ribosylated (PAR) proteins in Rb-null MEFs by Western blot, as a measure of Parp activity, we observed that PAR levels were not markedly changed in wild-type MEFs following cisplatin treatment (Fig. 4F, compare lane 2 to lane 1), and was completely repressed by pretreatment with the Parp inhibitor 3,4-dihydro-5 [4-(1-piperindyinyl)butoxy]-1(2H)-isoquinoline (DPQ; Fig. 4F, lane 3), consistent with the levels of PAR-conjugated proteins due to basal Parp activity. Cisplatin treatment markedly increased PAR levels in Rb-null MEFs (Fig. 4F, lane 6) compared with untreated Rb-null MEFs (Fig. 4F, lane 4) or to cisplatin-treated wild-type MEFs (Fig. 4F, lane 2). Importantly for the studies that will follow, we observed that pretreatment of Rb-null MEFs with DPQ for 30 minutes inhibited PAR conjugation of proteins in both untreated and cisplatin-treated Rb-null MEFs (Fig. 4F, lanes 5 and 7, respectively). Finally, we observed that serum starvation prevented increased PAR levels in cisplatin-treated Rb-null MEFs (Fig. 4F, lane 9), consistent with serum starvation limiting Parp activity in Rb-null MEFs. Intriguingly, serum starvation induces PAR levels in wild-type MEFs independent of cisplatin treatment (Supplementary Fig. S1), suggesting that wild-type MEFs undergo DNA damage when deprived of serum factors.
Parp Inhibition Protects against Cisplatin-Induced Cell Death
Given that serum starvation limited Parp activity in cisplatin-treated Rb-null MEFs and also protected against cisplatin-induced cell death, we set out to determine whether increased Parp activity explained the NAD+ depletion and cell death of cisplatin-treated Rb-null MEFs. DPQ and INH2BP (5-iodo-6-amino-1,2-benzopyrone), two of the most potent (IC50 <50 nmol/L) and selective Parp inhibitors available, were used to examine the response of Rb-null MEFs to cisplatin. We observed that both DPQ and INH2BP restored NAD+ levels in cisplatin-treated Rb-null MEFs (Fig. 5A), and protected significantly against cisplatin-induced cell death (Fig. 5B and F). Parp inhibitors and exogenous NAD+ also protected against cell death induced by treatment of Rb-null MEFs with etoposide or hydroxyurea (Fig. 5C), indicating that the observed effects of Parp inhibition were not unique to cisplatin.
FIGURE 5.
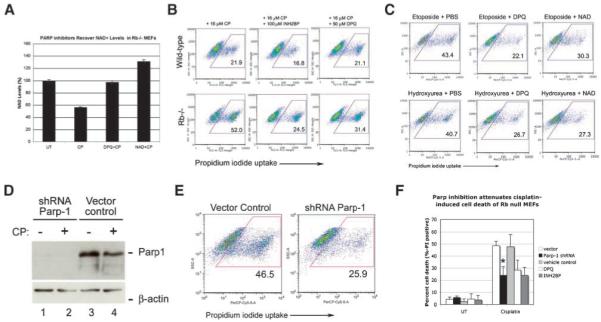
Blocking elevated Parp activity protects Rb-null MEFs from cell death. A. Determination of the effects of PARP inhibition or addition of exogenous NAD+ for NAD+ depletion in cisplatin-treated Rb-null MEFs at 16 h following cisplatin treatment. B. Flow cytometric analysis of the effect of the PARP inhibitors DPQ and INH2BP on cell death measured by propidium iodide uptake from wild-type and Rb-null MEFs treated with cisplatin for 24 h. C. Flow cytometric analysis of the effect of PARP inhibition with DPQ or NAD+ treatment on propidium iodide uptake from Rb-null MEFs treated with 20 μmol/L of etoposide or 2 mmol/L of hydroxyurea for 24 h. D. Western blot analysis to confirm the knockdown of Parp-1 by shRNA in untreated and cisplatin-treated Rb-null MEFs, but not in vector control-transduced MEFs. E. Flow cytometric analysis of propidium iodide uptake from Rb-null MEFs that had been transduced with control vector or with vector expressing shRNA to Parp-1, following treatment with 16 μmol/L of cisplatin for 24 h. F. Histogram of three separate experiments illustrating the protective effect of Parp-1 shRNA or chemical Parp inhibition against cell death of cisplatin-treated Rb-null MEFs indicates a statistically significant difference in levels of cell death induced by Parp-1 knockdown (*, P < 0.05).
To control for the potential off-target effects of Parp inhibitors, we knocked down Parp-1 in Rb-null MEFs using a previously validated small hairpin RNA to Parp-1 (18). Parp-1 is the major Parp family member activated in response to DNA damage (21). We confirmed Parp-1 knockdown by Western blotting (Fig. 5D, lanes 1 and 2) and showed that this significantly protected Rb-null MEFs from cisplatin-induced cell death (Fig. 5E and F). These results show that Rb-null MEFs are sensitized to cell death induced by DNA-damaging agents due to elevated Parp-1 activity.
PARP Inhibition Limits the Cytotoxic Effect of Cisplatin against Rb-Null Tumors Grown In vivo
Given the potential significance of these findings (done with primary mouse fibroblasts) for the proposed use of PARP inhibitors against human cancers, we also examined the role of PARP activity in the drug sensitivity of Rb-deficient human tumor cells. Saos-2 human osteosarcoma cells and other Rb-deficient human tumor cell lines are highly sensitive to killing by cisplatin (15) but can be protected against death by restoring pRb expression (Fig. 6A). Importantly for our current findings, PARP inhibitors and exogenous NAD+ protected cisplatin-treated Rb-deficient Saos-2 tumor cells from cell death (Fig. 6B and C), indicating that our findings are relevant to both primary MEFs and human tumor cells that lack the Rb tumor suppressor.
FIGURE 6.

PARP inhibitors attenuate DNA damage–induced death of tumor cells. A. The human Rb-deficient osteosarcoma tumor cell line Saos-2 was protected against cisplatin-induced killing by restoring pRb expression. B. Histogram of three separate experiments illustrating the protective effect of DPQ and NAD+ against cell death of cisplatin-treated Rb-deficient Saos-2 osteosarcoma tumor cells. C. Flow cytometric analysis of the effect of PARP inhibition with DPQ or NAD+ treatment on propidium iodide exclusion from Saos-2 tumor cells treated for 24 h with 16 μmol/L of cisplatin. D and E. Graphic representation of the effect of no treatment (UT, dark blue line), 5 mg/kg of cisplatin (CP, pink line), 10 mg/kg of DPQ (DPQ, yellow line), or combined treatment with 5 mg/mL of cisplatin and 10 mg/mL of DPQ (CP + DPQ, turquoise line) on the volume of wild-type (D) and Rb-null (E) teratocarcinomas in nude mice. The tumor volume difference on day 19 between wild-type tumors and Rb-null tumors treated with cisplatin was statistically significant (P < 0.003). The volume difference between tumors treated with cisplatin versus those treated with cisplatin plus DPQ was statistically significant for Rb-null tumors (P < 0.001) but not for wild-type tumors (P = 0.277). Points, mean tumor volume for each time point determined for 10 tumors per treatment (untreated or cisplatin treated) for each genotype (wild-type or Rb−/−); bars, SD. F. TUNEL staining for cell death–adjacent sections of cisplatin-treated wild-type tumors, cisplatin-treated Rb-null tumors and cisplatin + DPQ–treated Rb-null tumors.
To determine the physiologic significance of our findings for chemotherapy and tumor growth in vivo, we used wild-type and Rb-null embryonic stem cells to generate teratocarcinomas in nude mice. The advantage of this system is that syngeneic tumors can be grown in vivo and used to ask how Rb tumor suppressor gene status affects drug sensitivity. Wild-type and Rb-null teratocarcinomas develop with similar growth kinetics and morphology (23). Thus, we injected cohorts of tumor-grafted mice i.p. with either vehicle control, 5 mg/kg of cisplatin, 10 mg/kg of DPQ, or the combined treatment of 5 mg/kg of cisplatin plus 10 mg/kg of DPQ. The effect of the drugs on tumor growth was expressed as a percentage of the tumor volume after 19 days of treatment relative to that measured prior to treatment.
Consistent with our data obtained in vitro using MEFs and osteosarcoma tumor cell lines, we observed a greater inhibitory effect of cisplatin on the growth of Rb-null tumors in vivo (Fig. 6E, pink line) compared with untreated Rb-null tumors (Fig. 6E, dark blue line) than on the growth of wild-type tumors (Fig. 6D, compare pink line to dark blue line). The rate of growth of Rb-null tumors was more significantly impaired by cisplatin treatment compared with wild-type tumors, such that by day 9 following treatment, Rb-null tumors had stopped increasing in volume (Fig. 6E, pink line), whereas wild-type tumors continued to grow out to day 15 (Fig. 6D, pink line). Furthermore, when we examined cisplatin-treated tumors by terminal nucleotidyl transferase–mediated nick end labeling assay, which detects both necrotic cell death as well as apoptotic cell death (24), we observed greater levels of cell death in Rb-null tumors compared with wild-type tumors (Fig. 6F). When we examined the effect of the PARP inhibitor DPQ on tumor growth, we observed no overall effect from DPQ alone, irrespective of genotype (Fig. 6D and E, yellow line). However, when DPQ treatment was combined with cisplatin treatment, we noticed a marked attenuation of the inhibitory effect of cisplatin on Rb-null tumor growth (Fig. 6E, turquoise line), such that Rb-null tumors now showed similar cisplatin sensitivity to that of wild-type tumors (Fig. 6D, turquoise line) over the duration of the experiment (3 weeks). In particular, whereas cisplatin-treated Rb-null tumors had essentially stopped growing by 9 days of treatment (Fig. 6E, pink line), tumors treated with the combined dose of cisplatin and DPQ continued growing out to 15 days (Fig. 6E, turquoise line), as was seen for wild-type tumors treated with cisplatin alone (Fig. 6D, pink line). Furthermore, histologic analysis revealed that DPQ had significantly reduced cell death induced by cisplatin in Rb-null tumors (Fig. 6F). These results indicate that PARP inhibition diminished the cytotoxic effect of cisplatin on Rb-null tumors in vivo such that differential killing by cisplatin was no longer observed relative to wild-type tumors.
Discussion
The ability to preferentially kill tumor cells is a key goal in cancer therapy. We set out to determine the molecular basis by which loss of the Rb tumor suppressor leads to cell death in response to genotoxic stress, as a paradigm for understanding the selective killing of tumor cells by chemotherapeutic agents. Our work has shown that Rb-null cells are more sensitive to killing by cisplatin, etoposide, and hydroxyurea compared with wild-type control cells due to the elevated activity of Parp-1 and NAD+ depletion, and that this cell death was caspase-independent with features of necrosis.
Consistent with deregulated expression of E2F-dependent cell cycle genes and aberrant S phase entry being a major determinant of the sensitivity of Rb-null MEFs to cisplatin and other DNA-damaging agents, we observed that inducing cell cycle arrest through serum starvation inhibited cisplatin-induced cell death. Our observations do not rule out a role for pRb in direct repression of cell death genes that may come into play in developmental or differentiation processes, or in response to other types of stresses not tested here, when an acute response may be less critical.
We showed that Parp inhibitors or Parp-1 knockdown reduced the level of cisplatin-induced cell death in Rb-null MEFs down to that observed in similarly treated wild-type MEFs. This indicated that whereas elevated Parp-1 activity explains the differential sensitivity of Rb-null cells to genotoxic stress, it does not explain the incidence of cell death in cisplatin-treated wild-type MEFs or the residual death of Rb-null MEFs. Similarly, aphidicolin-induced cell cycle arrest reduced the incidence of cell death in cisplatin-treated Rb-null MEFs down to that of cisplatin-treated wild-type MEFs (data not shown). However, we noted that serum starvation reduced cell death in both wild-type and Rb-null MEFs down to basal levels, suggesting that serum starvation had a dual effect on survival that extended beyond the inhibition of cell cycle and Parp activity. Such effects of serum starvation on promoting survival may include the inhibition of mTOR and induction of autophagy, a process that has previously been shown to promote survival under conditions of nutrient stress (25).
Our data indicates that loss of the Rb tumor suppressor sensitizes cells to DNA damage–induced necrosis and, together with previous work reporting the induction of necrosis in Bax−/−;Bak−/− MEFs by alkylating agents (18), suggests that the ability to induce necrosis will be a major factor in determining the efficacy of anticancer drugs. Interestingly, Rb-null MEFs are not inherently defective for apoptosis, as were Bax−/−Bak−/− MEFs (18), because we show that they are sensitive to Fas-induced apoptosis and others have shown that Rb-null cells are sensitized to TNF-α–induced apoptosis (26). Rather, Rb-null MEFs showed a unique sensitivity to necrosis induced by key DNA-damaging agents as a consequence of elevated Parp-1 activity. Parp-1 is cleaved by caspases in response to apoptotic signals, and our data showing that caspase inhibition promoted necrotic cell death in response to DNA damage is consistent with the concept that Parp-1 activity determines the balance between apoptosis and necrosis in stressed populations of cells. Thus, it is possible that populations of Rb-null MEFs undergo either apoptosis or necrosis in response to DNA-damaging agents, and that the relative level of apoptosis compared with necrosis within the population is a function of how rapidly or effectively Parp-1 gets cleaved by caspases.
HMG-B1 is a chromatin component that is normally bound within the minor groove of genomic DNA in healthy cells, but under conditions of necrosis, it is released into the cytoplasm and extracellular milieu where it generates an inflammatory response (19). We showed that HMG-B1 is released from the nuclei of Rb-null MEFs in response to treatment with cisplatin, etoposide, or hydroxyurea but not from similarly treated wild-type MEFs. Together with a recent report identifying a role for Parp-1 in promoting the release of HMG-B1 from the nucleus (22), our data therefore suggests that loss of Rb sensitizes cells to DNA damage–induced necrosis by inducing Parp-1 activity that, among other events, leads to nuclear release of HMG-B1.
The use of PARP inhibitors has been proposed for the treatment of Brca-deficient tumors (27, 28), and although they may work well for tumor types that display defects in homologous recombination, such as BRCA-1–deficient or BRCA-2–deficient human breast and ovarian cancer, their use may be counterproductive in therapy against tumors with mutations that inactivate the Rb tumor suppressor. In summary, our work provides a mechanistic insight into how loss of pRb and defective cell cycle checkpoint control leads to cell death in response to genotoxic agents. This work has significance for understanding the clinical applications of chemotherapeutic drugs that exploit defects in cell cycle checkpoint control such as those found in most tumor cells.
Materials and Methods
Cell Culture and Transfection Protocols
Cells were cultured in DMEM, containing 10% or 0.1% fetal bovine serum as specified. MEFs from passages 1 to 4 were used for all experiments. All experiments were carried out in triplicate using at least two different isolates of Rb-null MEFs. Retroviral vectors (pBabe-puro) with the shRNA hairpins to Parp-1 were transfected into LinxE packaging cell line, and viral supernatant was used for the infection of duplicate passage 1 MEF cultures. Puromycin (6 μg/mL) was applied to one of the duplicate infected cultures to ensure an infection efficiency of >80% to 90%, and to avoid puromycin selection on the second duplicate culture that was used for experiments. DPQ, INH2BP, and Z-VAD-fmk (Z-Val-Ala-Asp-CH2F) were obtained from Calbiochem. Aphidicolin (Sigma) was added to cell cultures at 2 μg/mL, 48 h prior to cisplatin treatment.
Cell Staining and Analysis
Cell Death Assay
Cells were stained with 10 μg/mL of propidium iodide for 15 min prior to harvesting. All cells (adherent and floating) were harvested for analysis.
γH2AX Staining
Harvested cells were fixed with 70% ethanol at −20°C. After permeabilization with TST [TBS (pH 7.4), 4% fetal bovine serum, and 0.1% Triton X-100], cells were incubated with the diluted mouse monoclonal anti–γH2AX-FITC antibody (Upstate Biotechnology) for 2 h at room temperature, rinsed with TBS/2% fetal bovine serum and analyzed.
Cell Cycle Analysis
MEFs were cultured in BrdUrd (30 μg/mL, Sigma) for 4 h and BrdUrd incorporation analyzed in fixed cells using anti–BrdU-FITC (Clontech), 10 μg/mL propidium iodide and 10 μg/mL of RNase A.
Immunofluorescence
HMG-B1 antibodies were used as described previously (19).
Electron Microscopy
Cultured cell pellets were fixed in 2.5% glutaraldehyde, and sections analyzed using a Philips CM120 transmission electron microscope.
Antibodies and Western Blots
Anti-pRb (G3-245) and anti-PAR (551813) antibodies were from BD Transduction Laboratories, anti–β–actin (C-11) and anti-Parp (H-250) antibodies were from Santa Cruz Biotechnology, anti–HMG-B1 (18256) was from Abcam, and anti–α-tubulin (05-661) was from Upstate Biotechnology. Cell extracts were prepared in a high-salt extraction buffer [100 mmol/L HEPES (pH 7.4), 0.5 mol/L KCl, 5 mmol/L MgCl2, 0.5 mmol/L EDTA, and 20% glycerol] for pRb and Parp-1, and in radioimmunoprecipitation assay buffer (11) for PAR Western blot analyses. Densitometry was done using the Fluorchem 8900 from Alpha Innotech Corporation.
NAD, ATP, PARP Activity, and TNF-α Assays
NAD+ Assay
Cells were extracted in 0.5 N HClO4 and neutralized supernatants were mixed with the reaction buffer of 0.1 mmol/L 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide, 0.9 mmol/L of phenazine methosulfate, 13 units/mL of alcohol dehydrogenase, 100 mmol/L of nicotinamide, and 5.7% ethanol in 61 mmol/L of Gly-Gly buffer (pH 7.4). The A560 nm was determined immediately and again after 10 min.
ATP Assay
Cells were trypsinized, counted, and resuspended at 1,000 cells/μL in sterile double-distilled water. After boiling, the ATP concentration in cleared supernatant was determined using ATP Determination Kit (Molecular Probes A-22066).
TNF-α ELISA
Wild-type or Rb-null MEFs were treated with 16 μmol/L of cisplatin for 16 h, and then fresh medium without cisplatin was added for a further 24 h. Culture medium conditioned in this way was added to primary bone marrow macrophage cultures for a further 48 h and TNF-α production assayed by ELISA (R&D Systems).
Real-time PCR and Microarray Data
Relative quantitation of real-time PCR products were done using QuantiTect SYBR Green PCR and the Applied Biosystems 7900 Fast Real-time PCR System, and analyzed using the associated SDS 2.3 software. Samples were amplified in triplicate and normalized by subtracting CT values for 18S rRNA. The microarray data discussed in this publication have been deposited in National Center for Biotechnology Information’s Gene Expression Omnibus5 and are accessible through GEO Series Accession no. GSE6206.4
Mice and In vivo Tumor Studies
Timed matings of Rb heterozygous mice were set up to generate MEFs from E13.5 embryos. A total of 40 mice (6-8 wk old; Harlan Sprague-Dawley) were injected s.c. with 2 × 106 embryonic stem cells (wild-type embryonic stem cells on the left flank, Rb−/− embryonic stem cells on the right flank) to generate teratocarcinomas that were injected i.p. 2 wk later with vehicle control, 5 mg/kg of cisplatin (University of Chicago Hospital Pharmacy), 10 mg/kg of DPQ (Calbiochem), or a combined 5 mg/kg of cisplatin and 10 mg/kg of DPQ. Cisplatin was administered on days 0, 4, 8, 12, and 16, whereas DPQ was given on days −1, 0, 1, 4, 5, 8, 9, 12, 13, 16, and 17. Tumor volume (V = length × width × height / 8) was measured daily with electronic calipers and expressed as a percentage of tumor volume on day 0. Tumors were fixed in 10% neutral buffered formalin and sectioned for immunohistochemical staining.
Supplementary Material
Acknowledgments
We thank Craig Thompson and Dara Ditsworth for generously providing the shRNAs to Parp-1 and for advice on how best to perform PAR westerns. We are grateful to Xinmin Li and Jamie Zhou of the University of Chicago Functional Genomics Facility and Yimei Chen of the Electron Microscopy Core Facility.
Grant support: V Foundation Scholar award (K.F. Macleod) and the Cancer Research Foundation.
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Note: Supplementary data for this article are available at Molecular Cancer Research Online (http://mcr.aacrjournals.org/).
Current address for H. Liu: Institute for Stem Cell Biology and Regenerative Medicine, Stanford University, Stanford, CA 94304-1334.
References
- 1.Almasan A, Yin Y, Kelly R, et al. Deficiency of retinoblastoma protein leads to inappropriate S-phase entry, activation of E2F-responsive genes and apoptosis. Proc Natl Acad Sci U S A. 1995;92:5436–40. doi: 10.1073/pnas.92.12.5436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harrington EA, Bruce JL, Harlow E, Dyson N. pRB plays an essential role in cell cycle arrest induced by DNA damage. Proc Natl Acad Sci U S A. 1998;95:11945–50. doi: 10.1073/pnas.95.20.11945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Knudsen KE, Booth D, Naderi S, et al. RB-dependent S-phase response to DNA damage. Mol Cell Biol. 2000;20:7751–63. doi: 10.1128/mcb.20.20.7751-7763.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Knudsen ES, Knudsen KE. Tailoring to RB: tumour suppressor status and therapeutic response. Nat Rev Cancer. 2008;8:714–24. doi: 10.1038/nrc2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chau BN, Wang JYJ. Coordinated regulation of life and death by RB. Nat Rev Cancer. 2003;3:130–8. doi: 10.1038/nrc993. [DOI] [PubMed] [Google Scholar]
- 6.Lazzerini Denchi E, Helin K. E2F1 is crucial for E2F-dependent apoptosis. EMBO Rep. 2005;6:661–8. doi: 10.1038/sj.embor.7400452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Trimarchi J, Lees JA. Sibling rivalry in the E2F family. Nat Rev Mol Cell Biol. 2002;3:11–20. doi: 10.1038/nrm714. [DOI] [PubMed] [Google Scholar]
- 8.Cam H, Dynlacht BD. Emerging roles for E2F: beyond the G1/S transition and DNA replication. Cancer Cell. 2003;3:311–6. doi: 10.1016/s1535-6108(03)00080-1. [DOI] [PubMed] [Google Scholar]
- 9.Moroni MC, Hickman ES, Denchi EL, et al. Apaf-1 is a transcriptional target for E2F and p53. Nat Cell Biol. 2001;3:552–8. doi: 10.1038/35078527. [DOI] [PubMed] [Google Scholar]
- 10.Nahle Z, Polakoff J, Davuluri RV, et al. Direct coupling of the cell cycle and cell death machinery by E2F. Nat Cell Biol. 2002;4:859–64. doi: 10.1038/ncb868. [DOI] [PubMed] [Google Scholar]
- 11.Irwin M, Marin MC, Phillips AC, et al. Role for the p53 homologue p73 in E2F-1 induced apoptosis. Nature. 2000;407:645–8. doi: 10.1038/35036614. [DOI] [PubMed] [Google Scholar]
- 12.Hershko T, Ginsberg D. Up-regulation of Bcl-2 homology 3 (BH3)-only proteins by E2F-1 mediates apoptosis. J Biol Chem. 2004;279:8627–34. doi: 10.1074/jbc.M312866200. [DOI] [PubMed] [Google Scholar]
- 13.Herrera RE, Sah VP, Williams BO, Makela TP, Weinberg RA, Jacks T. Altered cell cycle kinetics, gene expression and G1 restriction point regulation in Rb-deficient fibroblasts. Mol Cell Biol. 1996;16:2402–7. doi: 10.1128/mcb.16.5.2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Foijer F, Wolthuis RMF, Doodeman V, Medema RH, te Riele H. Mitogen requirement for cell cycle progression in the absence of pocket protein activity. Cancer Cell. 2005;8:455–66. doi: 10.1016/j.ccr.2005.10.021. [DOI] [PubMed] [Google Scholar]
- 15.Deverman BE, Cook BL, Manson SR, et al. Bcl-XL deamidation is a critical switch in the regulation of the response to DNA damage. Cell. 2002;111:51–62. doi: 10.1016/s0092-8674(02)00972-8. [DOI] [PubMed] [Google Scholar]
- 16.Lowe SM, Ruley HE, Jacks T, Housman DE. p53-dependent apoptosis modulates the cytoxicity of anticancer agents. Cell. 1993;74:957–67. doi: 10.1016/0092-8674(93)90719-7. [DOI] [PubMed] [Google Scholar]
- 17.Macleod K, Hu Y, Jacks T. Loss of Rb activates both p53-dependent and -independent cell death pathways in the developing mouse nervous system. EMBO J. 1996;15:6178–88. [PMC free article] [PubMed] [Google Scholar]
- 18.Zong W, Ditsworth D, Bauer DE, Wang ZQ, Thompson CB. Alkylating DNA damage stimulates a regulated form of necrotic cell death. Genes Dev. 2004;18:1272–82. doi: 10.1101/gad.1199904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–5. doi: 10.1038/nature00858. [DOI] [PubMed] [Google Scholar]
- 20.Bouchard VJ, Rouleau M, Poirier GG. PARP-1, a determinant of cell survival in response to DNA damage. Exp Hematol. 2003;31:446–54. doi: 10.1016/s0301-472x(03)00083-3. [DOI] [PubMed] [Google Scholar]
- 21.Schreiber V, Dantzer F, Ame JC, De Murcia G. Poly(ADP-ribose): novel functions for an old molecule. Nat Rev Mol Biol. 2006;7:517–28. doi: 10.1038/nrm1963. [DOI] [PubMed] [Google Scholar]
- 22.Ditsworth D, Zong WX, Thompson CB. Activation of poly(ADP)-ribose polymerase (PARP-1) induces release of the pro-inflammatory mediator HMGB1 from the nucleus. J Biol Chem. 2007;282:17845–54. doi: 10.1074/jbc.M701465200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dannenberg JH, van Rossum A, Schuijff L, te Riele H. Ablation of the retinoblastoma gene family deregulates G1 control causing immortalisation and increased cell turnover under growth restricting conditions. Genes Dev. 2000;14:3051–64. doi: 10.1101/gad.847700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grasl-Kraupp B, Ruttkay-Nedecky B, Koudelka H, Bukowska K, Bursch W, Schulte-Hermann R. In situ detection of fragmented DNA (TUNEL assay) fails to discriminate among apoptosis, necrosis and autolytic cell death: a cautionary note. Hepatology. 1995;21:1465–68. doi: 10.1002/hep.1840210534. [DOI] [PubMed] [Google Scholar]
- 25.Sabatini DM. mTOR and cancer: insights into a complex relationship. Nat Rev Cancer. 2006;6:729–34. doi: 10.1038/nrc1974. [DOI] [PubMed] [Google Scholar]
- 26.Phillips AC, Ernst MK, Bates S, Rice NR, Vousden KH. E2F-1 potentiates cell death by blocking anti-apoptotic signaling pathways. Mol Cell. 1999;4:771, 81. doi: 10.1016/s1097-2765(00)80387-1. [DOI] [PubMed] [Google Scholar]
- 27.Bryant HE, Schultz N, Thomas HD, et al. Specific killing of BRCA2deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–7. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 28.Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–20. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.